

Abstract
The multidomain HIV-1 Vif protein recruits several cellular partners to achieve neutralization of the antiviral activity of APOBEC3 proteins. Vif neutralizes APOBEC-3G and APOBEC3F predominantly by forming an E3 ubiquitin ligase with Cullin5, ElonginB and ElonginC that targets these proteins for degradation by the ubiquitin-protea-some pathway. Vif associates with the Cullin5-ElonginB-ElonginC complex by binding directly to ElonginC via its SOCS-box motif and to Cullin5 via hydrophobic residues within a zinc-binding region formed by a conserved HCCH motif. The HIV-1 Vif-Cullin5-ElonginBC complex is then able to ubiquitinate the APOBEC3G factor bound to Vif by its N-terminal domain. In this review, we summarize the current knowledge about the structural determinants of Vif that allow it to interact with cellular and viral partners.
Keywords: Amino Acid Motifs; Gene Products, vif; genetics; metabolism; HIV-1; genetics; HIV-2; genetics; Humans; Protein Binding; Protein Structure, Tertiary; Simian immunodeficiency virus; genetics
Keywords: Vif protein, APOBEC3G, Cullin5, Elongin, reverse transcription, RNA
I. Introduction
The Human immunodeficiency virus (HIV) belongs to the Retroviridae family and, in contrast with simple retroviruses, it encodes for several proteins in addition to the three viral genes gag, pol and env, which constitute their structural and enzymatic repertoire. These proteins are usually subdivided into regulatory proteins such as Tat, Rev, and Nef, and auxiliary proteins including Vif (Virion Infectivity Factor), Vpr and Vpu. The HIV auxiliary genes, initially thought to be dispensable for viral replication, are now known to function as crucial enhancers of viral pathogenesis. Vif, Vpr and Vpu act as versatile adaptors that connect viral and cellular pathways, and lead to efficient viral replication, assembly and release (for a review, see [1]). HIV-1 Vif is a cytoplasmic, 23 kDa protein, expressed late in the viral cycle and has been known for a long time to be essential for viral replication. Vif is required in nonpermissive cells but is dispensable for replication in permissive cell lines [2–5]. In non-permissive cells, Vif-defective viruses (HIV-1Δvif) can produce virions, but they fail to complete reverse transcription and cannot successfully infect new cells [6,7]. It took almost 20 years to identify the function of Vif [8,9]. It actually counteracts the antiviral activity of recently identified members of the cellular cytidine deaminase family, APOBEC3B, APOBEC3F and APOBEC3G (for a review see [10]). In the absence of Vif, APOBEC3G is packaged into virions as a stable complex with the viral core [11,12]. This provokes the deamination of cytidine to uracil during the subsequent round of viral replication, leading to the production of non-functional proviruses. Vif interacts with and adapts APOBEC3G to an ElonginB-ElonginC-Cullin5 E3 ligase complex [13]. The bound APOBEC3G is then ubiquitinated and degraded by the proteasome, allowing viral replication. It has also been proposed that Vif can be down regulated APOBEC3G at the translational level [14–16].
Recent work suggest that DNA editing is not always necessary for antiviral activity and that APOBEC3G and APOBEC3F may exert their antiviral activity by other mechanisms than cytidine deamination [17,18]. To understand the function of a given protein at the molecular level, an appreciation of functional domains, motifs, and residues can be of tremendous help. With this aim, this review describes the general biochemical properties of Vif and then its interactions with cellular and viral partners from a structural point of view.
II. General Biochemical Properties of VIF and Definition of its Different Domains
To date, little structural data is available on HIV-1 Vif protein and, more importantly no three-dimensional structure exists. This lack of data is first due to the difficulty of expressing high levels of soluble recombinant protein using either prokaryotic or baculovirus expression systems. Vif is a highly basic protein (pI = 10.7) that is conserved in all lentiviruses, except Equine Infectious Anemia Virus [19]. The Vif proteins of closely related HIV-1 strains are highly conserved (e.g. 91% of identity between HIV-1 HXB2 and HIV- 1 MN isolates), while those of slightly more distantly related lentiviruses have diverged significantly (e.g. there is a maximum of 30% of identity between HIV-1 HXB2 and SIV isolates) [20]. Alignments of Vif sequences from HIV-1, HIV-2, and SIV subtypes (Fig. 1) highlight several highly conserved motifs, i.e. an N-terminal tryptophan-rich stretch, a conserved HCCH motif and a SOCS-box motif. The N-terminal tryptophan-rich stretch (residues 1 to 21) is highly conserved (Fig. 1). Interestingly, different tryptophans (Fig. 2a) are required to mediate the Vif recognition and suppression of the target molecules APOBEC3G and APOBEC3F [21]. This point will be discussed in section III. Amino acids 63–70 and 86–89 that are well conserved and predicted to be important for formation of β-strand structures (Fig. 1) are critical for maintaining a normal expression level of Vif and for viral infectivity [22]. In HIV-1, the conserved glutamic acid at position 88 and the tryptophan at position 89 are located within the charged central hydrophilic region 88EWRKKR93 that is thought to enhance steady-state expression of Vif in host cells [22]. The HCCH motif (residues 108–139) consists of two conserved His/Cys pairs flanking a predicted α-helix that contains a cluster of hydrophobic conserved residues (Figs. 1,2). This motif was shown to coordinate one Zn2+ ion through the universally conserved residues H108, C114, C133 and H139, and to bind directly to Cullin5 [23,24] (see section IV). The highly conserved 144SLQYLA149 motif [20] defines the so-called BC-box motif and is crucial for inactivation of APOBEC3 proteins [13]. This BC-box motif is responsible for the binding to ElonginC, which further targets APOBEC3 antiviral factors to the proteasome. There are high similarities between the conserved SLQ(Y/F)LA ΦΦΦΦ motif (Φ for hydrophobic residue) of Vif and the SOCS-box of SOCS protein (suppressor of cytokine signalling). This subject will be developed in section V and for purpose of clarity, this domain will be called SOCS-box. Vif is a component of cytoplasmic RNPs mediating viral RNA interactions with the Pr55Gag precursor and cellular factors [25–27] and Vif binds HIV-1 genomic RNA in vivo [25,27] and in vitro [25,28]. In addition, any mutation reducing the affinity of Vif for RNA diminishes viral replication in non-permissive cell lines, suggesting that RNA also plays a central role in Vif function [26,27]. This point will be issued in section VI. Vif is subject to intravirion processing by the HIV-1 protease [29]. Importantly, mutations in Vif that affect Pr-dependent processing also affect the ability of Vif to regulate viral infectivity, suggesting that intravirion processing of Vif is functionally relevant [29]. The processing site is located after L150 dowstream the highly conserved SOCS-box (Fig. 2b). The authors suggest that the Vif processing site is defined by structural constraints rather than by a specific amino acid sequence [30]. Post-translational phosphorylation of viral proteins by cellular kinases regulates HIV-1 infectivity. Many viral proteins are regulated by phosphorylation at different stages of the virus life cycle. Vif is phosphorylated in vitro and in vivo by cellular kinases and Vif phosphorylation is important for HIV-1 replication [31,32]. Four major phosphorylation sites were identified: T96, S144, T155 and T188, the three latter within the C-terminal domain of Vif (Fig. 2a). Importantly, T96 and S144 are highly conserved in all lentiviruses. Mutations of T96 resulted in significant loss of Vif activity and inhibition of HIV-1 replication [32]. Mutation of S144 to alanine resulted in loss of Vif activity, suggesting that phosphorylation at this site plays an important role in regulating HIV-1 replication and infectivity [31]. Synthetic Vif peptides corresponding to the local sequences of the phosphorylation sites were not phosphorylated by MAPK, suggesting that recognition of these sites by MAPK is likely to require structural determinants outside the phosphorylation sites [32]. Like several other proteins encoded by HIV-1, Vif proteins possess a strong tendency toward self-association. In relatively native conditions, Vif proteins form multimers in vitro, including dimers, trimers and tetramers [33]. This propensity for self association, from dimers to tetramers, has been also observed in cross-linking experiments [34]. This behavior was not limited to in vitro studies, as experiments based on co-immunoprecipitation or two-hybrid systems revealed Vif-Vif association within cells [33]. The domain mediating Vif self-association was first identified within the C-terminal domain of the protein in the proline-rich 156–164 region [33]. More precisely, the conserved 161PPLP164 domain in HIV-1 Vif protein (Fig. 2b) was shown to play a key role in multimerization [35]. Vif multimerization at its C-terminus via this motif is thought to release Pr55Gag, which binds to this region [35]. In HIV-1, Vif mulimerization is crucial for viral infectivity [35] and for preventing APOBEC3G incorporation into viral particles [36]. However, surprisingly, this multimerization site is not conserved in HIV-2 or SIV (Fig. 1). Some regions of HIV-1 Vif are predicted to be intrinsically disordered (solid lines in Fig. 1). Indeed, large segments of HIV-1 Vif secondary structure are predicted to be random coil (70–84, 130–143, 155–192), with however a more ordered secondary structure predicted for the N-terminus consisting of mostly β-sheets and one α-helix. Very recently, two models for the three-dimensional structure of HIV-1 Vif were presented [37,38]. Briefly, both models are based on predicted secondary structures shown in Fig. 1 and the search in the PDB (Protein Data Bank http://www.rcsb.org/pdb/home/home.do) for protein 3D structures that shares the same secondary structures. The second model [38], which seems to be more complete, incorporated the results of mutagenesis studies [39–41] that indicate that Vif contains at least two functionally crucial domains: an N-terminal domain that is important for binding to APOBEC3G, and a C-terminal region with a conserved SLQ(Y/F)LAΦΦΦΦ motif similar to SOCS-box that is responsible for its interaction with the ElonginB-ElonginC complex [13]. A three-dimensional model of HIV-1 Vif was constructed by homology modelling using two templates: the SOCS-box of VHL (von Hippel-Lindau tumor suppressor protein) as the template for the C-terminal domain, and the N-terminal domain of Narl, whose secondary structure presents strong similarity to the N-terminal domain of Vif [38]. While the two models roughly present the same secondary structure of Vif, the three-dimensional foldings are really divergent. The difficulties lie not only in organizing the secondary elements with respect to each other to access the 3D-fold but also to model the large loops present in Vif. Moreover, none of these models took into account the presence of the HCCH domain that is also crucial for Vif activity and that could be determinant for Vif folding. Each model can however provide specific details about mutation sites, but they should be used with caution, because of lack of structural data such as X-ray diffraction or NMR data.
Figure 1. Sequence Alignments of HIV/SIV Vif proteins.
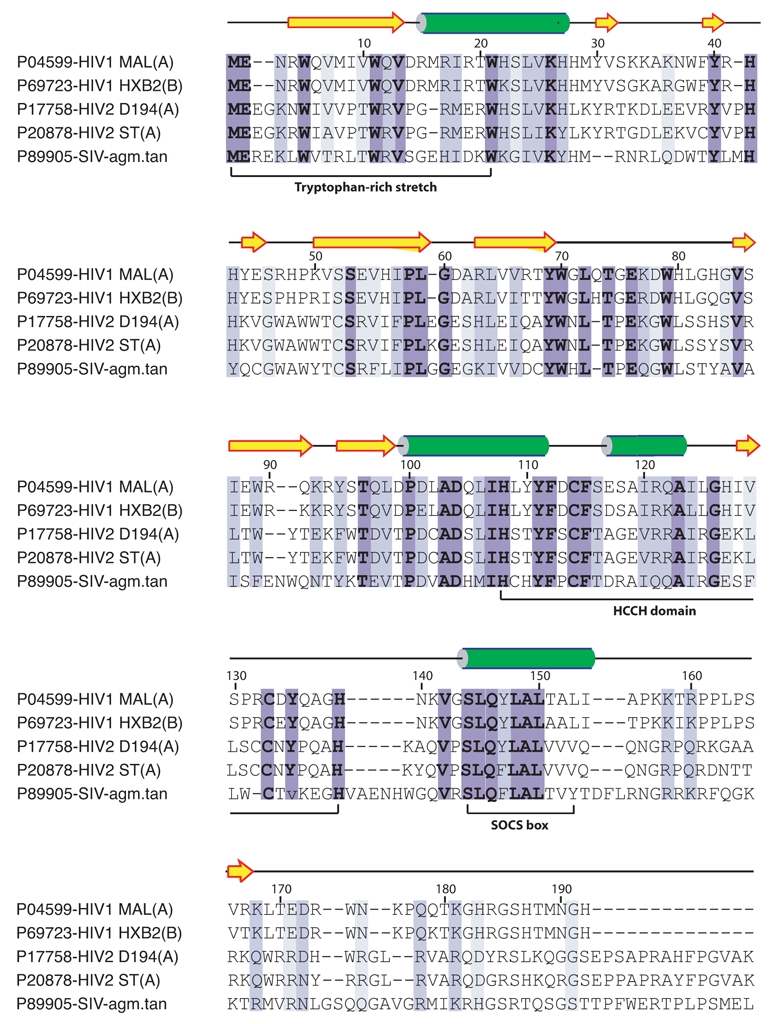
Highly conserved residues are on a dark blue background whereas residues with similar physico-chemical characters are on a light blue background. The accession number of each sequence in the Swiss-Prot database is indicated before its name. The predicted secondary structure of the HIV-1 HXB2 Vif protein using the PSI-PRED program [88] is shown below the sequences as arrows for β-strands and cylinder for α-helices. Intrinsically disordered sequences are indicated by solid lines.
Figure 2. The functional domains in HIV Vif.
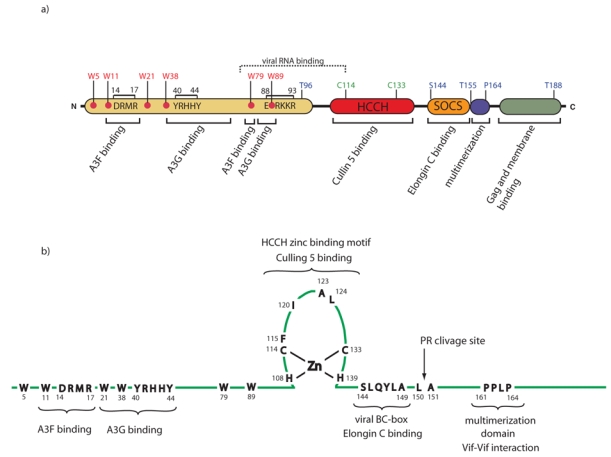
a) HIV-1 Vif contains several functional domains, including an APOBEC3F/G binding domain, a conserved zinc-binding hydrophobic HCCH motif and a downstream SOCS-box. Conserved Tryptophans important for APOBEC binding are indicated as well as Serine and Threonines that can be phosphorylated. b) Detailed architecture of the HCCH domain, of the multimerization domain and of the SOCS-box encompassing the viral BC-box 144SLQYLA149 plus 4 hydrophobic residues of HIV-1 Vif protein. Pr clivage site indicates the clivage site of the viral protease.
III. The N-Terminal Domain of VIF: Interaction with APOBEC3F and APOBEC3G
Seven members of the APOBEC3 family (APOBEC3A–H) have been described in primates with varying degrees of anti-viral activity, ranging from having no described activity to potent inhibitory effects against retroviruses and retrotransposons (for a review, see [10]). Binding of Vif to APOBEC3G/3F is essential for their degradation by the Vif-Cullin5 E3 ligase [9,16,42] and a detailed knowledge of the interactions between these two proteins is of great importance for the development of new drugs aiming at blocking Vif-mediated degradation of APOBEC3G. In 2004, several investigators showed that the Vif-APOBEC3G interaction is species specific [43–46]. Indeed, human, chimpanzee, rhesus macaque (mac) and African green monkey (agm) APOBEC3G proteins can inhibit Δvif HIV-1, while only the human and chimpanzee proteins are sensitive to HIV-1 Vif. Sequence analysis and mutagenesis showed that amino acid 128 (D in human and K in mac and agm) in APOBEC3G was responsible for Vif sensitivity. However, while the interaction between Vif and APOBEC3G was severely affected when amino acid 128 was mutated, it was not completely abrogated [46] and surrounding amino acids such as P129 and D130 [47] and region 54–124 [48] have been shown to modulate the APOBEC3G-Vif interaction. Unlike APOBEC3G, modification of amino acid 128 in human APOBEC3F did not change its recognition by HIV-1 or SIVagm Vif [49]. It should also be pointed out that both Vif and APOBEC3G are RNA-binding proteins [28,50] and that Vif-APOBEC3G interactions could be mediated by RNA although a recent study showed that RNA was not necessary for these proteins to interact [51].
Regarding the Vif protein, several lines of evidence suggested that the N-terminal region contains the main binding sites for APOBEC3G and APOBEC3F (Fig. 2a) [9,21,42,52–57], while a short sequence encompassing amino acids 169– 192 in the C-terminal has also been shown to mediate Vif- APOBEC3G interactions [53]. A previous study reported that single-amino-acid changes in Vif sequences isolated from HIV-1-infected patients could be sufficient to prevent APOBEC3 neutralization [52]. Since then, a number of mutagenesis studies revealed that substitutions of highly conserved residues in the N-terminus of Vif differentially affect the interactions of this protein with APOBEC3G and APOBEC3F interactions [21,52,55–57]. For instance, residues 85 to 99 [53] and 40YRHHY44 of Vif are important for binding to APOBEC3G [56,57] whereas the 14DRMR17 residues are crucial for binding to APOBEC3F (Fig. 2a) [55,57]. A similar phenomenon was also observed with conserved tryptophan residues located in the N-terminus of Vif: W11 and W79 are critical for interaction with APOBEC3F while all other tryptophan residues (W5, W21, W38 and W89) are important for Vif activity against APOBEC3G [21]. In addition to these residues, other amino acids in Vif are likely to contribute to APOBEC3G or APOBEC3F binding. It has been shown that I9 [54], K22, E45, and N48 [52,57] substitutions suppressed Vif function against APOBEC3G but not APOBEC3F, but that Q12 [52,57] reduced the ability of Vif to neutralize APOBEC3F. Taken together, these data suggest that Vif contains nonlinear binding sites for APOBEC3G and APOBEC3F in its N-terminus region (Fig. 2a) and that Vif- APOBEC3G interaction is dependent on electrostatic interactions. Indeed, Schrofelbauer and co-workers [55] showed that the defect of interaction between the D128K mutant of human APOBEC3G and HIV-1 Vif could be compensated by substituting R15 and R17 residues by neutral or negatively charged residues. These Vif mutants were also able to interact with macAPOBEC3G and agmAPOBEC3G, which are naturally positively charged at position 128 (K). Moreover, the substitution of 14DRMR17 by SERQ, the exact amino acid sequence found in agmVif, or by SEMQ, was sufficient to allow functional interaction of HIV-1 Vif with mac and agmAPOBEC3G, as well as human APOBEC3G and D128K-APOBEC3G. Thus, the fact that region 14–17 of HIV-1 Vif removes the species restriction imposed by residue 128 of APOBEC3G suggests a direct interaction between these two regions. However, it does not rule out any conformational rearrangement induced by mutations and the fact that the affinity between Vif and APOBEC3G and APOBEC3F could be increased by additional interactions [21,55–57].
IV. HCCH Zn Binding Domain of VIF: a CULLIN5 Binding Domain
Vif hijacks the cellular Cullin5-Ring E3 ubiquitin ligase to degrade APOBEC3 proteins (Fig. 3). Vif was shown to recruit Cullin5 in concert with ElonginB and C and Rbx1 to form an E3 ubiquitin ligase complex [13,40,58] similar to ElonginB/C-Cul2-SOCS-box complexes [59,60]. Vif proteins lack a clear Cullin5-box, and so the molecular basis of the recruitment of Cullin5 was first unclear. The first residues shown to interact with Cullin5 but not with ElonginB/C were the highly conserved C114 and C133, upstream the SOCS-box [13] (Fig. 2a). Indeed, mutation of each cysteine residue individually into a serine residue severely impaired the ability of Vif to interact with Cullin5, but not with ElonginB/C. The two cysteine residues were then shown to belong to a completely conserved HCCH motif with the sequence H108-X5-C114-X18-C133-X5-H139 (Fig. 2b). The HCCH sequence and spacing are highly conserved in Vif proteins and are critical for Vif function [61]. All individual residues of this HCCH motif were shown to be necessary for interaction with Cullin5 [62]. Indeed, individual substitutions in each of the His or Cys abolished the interaction with Cullin5 (in SIV-primate) leading the authors to the conclusion that not only the two cysteines but also the two histidines contribute to the interaction [62]. All conserved amino acids of this motif (between H108-H139) except G126 were found to be critical for assembly of the Vif-Cullin5 E3 ubiquitin ligase [61]. This HCCH motif was subsequently shown to form a structural domain that has the potential to bind zinc, making Vif a zinc-binding protein [23,24]. This HCCH motif is reminiscent of a zinc-finger like structure. However, the arrangement of the His and Cys residues and the spacing between them in Vif appear to be unique when compared to other known classes of zinc-binding domains [63]. Here again, it was shown that each individual zinc-coordinating residue was essential for Cullin5 association and APOBEC3G degradation but not for ElonginB/C binding [23,24]. Moreover, a highly conserved stretch of hydrophobic residues F115-X4-I/V120-X2-A123-I/L124 (Figs. 1,2) within the Vif zinc-binding domain is required for the interaction with Cullin5 [23]. F115 is present in most lentiviral Vif proteins, with W occasionally seen, and F115A, I120S and AL123/124SS mutants lost the ability to interact with Cullin5 [23]. The hydrophobic motif within the zinc-binding domain of Vif forms a critical functional interface with Cullin5. Disruption of this motif results in a defective Vif that is unable to recruit Cullin5 and consequently has lost the ability to degrade APOBEC3G [23]. Interestingly, it has been reported that HIV-1 Vif protein, as well as a minimalist HCCH peptide mimicking the HCCH domain of Vif, changes its conformation upon zinc binding [64]. Moreover zinc binding leads to rapid protein aggregation that is efficiently reversed upon chelating agent treatment like EDTA [64]. Zinc binding generates a conformation capable of forming high order protein assemblies. This self-association property of Vif protein recalls the capacity of Vif to form multimers, but it does not involve the same domain and the multimerization via the PPLP domain lead up to tetramers and not to aggregation. The authors proposed that zinc binding alters the native protein conformation to expose a protein-protein interaction domain. The resulting conformation would display surface side chain residues to the solvent, and could mediate protein-protein interaction, for example with Cullin5 [64]. A recent study demonstrates that the arrangement of the zinc coordination residues, HCCH, and the maintenance of both the conserved residues and the spacing within the HCCH motif are critical for Vif function. Indeed deletion of non conserved residues in each portion of the HCCH motif affected recruitment of Cullin5, APOBEC3G degradation and viral infectivity [61]. It has also been shown that chelation of zinc in cells by a membrane-permeable zinc chelator prevents Vif function in infectivity assays, allowing the virus to become sensitive to the antiviral activity of APOBEC3G [65]. To conclude, the HCCH zinc-binding motif facilitates Vif-Cullin5 binding by playing a structural role in positioning hydrophobic residues for direct contact with Cullin5.
Figure 3. Function of Vif.
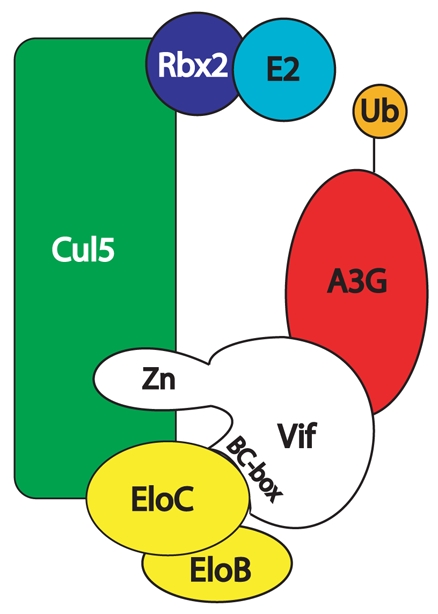
Vif specifically recruits APOBEC3G into a Cullin5 E3-ligase complex for ubiquitination and subsequent proteasome degradation. Vif associates with the Cullin5-ElonginB-ElonginC complex by binding directly to ElonginC via its SOCSbox motif and to Cullin5 via hydrophobic residues within a zinc-binding region formed by a conserved HCCH motif. The HIV-1 Vif-Cullin5-ElonginBC complex is then able to ubiquitinate the APOBEC3G factor bound to Vif by its N-terminal domain.
V. SOCS-BOX Domain of VIF: an ELONGIN B/C Binding Domain
It was known for a long time that substituting alanines for the 144SLQ149 Vif motif causes a loss of HIV-1 infectivity [66]. The molecular explanation has been discovered later [40], when this mutation was shown to affect formation of the Vif-Cullin5-ElonginBC complex. Indeed, the ElonginBElonginC heterodimer is known to interact with SOCS-box containing proteins [67,68]. Alignment of Vif proteins from primate and non primate lentiviruses led to the recognition of a highly conserved motif 144(S/T)LQ(F/Y/R)LA149 [20], homologous to a BC-box sequence conserved in proteins that bind to ElonginB-ElonginC heterodimers [69]. More recently, the conserved SLQ(Y/F)LAΦΦΦΦ motif of Vif was described to possess striking resemblance with the SOCSbox of SOCS proteins [13]. This putative SOCS-box of Vif was then shown to mediate its interaction with ElonginC [13,58]. A major difference between the SOCS-box-like motif in HIV/SIV Vif and the consensus SOCS-box motif [69] is the lack of a highly conserved C, replaced by A149 in Vif. This highly conserved C is critical for interaction with ElonginC [67]. In Vif, both A and C at position 149 can mediate the interaction with ElonginC [13]. However, longer side chain residues at this position, like L or T, prevent binding of ElonginC and degradation of APOBEC3G [13].
Amino acids 145–155 are predicted to form an α-helix (Fig. 1). Using the crystal structure of VHL-ElonginC [70] as a model, Yu et al. [13] have shown that L145, A149, A152 and L153 of Vif could form a hydrophobic cluster with spacing similar to hydrophobic residues of VHL. Importantly, these HIV-1 Vif hydrophobic residues are predicted to be on the same face of the α-helix and could fit into the hydrophobic pocket of ElonginC.
To sum up, the multidomain Vif protein recruits several cellular partners to achieve the degradation of APOBEC3 proteins. Vif neutralizes the antiviral activity of APOBEC-3G and APOBEC3F predominantly by forming an E3 ubiquitin ligase with Cullin5, ElonginB and ElonginC that targets these proteins for degradation by the ubiquitin-protea-some pathway. Vif associates with the Cullin5-ElonginB-ElonginC complex by binding directly to ElonginC via its SOCS-Box motif and to Cullin5 via hydrophobic residues within a zinc-binding region formed by a conserved HCCH motif (Fig. 3). The HIV-1 Vif-Cullin5-ElonginBC complex is then able to ubiquitinate the APOBEC3G factor bound to Vif by its N-terminal domain [71].
VI. Interaction with the Genomic RNA
In infected cells, HIV-1 Vif co-localizes with the Pr55Gag precursor in membrane-free cytoplasmic complexes that were proposed to be intermediate assembly complexes [72]. On the other hand, Pr55Gag precursors associate with HIV-1 genomic RNA (gRNA) at a perinuclear/centrosomal site [73]. In addition, it was shown that Vif specifically binds HIV-1 gRNA in vitro, and that it binds this RNA in the cytoplasm of virus producing cells to form a 40S mRNP complex that contains neither the Pr55Gag precursor nor the mature nucleocapsid protein (NCp7) [26]. These results suggest that the HIV-1 gRNA may be engaged in different complexes. However, the same authors showed that in vitro, the affinity of Vif for the HIV-1 gRNA significantly decreased in the presence of Pr55Gag, while the affinity of the latter protein for HIV-1 RNA was not affected by Vif. Recently, we found that Vif preferentially and cooperatively binds to the 5′ region of the HIV-1 gRNA, and that this region contains very high affinity Vif binding sites [28,74]. Therefore, one can assume that some Vif proteins would remain associated to the gRNA even in the presence of Gag. Indeed, by studying the cell-free assembly of immature HIV-1 capsids, Lingappa and co-workers observed intermediate assembly complexes containing the Gag and Gag-Pol precursors, as well as Vif and a cellular factor, HP68, which is absolutely required to complete assembly [75]. In line with these results, it has been reported that incorporation of Vif into viral particles is mediated (at least in part) by interactions with the gRNA [27]. Interactions of Vif with Gag and Gag-Pol likely also contribute to this process [76].
A strong affinity RNA binding site was identified in the N-terminal region of Vif (amino acids 1–64), while the Cterminal region, which harbors many positively charged residues, only weakly contributed to RNA binding [26]. Within the N-terminal region of Vif, substitutions W11A, Y30A, and Y40A strongly reduced binding to both poly(G) and an RNA probe corresponding to nucleotides 5104 to 5287 of the HIV-1 genome. In addition, viruses harboring any of these substitutions were non-infectious in H9 T cells, suggesting that RNA binding is crucial for Vif function [26]. Moreover, viruses harboring mutations in the zinc finger domains of NCp7 that fail to package gRNA do not package Vif, and Vif is not encapsidated in a HIV-1 RNA-packaging mutant [27]. These authors also showed that deletions of the C-terminal domain or of the viral SOCS-box had no effect on Vif packaging. Surprisingly in light of the results of Zhang et al. mentioned above, Khan and co-workers found that deletion of residues 23 to 43 increased the efficiency of Vif packaging twofold [27]. On the other hand, deletion of amino acids 75 to 114 completely abolished packaging of Vif. However, as this deletion encompasses part of the zincbinding motif (Fig. 2b), this result may be the indirect consequence of a major conformational change of Vif (see section IV).
The main HIV-1 RNA binding protein is the mature nucleocapsid protein NCp7 (and the NC domain embedded in the Gag precursor), which is a potent RNA chaperone (For reviews see [77,78]). Recent studies pointed at significant differences and similarities between Vif and NCp7. Whereas the binding affinity of NCp7 for RNA strongly decreases as the salt concentration increases [77,79], the Vif/RNA complexes resist to high salt concentrations [26,28,74]. Indeed, at the physiological ionic conditions, Vif binds stronger to gRNA than NCp7 [28,74]. NCp7 binds to any nucleic acids with significant affinity, but it displays some preference for UG or TG-rich sequences and for GNG sequences within single stranded loops. The RNA and DNA sites displaying the highest affinity for Vif are single-stranded G-rich and Crich sequences [74] and poly(G) is the homopolymer that binds Vif with the highest affinity [26]. The determinants of specific binding of Vif to nucleic acids remain to be determined, but Vif binding seems to be more specific than NCp7 binding, as we identified 500 nucleotide RNA fragments that weakly bind Vif [28,74].
Interestingly, we recently found that Vif promotes annealing of tRNALys,3 to the primer binding site (PBS), favors dimerization of the HIV-1 gRNA, decreases pausing of reverse transcriptase and enhances the first strand transfer taking place during reverse transcription, indicating that Vif is a bona fide RNA chaperone [80]. Even though the chaperone activity of Vif is less pronounced than that of NCp7, Vif has dominant effects on NCp7. Indeed, Vif inhibits some, but not all, NCp7-mediated functions including tRNALys,3 annealing and conversion of the loose gRNA dimer into a tight dimer [80]. These inhibitory effects of Vif are likely relieved at the last stages of virion assembly, as only limited amounts of Vif are packaged [81–84], and thus Vif could act as a temporal regulator of NCp7 activity. Recent reports indicated that deamination is only one of the mechanisms contributing to the antiviral activity of APOBEC3G and APOBEC3F. Some authors reported that these proteins also inhibit NCp7- mediated tRNALys,3 annealing [85], and DNA strand transfers during reverse transcription [86], while others observed an effect on DNA elongation [87]. These results may explain the need for redundant RNA chaperone activities in cells expressing APOBEC3G and APOBEC3F.
CONCLUSIONS
In conclusion, Vif is a multidomain and multifunctional protein that therefore represent a viable target for both therapeutic and preventive interventions. However, there are currently no drugs against this protein in clinical trials. To date, its function and structural features have been fairly well investigated. However, there still exist numerous aspects that need to be resolved. A three-dimensional structure of Vif, alone if Vif is not intrinsically disordered, or in complex with cellular partner(s) would greatly help us to understand its behavior at the molecular level.
Acknowledgments
JCP and RM wish to thank Simon Henriet, Gaelle Mercenne, Serena Bernacchi, and Lucile Sinck who participated in the work performed in the Marquet’s Lab described in this review. The authors thank the ‘Agence Nationale de Recherche sur le SIDA’ (ANRS) and the CNRS for financial support in their respective laboratories.
ABBREVIATIONS
- APOBEC
APOlipoprotein B mRNA-Editing enzyme Catalytic polypeptide-like domain
- SOCS
Suppressor of cytokine signaling
- gRNA
Genomic RNA
- PDB
Protein Data Bank http//www.rcsb.org/pdb/home/home.do
- VHL
Von Hippel-Lindau tumor suppressor protein
References
- 1.Strebel K. HIV accessory genes Vif and Vpu. Adv Pharmacol. 2007;55:199–232. doi: 10.1016/S1054-3589(07)55006-4. [DOI] [PubMed] [Google Scholar]
- 2.Fan L, Peden K. Cell-free transmission of Vif mutants of HIV-1. Virology. 1992;190:19–29. doi: 10.1016/0042-6822(92)91188-z. [DOI] [PubMed] [Google Scholar]
- 3.Gabuzda DH, Lawrence K, Langhoff E, et al. Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J Virol. 1992;66:6489–6495. doi: 10.1128/jvi.66.11.6489-6495.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borman AM, Quillent C, Charneau P, Dauguet C, Clavel F. Human immunodeficiency virus type 1 Vif- mutant particles from restrictive cells: role of Vif in correct particle assembly and infectivity. J Virol. 1995;69:2058–2067. doi: 10.1128/jvi.69.4.2058-2067.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Courcoul M, Patience C, Rey F, et al. Peripheral blood mononuclear cells produce normal amounts of defective Vif- human immunodeficiency virus type 1 particles which are restricted for the preretrotranscription steps. J Virol. 1995;69:2068–2074. doi: 10.1128/jvi.69.4.2068-2074.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sova P, Volsky DJ. Efficiency of viral DNA synthesis during infection of permissive and non-permissive cells with vif-negative human immunodeficiency virus type 1. J Virol. 1993;67:6322–6326. doi: 10.1128/jvi.67.10.6322-6326.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Schwedler U, Song J, Aiken C, Trono D. Vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J Virol. 1993;67:4945–4955. doi: 10.1128/jvi.67.8.4945-4955.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 9.Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9:1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- 10.Holmes RK, Malim MH, Bishop KN. APOBEC-mediated viral restriction: not simply editing? Trends Biochem Sci. 2007;32:118–128. doi: 10.1016/j.tibs.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 11.Khan MA, Kao S, Miyagi E, et al. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J Virol. 2005;79:5870–5874. doi: 10.1128/JVI.79.9.5870-5874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soros VB, Yonemoto W, Greene WC. Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog. 2007;3:e15. doi: 10.1371/journal.ppat.0030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 2004;18:2867–2872. doi: 10.1101/gad.1250204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kao S, Khan MA, Miyagi E, et al. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J Virol. 2003;77:11398–11407. doi: 10.1128/JVI.77.21.11398-11407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mariani R, Chen D, Schrofelbauer B, et al. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114:21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- 16.Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- 17.Newman EN, Holmes RK, Craig HM, et al. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol. 2005;15:166–170. doi: 10.1016/j.cub.2004.12.068. [DOI] [PubMed] [Google Scholar]
- 18.Holmes RK, Koning FA, Bishop KN, Malim MH. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem. 2007;282:2587–2595. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- 19.Kawakami T, Sherman L, Dahlberg J, et al. Nucleotide sequence analysis of equine infectious anemia virus proviral DNA. Virology. 1987;158:300–312. doi: 10.1016/0042-6822(87)90202-9. [DOI] [PubMed] [Google Scholar]
- 20.Oberste MS, Gonda MA. Conservation of amino-acid sequence motifs in lentivirus Vif proteins. Virus Genes. 1992;6:95–102. doi: 10.1007/BF01703760. [DOI] [PubMed] [Google Scholar]
- 21.Tian C, Yu X, Zhang W, et al. Differential requirement for conserved tryptophans in human immunodeficiency virus type 1 Vif for the selective suppression of APOBEC3G and APOBEC3F. J Virol. 2006;80:3112–3115. doi: 10.1128/JVI.80.6.3112-3115.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujita M, Akari H, Sakurai A, et al. Expression of HIV-1 accessory protein Vif is controlled uniquely to be low and optimal by proteasome degradation. Microbes Infect. 2004;6:791–798. doi: 10.1016/j.micinf.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 23.Xiao Z, Ehrlich E, Yu Y, et al. Assembly of HIV-1 Vif-Cul5 E3 ubiquitin ligase through a novel zinc-binding domain-stabilized hydrophobic interface in Vif. Virology. 2006;349:290–299. doi: 10.1016/j.virol.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 24.Mehle A, Thomas ER, Rajendran KS, Gabuzda D. A zinc-binding region in Vif binds Cul5 and determines cullin selection. J Biol Chem. 2006;281:17259–17265. doi: 10.1074/jbc.M602413200. [DOI] [PubMed] [Google Scholar]
- 25.Dettenhofer M, Cen S, Carlson BA, Kleiman L, Yu XF. Association of human immunodeficiency virus type 1 Vif with RNA and its role in reverse transcription. J Virol. 2000;74:8938–8945. doi: 10.1128/jvi.74.19.8938-8945.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang H, Pomerantz RJ, Dornadula G, Sun Y. Human immunodeficiency virus type 1 Vif protein is an integral component of an mRNP complex of viral RNA and could be involved in the viral RNA folding and packaging process. J Virol. 2000;74:8252–8261. doi: 10.1128/jvi.74.18.8252-8261.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khan MA, Aberham C, Kao S, et al. Human immunodeficiency virus type 1 Vif protein is packaged into the nucleoprotein complex through an interaction with viral genomic RNA. J Virol. 2001;75:7252–7265. doi: 10.1128/JVI.75.16.7252-7265.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henriet S, Richer D, Bernacchi S, et al. Cooperative and specific binding of Vif to the 5′ region of HIV-1 genomic RNA. J Mol Biol. 2005;354:55–72. doi: 10.1016/j.jmb.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 29.Khan MA, Akari H, Kao S, et al. Intravirion processing of the human immunodeficiency virus type 1 Vif protein by the viral protease may be correlated with Vif function. J Virol. 2002;76:9112–9123. doi: 10.1128/JVI.76.18.9112-9123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hutoran M, Britan E, Baraz L, et al. Abrogation of Vif function by peptide derived from the N-terminal region of the human immunodeficiency virus type 1 (HIV-1) protease. Virology. 2004;330:261–270. doi: 10.1016/j.virol.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 31.Yang X, Goncalves J, Gabuzda D. Phosphorylation of Vif and its role in HIV-1 replication. J Biol Chem. 1996;271:10121–10129. doi: 10.1074/jbc.271.17.10121. [DOI] [PubMed] [Google Scholar]
- 32.Yang X, Gabuzda D. Mitogen-activated protein kinase phosphorylates and regulates the HIV-1 Vif protein. J Biol Chem. 1998;273:29879–29887. doi: 10.1074/jbc.273.45.29879. [DOI] [PubMed] [Google Scholar]
- 33.Yang S, Sun Y, Zhang H. The multimerization of human immunodeficiency virus type I Vif protein: a requirement for Vif function in the viral life cycle. J Biol Chem. 2001;276:4889–4893. doi: 10.1074/jbc.M004895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Auclair JR, Green KM, Shandilya S, et al. Mass spectrometry analysis of HIV-1 Vif reveals an increase in ordered structure upon oligomerization in regions necessary for viral infectivity. Proteins. 2007;69:270–284. doi: 10.1002/prot.21471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang B, Gao L, Li L, et al. Potent suppression of viral infectivity by the peptides that inhibit multimerization of human immunodeficiency virus type 1 (HIV-1) Vif proteins. J Biol Chem. 2003;278:6596–6602. doi: 10.1074/jbc.M210164200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller JH, Presnyak V, Smith HC. The dimerization domain of HIV-1 viral infectivity factor Vif is required to block APOBEC3G incorporation with virions. Retrovirology. 2007;4:81. doi: 10.1186/1742-4690-4-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balaji S, Kalpana R, Shapshak P. Paradigm development: comparative and predictive 3D modeling of HIV-1 Virion Infectivity Factor (Vif) Bioinformation. 2006;1:290–309. doi: 10.6026/97320630001290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lv W, Liu Z, Jin H, et al. Three-dimensional structure of HIV-1 VIF constructed by comparative modeling and the function characterization analyzed by molecular dynamics simulation. Org Biomol Chem. 2007;5:617–626. doi: 10.1039/b612050d. [DOI] [PubMed] [Google Scholar]
- 39.Liu B, Yu X, Luo K, Yu Y, Yu XF. Influence of primate lentiviral Vif and proteasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G. J Virol. 2004;78:2072–2081. doi: 10.1128/JVI.78.4.2072-2081.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu X, Yu Y, Liu B, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 41.Mehle A, Strack B, Ancuta P, et al. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004;279:7792–7798. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- 42.Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol. 2003;13:2009–2013. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 43.Bogerd HP, Doehle BP, Wiegand HL, Cullen BR. A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc Natl Acad Sci USA. 2004;101:3770–3774. doi: 10.1073/pnas.0307713101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mangeat B, Turelli P, Liao S, Trono D. A single amino acid determinant governs the species-specific sensitivity of APOBEC3G to Vif action. J Biol Chem. 2004;279:14481–14483. doi: 10.1074/jbc.C400060200. [DOI] [PubMed] [Google Scholar]
- 45.Schrofelbauer B, Chen D, Landau NR. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif) Proc Natl Acad Sci USA. 2004;101:3927–3932. doi: 10.1073/pnas.0307132101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu H, Svarovskaia ES, Barr R, et al. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc Natl Acad Sci USA. 2004;101:5652–5657. doi: 10.1073/pnas.0400830101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huthoff H, Malim MH. Identification of amino acid residues in APOBEC3G required for regulation by human immunodeficiency virus type 1 Vif and Virion encapsidation. J Virol. 2007;81:3807–3815. doi: 10.1128/JVI.02795-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J, Potash MJ, Volsky DJ. Functional domains of APOBEC3G required for antiviral activity. J Cell Biochem. 2004;92:560–572. doi: 10.1002/jcb.20082. [DOI] [PubMed] [Google Scholar]
- 49.Liu B, Sarkis PT, Luo K, Yu Y, Yu XF. Regulation of Apobec3F and human immunodeficiency virus type 1 Vif by Vif-Cul5- ElonB/C E3 ubiquitin ligase. J Virol. 2005;79:9579–9587. doi: 10.1128/JVI.79.15.9579-9587.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwatani Y, Takeuchi H, Strebel K, Levin JG. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J Virol. 2006;80:5992–6002. doi: 10.1128/JVI.02680-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gallois-Montbrun S, Kramer B, Swanson CM, et al. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J Virol. 2007;81:2165–2178. doi: 10.1128/JVI.02287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simon V, Zennou V, Murray D, et al. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV- 1 diversification. PLoS Pathog. 2005;1:e6. doi: 10.1371/journal.ppat.0010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santa-Marta M, Aires da Silva F, Fonseca A, Goncalves J. HIV-1 Vif can directly inhibit APOBEC3G-mediated cytidine deamination by using a single amino acid interaction and without protein degradation. J Biol Chem. 2005;280:8765–8775. doi: 10.1074/jbc.M409309200. [DOI] [PubMed] [Google Scholar]
- 54.Wichroski MJ, Ichiyama K, Rana TM. Analysis of HIV-1 viral infectivity factor-mediated proteasome-dependent depletion of APOBEC3G: correlating function and subcellular localization. J Biol Chem. 2005;280:8387–8396. doi: 10.1074/jbc.M408048200. [DOI] [PubMed] [Google Scholar]
- 55.Schrofelbauer B, Senger T, Manning G, Landau NR. Mutational alteration of human immunodeficiency virus type 1 Vif allows for functional interaction with nonhuman primate APOBEC3G. J Virol. 2006;80:5984–5991. doi: 10.1128/JVI.00388-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mehle A, Wilson H, Zhang C, et al. Identification of an APOBEC3G binding site in human immunodeficiency virus type 1 Vif and inhibitors of Vif-APOBEC3G binding. J Virol. 2007;81:13235–13241. doi: 10.1128/JVI.00204-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Russell RA, Pathak VK. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J Virol. 2007;81:8201–8210. doi: 10.1128/JVI.00395-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004;18:2861–2866. doi: 10.1101/gad.1249904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deshaies RJ. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol. 1999;15:435–467. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- 60.Pintard L, Willems A, Peter M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. EMBO J. 2004;23:1681–1687. doi: 10.1038/sj.emboj.7600186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiao Z, Xiong Y, Zhang W, et al. Characterization of a novel Cullin5 binding domain in HIV-1 Vif. J Mol Biol. 2007;373:541–550. doi: 10.1016/j.jmb.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 62.Luo K, Xiao Z, Ehrlich E, et al. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc Natl Acad Sci USA. 2005;102:11444–11449. doi: 10.1073/pnas.0502440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krishna SS, Majumdar I, Grishin NV. Structural classification of zinc fingers: survey and summary. Nucleic Acids Res. 2003;31:532–550. doi: 10.1093/nar/gkg161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Paul I, Cui J, Maynard EL. Zinc binding to the HCCH motif of HIV-1 virion infectivity factor induces a conformational change that mediates protein-protein interactions. Proc Natl Acad Sci USA. 2006;103:18475–18480. doi: 10.1073/pnas.0604150103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiao Z, Ehrlich E, Luo K, Xiong Y, Yu XF. Zinc chelation inhibits HIV Vif activity and liberates antiviral function of the cytidine deaminase APOBEC3G. FASEB J. 2007;21:217–222. doi: 10.1096/fj.06-6773com. [DOI] [PubMed] [Google Scholar]
- 66.Simon JH, Sheehy AM, Carpenter EA, Fouchier RA, Malim MH. Mutational analysis of the human immunodeficiency virus type 1 Vif protein. J Virol. 1999;73:2675–2681. doi: 10.1128/jvi.73.4.2675-2681.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamura T, Sato S, Haque D, et al. The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev. 1998;12:3872–3881. doi: 10.1101/gad.12.24.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang JG, Farley A, Nicholson SE, et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci USA. 1999;96:2071–2076. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kile BT, Schulman BA, Alexander WS, et al. The SOCS box: a tale of destruction and degradation. Trends Biochem Sci. 2002;27:235–241. doi: 10.1016/s0968-0004(02)02085-6. [DOI] [PubMed] [Google Scholar]
- 70.Stebbins CE, Kaelin WG, Jr, Pavletich NP. Structure of the VHLElonginC- ElonginB complex: implications for VHL tumor suppressor function. Science. 1999;284:455–461. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- 71.Kobayashi M, Takaori-Kondo A, Miyauchi Y, Iwai K, Uchiyama T. Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J Biol Chem. 2005;280:18573–18578. doi: 10.1074/jbc.C500082200. [DOI] [PubMed] [Google Scholar]
- 72.Simon JH, Carpenter EA, Fouchier RA, Malim MH. Vif and the p55(Gag) polyprotein of human immunodeficiency virus type 1 are present in colocalizing membrane-free cytoplasmic complexes. J Virol. 1999;73:2667–2674. doi: 10.1128/jvi.73.4.2667-2674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Poole E, Strappe P, Mok HP, Hicks R, Lever AM. HIV-1 Gag- RNA interaction occurs at a perinuclear/centrosomal site; analysis by confocal microscopy and FRET. Traffic. 2005;6:741–755. doi: 10.1111/j.1600-0854.2005.00312.x. [DOI] [PubMed] [Google Scholar]
- 74.Bernacchi S, Henriet S, Dumas P, Paillart JC, Marquet R. RNA and DNA binding properties of HIV-1 Vif protein: a fluorescence study. J Biol Chem. 2007;282:26361–26368. doi: 10.1074/jbc.M703122200. [DOI] [PubMed] [Google Scholar]
- 75.Zimmerman C, Klein KC, Kiser PK, et al. Identification of a host protein essential for assembly of immature HIV-1 capsids. Nature. 2002;415:88–92. doi: 10.1038/415088a. [DOI] [PubMed] [Google Scholar]
- 76.Bardy M, Gay B, Pebernard S, et al. Interaction of human immunodeficiency virus type 1 Vif with Gag and Gag-Pol precursors: coencapsidation and interference with viral protease-mediated Gag processing. J Gen Virol. 2001;82:2719–2733. doi: 10.1099/0022-1317-82-11-2719. [DOI] [PubMed] [Google Scholar]
- 77.Levin JG, Guo J, Rouzina I, Musier-Forsyth K. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog Nucleic Acid Res Mol Biol. 2005;80:217–286. doi: 10.1016/S0079-6603(05)80006-6. [DOI] [PubMed] [Google Scholar]
- 78.Tisne C. Structural bases of the annealing of primer tRNA(3Lys) to the HIV-1 viral RNA. Curr HIV Res. 2005;3:147–156. doi: 10.2174/1570162053506919. [DOI] [PubMed] [Google Scholar]
- 79.Barraud P, Gaudin C, Dardel F, Tisne C. New insights into the formation of HIV-1 reverse transcription initiation complex. Biochimie. 2007;89:1204–1210. doi: 10.1016/j.biochi.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 80.Henriet S, Sinck L, Bec G, et al. Vif is a RNA chaperone that could temporally regulate RNA dimerization and the early steps of HIV-1 reverse transcription. Nucleic Acids Res. 2007;35:5141–5153. doi: 10.1093/nar/gkm542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu H, Wu X, Newman M, et al. The Vif protein of human and simian immunodeficiency viruses is packaged into virions and associates with viral core structures. J Virol. 1995;69:7630–7638. doi: 10.1128/jvi.69.12.7630-7638.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Camaur D, Trono D. Characterization of human immunodeficiency virus type 1 Vif particle incorporation. J Virol. 1996;70:6106–6111. doi: 10.1128/jvi.70.9.6106-6111.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dettenhofer M, Yu XF. Highly purified human immunodeficiency virus type 1 reveals a virtual absence of Vif in virions. J Virol. 1999;73:1460–1467. doi: 10.1128/jvi.73.2.1460-1467.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kao S, Akari H, Khan MA, et al. Human immunodeficiency virus type 1 Vif is efficiently packaged into virions during productive but not chronic infection. J Virol. 2003;77:1131–1140. doi: 10.1128/JVI.77.2.1131-1140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guo F, Cen S, Niu M, et al. The interaction of APOBEC3G with human immunodeficiency virus type 1 nucleocapsid inhibits tRNA3Lys annealing to viral RNA. J Virol. 2007;81:11322–11331. doi: 10.1128/JVI.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li XY, Guo F, Zhang L, Kleiman L, Cen S. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J Biol Chem. 2007;282:32065–32074. doi: 10.1074/jbc.M703423200. [DOI] [PubMed] [Google Scholar]
- 87.Iwatani Y, Chan DS, Wang F, et al. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007 doi: 10.1093/nar/gkm750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jones DT. Protein secondary structure prediction based on positionspecific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]