

Abstract
Motivation: Genomic instability in cancer leads to abnormal genome copy number alterations (CNA) that are associated with the development and behavior of tumors. Advances in microarray technology have allowed for greater resolution in detection of DNA copy number changes (amplifications or deletions) across the genome. However, the increase in number of measured signals and accompanying noise from the array probes present a challenge in accurate and fast identification of breakpoints that define CNA. This article proposes a novel detection technique that exploits the use of piece wise constant (PWC) vectors to represent genome copy number and sparse Bayesian learning (SBL) to detect CNA breakpoints.
Methods: First, a compact linear algebra representation for the genome copy number is developed from normalized probe intensities. Second, SBL is applied and optimized to infer locations where copy number changes occur. Third, a backward elimination (BE) procedure is used to rank the inferred breakpoints; and a cut-off point can be efficiently adjusted in this procedure to control for the false discovery rate (FDR).
Results: The performance of our algorithm is evaluated using simulated and real genome datasets and compared to other existing techniques. Our approach achieves the highest accuracy and lowest FDR while improving computational speed by several orders of magnitude. The proposed algorithm has been developed into a free standing software application (GADA, Genome Alteration Detection Algorithm).
Availability: http://biron.usc.edu/~piquereg/GADA
Contact: jpei@chop.swmed.edu and rpique@ieee.org
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Copy number alterations (CNA) involving deletion or replication of entire chromosomes or chromosomal regions are known to occur in numerous genetic disorders (i.e. Down's syndrome, Klinefelter's syndrome), while replications of multiple chromosomes leading to states of hyperploidy are well known in cancer biology (Albertson et al., 2003). Similarly, regional CNA have been demonstrated in tumors, and linked to leading them to develop aggressive behavior. Examples include loss of RB tumor suppressor in retinoblastoma or MYCN proto-oncogene amplification in neuroblastoma. Recently, large numbers of polymorphic CNA have also been described in the human genome (Redon et al., 2006). Array-based technologies use genetic material as sensors or probes to estimate copy number for the intended genomic regions. The resolution for detection of CNA depends on the number and type of probes placed on these arrays. Comparative genomic hybridization (CGH, Kallioniemi et al. 1992) is one of the earlier array platforms that uses large insert DNA fragments (kilobases) as probes in measuring DNA copy number. These probes, numbering typically in thousands, allow co-hybridization to take place between a fluorescently tagged genome of interest and a normal reference genome. The relative intensity at a given probe is directly proportional to the copy number for that region. More recently, platforms using short oligonucleotide probes (≤60 bases), which allow placement of hundreds of thousands of probes on an array, have become more widely used (Pollack et al., 1999) The majority of these arrays use oligonucleotides that also probe for regions with genotype polymorphisms thus providing both copy number and genotype information (Huang et al., 2004; Peiffer et al., 2006). The increase in the probe density poses computational challenges to accurately and efficiently assess DNA copy number and identify altered regions.
Several algorithms have been proposed to detect CNA (Broet and Richardson, 2006; Fridlyand et al., 2004; Huang et al., 2004; Huang et al., 2005; Hsu et al., 2005; Lipson et al., 2006; Marioni et al., 2006; Nannya et al., 2005; Olshen et al., 2004; Picard et al., 2005; Pollack et al., 1999; Zhao et al., 2004). Most of these algorithms rely on a fundamental characteristic, namely, that a genome is composed of relatively long segments, DNA sequences, that have a constant number of copies present. The genomic segments can be represented by m probes mapping to a specific position on the genome having cm copies. The copy numbers cm can be ordered and arranged as vectors c that have two key characteristics; (i) they are piecewise constant (PWC) with very small number of breakpoints relative to the number of probes; and, (ii) they have discrete values (DIS) (i.e. copy numbers can only be 0, 1, 2, 3, …). However, these properties cannot be directly observed in the log-intensities ym measured with microarrays, due to contamination by biological and technical noise; thus a widely used model is:
| (1) |
where xm represents the average log intensity, and εm is an additive zero-mean white random process (Fig. 1).
Fig. 1.

Graphical representation of the observation model 1 using a chromosome section with two alterations as an example (simulated data). The underlying mean hybridization intensity xm is piece wise constant (PWC) with breakpoints 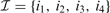 that mark the starting probe of each segment, and amplitudes a = (a0,a1,a2,a3,a4) that depend on the underlying number of copies (DIS). The observed probe hybridization intensities ym do not follow this expected behavior due to degradation by hybridization noise εm.
that mark the starting probe of each segment, and amplitudes a = (a0,a1,a2,a3,a4) that depend on the underlying number of copies (DIS). The observed probe hybridization intensities ym do not follow this expected behavior due to degradation by hybridization noise εm.
Most techniques exploit the assumption that xm ∝ log2(cm) and that properties PWC and DIS, as introduced above, are met. For example, one of the first and simplest techniques to exploit PWC consisted of applying a smoothing filter followed by a threshold (Huang et al., 2004; Pollack et al., 1999). This has been improved upon by more specialized techniques such as wavelets (Hsu et al., 2005), segmentation (Lipson et al., 2006; Olshen et al., 2004; Picard et al., 2005) or penalized least-squares (Huang et al., 2005). Additionally, hidden Markov models (HMM) (Fridlyand et al., 2004; Marioni et al., 2006; Nannya et al., 2005; Zhao et al., 2004) and Bayesian methods (Broet and Richardson, 2006) exploit both PWC and DIS by assuming that each observation ym comes from a probe in a particular hidden copy number state cm to be inferred. Exploiting DIS can be difficult in the case of specimens containing a heterogeneous population of cells with respect to DNA copy numbers, which typically occurs in the case of tumor samples, where xm=log2(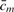 ) would correspond to the average copy number in the mixture.
) would correspond to the average copy number in the mixture.
Among all the previous methods, circular binary segmentation (CBS) by Olshen et al. 2004 was found one of the most accurate methods for CNA detection by two independent comparative studies (Lai et al., 2005; Willenbrock and Fridlyand, 2005) but was also one of the slowest. These studies used synthetic datasets where the CNA occur at known positions, the probes are uniformly spaced, and the hybridization noise is generated according to a white Gaussian distribution. More recently, new approaches (Engler et al., 2006; Rueda and Diaz-Uriarte, 2007; Shah et al., 2006) have extended previously proposed methods in order to target specific scenarios not considered by the CBS approach, e.g. presence of outliers (Shah et al., 2006), non-uniform probe spacing (Rueda and Diaz-Uriarte, 2007) and chromosomes with a reduced number of probes and non-uniform variance (Engler et al., 2006). In this article we focus on the default conditions and metrics proposed by Willenbrock and Fridlyand (2005) under which our results show that these new algorithms do not give better accuracy than that of CBS. The performance of these algorithms under different conditions that may arise on specific microarray platforms should be investigated in future work. Recently, the computational performance of CBS algorithm has significantly improved with a new approximate version (Venkatraman and Olshen, 2007) with no significant loss of performance. However, the run-times of this new version and the other new algorithms are still very high, especially when applied to the new high density array platforms.
In this article, we propose a novel modeling of genomic data using PWC vectors that can be efficiently exploited to build algorithms for CNA detection with a very significant gain in computational speed. We also propose a new approach that we called GADA (Genome Alteration Detection Algorithm) for CNA detection from array data that combines the sparse Bayesian learning (SBL) technique introduced by Tipping (2001) and a backward elimination (BE) procedure that can efficiently adjust the accuracy trade-off between sensitivity and the FDR.
We evaluated our algorithm using the simulated array-CGH dataset proposed by Willenbrock and Fridlyand (2005), where the underlying positions of copy number changes are known and can be used as a benchmark to compare algorithms accuracies. We also evaluated the performance of three algorithms (Engler et al., 2006; Rueda and Diaz-Uriarte, 2007; Shah et al., 2006) that appeared after the Willenbrock and Fridlyand (2005) comparative study, and the newer CBS implementation (Venkatraman and Olshen, 2007). Using that benchmark dataset our GADA approach obtained one of the best accuracies, and the best performance in terms of computational speed, followed by CBS. Additionally we compared the results of our algorithm and CBS on data generated from several array types from two commercial manufacturers (Affymetrix and Illumina) using DNA from four different neuroblastoma cell lines. Our results indicate that our algorithm can analyze data efficiently from high density platforms and provide an accuracy similar or better than that of state of the art algorithms, but with reduced computation costs. On the new large array platforms, our algorithm is two orders of magnitude faster than CBS (Olshen et al., 2004).
2 MATERIALS AND METHODS
For all analyses, we employed a 2.8 Ghz Pentium Processor. The SBL algorithm has been implemented in C and is called from Matlab version 7.0 (Mathworks, Natick, MA). The SBL algorithm is also implemented as a software package named GADA available at (http://biron.usc.edu/~piquereg/GADA). For comparison analysis, we used the latest implementation of CBS (Venkatraman and Olshen, 2007) developed in Fortran (available in R from the Bioconductor package DNAcopy).
2.1 Neuroblastoma genomic data from array platforms
Four neuroblastoma cell lines, two with known MYCN oncogene amplification (SK-N-BE2, SMS-KAN) and two lacking MYCN amplification (LAN-6, CHLA-20) were grown in RPMI medium with 10% FCS to confluence prior to extraction of DNA using STAT60 (Tel-Test, Inc.). The same stock of DNA was used to perform whole genome analysis for CNA using Affymetrix SNP arrays 50K Xba, 250K Sty, and 250K Nsp and Illumina GoldenGate 550K SNP array based on their respective protocols. The raw data obtained from the Affymetrix platform arrays were normalized using routines employed in Copy Number Analysis Tool version 3.0 in which log2ratios of the intensity of the probes were calculated after fitting a regression model generated from a normal set of diploid samples. The Illumina platform data were normalized and summarized using the BeadStudio Genotype analysis software and the log-R-ratio data were exported for further analyses. Data from 60 NCI cell lines generated using Affymetrix 50K Hind and 50K Xba (Garraway et al., 2005) were also used to assess the computational speed of the algorithm (GEO accession: GSE2520).
2.2 Simulated CGH data
The datasets used to compare the algorithms' rates of accuracy (sensitivity and FDR) are those proposed by Willenbrock and Fridlyand (2005). To further assess these metrics in CNA occurring in genomes with differing complexities, we generated six additional simulated datasets containing 200 genomes each with 20 chromosomes. All datasets were generated in Matlab forming chromosomes of length 200 probes and sampling the CNA from the same empirical distribution used by Willenbrock and Fridlyand (2005), but were categorized by the number and length of CNA. These categories include: (1) no breakpoints, (2) only one breakpoint at any position uniformly distributed, (3-6) are generated as in Willenbrock and Fridlyand (2005) but categorized by the number of breakpoints and the length of the altered segments. Chromosomes with few (many) breakpoints have [2–4] ([5–10]) breakpoints. Large (small) alterations are generated by sampling the altered segments with lengths within the range [10–150] ([1–9]).
Table 1 shows definitions of the accuracy metrics used in the analyses of simulated data. These include sensitivity (expected recall) and FDR (1-expected precision) in locating copy number changes. A breakpoint is claimed to have been detected correctly only if it is placed within a distance of δ probes from the true breakpoint. In evaluating the performance of the algorithms, an algorithm was indicated to perform better if (1) the algorithm's FDR was smaller with same sensitivity, or (2) if its sensitivity was higher with same FDR, or (3) if both the FDR was lower and the sensitivity higher compared to the other algorithm. All other cases were considered uninformative (e.g. similar FDR and sensitivity or discordant FDR and sensitivity). For each sample in a given simulated dataset, the performance (FDR and sensitivity) of the algorithms was measured. The proportion of times that an algorithm performed better was obtained using only the informative cases. The two-sample test for binomial proportions (or McNemar's test) was used then to assess differences in the performance of the algorithms.
Table 1.
Possible outcomes for each candidate breakpoint position
| Breakpoint | Not detected | Detected |
|---|---|---|
| Present | FN | TP |
| Not present | TN | FP |
| Performance metrics: | ||
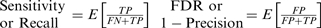 | ||
A True Positive (TP) only occurs if the breakpoint that has been detected is within a distance of δ probes form a true breakpoint. If there are more than one breakpoint detected within this vicinity, only the closest one is considered TP and the remainders are False Positives (FP). The true breakpoint positions that are not detected are False Negatives (FN). The regions without a breakpoint where no breakpoints have been detected are True Negatives (TN). M is the number of candidate breakpoints (i.e. number of probes = TP+FP+TN+FN). The number of breakpoints falling in each of these categories are random numbers obtained on each simulated sample; thus expected values can be obtained for False Discovery Rate (FDR = 1− Precision) and Sensitivity (Recall) by taking the average over all the simulated samples.
Concordance between algorithms was measured as 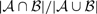 (Kosko, 2004); where
(Kosko, 2004); where 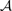 and
and 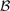 are the breakpoint sets returned by each algorithm. Breakpoints belong to the intersection (i.e. are considered to be the same), if they are separated by less than δ = 2 probes.
are the breakpoint sets returned by each algorithm. Breakpoints belong to the intersection (i.e. are considered to be the same), if they are separated by less than δ = 2 probes.
2.3 PWC vectors representation of Genomic data
Our first major contribution is the development of a compact description for the copy number along the chromosome using PWC vectors (green signal in Fig. 1). Using simple linear algebra, any PWC vector x with K breakpoints 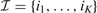 can be compactly represented by a linear combination of K step vectors fi (each with a single breakpoint i in
can be compactly represented by a linear combination of K step vectors fi (each with a single breakpoint i in 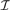 , see Fig. 2) plus a constant vector f0.
, see Fig. 2) plus a constant vector f0.
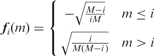 |
(2) |
| (3) |
Fig. 2.

Step vector fi with a breakpoint between probe i and i + 1 as defined in Equation (2). Notice that the step vectors have been normalized to have unit norm, 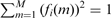 , and average zero for i > 0,
, and average zero for i > 0, 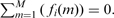
Therefore, in matrix notation we can write this linear combination as:
| (4) |
where the columns of F are the step functions (F=[ f0, f1,…, fM−1]); and, w is a sparse vector, i.e. there are only K + 1 non-zero components. Equivalently, we can remove the components of w that are zero and write:
| (5) |
where 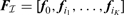 and
and 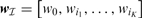 . This representation has three very important properties that are rigorously proved in Supplementary Section 1. First, the columns of F form a basis that can be used to represent any arbitrary vector. Second, it has a nested structure, and for each additional breakpoint i that the PWC vector may contain, we only require an additional weight wi to be non-zero. Third, any arbitrary PWC vector with exactly K breakpoints can be represented with K + 1 non-zero components which is proved to be the minimum possible amount; i.e. maximal sparseness.
. This representation has three very important properties that are rigorously proved in Supplementary Section 1. First, the columns of F form a basis that can be used to represent any arbitrary vector. Second, it has a nested structure, and for each additional breakpoint i that the PWC vector may contain, we only require an additional weight wi to be non-zero. Third, any arbitrary PWC vector with exactly K breakpoints can be represented with K + 1 non-zero components which is proved to be the minimum possible amount; i.e. maximal sparseness.
To the best of our knowledge, we are the first to explicitly propose this representation in the context of genome copy number variations (Pique-Regi et al., 2007) and to exploit its properties to develop a highly accurate and efficient detection technique that will be detailed in the following sections.
2.4 Formulation of breakpoint detection problem
The compact representation developed in the previous section can be used to facilitate estimating x from a degraded observation y generated as in model (1):
| (6) |
where x has been replaced by its representation in terms of the basis vectors, Fw. Since the number of copy number changes is very small compared to the number of probes, K ≪ M, then x = Fw has a sparse representation in the F basis, while the noise ε is not sparse in this representation. Under this scenario, the problem is formulated as that of finding ◯ = Fŵ that is closest to the observed y subject to having only K non-zero components of ŵ.
| (7) |
Different measures of closeness e(.) and sparseness s(.) can be used. For closeness, we will use the least squares error measure in this article, since it is the most widely used for approximation and will facilitate comparison among algorithms, although it may be sensitive to outliers. For measuring sparseness we are especially interested in the l0 norm (i.e. the number of wm ≠ 0), which best models the biological property that K ≪ M without imposing any restriction on the specific values of wm.
Then, the optimization with these measures can be rewritten as follows:
| (8) |
where the lp norm and the l0 pseudo-norm are defined as:
| (9) |
and with λ > 0 as a trade-off parameter between goodness of fit and sparseness.
Finding a solution for the problem of (8) would require solving 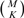 least squares problems. This approach is intractable for chromosome lengths M and number of discontinuities K that are typical for our application. There exist several popular sub-optimal approaches (Chen et al., 1998; Hastie et al., 2001; Mallat and Zhang, 1993; Seber and Lee, 2003; Patil et al., 1993) that use a greedy search strategy or that replace the l0 by an l1. However, as discussed in Supplementary Section 2, the performance of these methods is severely limited by the high collinearity (lack of orthogonality) between the columns of F (Donoho et al., 2006), as compared to sparse Bayesian learning (see next Section) for the specific application of CNA detection (Pique-Regi et al., 2007).
least squares problems. This approach is intractable for chromosome lengths M and number of discontinuities K that are typical for our application. There exist several popular sub-optimal approaches (Chen et al., 1998; Hastie et al., 2001; Mallat and Zhang, 1993; Seber and Lee, 2003; Patil et al., 1993) that use a greedy search strategy or that replace the l0 by an l1. However, as discussed in Supplementary Section 2, the performance of these methods is severely limited by the high collinearity (lack of orthogonality) between the columns of F (Donoho et al., 2006), as compared to sparse Bayesian learning (see next Section) for the specific application of CNA detection (Pique-Regi et al., 2007).
2.5 Sparse Bayesian learning (SBL)
The optimization problem defined in Equation (8) can be formulated from a Bayesian estimation point of view, as was done by Wipf and Rao (2004), for the case where F is an arbitrary matrix, and solved using SBL (Tipping, 2001), an empirical Bayes approach. Following Wipf and Rao (2004), the problem in Equation (8) can be cast as a maximum a posteriori (MAP) estimate:
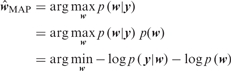 |
(10) |
where the observation model p (y|w) specifies the goodness of fit measure e(.) and the prior distribution for the weights p (w) specifies the sparseness measure s(.) in Equation (7).
In SBL (Tipping, 2001), the observation model is assumed normal (leading to a mean square error as a measure of fit)
| (11) |
and the prior distribution for the weights is specified as a hierarchical prior:
| (12) |
where α is a vector of hyperparameters that are distributed according to a gamma distribution:
| (13) |
This prior has several useful features. First, given the hyperparameters α, the conditional posterior weight distribution (14) is normal:
| (14) |
and following Tipping (2001), p(w|y) can be correctly approximated by point estimates as 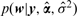 ; thus, the MAP is given by the posterior mean ŵ = μ, (replacing σ, and α by their point estimates, i.e. an empirical Bayes approach):
; thus, the MAP is given by the posterior mean ŵ = μ, (replacing σ, and α by their point estimates, i.e. an empirical Bayes approach):
| (15) |
Second, by treating the weights w as hidden variables, the maximum likelihood estimation for the hyperparameters α can be obtained by the EM algorithm McLachlan and Krishnan (1997); for each step l until convergence:
| (16) |
| (17) |
Finally, although this hierarchical prior does not appear to encourage sparseness, it has been demonstrated that indeed it has very good sparseness properties (Tipping, 2001; Wipf and Rao, 2004). This behavior can be unveiled by finding the marginal ‘effective’ prior, p(w), which is is an i.i.d. t-distribution with 2a degrees of freedom and a scale parameter of 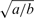 (see Supplementary Section 3.1). When b → 0 and a is small, this distribution peaks very sharply at zero, and has very thick flat tails that decay at (1+2a) rate in log-scale:
(see Supplementary Section 3.1). When b → 0 and a is small, this distribution peaks very sharply at zero, and has very thick flat tails that decay at (1+2a) rate in log-scale:
| (18) |
Thus, as shown in Figure 3, with this prior we obtain a sparseness cost that more closely approximates the desired l0 norm. In other words, this prior forces a very large number of weights to be zero while the non-zero weights are free to take any value (in Figure 3 the sparseness penalty is almost constant for any r > 0), which matches well our underlying biological knowledge for copy number changes.
Fig. 3.

SBL and l1 sparseness metrics compared to the desired l0 norm (dotted line). Each curve represents the sparseness metric for an arbitrary vector w with only K = 1, … , 5 non-zero coefficients at any position. All the non-zero weights are given the same magnitude r for different values of r on the x axis Ideally, we would like the sparseness metric to be inversely proportional to the the l0 norm, which will be equal to the number of non-zero components (K) regardless of the value of the components themselves (i.e. r). Note that the SBL metric approximates better the l0 norm, while l1 norm deviates significantly from this ideal behavior.
Although, the model contains several hyperparameters, the α and σ parameters are estimated from the data while b is set to zero (uninformative prior). Thus, sparseness is adjusted solely by the a parameter (Section 2.6 and Supplementary Section 3.1)
2.6 Implementation of SBL to find copy number alterations
To the best of our knowledge this is the first time that SBL has been employed to find copy number alterations, and more specifically with the PWC representation that we propose, where F has a very special structure. One of our contributions (Pique-Regi et al., 2007) is the observation that SBL can function well in our situation where significant collinearity exists, unlike other standard methods in Supplementary Section 2.
Additionally, SBL computational performance can be optimized for our PWC representation by exploiting the nested structure property. Direct computation of Equations (15) and (17) for an arbitrary F would require O(M3) operations (Tipping, 2001; Wipf and Rao, 2004). However, for our particular F in (2), 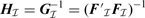 is, for all possible
is, for all possible 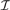 , a symmetric tridiagonal matrix, with main diagonal
, a symmetric tridiagonal matrix, with main diagonal
| (19) |
and upper/lower diagonal
| (20) |
This structure can be used to efficiently compute Σmm and μm for each EM step (16) in O(M) steps (see 9–14 in Algorithm 1).
Additional computational savings are achieved through removal of columns of F that correspond to the breakpoints whose weights w are very likely to become zero (lines 15–19 in Algorithm 1). This approach was used by Tipping (2001) for the general F case, but, when combined with the tridiagonal structure exploited here, each EM step is solved more rapidly; complexity is 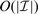 , so that the speed increases as the number of remaining breakpoints
, so that the speed increases as the number of remaining breakpoints 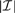 decreases.
decreases.
In our implementation, σ2 is estimated from the data. The parameter σ2 in the previous work (Tipping, 2001; Wipf and Rao, 2004) is usually jointly estimated by the EM algorithm. However, since each chromosome in the genome is analyzed independently, and σ2 is assumed to be the same for all chromosomes, it is more robust to estimate σ2 for the entire genome before applying the EM algorithm in each chromosome. In this article, σ2 is estimated as
| (21) |
in which the difference ym − ym − 1 removes the baseline PWC component and is distributed as 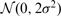 except for the breakpoints, which can be removed in the sum by replacing the mean by a trimmed mean. Similar estimates have also been widely employed in signal denoising approaches (Dragotti and Vetterli, 2002).
except for the breakpoints, which can be removed in the sum by replacing the mean by a trimmed mean. Similar estimates have also been widely employed in signal denoising approaches (Dragotti and Vetterli, 2002).

Finally, the EM algorithm is guaranteed to improve the solution after each step and will always converge (Wipf and Rao, 2004), but it may converge to a local minimum instead of the global minimum. However, these local minima are indeed always sparse (see Theorem 2 in Wipf and Rao 2004). The degree of sparseness in the SBL algorithm is controlled by the parameter a, as can be seen from Equation (18) and Supplementary Figure 8, whereby an increase in a causes a sharper peak at zero with faster tail decay and leads to a sparser solution. The a parameter also controls the convergence rate of the EM algorithm, with larger a leading to faster convergence. However, larger values of a are not always desirable and lead to suboptimal placement of breakpoints because of rapid convergence of the EM algorithm to a local minimum. The EM local minimum problem can be corrected by checking the statistical evidence for each breakpoint after obtaining a set of breakpoints at an appropriate a level. The statistical significance test can be performed by a backward elimination procedure described next section, which also allows more flexibility in setting the final desired degree of sparseness.
2.7 Breakpoint ranking by Backward elimination
Not all breakpoints found by SBL have the same statistical significance since noise may make areas without any underlying alteration appear similar to those areas corresponding to actual alterations. Some breakpoints mark the separation between two long segments (i.e. such that each segment includes many probes) and are such that the difference between the estimated amplitudes of the two segments is large. Such breakpoints are more likely to correspond to true underlying changes in copy number, and therefore will have a higher statistical score 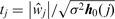 (see Supplementary Section 4). This score depends on the two contiguous breakpoints, and thus significance scores will change every time a breakpoint is removed (i.e. two segments are merged).
(see Supplementary Section 4). This score depends on the two contiguous breakpoints, and thus significance scores will change every time a breakpoint is removed (i.e. two segments are merged).

Instead of testing all the possible breakpoint combinations (i.e. segmentations), we have adopted a sub-optimal backward elimination (BE) strategy, in which we recursively eliminate the breakpoint with lowest statistical evidence tj. Although the procedure is suboptimal, since we may eliminate breakpoints that would be more significant in a later stage, it is generally seen as much less sensitive than forward selection (Kohavi and John, 1997). The BE procedure can be stopped when all the remaining breakpoints have tj higher than a specified T, the BE critical value. Moreover, with 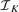 being the breakpoint set obtained from SBL, the procedure creates a sequence of nested subsets
being the breakpoint set obtained from SBL, the procedure creates a sequence of nested subsets 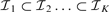 , which are obtained backwards, and such that successive subsets differ only in one discontinuity: this directly provides a breakpoint ranking. This ranking r is obtained efficiently by Algorithm 2 in
, which are obtained backwards, and such that successive subsets differ only in one discontinuity: this directly provides a breakpoint ranking. This ranking r is obtained efficiently by Algorithm 2 in 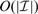 , where we exploit the fact that removing one discontinuity at a time only affects the two neighboring breakpoints (lines 9 and 12).
, where we exploit the fact that removing one discontinuity at a time only affects the two neighboring breakpoints (lines 9 and 12).
Therefore, with the ranking of breakpoints r, we can adjust the final breakpoint list to any critical value of T with no additional computational cost. This provides great flexibility in adjusting the final breakpoint set. The expected false discovery rate (FDR) is monotonically decreasing with T, thus we can obtain a list of breakpoints with lower FDR by increasing threshold T (see Supplementary Section 4.1).
2.8 GADA approach to CNA detection
The final proposed method to detect CNA, which we call GADA, is a two step approach. First, we apply SBL, which will provide a set of breakpoints with a specified initial level of sparseness controlled by the prior hyperparameter a. Then, the second step ranks the breakpoints provided by SBL by using a BE procedure, where the critical value T is used to establish the final degree of desired sparseness. The combination of these two approaches provides greater accuracy and flexibility.
First, it provides greater accuracy because each step minimizes the impact of the assumptions made by the other. SBL provides a better search strategy because effective removal of breakpoints is accomplished in several EM iterations. However, the breakpoint set detected by SBL may still include some spurious breakpoints (see Section 2.6). These ‘false’ breakpoints are then removed using the BE procedure (Section 2.7). The BE approach is greedy and fast, and it benefits from starting from a smaller set of breakpoints provided by the SBL, since fewer errors will accumulate with a smaller set (Supplementary Section 4).
Second, it provides greater flexibility in adjusting the final breakpoint set. Both a and T can adjust sparseness in an equivalent way. We have shown that breakpoints obtained with higher sparseness settings in SBL (i.e. larger a values) tend to be subsets of those obtained with lower sparseness settings when evaluated using the same T value in BE (Supplementary Section 6.1). Moreover, adjusting T can be done at no additional computational cost. Thus, SBL will be used with a small a, that gives a high initial sensitivity, and BE adjusts the final level of FDR.
The foreseeable usage by a practitioner of the GADA approach in detecting CNA would start analyzing a large collection of microarray samples with a small initial a. This a can be obtained by analyzing a small subset of samples and/or chromosomes. However, we have found by analyzing simulated and real datasets on platforms ranging from 50 K to 550 K probes that a = 0.2 is small enough to give the necessary initial level of sensitivity (see Supplementary Section 6). Following analysis of samples with SBL, the user can adjust T to obtain the final breakpoint set. A significance value α = P (|t| > T|w = 0) can be computed if the array noise is considered normal (t ∼ N(0,1)), or estimated using a resampling procedure. Any of the procedures that are typically used to control for FDR are not recommended for adjusting T because they do not take into account the dependence structure among the breakpoints. However, if replicate samples are available, the FDR can be estimated at a given T.
Finally, the SBL and BE procedures provide a segmentation, i.e. the representation of the data in a set of segments defined by their amplitudes and breakpoint positions. As in other segmentation procedures like DNAcopy (Olshen et al., 2004) and CGHseg (Picard et al., 2005) an additional step is required to classify the different segments amplitudes into a copy number (0, 1, 2, 3, 4, …) or alteration status (Non-Altered, Gain and Loss). There already exist several thresholding approaches (Huang et al., 2004; Pollack et al., 1999) and the MergeLevels approach (Willenbrock and Fridlyand, 2005) that can be used to accomplish this task (see Supplementary Section 5)
3 EXPERIMENTAL RESULTS
3.1 Performance comparisons in simulation dataset
We evaluated the performance of the proposed algorithm and compared the results with other published algorithms that are publicly available; including CBS (Olshen et al., 2004; Venkatraman and Olshen, 2007), SWARRAY (Komura et al., 2006), HMM (Fridlyand et al., 2004), RHMM (Shah et al., 2006), PL (Engler et al., 2006), RJaCGH (Rueda and Diaz-Uriarte, 2007) and GLAD (Hupe et al., 2004). We employed a simulated array-CGH dataset introduced by Willenbrock and Fridlyand (2005) with known CNA positions, where the accuracy in detecting breakpoints was measured in terms of sensitivity (expected recall) and FDR (1- expected precision) as defined in Section 2.2.
The performance in terms of accuracy for all the analyzed algorithms (using the default parameters) is reported in Figure 4. Three of the methods, CBS, HMM and GLAD were previously analyzed by Willenbrock and Fridlyand (2005) and results are identical to those reported previously. The faster new CBS (Venkatraman and Olshen, 2007) was also evaluated with results matching those from the previous implementation (Olshen et al., 2004). For RJaCGH, due to the long computational running time of the algorithm (> 1 day), the segmentation results were obtained directly from the authors and then evaluated with the metrics employed in this article. GADA, CBS, RJaCGH and RHMM are the most accurate algorithms either in terms of sensitivity or FDR; while the remaining algorithms clearly show poorer accuracy in both metrics. Among these top four algorithms, considering the times required to analyze the entire dataset, GADA (48 s) is fastest, followed by CBS (625 s), RHMM (41 min) and RJaCGH (> 1 day).
Fig. 4.

Median sensitivity and FDR for detecting known copy number changes within a probe window of δ length (δ = 0 − 3). The results are obtained using the default parameters settings in each algorithm (in GADA this is a = 0.2 and T = 4). The median and the interquartile range (IQR) are taken across the 500 samples.
In Figure 5, the parameters that control the trade-off between sensitivity and FDR are adjusted in GADA and CBS to generate the precision versus recall operation curves (PROC). The single operating points generated by RJaCGH and RHMM algorithms (using their default parameters) are also shown for comparison. The results show no significant differences in performance among these four algorithms. The GADA results presented in this section are also not sensitive to different choices of the a parameter. Supplementary Figure 10 shows that essentially the same results as in Figure 5 are obtained for a range of a parameters. As discussed in Section 2.8, GADA is a two step procedure controlled by two parameters a and T. Setting a higher a simply makes the PROC curve shorter (i.e. it start further to the left and to the bottom) since all the breakpoints that would be removed by BE are instead eliminated in the SBL step. It should also be noted that RJaCGH, RHMM and PL, are reported to have a better accuracy than CBS in situations different than the ones evaluated by the employed dataset, which may include: non-uniform probe spacing, chromosomes with a reduced number of probes, non-uniform variance, and presence of outliers. Future research should study the impact of these situations on GADA performance, as well possible extensions to GADA in order to handle them.
Fig. 5.

PROC operational curves for the mean sensitivity versus false discovery rate in detecting real copy number changes within a δ = 2 probe length window in the dataset introduced by Willenbrock and Fridlyand (2005) (averages taken across the 500 samples). RJaCGH and RHMM results are obtained using the default parameter settings and provide a single point. CBS operation points are obtained by varying the α, while GADA operating points were obtained by varying the T parameters with the default a = 0.2.
In what follows we focus on comparing GADA to CBS, the baseline algorithm that most of the recent approaches use for comparison. The newer algorithms are not included in this analysis as they do not show significant improvements over CBS using the standard evaluation methods designed by Willenbrock and Fridlyand (2005) and have considerably slower running-times.
The simulated dataset used by Willenbrock and Fridlyand (2005) represents a mixture of simulated genomes with respect to the number of breakpoints and size of the CNA. We observed that the majority of the simulated genomes have few breakpoints with large altered regions (data not shown). To further assess the performance of GADA and CBS on genomes with complex patterns of CNA, typical of those observed in tumors, we generated six additional simulated datasets. These datasets contained varying complexity of CNA and were derived using the same procedure proposed by Willenbrock and Fridlyand (2005) (see Section 2 for details). The datasets included both ‘quiet’ genomes (0–1 breakpoint) and complex genomes involving few or multiple breakpoints resulting in small or large CNA regions. The performances of GADA and CBS on these six datasets are provided in Table 2. Both algorithms work well for finding a small number of discontinuities within large segments, but there is significant evidence of advantage of GADA over CBS for the more complex cases. However, the magnitude of the overall differences in sensitivity and FDR between GADA and CBS are relatively small (< 3%); and the main advantage of our approach is in its flexibility and computational speed when analyzing large density arrays.
Table 2.
Sensitivity and FDR dependence on the number of breakpoints and segment length
| Num. of breaks | Segment length | FDR % | Sensitivity % | P -value | ||||
|---|---|---|---|---|---|---|---|---|
| GADA | CBS | (< , >) | GADA | CBS | (> , <) | |||
| 0 | — | 0.00 | 0.00 | (19,54) | — | — | — | — |
| 1 | Any | 5.00 | 5.00 | (76,62) | 95.00 | 95.00 | (43,41) | (74,52) 0.025 |
| Few | Large | 4.04 | 5.61 | (91,70) | 95.96 | 97.83 | (24,95) | (60,69) 0.21 |
| Few | Small | 3.85 | 3.48 | (84,77) | 80.39 | 77.78 | (129,30) | (94,34) 6E−8 |
| Many | Large | 2.97 | 5.56 | (162,30) | 95.28 | 96.23 | (50,92) | (100,30) 4E−10 |
| Many | Small | 2.15 | 2.84 | (119,62) | 77.23 | 76.07 | (155,38) | (114,20) 2E−16 |
All experiments consist of 200 samples with 20 chromosomes containing 200 probes. Each row represents a set of samples with different genomic complexity as described in the Methods section. For all the cases, the GADA algorithm is set to T = 4.0, and CBS to α = 0.01, since this provides comparable performance points in the PROC curves, and allows comparison to other cases. The median sensitivity and False Discovery Rate % in breakpoint detection within two probes δ = 2 are evaluated. The FDR and sensitivity of GADA and CBS are also compared for each sample in a given dataset and the number of times where FDR and sensitivity are smaller or larger (< , >) between the two algorithms are reported. The rightmost column counts the number of times one algorithm (GADA,CBS) is performing better than the other both in terms of FDR or Sensitivity and a p-value is computed as described in the Section 2.2. Results indicate that GADA has a lower FDR when the number of breakpoints is large, and a higher sensitivity for small segments. The results are consistent for other choices of δ = 0,1,3 (data not shown).
3.2 Computational speed in commercial microarray platforms
We recorded the time required to analyze by GADA and CBS copy number data generated on Affymetrix or Illumina platforms from neuroblastoma cell lines or NCI cell lines. Results are summarized in Table 3. The GADA algorithm was on average 100 times faster than the latest implementation of CBS. The GADA algorithm provides an additional advantage by identifying all breakpoints corresponding to all the operating points of the PROC curve within the time frames shown in Table 3. This allows real-time control of the final adjustment of the representation of CNA regions corresponding to different choices of the critical value T with no additional computational time; while in the current implementation of CBS, the entire procedure needs to be repeated to obtain set of breakpoints at a different value of the α parameter.
Table 3.
Average analysis time (seconds) for Affymetrix and Illumina microarrays
| 50 K | 100 K | 250 K | 500 K | Illumina | |
|---|---|---|---|---|---|
| GADA | 1.5 | 2.98 | 7.10 | 15.95 | 20.49 |
| CBS | 197.7 | 444.90 | 597.72 | 1262.40 | 2665.00 |
Average time required to analyze the data in seconds per chip (only the time spent by the detection algorithm is counted). The 100 K and the 500 K columns correspond to the analysis of the combination of the two 50 K (Hind/Xba) and two 250 K (Nsp/Sty) chips, respectively.
The computational complexity of SBL has been greatly optimized by exploiting the properties of the PWC representation as described in Methods Section 2.6. The EM algorithm converges very fast, and each EM step is solved in a linear number of operations O(M), resulting in an overall running time that, as confirmed in Table 3, increases linearly with the array size M. In contrast, the computational complexity of CBS is composed of two parts; the circular binary segmentation optimization O(M2); and, the hybrid permutation test (Venkatraman and Olshen, 2007) that decides whether or not to proceed with the recursive segmentation O(MP) (P is the number of permutations). The hybrid permutation test in CBS has improved the previous implementation (Olshen et al., 2004) which required O(M2 P); however, the overall complexity is still limited by the circular segmentation O(M2).
3.3 Comparison of neuroblastoma CNA detection using different array platforms
The DNA from two neuroblastoma cell lines with (SK-N-BE2, SMS-KAN) and without (CHLA-20, LAN-6) MYCN oncogene amplification were analyzed for DNA copy number alterations. Three Affymetrix genotyping arrays (50K Xba, 250K Nsp, 250K Sty) and Illumina's humanhap550 genotyping beadchip were used to generate the copy number data. A total of 105 breakpoints were identified for at least two of the platforms using the SBL algorithm and were used for further analysis (Supplementary Tables 6 and 7). Figure 6 shows graphical output of the algorithm on representative chromosomes where significant CNA are known to be associated with neuroblastoma.
Fig. 6.

Inferred copy numbers from neuroblastoma cell-lines SK-N-BE2, SMS-KAN, LAN-6 and CHLA-20. Cell-lines were analyzed using Affymetrix's genotyping arrays 50K Xba, 250K Nsp and 250K Sty and Illumina's humanhap550 genotyping beadchip. The output of our software GADA(SBL) used the critical value of T = 4.8 and is compared to DNAcopy (CBS) with α = 0.01. T was adjusted to the point where an increase on T removed concordant CNA between samples and platforms, and a decrease on T did not provide additional concordant CNA regions. Blue color tones indicate loss of genetic material, and red color tones amplification.
Of the 105 breakpoints identified, 68 (65%) were identified on all platforms using GADA (Supplementary Table 6). The lowest density platform Xba, detected 78 (75%) of the 105 breakpoints, while the highest density platforms detected all (100%) the breakpoints. The detected alterations include the correct identification of the MYCN oncogene in the two cell lines with known MYCN amplification status and other common alterations found in neuroblastoma genome: loss of proximal region of 1p, gain of 17q, loss of distal region of 11q. Although the SK-N-BE2 showed copy number of two for chromosome 1p (Fig. 6), genotype information revealed loss of heterozygosity (LOH) in this region (i.e. uniparental disomy - data not shown) with gain of 1q not reflecting any significant change in the rate of heterozygosity. There was also no gain of 17q in this cell line but there was loss of 17p and LOH for this region. Finally, we compared GADA and CBS detection performance in this real dataset. The concordance rate between GADA and CBS for breakpoints that were detected by at least two platforms was 93% (Array specific concordances: Xba 97%, Nsp 90%, Sty 98%, Nsp+Sty 90%, Illumina 95%). There was also no significant difference between CBS and GADA in the distribution of distances for concordant breakpoints identified across the array platforms (Supplementary Table 8).
4 CONCLUSIONS
In this article we have introduced a new representation for genome copy number data and methodologies to detect CNA. The proposed PWC representation provides very useful properties such as sparseness, embeddedness and computational efficiency. This representation was exploited using a novel combination of two algorithms. The first one is based on SBL, and the second one is a stepwise BE procedure. Combination of these approaches result in an accurate and fast methodology, which we call GADA, to detect CNA. To the best of our knowledge, this is the first report that applies SBL to detect copy number changes or to estimate PWC representations in any application.
In simulated datasets, the GADA approach obtained the best performance in accurately detecting CNA when compared to other approaches. We have also demonstrated its applicability to two different commercial microarray platforms (Affymetrix and Illumina). The fast computational speeds obtained in analyzing these large arrays should allow further development of our algorithm in analyzing large cohorts of samples.
Although inclusion of allele specific copy number data has not been addressed in this work, the Bayesian framework in our algorithm could be extended to include the genotype data to improve placement of breakpoint positions. The genotype data and population heterozygosity frequencies could be used to jointly estimate loss of heterozygosity and allele specific copy number alterations. The advantage of such an approach is evident in our analyzed data of tumor cell lines with copy neutral LOH of chromosome 1p.
The performance of the proposed GADA approach has been studied and evaluated assuming that hybridization noise is additive white Gaussian (Willenbrock and Fridlyand, 2005). However, real microarray probe hybridization intensities may be affected by a wide range of platform specific effects like regional trends, non-uniform variance and outliers. Normalization of the microarray probe intensities can correct or minimize the impact of some of these effects in a pre-processing step to ensure that the data follows closely the model. Additionally, there exist several statistical tests (e.g. White test, Breusch-Pagan test or Kolmogorov-Smirnov) that could be performed on the residuals of the resulting segmentations to check for presence of the effects ignored by the model. Future research should evaluate the impact on the accuracy of GADA based on these different possible departures from the assumed model, and consider how these departures could be included in the Bayesian approach that has been described in this article.
The statistical and signal processing approaches introduced in this article are implemented in the GADA software for identification of CNA in tumor samples.
Supplementary Material
ACKNOWLEDGEMENT
We would like to acknowledge Dr H. Willenbrock for providing the simulated dataset of (Willenbrock and Fridlyand, 2005), Dr A.B. Olshen for discussion about CBS, and Dr R. Diaz-Uriarte for assistance with RJaCGH. We also thank Dr R. Sposto for reviewing the manuscript and providing useful comments. This research has been supported in part by grants K12-CA60104 from the National Institute of Health's Child Health Research Career Development Award Program and the Pre-Institute Award from the Pediatric Brain Tumor Foundation (S.A.) and by CA60104 from the National Cancer Institute and the Neuroblastoma Children's; Cancer Society (R.C.S.).
Conflict of Interest: none declared.
REFERENCES
- Albertson DG, et al. Chromosome aberrations in solid tumors. Nat. Genet. 2003;34:369–376. doi: 10.1038/ng1215. [DOI] [PubMed] [Google Scholar]
- Broet P, Richardson S. Detection of gene copy number changes in CGH microarrays using a spatially correlated mixture model. Bioinformatics. 2006;22:911–918. doi: 10.1093/bioinformatics/btl035. [DOI] [PubMed] [Google Scholar]
- Chen SS, et al. Atomic decomposition by basis pursuit. SIAM J. Sci. Comput. 1998;20:33–61. [Google Scholar]
- Donoho D, et al. Stable recovery of sparse overcomplete representations in the presence of noise. IEEE T. Inform. Theory. 2006;52:6–18. [Google Scholar]
- Dragotti P, Vetterli M. Wavelet footprints: Theory, algorithms, and applications. IEEE T. Signal Proces. 2002;51:1306–1323. [Google Scholar]
- Engler DA, et al. A pseudolikelihood approach for simultaneous analysis of array comparative genomic hybridizations. Biostatistics. 2006;7:399–421. doi: 10.1093/biostatistics/kxj015. [DOI] [PubMed] [Google Scholar]
- Fridlyand J, et al. Hidden markov models approach to the analysis of array cgh data. J. Multivariate Anal. 2004;90:132–153. [Google Scholar]
- Garraway LA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- Hastie T, et al. The Elements of Statistical Learning. New York, NY: Springer; 2001. [Google Scholar]
- Hsu L, et al. Denoising array-based comparative genomic hybridization data using wavelets. Biostatistics. 2005;6:211–226. doi: 10.1093/biostatistics/kxi004. [DOI] [PubMed] [Google Scholar]
- Huang J, et al. Whole genome DNA copy number changes identified by high density oligonucleotide arrays. Hum. Genomics. 2004;1:287–299. doi: 10.1186/1479-7364-1-4-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, et al. Detection of DNA copy number alterations using penalized least squares regression. Bioinformatics. 2005;21:3811–3817. doi: 10.1093/bioinformatics/bti646. [DOI] [PubMed] [Google Scholar]
- Hupe P, et al. Analysis of array cgh data: from signal ratio to gain and loss of dna regions. Bioinformatics. 2004;20:3413–3422. doi: 10.1093/bioinformatics/bth418. [DOI] [PubMed] [Google Scholar]
- Kallioniemi A, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258:818–821. doi: 10.1126/science.1359641. [DOI] [PubMed] [Google Scholar]
- Kohavi R, John GH. Wrappers for feature subset selection. Artif. Intell. 1997;97:273–324. [Google Scholar]
- Komura D, et al. Genome-wide detection of human copy number variations using high-density DNA oligonucleotide arrays. Genome Res. 2006;16:1575–1584. doi: 10.1101/gr.5629106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosko B. Probable equivalence, superpower sets, and superconditionals. Int. J. Intell. Sys. 2004;19:1151–1171. [Google Scholar]
- Lai WR, et al. Comparative analysis of algorithms for identifying amplifications and deletions in array cgh data. Bioinformatics. 2005;21:3763–3770. doi: 10.1093/bioinformatics/bti611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson D, et al. Efficient calculation of interval scores for DNA copy number data analysis. J. Comput. Biol. 2006;13:215–228. doi: 10.1089/cmb.2006.13.215. [DOI] [PubMed] [Google Scholar]
- Mallat S, Zhang Z. Matching pursuits with time-frequency dictionaries. IEEE. Trans. Signal proces. 1993;41:3397–3415. [Google Scholar]
- Marioni JC, et al. BioHMM: a heterogeneous hidden markov model for segmenting array CGH data. Bioinformatics. 2006;22:1144–1146. doi: 10.1093/bioinformatics/btl089. [DOI] [PubMed] [Google Scholar]
- McLachlan GJ, Krishnan T. The EM Algorithm and Extensions. New York, NY: Wiley; 1997. [Google Scholar]
- Nannya Y, et al. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–6079. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- Olshen AB, et al. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- Pati Y, et al. Orthogonal matching pursuit: Recursive function approximation with applications to wavelet decomposition. Proceedings of the 27th Annual Asilomar Conference on Signals, Systems, and Computers; Pacific Grove, CA. 1993. [Google Scholar]
- Peiffer DA, et al. High-resolution genomic profiling of chromosomal aberrations using infinium whole-genome genotyping. Genome Res. 2006;16:1136–1148. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, et al. A statistical approach for array CGH data analysis. BMC Bioinformatics. 2005;6:27. doi: 10.1186/1471-2105-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pique-Regi R, et al. Wavelet footprints and sparse bayesian learning for DNA copy number change analysis. Proceedings International Conference on Acoustics, Speech, and Signal Processing.2007. [Google Scholar]
- Pollack JR, et al. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat. Genet. 1999;23:41–46. doi: 10.1038/12640. [DOI] [PubMed] [Google Scholar]
- Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda OM, Diaz-Uriarte R. Flexible and accurate detection of genomic copy-number changes from acgh. PLoS Comput. Biol. 2007;3:e122. doi: 10.1371/journal.pcbi.0030122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seber GAF, Lee AJ. Linear Regression Analysis. second edition. New York: John Wiley; 2003. [Google Scholar]
- Shah SP, et al. Integrating copy number polymorphisms into array cgh analysis using a robust hmm. Bioinformatics. 2006;22:e431–e439. doi: 10.1093/bioinformatics/btl238. [DOI] [PubMed] [Google Scholar]
- Tipping ME. Sparse bayesian learning and the relevance vector machine. J. Mach. Learn. Res. 2001;1:211–244. [Google Scholar]
- Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23:657–663. doi: 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- Willenbrock H, Fridlyand J. A comparison study: applying segmentation to array CGH data for downstream analyses. Bioinformatics. 2005;21:4084–4091. doi: 10.1093/bioinformatics/bti677. [DOI] [PubMed] [Google Scholar]
- Wipf D, Rao B. Sparse Bayesian learning for basis selection. IEEE. Trans. Signal Proces. 2004;52:2153–2164. [Google Scholar]
- Zhao X, et al. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 2004;64:3060–3071. doi: 10.1158/0008-5472.can-03-3308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.