

Conspectus
Modern chemistry emerged from the quest to describe the three-dimensional structure of molecules: van’t Hoff’s tetravalent carbon placed symmetry and dissymmetry at the heart of chemistry. In this Account, we explore how modern theory, synthesis, and spectroscopy can be used in concert to elucidate the symmetry and dissymmetry of molecules and their assemblies. Chiroptical spectroscopy—including optical rotatory dispersion (ORD), electronic circular dichroism (ECD), vibrational circular dichroism (VCD), and Raman optical activity (ROA)—measures the response of dissymmetric structures to electromagnetic radiation. This response can in turn reveal the arrangement of atoms in space, but deciphering the molecular information encoded in chiroptical spectra requires an effective theoretical approach. Although important correlations between ECD and molecular stereochemistry have existed for some time, a battery of accurate new theoretical methods that link a much wider range of chiroptical spectroscopies to structure have emerged over the last decade. The promise of this field is considerable: theory and spectroscopy can assist in assigning the relative and absolute configurations of complex products, in revealing the structure of non-covalent aggregates, in defining metrics for molecular diversity based upon polarization response, and in designing chirally imprinted nanomaterials. The physical organic chemistry of chirality is fascinating in its own right: defining atomic and group contributions to optical rotation (OR) is now possible. Although the common expectation is that chiroptical response is determined solely by a chiral solute’s electronic structure in a given environment, chiral imprinting effects on the surrounding medium and molecular assembly can, in fact, dominate the chiroptical signatures.
The theoretical interpretation of chiroptical markers is challenging because the optical properties are subtle, which results from the strong electric dipole and the weaker electric quadrupole and magnetic dipole perturbations of the electromagnetic field. Moreover, OR arises from a combination of nearly canceling contributions to the electronic response. Indeed, the challenge posed by the chiroptical properties delayed the advent of even qualitatively accurate descriptions for some chiroptical signatures until the last decade when, for example, prediction of the observed sign of experimental OR became accessible to theory.
The computation of chiroptical signatures, in close coordination with synthesis and spectroscopy, provides a powerful framework to diagnose and to interpret the dissymmetry of chemical structures and molecular assemblies. Chiroptical theory now produces new schemes to elucidate structure, to describe the specific molecular sources of chiroptical signatures, and to assist in our understanding of how dissymmetry is templated and propagated in the condensed phase.
Introduction
Chiroptical effects were first reported about 200 years ago. Modern theoretical descriptions of the most familiar chiroptical signature, optical rotation (OR),1 emerged just a few years after the disclosure of the Schrödinger equation in 1926. Yet, reliable methods to predict OR are just a decade old. The first accurate ab initio calculations of OR used static-field linear-response methods2 and time-dependent response methods.3 Success in assigning the absolute configuration of small molecules and natural products using these theoretical methods immediately followed,3 as did further advances in electronic structure methods.4,5
The elucidation of molecular structure is a worthy goal for theory, but reliable chiroptical algorithms provide a much richer cache. This account focuses on contributions from our group in elucidating molecular structure, and in understanding how a combined spectroscopic-synthetic-theoretical program can be used to analyze and correlate complex structures. Optical rotatory dispersion (ORD), electronic circular dichroism (ECD),6 vibrational circular dichroism (VCD),6 and Raman optical activity (ROA)7 have proven to be of particular value in this regard.
We were first drawn to this problem by a desire to understand the relationship between chemical structures and OR, with an eye on using this understanding to solve challenges in the assignment of absolute configuration. Our studies immediately showed how strongly geometry impacts OR, motivating studies of molecular assembly and solvation. Investigations of complementary probes of dissymmetry followed. Here, we summarize the physical origins of chiroptical phenomena, and then describe how molecular substitution, geometry, assembly and solvation, influence the chiroptical response. In fact OR depends strongly on all of these factors. Chiroptical spectroscopies probe different aspects of stereochemistry, aggregation, and solvation. OR interrogates overall (thermally averaged) molecular structure because it is derived from the sum of all mixed electric dipole-magnetic dipole transitions from ground to excited states. ECD, in contrast, probes the electric dipole-magnetic dipole transition strength (rotational strength) between the ground and specific excited states. Vibrational chiroptical spectroscopies (VCD and ROA) further couple the electronic response properties to specific vibrational modes of motion. We have used Monte Carlo (MC) and molecular dynamics (MD) simulations in combination with experimental and simulated ROA data to assess ensembles of solution structures, particularly those of peptides.
Origins of OR, ECD, VCD and ROA
Chiroptical spectroscopy cannot be formulated in the familiar electric-dipole approximation. Indeed, the spatial variation of the incident fields on the molecular length scale “senses” the molecular dissymmetry. Deciphering molecular structure information encoded in the chiroptical response requires three electronic response tensors: the electronic polarizability (α), the electric dipole-magnetic dipole polarizability (G′′), which takes center stage, and the electric dipole-electric quadrupole (A) tensors.1,8,9
OR is recognized as the rotation of plane-polarized light as it passes through an optically active medium, and the ORD represents the variation in OR of an optically active medium with wavelength. The optical rotatory parameter (β) is an isotropic average, proportional to the trace of the G′′ tensor. Coupled-cluster (CC), time dependent density functional theory (TD-DFT), and Hartree-Fock (HF) methods have been used to compute OR and ORD;4 the computational efficiency of TD-DFT methods favors their use in many instances.5
ECD and VCD represent the differential absorption of right- and left-circularly polarized light associated with electronic and vibrational excitations of chiral molecules6, respectively. The ECD signal is proportional to the rotational strength R, the dot product of the electric dipole and magnetic dipole transition moments. ECD calculations based on empirical models, QM treatments, and mixed methods have been used to describe ECD spectra of small organic compounds, polypeptides and globular proteins.6 Recently, CC and TD-DFT methods have also been applied to ECD and VCD.4 ECD relies on the presence of electronic transitions, often n->π* or π->π* transitions, and probes dissymmetry in the molecular array situated proximal to the transition.
ROA measures the different intensity of right- and left-circularly polarized Raman scattered light, and reports on chirality associated with vibrational modes.7 The interference among light waves scattered via α, G′′, and A produces a dependence of the scattered intensity on the circular polarization of the incident light. The ROA signal is calculated from α, G′′, A.7 As with OR, ORD, ECD and VCD, ROA computations based on TD-DFT are of particular recent interest.10
In our group, we use OR, ORD, ECD and ROA to determine: (1) the relative and absolute configuration of natural products and synthetic compounds, (2) the conformer distribution of chiral molecules in solution, (3) the structure of non-covalent molecular assemblies, (4) the structure of molecule-solvent assemblies, including chiral solvent imprints, and (5) the nature of chirally imprinted nanoclusters.
Probing molecular dissymmetry using chiroptical spectroscopy
1. Configuration and conformational analysis of molecules
Conventional methods to determine the absolute configuration (AC) of chiral substances include X-ray diffraction, ECD, nuclear magnetic resonance (NMR) analysis of Mosher esters and related covalent derivatives,11 total synthesis,12 and chemical degradation.13 These approaches are often time consuming, costly, and may require significant amounts of analyte. The comparison of computed and experimental OR data, in contrast, can provide a streamlined approach that, at a minimum, significantly reduces the number of possible stereoisomers.
We reported the first ab initio theoretical AC assignment of a natural product, hennoxazole A, by computing the molar rotation of its fragments and using van’t Hoff’s superposition principle to reach a final assignment.3 This approach divided the flexible natural product into weakly interacting fragments, computed the Boltzmann-averaged molar ORs for each fragment, and summed the contributions to obtain a composite value for comparison with experimental ORs. The hennoxazole analysis clearly demonstrated the hypersensitivity of OR calculations to geometry. Thus, reliable predictions of the OR for flexible structures require extensive thermal conformational averaging. Prior experimental studies had already validated the additivity of fragment OR contributions for this natural product.14
The computation of OR was successfully extended to determine the AC of the natural product pitiamide A.15 Subsequently, we also combined the use of NMR NOEs, chemical shifts, and J-coupling measurements with experimental and computational OR and ECD data to determine the AC of the marine natural product bistramide C (Figure 1).16 This study demonstrated that the judicious use of electronic structure analysis and spectroscopic data enabled a successful AC assignment even of a large and conformationally flexible natural product with multiple unassigned stereocenters. Other groups have also noted the utility of applying multiple chiroptical methods (including ORD, ECD, VCD, and ROA) to determine AC.17
Figure 1.
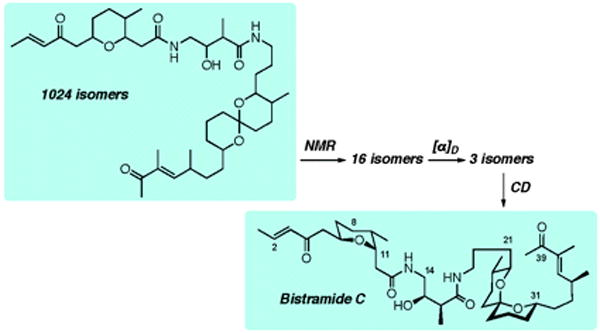
The absolute configuration of bistramide C was determined using NMR NOEs, J-couplings with TD-DFT calculations of molar optical rotations and circular dichroism (CD).16 From a total of 1024 stereoisomers that are theoretically possible for this structure, a pool of 16 and then 3 stereoisomers were identified as potential structures using the NMR and αD data. Finally, out of the 3 possible stereoisomers, the absolute configuration of (+)-(6R,9S,11S,15R,16S,22R,23S, 27S,31S,34S)-bistramide C was deduced from its CD spectrum.
The reliability of computational methods for predicting OR was further validated by assigning the previously unknown configuration of a series of small organic molecules. For example, we used TD-DFT OR calculations to assign the absolute configurations of trans-(S)- and trans-(R)-ladderanes.18 Subsequently, Mascitti and Corey carried out an enantioselective synthesis of pentacycloanammoxic acid with a trans-(S)-ladderane core.19 The product was found to have a positive [α]D, consistent with our predictions and a general rule for this novel compound class based entirely on computational analysis.
Beyond its practical applications, the development of computational methods that successfully describe chiroptical response is also establishing more intuitive links between OR and structure. This is not a trivial task since there is no intuitive or reliable fragment based rule to predict either the sign or magnitude of the OR for even the simplest molecules, despite earlier attempts to establish such correlations.20-22 This gap in knowledge exists even though tens of thousand of experimental OR measurements have been recorded. OR may be the most frequently measured physical constant for which rudimentary structure-property rules remain yet unknown.
One approach to understanding the origins of OR is to define chemical group contributions. We have developed algorithms that map atomic and functional group contributions to OR and thus provide insight into geometric effects and damping of the dissymmetric footprint with distance from the stereocenter.23 Atomic contribution maps diagnose the sensitivity of OR to molecular geometry, and thus assist in making reliable predictions of molar rotations for flexible molecules.24 This sensitivity is highlighted in a recent study of Vaccaro and co-workers that described a strong dependence of computed OR on molecular conformations in 2-substituted butanes,25 similar to earlier treatment of OR in twisted “molecular wire potentials”.26
The increasing availability of commercial ROA spectrometers and the corresponding computational methods have improved the utility of ROA spectroscopy for the stereochemical and conformational analysis of chiral molecules.27 We have showcased the benefits of ROA computations by predicting: (1) the AC of the synthetic musk (4S)-Galaxolide®, which is composed of a mixture of (4S,7S)- and (4S,7R)-epimers,28 and (2) the distribution of dominant N-acetylalanine-N’-methylamide (Ala dipeptide model) conformations in aqueous solution.29
The conformational flexibility of the alanine dipeptide model has been probed using multiple spectroscopic methods, including ROA. Despite these extensive studies, a consensus aqueous solution structure for this compound has not yet emerged. For example, the 13C NMR spectra of the alanine dipeptide model in water and in liquid-crystalline media suggest that its structure in both media is dominated by left-handed PPII helical conformations.30,31 Following these NMR studies, Mehta et al. concluded based on 13C NMR spectra that the alanine dipeptide model adopts a mixture of PPII and compact right-handed αR helical conformations in water.32 Kim et al. probed the conformational heterogeneity of the alanine dipeptide model in aqueous solution using 2D-IR and reported the presence of PPII-like conformations,33 ruling out αR helical conformations. Raman spectroscopy, however, suggests that PPII, αR, and the intra-molecular hydrogen bonded C7eq conformations co-exist in aqueous media.34
The significant variation of the proposed alanine dipeptide model conformations warrants further analysis, and supports the utility of closely linking chiroptical simulations to experimental data. Indeed, we have analyzed the ROA spectra of the alanine dipeptide model in H2O and D2O using TD-DFT on Monte Carlo (MC) sampled geometries,29 examining the consistency of MC generated structures and the corresponding simulated spectra with experimental ROA data. Our analysis of the Raman and ROA spectra of this compound indicates that αR and PPII conformations are predominant in aqueous solution (Figure 2). Therefore, it is inappropriate to ascribe all of the experimental signals to one dominant conformation, namely the PPII, as suggested by previous spectroscopic studies.30,31,33 This approach of simulating spectra from an extensively sampled ensemble of theoretical structures and pruning them to a subset that is consistent with experimental spectra is growing in popularity in the biophysical context.35
Figure 2.
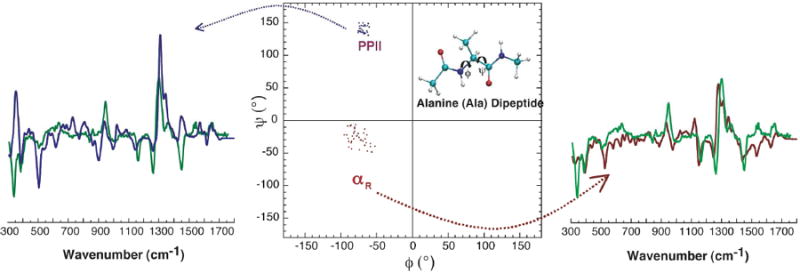
Measured (green) and computed Raman optical activity spectra derived from the extended left-handed PPII (blue) and the compact right-handed αR (red) helical conformations of an alanine dipeptide model in aqueous solution. The fact that the simulated spectra account for the key experimental spectral features indicates that the dipeptide model assumes these conformations in aqueous solution.29
2. Chiroptical signatures of intra- and intermolecular interactions
Analysis of atomic and chemical group contributions to OR indicates that chemical groups far from the stereogenic center(s) can dominate the chiroptical effect. We found that the large difference in the OR of the marine natural products calyculin A and B, which differ only by the (E)- vs (Z)-configuration of a terminal double bond in a tetraene unit distant from the stereocenters, arise from the polarizability of the terminal cyanide (CN) substituent, and are not due to the dissymmetric twisting of the tetraene unit or the direct perturbation of the stereogenic carbons.36 Figure 3 shows the Boltzmann-weighted atomic contribution maps for the [α]D of calyculin fragments with (E)- and (Z)-configurations. These maps demonstrate that π-mediated intramolecular electronic communication between the stereocenter and a distant CN group dominates the OR. Other polarizable groups with cis/trans-derivatives such as enones and enoates coupled via conjugated π-electron pathways to stereogenic centers are likely to display similar behavior. The nature of the response depends on the substituent dipole moment as well as the strength of its electronic coupling to the stereocenter.
Figure 3.

Boltzmann-weighted atomic contribution maps (BWAMs) of the optical rotation (OR) of calyculin A (A) and B (B) fragments.36 The atoms are colored according to their contributions to the OR, which highlights the large effects arising from the CN group and the two adjacent carbon atoms. The difference BWAM (C) of the calyculin A and B fragments also clearly visualizes the dominance of the CN group in optical activity.
Intermolecular interactions also significantly influence the OR of chiral molecules. For example, we found that the [α]D of (R)-pantolactone is controlled by intermolecular hydrogen bonding, and the equilibrium between monomers and dimers. Thus, the OR of (R)-pantolactone in carbon tetrachloride at room temperature arises from a combination of monomer and dimer structures (Figure 4).37
Figure 4.
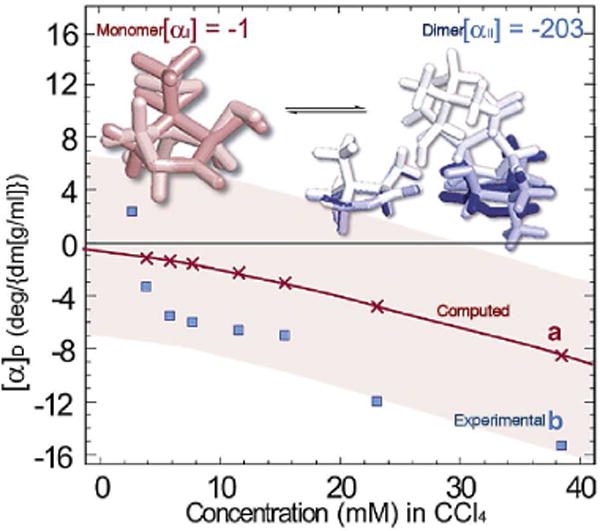
The concentration-dependent specific rotation, [α]D, of (R)-pantolactone in carbon tetrachloride (CCl4) at 26 °C.37 The computed [α]D= χI[αI] + χII[αII] (red line) values are based on the theoretically obtained thermally averaged specific rotation of the monomer ([αI]) and dimer ([αII]) species (top structures). The mole fractions χI and χII of the monomer and dimer species were determined with the experimental Keq = 8.9 ± 0.6 M-1 at 26 °C.
The chiroptical response of molecules in solution can be opposite in sign compared to the gas phase.38 Specifically, 1) chiral solvent imprinting,39 2) perturbation of the solute electronic structure by solvation, including association of solvent molecules,40 3) solute clustering or assembly,37 and 4) shifts in the solute conformational distribution24 have all been shown to exert substantial changes on the OR.
Methyloxirane represents a particularly dramatic case of solvent effects on OR, and its properties have challenged some of the highest-level computational methods.41 In the long-wavelength regime, (S)-methyloxirane displays a positive ORD spectrum in water and a negative spectrum in benzene.42 Figure 5 shows that the hydrogen bonded methyloxirane-water adduct dominates the ORD in water.40 For methyloxirane in benzene, the chiral imprint in the surrounding benzene cluster dominates the ORD spectrum.39 Remarkably, removing the methyloxirane and computing the ORD of the imprinted benzene essentially reproduces the observed ORD spectra.
Figure 5.
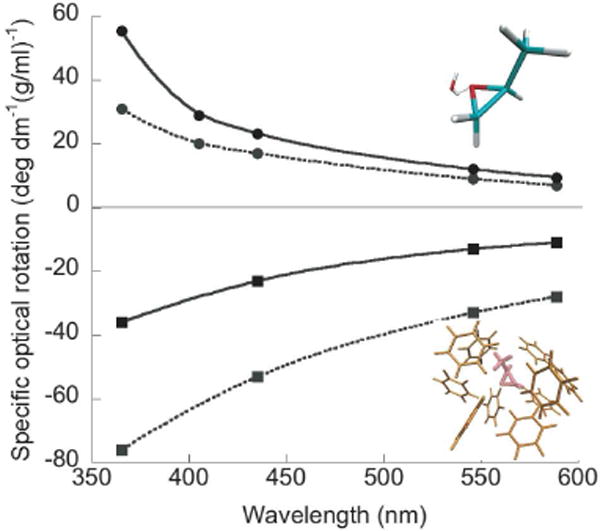
Measured (dashed line) and computed (solid line) ORD of (S)-methyloxirane in water (●) and benzene (■). The hydrogen bonded methyloxirane-water adduct (top structure) dominates the ORD in aqueous solution.40 In contrast, the chiral benzene cluster (bottom structure) dominates the ORD in benzene.39,45
In order to understand the physical origins of the positive OR of methyloxirane in water, we computed40 the specific rotation ([α]D) of the (S)-methyloxirane-water adducts at 589 nm as a function of water angular positions. Figure 6 shows the angle histogram for (S)-methyloxirane-water adducts, which illustrates that positive [α]D values for methyloxirane in water arise from water anti-configurations (where water is on the opposite face of the oxirane ring from the methyl group). Xu and coworkers recently studied methyloxirane in water using VCD, OR, and simulation.43 They concluded that the methyloxirane-water binary complex dominates the chiroptical signature in aqueous media at room temperature, and the anti-conformation is preferred over the syn-conformation, consistent with our predictions.40
Figure 6.
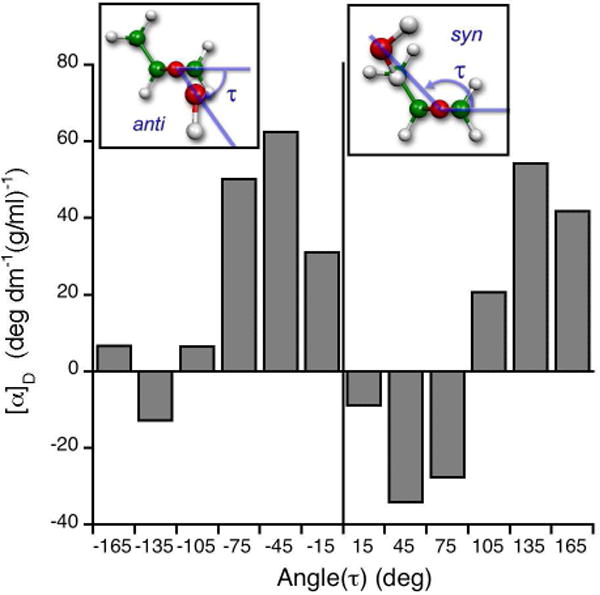
Distribution of optical rotation of (S)-methyloxirane-water adduct at 589 nm, [α]D, as a function of water molecule position defined by the angle τ.40 The inserts show structures with water molecules on the same or opposite face of the oxirane ring from the methyl group, defined as the syn-or anti-configurations, respectively. The anti-configuration dominates the positive [α]D of (S)-methyloxirane in water.
The ORD signature of methyloxirane in benzene contrasts dramatically with its behavior in water. In benzene, the ORD arises from the chiral solvent imprint, not from the solvated parent structure.39 The methyloxirane-benzene system demonstrates that an achiral solvent, perturbed by a chiral solute, may dominate the chiroptical response. Indeed, chiral solutes are known to induce chiral solvent ordering that contributes to the chiroptical response. In complementary studies, Fidler et al. showed that dissymmetric solvent organization around a chiral solute accounts for 10-20% of the total CD intensity attributable to an optically active chromophore.44 Yet, the dominance of induced chirality on the OR signature was shown for the first time by the theoretical dissection of methyloxirane’s ORD spectrum in benzene.45 More recently, Wang and Cann employed MD simulations to study the extent of chirality transfer from a chiral solute to solvent.46 They concluded that chirality transfer is most pronounced for solvents that are hydrogen bonded to solutes, and that the dissymmetric solvent shell at specific positions contributes strongly to the chirality transfer.
Continuum and point charge solvent models for computing the chiroptical response in the condensed phase do not describe induced chirality. The absence of a priori knowledge of solvent-solute interactions, or their influence on the chiroptical signature, necessitates an exploration of thermally averaged solute-solvent clusters and a comparison with simpler solvent studies, as shown by theoretical investigations of solvent effects on methyloxirane OR.39,40
3. Chirality of nanoclusters
The optical activity of metal complexes47 and metal nanoclusters is of particular interest. For example, the chirality of nanoclusters with chiral adsorbates is the subject of intense study because of potential applications in enantioselective catalysis,48 enantiodiscrimination,49 enantioselective crystallization50 and photonics.51 Theory indicates that the chiroptic response of metal clusters with chiral adsorbates may arise from the intrinsically chiral core structure of a metal cluster, a chiral surface or chiral adsorbates.52 We used a simple dissymmetrically-perturbed particle-in-a-box model to examine the influence of dissymmetric adsorbates on the chiroptical signatures of nanoclusters. CD was predicted to be induced in a nanoparticle by a chiral monolayer as in glutathione-passivated gold nanoclusters.53 An “inside-out” analogue of the solution chiral imprints described above, our analysis indicates that the chiroptical response of the chiral monolayer protected cluster can arise from imprinting the adsorbate structure on the nanoparticle electronic charge distribution (Figure 7). Gautier and Bürgi recently showed (using thiolate-for-thiolate ligand exchange experiments on gold nanoparticles) that the optical activity of gold nanoparticles is indeed dictated by the chirality of the adsorbed thiolates,54 consistent with our predictions.
Figure 7.
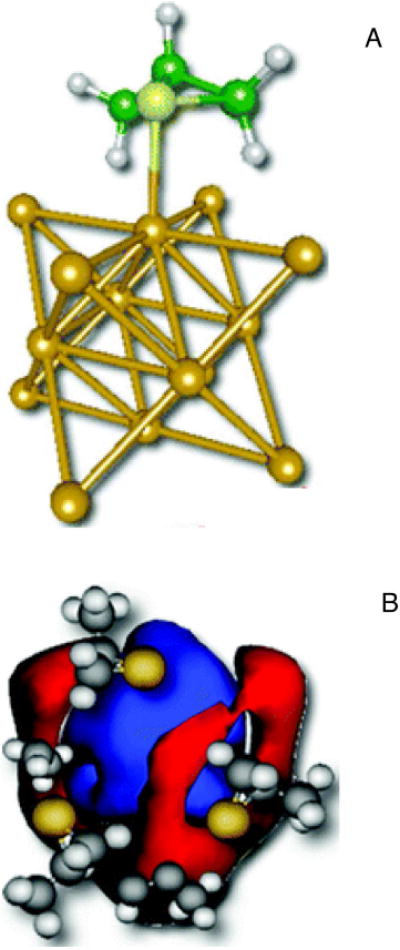
Schematic representations (A) of a chiral adsorbate (R-methylthiirane; green) and a gold cluster (yellow).53 Induced chiral-image charge densities using particle-in-a-box model illustrate (B) that the negatively charged sulfur groups (yellow) of the adsorbate dissymmetrically perturb the electron density in the box, leaving positive image charges in the central region (blue), and negative charges (red) on the edge of the box.
New Horizons
While our studies of chiroptical signatures began with the “simple” challenge of describing the sign and magnitude of OR, our explorations have found that the influence of molecular geometry, self-assembly, solvent interactions and binding, and solvent imprinting can dominate the chiroptical signature. Indeed, our theoretical studies of methyloxirane’s imprint in a benzene solution show that this solute’s chiral solvation shell can dominate the measured OR. It will be interesting to explore whether chirality transfer from a solute to a chiral solvent is a general effect, i.e. if it can be observed and controlled in other contexts. The experimental possibilities for observing these phenomena are improving. For example, Xu and coworkers recently demonstrated that chirality transfer from methyloxirane to hydrogen-bonded water could be determined with VCD.43 Indeed, the dissymmetric distribution of molecules surrounding chiral solutes is used in molecular imprinting techniques to synthesize polymers with molecular recognition capabilities.55 Hoshino et al. recently showed that polymerization of monomers in the presence of the bee toxin melittin produced imprinted polymer nanoparticles with a high affinity for mellitin.56 Thus, polymerization “locks in” the solvent imprint of melittin, producing polymers with little affinity for other peptides.56 One may imagine that asymmetric organic synthesis and chiral chromatographic separation could be impacted similarly by imprinting effects.
Chiral adsorbates on metal surfaces were recently shown to “imprint” the quantum dynamics of electron transfer. The “current” associated with electronic excitation via circularly polarized light interacts differently with left- and right-handed chiral adsorbates, producing distinctive electron transfer yields. Remarkably, both the electron and its angular momentum can be transmitted through a chiral medium.57 Since this chirality effect depends on the transport mechanism (e.g., tunneling vs. resonant transport), the signature promises to diagnose electron transport mechanism.
Intrinsically disordered proteins are implicated as having important biological functions in cellular regulation and signaling, and their structural characterization is of intense interest.58,59 Recent studies suggest that ROA can be used to characterize backbone conformations of unfolded polypeptides and proteins in aqueous solution,60 especially when an assignment based on ECD is difficult.61 We have shown that ROA calculations based on DFT and MC simulation can be used to identify populated conformations of the peptide backbone in aqueous solution.29
Mounting evidence indicates that water is an integral part of protein structure, dynamics and function.62 For example, protein folding kinetics are determined by both protein and the interacting water shell.63 ROA studies of polypeptides and proteins also indicate that water facilitates conformational fluctuations.64 Thus, ROA may be used to identify the chiroptical fingerprints of organized water layers.
It has now been thoroughly documented that theoretical predictions of ORD, ECD and ROA spectra depend strongly on the QM methods employed,65 and that the predicted spectra are strongly geometry dependent. This does not really come as a surprise. Chiroptical signatures are small because of the weakness of the magnetic dipole interactions and, in the case of ORD, because the trace of G′′ is 1-3 orders of magnitude smaller than contributing G′ tensor elements.66 Response properties are well understood to depend strongly on basis set (influencing the ability of electrons to polarize in applied fields), the description of electron correlation (influencing the position of resonances in the electronic response), solvent, molecular geometry, and molecular vibrations. In contrast, anisotropic Rayleigh OA (RayOA) is not determined by the trace of G′ and is attractively robust to changes in the level of theory.67 As such, RayOA promises to provide an elegant probe of molecular structure.
RayOA is the difference in the Rayleigh scattered intensity for right and left circular polarized light. Figure 8 shows the computed frequency-dependent depolarized right-angle RayOA curves computed for (+)-(5S, 11S)-Tröger’s base, using Hartree Fock (HF) and DFT methods with different functionals. 67 The predicted RayOA signal has the same sign and nearly identical frequency dependence in the 400 cm-1 to 700 cm-1 range. The robustness of the sign to the QM method suggests a particularly valuable place for RayOA spectroscopy if the appropriate instrumentation can be developed.
Figure 8.
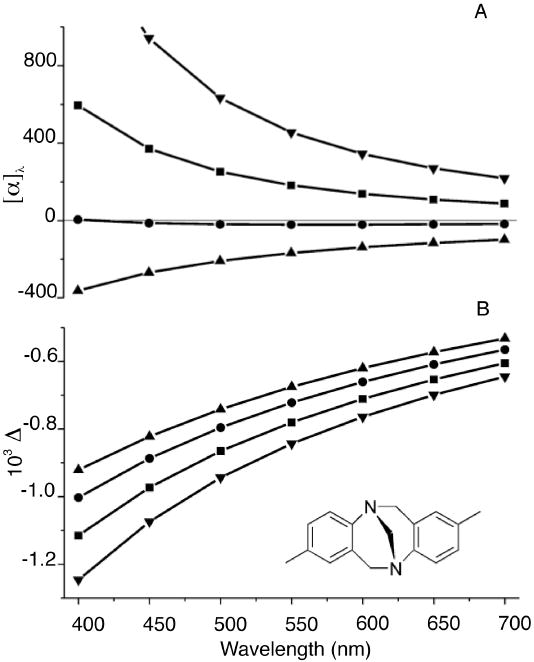
ORD (A) and depolarized right-angle ICP RayOA curves (B) of (+)-(5S, 11S)- Tröger’s base calculated using HF (▲) and DFT with B3LYP (■), BHLYP (●), BLYP (▼) functionals. Calculations used the 6-31G* basis set with the B3LYP/6-311G** optimized geometry. The predicted ORD curves show significant variation depending on the choice of the QM method. In contrast, the RayOA curves are uniformly negative in sign and possess nearly identical curvatures.68
Chiroptical response properties can be a starting point for establishing structure-activity correlations. For example, an olfactophore model68 describes the influence of a set of molecular features -- such as hydrophobic groups, and hydrogen-bond donors and acceptors -- that could be critical determinants of odor. Figure 9 show that the values of the anisotropic RayOA invariants βG2 and βA2 (calculated from the α, G′ and A tensors)67 can be used to differentiate between musky odorants and odorless compounds. Thus, dynamic molecular property tensors that contain information regarding both molecular topology and electronic structure may be useful as descriptors for structure-odor correlations. Chirality descriptors based on the α, G′ and A response tensors may therefore also provide key descriptors for biological activities and help to facilitate the computer-aided design of new odorants and drugs. The fundamental tools are now at hand to elucidate and to manipulate the chiroptical response and chiral imprinting of significant molecular assemblies. This emerging level of control seems likely to present extraordinary new opportunities for addressing challenges in asymmetric catalysis, nanomaterials, photoinduced redox processes, molecular templating/imprinting, and molecular stereochemistry.
Figure 9.
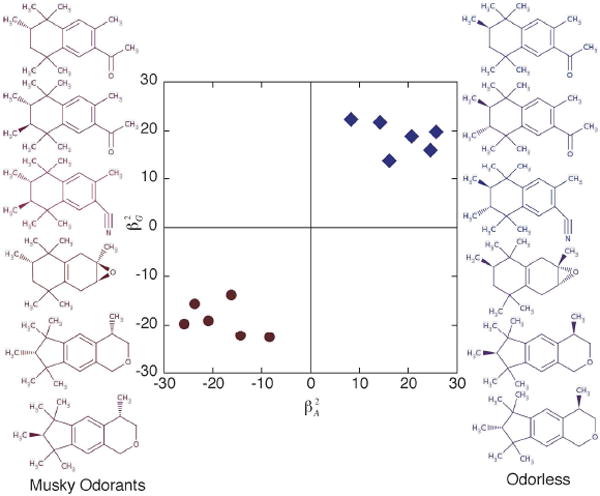
Chirality descriptors for an olfactophore model of structure-odor correlations. βG2 and βA2 are RayOA invariants derived from the dynamic molecular tensor properties G′ and A.68 The musky odorants (red) have negative βG2 and βA2. In contrast, the odorless compounds have positive βG2 and βA2. Thus, dynamic molecular tensor properties may be used as descriptor of structure-odor correlations.
Acknowledgments
We thank Rama K. Kondru, Michael-Rock Goldsmith, and Gérard Zuber for invaluable contributions to these projects. G. Zuber prepared Figure 9. We thank the National Science Foundation (NSF-CHE-0718043) and the National Institutes of Health, through a Center of Excellence in Chemical Methodologies and Library Development (P50GM067082), for financial support.
Biographies
Parag Mukhopadhyay earned a B.Sc. with honors in chemistry (University of Delhi), M.Sc. in chemistry (Indian Institute of Technology, Delhi), and M.Sc. in biological sciences (University of Calgary). He received a Ph.D. in chemistry from Duke University and is currently a post-doctoral associate at the Rockefeller University.
Peter Wipf was born in Aarau, Switzerland and studied chemistry at the University of Zürich. Following a postdoctoral appointment at the University of Virginia, he started his academic career at the University of Pittsburgh, where he holds the University Professor Chair in Chemistry.
David Beratan was born in Evanston, IL, and studied chemistry at Duke (BS) and Caltech (PhD). Following appointments at the Jet Propulsion Laboratory, he moved to the University of Pittsburgh and later to Duke University, where he is the R.J. Reynolds Professor of Chemistry and Professor of Biochemistry.
References
- 1.Kauzmann W. Quantum chemistry: An introduction. Academic Press; New York: 1957. [Google Scholar]
- 2.Polavarapu PL. Ab initio molecular optical rotations and absolute configurations. Mol Phys. 1997;91:551–554. [Google Scholar]
- 3.Kondru RK, Wipf P, Beratan DN. Theory-assisted determination of absolute stereochemistry for complex natural products via computation of molar rotation angles. J Am Chem Soc. 1998;120:2204–2205. [Google Scholar]
- 4.Crawford TD. Ab initio calculation of molecular chiroptical properties. Theor Chem Acc. 2006;115:227–245. [Google Scholar]
- 5.Autschbach J. Computation of optical rotation using time-dependent density functional theory. Comp Lett. 2007;3:131–150. [Google Scholar]
- 6.Berova N, Nakanishi K, Woody RW. Circular dichroism: principles and applications. Wiley-VCH; New York: 2000. [Google Scholar]
- 7.Barron LD. Molecular light scattering and Raman optical activity. 2. Cambridge University Press; Cambridge: 2004. [Google Scholar]
- 8.O’Loane JK. Optical-activity in small molecules, non-enantiomorphous crystals, and nematic liquid-crystals. Chem Rev. 1980;80:41–61. [Google Scholar]
- 9.Atkins PW, Friedman RS. Molecular Quantum Mechanics. Oxford University Press; Oxford: 1997. [Google Scholar]
- 10.Reiher M, Liegeois V, Ruud K. Basis set and density functional dependence of vibrational Raman optical activity calculations. J Phys Chem A. 2005;109:7567–7574. doi: 10.1021/jp052123h. [DOI] [PubMed] [Google Scholar]
- 11.Dale JA, Mosher HS. Nuclear magnetic resonance enantiomer reagents. Configurational correlations via nuclear magnetic resonance chemical-shifts of diastereomeric mandelate, O-methylmandelate, and α-methoxy-α-trifluoromethylphenylacetate (MTPA) esters. J Am Chem Soc. 1973;95:512–519. [Google Scholar]
- 12.Eliel EL, Wilen SH, Mander LN. Stereochemistry of organic compunds. Wiley-VCH; New York: 1994. [Google Scholar]
- 13.Nicolaou KC, Snyder SA. Chasing molecules that were never there: Misassigned natural products and the role of chemical synthesis in modern structure elucidation. Angew Chem Int Ed. 2005;44:1012–1044. doi: 10.1002/anie.200460864. [DOI] [PubMed] [Google Scholar]
- 14.Wipf P, Lim S. Total synthesis and structural studies of the antiviral marine natural product hennoxazole A. Chimia. 1996;50:157–167. [Google Scholar]
- 15.Ribe S, Kondru RK, Beratan DN, Wipf P. Optical rotation computation, total synthesis, and stereochemistry assignment of the marine natural product pitiamide A. J Am Chem Soc. 2000;122:4608–4617. [Google Scholar]
- 16.Zuber G, Goldsmith MR, Hopkins TD, Beratan DN, Wipf P. Systematic assignment of the configuration of flexible natural products by spectroscopic and computational methods: The bistramide C analysis. Org Lett. 2005;7:5269–5272. doi: 10.1021/ol052154v. [DOI] [PubMed] [Google Scholar]
- 17.Polavarapu PL. Why is it important to simultaneously use more than one chiroptical spectroscopic method for determining the structures of chiral molecules? Chirality. 2008;20:664–672. doi: 10.1002/chir.20475. [DOI] [PubMed] [Google Scholar]
- 18.Zuber G, Goldsmith MR, Beratan DN, Wipf P. Assignment of the absolute configuration of [n]-ladderanes by TD-DFT optical rotation calculations. Chirality. 2005;17:507–510. doi: 10.1002/chir.20190. [DOI] [PubMed] [Google Scholar]
- 19.Mascitti V, Corey EJ. Enantioselective synthesis of pentacycloanammoxic acid. J Am Chem Soc. 2006;128:3118–3119. doi: 10.1021/ja058370g. [DOI] [PubMed] [Google Scholar]
- 20.Brewster JH. A useful model of optical activity. I. Open chain compunds. J Am Chem Soc. 1959;81:5475–5483. [Google Scholar]
- 21.Lowe G. The absolute configuration of allenes. Chem Comm. 1965;17:411–413. [Google Scholar]
- 22.Hudson CS. The significance of certain numerical relations in the sugar group. J Am Chem Soc. 1909;31:66–86. [Google Scholar]
- 23.Kondru RK, Wipf P, Beratan DN. Atomic contributions to the optical rotation angle as a quantitative probe of molecular chirality. Science. 1998;282:2247–2250. doi: 10.1126/science.282.5397.2247. [DOI] [PubMed] [Google Scholar]
- 24.Kondru RK, Wipf P, Beratan DN. Structural and conformational dependence of optical rotation angles. J Phys Chem A. 1999;103:6603–6611. [Google Scholar]
- 25.Wiberg KB, Wang YG, Vaccaro PH, Cheeseman JR, Luderer MR. Conformational effects on optical rotation. 2-substituted butanes. J Phys Chem A. 2005;109:3405–3410. doi: 10.1021/jp040724n. [DOI] [PubMed] [Google Scholar]
- 26.Kondru RK, Lim S, Wipf P, Beratan DN. Synthetic and model computational studies of molar rotation additivity for interacting chiral centers: A reinvestigation of van’t Hoff’s principle. Chirality. 1997;9:469–477. doi: 10.1002/(SICI)1520-636X(1997)9:5/6<469::AID-CHIR13>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 27.Haesler J, Schindelholz I, Riguet E, Bochet CG, Hug W. Absolute configuration of chirally deuterated neopentane. Nature. 2007;446:526–529. doi: 10.1038/nature05653. [DOI] [PubMed] [Google Scholar]
- 28.Zuber G, Goldsmith MR, Beratan DN, Wipf P. Towards Raman optical activity calculations of large molecules. ChemPhysChem. 2005;6:595–597. doi: 10.1002/cphc.200400544. [DOI] [PubMed] [Google Scholar]
- 29.Mukhopadhyay P, Zuber G, Beratan DN. Characterizing aqueous solution conformation of a peptide backbone using Raman optical activity computations. Biophys J. 2008;95:5574–5586. doi: 10.1529/biophysj.108.137596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weise CF, Weisshaar JC. Conformational analysis of alanine dipeptide from dipolar couplings in a water-based liquid crystal. J Phys Chem A. 2003;107:3265–3277. [Google Scholar]
- 31.Poon CD, Samulski ET, Weise CF, Weisshaar JC. Do bridging water molecules dictate the structure of a model dipeptide in aqueous solution? J Am Chem Soc. 2000;122:5642–5643. [Google Scholar]
- 32.Mehta MA, Fry EA, Eddy MT, Dedeo MT, Anagnost AE, Long JR. Structure of the alanine dipeptide in condensed phases determined by C-13 NMR. J Phys Chem B. 2004;108:2777–2780. [Google Scholar]
- 33.Kim YS, Wang J, Hochstrasser RM. Two-dimensional infrared spectroscopy of the alanine dipeptide in aqueous solution. J Phys Chem B. 2005;109:7511–7521. doi: 10.1021/jp044989d. [DOI] [PubMed] [Google Scholar]
- 34.Takekiyo T, Imai T, Kato M, Taniguchi Y. Temperature and pressure effects on conformational equilibria of alanine dipeptide in aqueous solution. Biopolymers. 2004;73:283–290. doi: 10.1002/bip.10548. [DOI] [PubMed] [Google Scholar]
- 35.Vila JA, Aramini JM, Rossi P, Kuzin A, Su M, Seetharaman J, Xiao R, Tong L, Montelione GT, Scheraga HA. Quantum chemical C-13(alpha) chemical shift calculations for protein NMR structure determination, refinement, and validation. Proc Natl Acad Sci USA. 2008;105:14389–14394. doi: 10.1073/pnas.0807105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kondru RK, Beratan DN, Friestad GK, Smith AB, III, Wipf P. Chiral action at a distance: Remote substituent effects on the optical activity of calyculins A and B. Org Lett. 2000;2:1509–1512. doi: 10.1021/ol005634p. [DOI] [PubMed] [Google Scholar]
- 37.Goldsmith MR, Jayasuriya N, Beratan DN, Wipf P. Optical rotation of noncovalent aggregates. J Am Chem Soc. 2003;125:15696–15697. doi: 10.1021/ja0376893. [DOI] [PubMed] [Google Scholar]
- 38.Wilson SM, Wiberg KB, Cheeseman JR, Frisch MJ, Vaccaro PH. Nonresonant optical activity of isolated organic molecules. J Phys Chem A. 2005;109:11752–11764. doi: 10.1021/jp054283z. [DOI] [PubMed] [Google Scholar]
- 39.Mukhopadhyay P, Zuber G, Wipf P, Beratan DN. Contiribution of a solute’s chiral solvent imprint to optical rotation. Angew Chem Int Ed. 2007;46:6450–6452. doi: 10.1002/anie.200702273. [DOI] [PubMed] [Google Scholar]
- 40.Mukhopadhyay P, Zuber G, Goldsmith MR, Wipf P, Beratan DN. Solvent effect on optical rotation: A case study of methyloxirane in water. ChemPhysChem. 2006;7:2483–2486. doi: 10.1002/cphc.200600477. [DOI] [PubMed] [Google Scholar]
- 41.Kongsted J, Pedersen TB, Strange M, Osted A, Hansen AE, Mikkelsen KV, Pawlowski F, Jorgensen P, Hattig C. Coupled cluster calculations of the optical rotation of S-propylene oxide in gas phase and solution. Chem Phys Lett. 2005;401:385–392. [Google Scholar]
- 42.Kumata Y, Furukawa J, Fueno T. Effect of solvents on the optical rotation of propylene oxide. Bull Chem Soc Jpn. 1970;43:3920–3921. [Google Scholar]
- 43.Losada M, Nguyen P, Xu YJ. Solvation of propylene oxide in water: Vibrational circular dichroism, optical rotation, and computer simulation studies. J Phys Chem A. 2008;112:5621–5627. doi: 10.1021/jp801996m. [DOI] [PubMed] [Google Scholar]
- 44.Fidler J, Rodger PM, Rodger A. Chiral solvent structure around chiral molecules: Experimental and theoretical-study. J Am Chem Soc. 1994;116:7266–7273. [Google Scholar]
- 45.Neugebauer J. Induced chirality in achiral media - How theory unravels mysterious solvent effects. Angew Chem Int Ed. 2007;46:7738–7740. doi: 10.1002/anie.200702858. [DOI] [PubMed] [Google Scholar]
- 46.Wang SH, Canna NM. A molecular dynamics study of chirality transfer: The impact of a chiral solute on an achiral solvent. J Chem Phys. 2008;129:054507–054517. doi: 10.1063/1.2958911. [DOI] [PubMed] [Google Scholar]
- 47.Autschbach J. Density functional theory applied to calculating optical and spectroscopic properties of metal complexes: NMR and optical activity. Coord Chem Rev. 2007;251:1796–1821. [Google Scholar]
- 48.Bonnemann H, Braun GA. Enantioselective hydrogenations on platinum colloids. Angew Chem Int Ed. 1996;35:1992–1995. [Google Scholar]
- 49.Bieri M, Gautier C, Buergi T. Probing chiral interfaces by infrared spectroscopic methods. PCCP. 2007;9:671–685. doi: 10.1039/b609930k. [DOI] [PubMed] [Google Scholar]
- 50.Dressler DH, Mastai Y. Chiral crystallization of glutamic acid on self assembled films of cysteine. Chirality. 2007;19:358–365. doi: 10.1002/chir.20389. [DOI] [PubMed] [Google Scholar]
- 51.Hu X, An Q, Li G, Tao S, Liu J. Imprinted photonic polymers for chiral recognition. Angew Chem Int Ed. 2006;45:8145–8148. doi: 10.1002/anie.200601849. [DOI] [PubMed] [Google Scholar]
- 52.Behar-Levy H, Neumann O, Naaman R, Avnir D. Chirality induction in bulk gold and silver. Adv Mat. 2007;19:1207–1211. [Google Scholar]
- 53.Goldsmith MR, George CB, Zuber G, Naaman R, Waldeck DH, Wipf P, Beratan DN. The chiroptical signature of achiral metal clusters induced by dissymmetric adsorbates. PCCP. 2006;8:63–67. doi: 10.1039/b511563a. [DOI] [PubMed] [Google Scholar]
- 54.Gautier C, Burgi T. Chiral inversion of gold nanoparticles. J Am Chem Soc. 2008;130:7077–7084. doi: 10.1021/ja800256r. [DOI] [PubMed] [Google Scholar]
- 55.Zimmerman SC, Lemcoff NG. Synthetic hosts via molecular imprinting - are universal synthetic antibodies realistically possible? Chem Comm. 2004:5–14. doi: 10.1039/b304720b. [DOI] [PubMed] [Google Scholar]
- 56.Hoshino Y, Kodama T, Okahata Y, Kenneth JS. Peptide Imprinted Polymer Nanoparticles: A Plastic Antibody. J Am Chem Soc. 2008;130:15242–15243. doi: 10.1021/ja8062875. [DOI] [PubMed] [Google Scholar]
- 57.Skoutris SS, Beratan DN, Naaman R, Nitzan A, Waldeck DH. Chiral control of electron transmission through molecules. Phys Rev Lett. 2008;101:238103–238107. doi: 10.1103/PhysRevLett.101.238103. [DOI] [PubMed] [Google Scholar]
- 58.Wright PE, Dyson HJ. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J Mol Bio. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 59.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 60.Barron LD, Blanch EW, Hecht L. Unfolded proteins studied by Raman optical activity. Adv Protein Chem. 2002;62:51–90. doi: 10.1016/s0065-3233(02)62005-4. [DOI] [PubMed] [Google Scholar]
- 61.Glattli A, Daura X, Seebach D, van Gunsteren WF. Can one derive the conformational preference of a beta-peptide from its CD spectrum? J Am Chem Soc. 2002;124:12972–12978. doi: 10.1021/ja020758d. [DOI] [PubMed] [Google Scholar]
- 62.Helms V. Protein dynamics tightly connected to the dynamics of surrounding and internal water molecules. ChemPhysChem. 2007;8:23–33. doi: 10.1002/cphc.200600298. [DOI] [PubMed] [Google Scholar]
- 63.Kim SJ, Born B, Havenith M, Gruebele M. Real time detection of protein-water dynamics upon protein folding by terahertz absorption spectroscopy. Angew Chem Int Ed. 2008;47:6486–6489. doi: 10.1002/anie.200802281. [DOI] [PubMed] [Google Scholar]
- 64.Barron LD, Hecht L, Wilson G. The lubricant of life: A proposal that solvent water promotes extremely fast conformational fluctuations in mobile heteropolypeptide structure. Biochemistry. 1997;36:13143–13147. doi: 10.1021/bi971323j. [DOI] [PubMed] [Google Scholar]
- 65.Stephens PJ, Devlin FJ, Cheeseman JR, Frisch MJ. Calculation of optical rotation using density functional theory. J Phys Chem A. 2001;105:5356–5371. [Google Scholar]
- 66.Giorgio E, Viglione RG, Zanasi R, Rosini C. Ab initio calculation of optical rotatory dispersion (ORD) curves: A simple and reliable approach to the assignment of the molecular absolute configuration. J Am Chem Soc. 2004;126:12968–12976. doi: 10.1021/ja046875l. [DOI] [PubMed] [Google Scholar]
- 67.Zuber G, Wipf P, Beratan DN. Exploring the optical activity tensor by anisotropic Rayleigh optical activity scattering. ChemPhysChem. 2008;9:265–271. doi: 10.1002/cphc.200700660. [DOI] [PubMed] [Google Scholar]
- 68.Kraft P, Bajgrowicz JA, Denis C, Frater G. Odds and trends: Recent developments in the chemistry of odorants. Angew Chem Int Ed. 2000;39:2981–3010. doi: 10.1002/1521-3773(20000901)39:17<2980::aid-anie2980>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]