

Abstract
Metastatic bone cancer causes severe pain that is primarily treated with opioids. A model of bone cancer pain in which the progression of cancer pain and bone destruction is tightly controlled was used to evaluate the effects of sustained morphine treatment. In cancer-treated mice, morphine enhanced, rather than diminished, spontaneous, and evoked pain; these effects were dose-dependent and naloxone-sensitive. SP and CGRP positive DRG cells did not differ between sarcoma or control mice, but were increased following morphine in both groups. Morphine increased ATF-3 expression only in DRG cells of sarcoma mice. Morphine did not alter tumor growth in vitro or tumor burden in vivo but accelerated sarcoma-induced bone destruction and doubled the incidence of spontaneous fracture in a dose- and naloxone-sensitive manner. Morphine increased osteoclast activity and upregulated IL-1β within the femurs of sarcoma-treated mice suggesting enhancement of sarcoma-induced osteolysis. These results indicate that sustained morphine increases pain, osteolysis, bone loss, and spontaneous fracture, as well as markers of neuronal damage in DRG cells and expression of pro-inflammatory cytokines. Morphine treatment may result in “add-on” mechanisms of pain beyond those engaged by sarcoma alone. While it is not known whether the present findings in this model of osteolytic sarcoma will generalize to other cancers or opioids, the data suggest a need for increased understanding of neurobiological consequences of prolonged opioid exposure which may allow improvements in the use of opiates in the effective management of cancer pain.
Keywords: Cancer pain, Opioid hyperalgesia, Cancer, Osteoclast, IL-1, Opiate receptors
1. Introduction
Common cancers, such as breast and prostate, metastasize to bone eliciting osteolytic and osteoblastic reactions associated with incapacitating bone pain and fracture (Coleman, 1997, 2001; Luger et al., 2001). Advanced cancer pain is described as “moderate to severe” in approximately 40–50% and as “very severe or excruciating” in 25–30% of the patients (Ripamonti and Dickerson, 2001). Bone cancer pain is continuous and can be exacerbated by episodes of breakthrough pain (Clohisy and Mantyh, 2003). Cancer pain management follows the guidelines outlined in the WHO Ladder Approach for Relief of Cancer Pain that suggest adjusting the strength of the prescribed analgesics according to pain intensity (WHO, 1986; Blum et al., 2003; Mercadante and Fulfaro, 2005).
Opioids are recommended for treatment of moderate to severe cancer pain, with morphine most frequently used (Mercadante et al., 2005). In animal models of bone cancer, acute morphine administration alleviates pain (Luger et al., 2002; Menendez et al., 2003a, b, 2005; Baamonde et al., 2005; Urch et al., 2005). However, the chronic nature of cancer pain often requires prolonged opioid administration through controlled release tablets, repeated bolus injections, or transdermal patches (Heiskanen and Kalso, 1997; Warfield, 1998; Mercadante, 1999a, b; Allan et al., 2001; Hanks et al., 2001). Moreover, many (but not all) cancer patients require opioid dose escalation to maintain adequate pain relief due to the diminished analgesia with repeated opioid administration (i.e., “analgesic tolerance”), the advancement of the disease resulting in greater pain and therefore requiring more opioid, or both (Mercadante and Portenoy, 2001; Blum et al., 2003).
Clinical studies have reported that opioids administered through different routes of administration (transdermal, oral, i.th., i.v.) can unexpectedly produce hyperalgesia and allodynia, particularly during rapid opioid dose escalation (De Conno et al., 1991; Sjogren et al., 1993, 1994; Jacobsen et al., 1995; Mercadante et al., 2003; Chu et al., 2006), a phenomenon described as an Emerging Iatrogenic Syndrome (Mercadante et al., 2003). Preclinical studies have unexpectedly demonstrated that opioids can paradoxically enhance pain (Gardell et al., 2002; Ossipov et al., 2004; King et al., 2005a, b). Structurally distinct opioids including nonpeptidic agonists (e.g. morphine, oxymorphone, fentanyl) as well as peptides acting at μ-opioid receptors (e.g. DAMGO) have been shown to produce hyperalgesia (Laulin et al., 1998, 1999; Vanderah et al., 2000; Gardell et al., 2002, 2006; Celerier et al., 2006; Pud et al., 2006). It is presently unknown how opioid-induced pronociceptive changes might influence pain or disease progression in bone cancer.
In the present study, osteolytic sarcoma cells were injected and sealed into mouse femurs for evaluation of possible effects of sustained morphine infusion on pain, tumor burden, bone loss and spontaneous fracture under tightly controlled conditions. Sustained morphine administration elicited pronociceptive neuroplastic adaptations and enhanced, rather than reduced, sarcoma- induced pain in a dose-dependent and naloxonesensitive manner. Surprisingly, morphine infusion also accelerated sarcoma-induced bone loss and enhanced spontaneous fracture in a dose-dependent and naloxone- sensitive manner.
2. Methods
2.1. Surgical procedures and drug treatment
2.1.1. Strain of mouse
Male adult C3H/HeJ normal mice, weighing 20–25 g (Jackson Laboratories, Bar Harbor, ME), were chosen for histo-compatability with the NCTC 2472 tumor line [American Type Culture Collection (ATCC), Rockville, MD], previously shown to form lytic lesions in bone after intramedullary injection (Clohisy et al., 1995, 1996; Schwei et al., 1999; Luger et al., 2001).
2.1.2. Murine CCL-11 cells
Murine CCL-11 (NCTC clone 2472) sarcoma cells were maintained in NCTC 135 media containing 10% horse sera, passaged every 4 days, and harvested between 2 and 12 passages.
2.1.3. Surgery
Baseline spontaneous and evoked pain behaviors (described below), and radiograph images of the femur were obtained. Animals were anesthetized with ketamine/xylazine and an arthrotomy was performed exposing the condyles of the distal femur. A hole was drilled into the femur for the injection needle. Radiograph images (Faxitron) were taken on two planes to verify needle placement inside the intramedullary space of the femur. Sarcoma cells, 106 in 5 μl of alpha minimal essential medium (α MEM) containing 1% bovine serum albumin (BSA) or 5 μl of alpha MEM containing 1% BSA alone (control), were injected into the intramedullary space of the mouse femur of the right leg and the injection site sealed with dental amalgam.
2.1.4. Drug treatment
Mice were anesthetized with 0.5% halothane in O2 and primed osmotic minipumps (Alzet # 1007D; 0.5 μl/hour across 7 days) filled with morphine sulfate (50, 25, 7.5, 2.5 mg/ml) dissolved in saline or with saline were implanted (s.c.) 7 days following injection of CCL-11 cells into the femur. The concentrations of morphine sulfate resulted in daily doses of 20, 10, 3, or 1 mg/kg/day. These doses were chosen from, and were within, the ranges used for bolus and infusion protocols in mice (Luger et al., 2002; Juni et al., 2006) and did not produce overt signs of sedation or disruption of motor function. Blood morphine levels for the 10 and 20 mg/kg/day doses of morphine delivered through minipumps have been reported at 48-h post-pump implantation (Feng et al., 2006). Morphine sulfate (C34H40N2O10S; molecular weight 668.75) was obtained as a generous gift from National Institute on Drug Abuse Drug Supply Program (RTI Batch No. 8981-52; Ref. No. 10678).
Naloxone hydrochloride dihydrate (Sigma) was dissolved in saline (25 mg/ml) and administered (s.c.) via osmotic minipumps. Mice receiving both naloxone and morphine had two separate minipumps, one per compound, implanted. Pilot experiments demonstrated no effect of implantation of a second minipump delivering saline on morphine activity and the second minipump did not disrupt animal behavior or weight gain or produce overt signs of discomfort.
2.2. Behavioral pain measures
Each mouse was tested for movement-evoked pain, spontaneous pain behaviors (flinching and guarding), and tactile hypersensitivity, in that order, prior to treatment (BL) and 6, 10, and 12 days following surgery. For the control–saline, control –morphine, sarcoma–saline, and sarcoma–morphine groups, we used 10–12 mice per group. For the naloxone studies, 8–10 animals were used for each group (sarcoma–saline, sarcoma–morphine, sarcoma–naloxone, and sarcoma–morphine/naloxone). All pain measures were conducted by the same experimenter who was blinded to the treatments.
2.2.1. Thermal antinociception
Nociceptive testing was performed by utilizing the 52 °C hot plate (HP) test. HP test was performed by placing the mouse on a heated surface and determining the latency until a nociceptive response, demonstrated by licking or withdrawing of a hindpaw or attempts to jump out of the enclosure, was evident. The HP latencies were determined once before pump implantation, on day 6 after the surgery, and 24 h after the implantation of osmotic minipumps. A cut-off latency of 30 s was used to prevent tissue damage.
2.2.2. Spontaneous pain
Mice were placed in raised plexiglass chambers with a wire grid floor and allowed to acclimate to the chamber for 1 h. Guarding and flinching behaviors were measured during a 2 min observation period. The number of flinches was counted, and the time spent guarding the foot (the foot is lifted off of the floor) was measured.
2.2.3. Movement-evoked pain
Limb use was assessed as previously described (Luger et al., 2001). The mouse was placed in an empty mouse pan and observed while walking across the pan in a continuous motion. Limping and/or guarding behavior of the right (sarcoma treated) hindlimb was rated on the following scale: 0 = complete lack of use, 1 = partial non-use, 2 = limping and guarding, 3 = limping, 4 = normal walking.
2.2.4. Tactile hypersensitivity
Paw withdrawal thresholds in response to probing with calibrated von Frey filaments were determined in the manner described by Chaplan et al. (1994). Mice were kept in suspended cages with wire mesh floors and the von Frey filament applied perpendicularly to the plantar surface of the ipsilateral paw until it buckled slightly and was held for 3–6 s. A positive response was indicated by a sharp withdrawal of the paw. An initial probe equivalent to 2 g was applied and if the response was negative, the stimulus was incrementally increased until a positive response was obtained, then decreased until a negative result was obtained. This up–down method was repeated until three changes in behavior were determined, and the pattern of positive and negative responses was tabulated. The 50% paw withdrawal threshold was determined by the non-parametric method of Dixon (1980).
2.3. Determination of bone destruction
Radiographs were taken following behavioral testing using a Faxitron machine with images captured by a digital camera. Bone loss was rated by an experimenter blinded to treatment according to a 4-point scale: 0 = normal, 1 = bone loss observed with no fracture, 2 = full-thickness unicortical bone loss indicating unicortical bone fracture, 3 = full-thickness bicortical bone loss indicating bicortical bone fracture. This scale was modified from 0 to 5 scale previously described (Luger et al., 2001).
2.4. Immunohistochemistry, and histological analysis of tumor burden and osteoclastogenesis
2.4.1. Dorsal root ganglia
Mice received an overdose of ketamine HCL/xylazine (1 ml/kg) and were perfused transcardially with 0.1 M PBS followed by 10% neutral-buffered formalin (Sigma, St. Louis, MO, USA). DRG (L4) and ipsilateral femura were removed and postfixed overnight in 10% neutral-buffered formalin. DRG tissue was cryoprotected with 20% sucrose in 0.1 M PBS. Frozen DRG sections of 10 μm were washed in 0.1 PBS and then incubated with rabbit anti-SP antiserum or rabbit anti-CGRP antiserum (1:40,000; Bachem/Peninsula Labs), or rabbit anti-ATF3 antiserum (1:5000; Santa Cruz Biotechnology). Sections were then incubated in Alexa Fluor 568 goat anti-rabbit IgG or Alexa Fluor 525 (diluted 1:1000).
2.4.2. Image analysis and quantification
Fluorescence images of DRG sections were acquired with a Nikon E800 fluorescence microscope outfitted with filter sets for Cy3 and FITC and a Hamamatsu C5810 color CCD camera and its proprietary Image Processor software (Hamamatsu Photonic System, Bridgewater, NJ, USA). Stained cells were counted on 8–10 randomly selected L4 DRG sections from three animals per each condition. The results are expressed as a percentage of the estimated total number of neurons from these sections.
2.4.3. Femora
Femora were collected on day 10 after the surgery, rinsed in water and placed in Decal solution (RDO-Apex, Aurora, IL) for 1 h for decalcification and embedded in paraffin for sectioning. Femora were cut in the frontal plane 3-μm thick and stained with hematoxylin and eosin (H&E) to visualize normal marrow elements and cancer cells under bright field microscopy on a Nikon E800 at 4× magnification. Tumor or marrow areas within the femur (3–5 bones per treatment) were measured in mm2 between the epiphyseal plates using Metamorph imaging software.
To determine the number of osteoclasts, femurs were stained with Tartrate-resistant acid phosphatase (TRAP) kit (Sigma) as TRAP is commonly used as a marker of osteoclasts in bone as previously reported (Halleen et al., 1999; Takano et al., 2004, 2006; Beeton et al., 2006; Itoh et al., 2006; Ren et al., 2006; Wan et al., 2006). Images were analyzed under bright field microscopy on a Nikon E800 at 4× and 10× magnification. For all groups, osteoclasts were counted at the metaphysis of the femur, as bone destruction occurs mainly in this area (Honore et al., 2000a). Counts were performed across several (3–5) bones per treatment. Metamorph imaging software was used to quantify the results. The results are expressed as the mean number of osteocalsts per mm2 of intramedullary space.
2.5. In vitro analysis of sarcoma cell growth
Murine CCL-11 (NCTC clone 2472) sarcoma cells were maintained in NCTC 135 media containing 10% horse serum, 100 U/ml penicillin and 0.1 mg/ml streptomycin in a humidified atmosphere with 95% air/5% CO2. To evaluate the effect of morphine on cell growth, cells were treated with morphine for 2, 4, or 6 days, and for each time-point, the effect of morphine was evaluated over a concentration range of 10 nM to 100 μM. For each assay, cells were seeded at 1000 cells/well in 96-well plates and cultured for a total of 4 days, at which time cells reached a confluency of 50–70%. During the 4-day culture, cells were treated with various concentrations of morphine (8 wells used per concentration) on the same 96-well plates and separate plates were used for different duration of treatment. For the 2-day treatment, morphine was added to the cells 2 days after plating; for the 4-day treatment, morphine was added to the cells at the time of plating. For the 6-day treatment, cells were treated, in separate 75 cm2 flasks, with the various concentrations of morphine 2 days prior to seeding onto the 96-well plates, and the morphine treatment continued for four additional days after plating. Untreated control cells were included on each plate and assayed in parallel. BrdU was added to all the wells on day 3, 24 h prior to the termination of morphine treatment and the ELISA. BrdU was analyzed using Cell Proliferation ELISA, BrdU (colorimetric) kit according to the manufacturer’s instructions (Roche Applied Sciences, Cat # 11647229). The optical density at 450 nm (OD450) was read by a plate reader (Multiskan Ascent, Thermo Electron) as required for assays using the stop solution. For data analysis, the mean OD450 (n = 8) in the morphine-treated cells is expressed as a percent of that in the untreated control done in parallel. Normalized data from three independent experiments are expressed as means ± SEM.
2.6. IL-1β analysis in intramedullary exudate
Mice received an overdose of ketamine HCL/xylazine (1 ml/kg) and decapacitated. Femurs (5–6 per condition) were dissected and the intramedullary space flushed with 1 ml saline. Samples were analyzed using IL-1β ELISA kit according to the manufacturer’s instructions (Invitrogen). Protein quantification was done using BCA (bicinchoninic acid) assay (Pierce). Results are expressed as pg/mg protein.
2.7. Statistical analysis
Statistical comparisons between treatment groups were done using ANOVA. Pairwise comparisons were made with Student’s t-test, multiple comparisons between groups were done using Newman–Keuls Multiple Comparison Test. For the rating assays, limb use and bone loss, statistical comparisons were made with the Mann–Whitney test. Dose–response effects were done with linear regression analysis of the linear portion of the log dose–response curve. For all analysis, significance was set at p < 0.05.
3. Results
3.1. Morphine infusion induces antinociception
Delivery of morphine (1, 3, 10, 20 mg/kg/day) by osmotic minipumps implanted 7 days after intra-femoral injection of cancer cells or control media elicited dose-dependent thermal antinociception when measured 24 h after pump implantation. There were no significant differences in morphine’s antinociceptive effects between control- and sarcoma-treated mice. Hot plate latencies for control animals increased from baseline latencies of 11.96 ± 0.3 s pre-pump to 13.66 ± 0.7, 14.90 ± 0.41, 15.38 ± 0.63, and 17.52 ± 0.66 s at 24-h post-pump implantation for doses of 1, 3, 10 and 20 mg/kg/day, respectively. Hot plate latencies for sarcoma animals increased from 11.86 ± 0.27 to 12.04 ± 0.36, 14.45 ± 0.7, 16.12 ± 0.56, and 18.94 ± 1.16 s at 24-h post-pump implantation for doses of 1, 3, 10 and 20 mg/kg/day, respectively. Implantation of saline pumps failed to significantly alter hot plate latencies in either group. Of note, the highest dose of morphine (20 mg/kg delivered over 24 h) produced sub-maximal antinociception.
3.2. Sustained morphine enhances sarcoma-induced spontaneous pain
Behavioral analyses of spontaneous pain were employed to assess the effects of morphine administration on bone cancer-induced pain. In agreement with previous studies, mice injected with osteolytic sarcoma cells developed spontaneous pain by 12 days post-surgery (Luger et al., 2001). Morphine infusion beginning on day 7 after administration of cancer cells or control media enhanced sarcoma-induced flinching behavior at day 10, and this effect persisted throughout the course of the experiment (Fig. 1a). A higher percentage of sarcoma- treated mice that received morphine displayed guarding behavior when compared to sarcoma mice receiving saline infusion at day 10 (55% vs. 40% of all animals, respectively) and day 12 (82% vs. 60% of all animals, respectively). Sarcoma-treated mice that received sustained morphine also spent more time guarding the sarcoma-treated hindpaw compared to sarcoma- treated mice receiving saline infusion 12 days following sarcoma injection (Fig. 1b). Mice injected with control media and treated with sustained morphine exposure did not exhibit flinching or guarding behaviors throughout the study. These data indicate that sustained morphine exposure does not elicit spontaneous pain in control mice, but increases sarcoma-evoked spontaneous pain.
Fig. 1.

Sarcoma cells (sarcoma) or control media (control) were injected into the intramedullary space of the femur and behavior was tested beginning 6 days later. Saline or morphine osmotic minipumps were implanted 7 days after sarcoma/media injection and were tested 10 and 12 days after injection (3 and 5 days after minipump implantation). (a) Sarcoma-treated mice with morphine infusion showed increased flinching compared to all other treatment groups on test days 10 and 12. (b) All sarcoma-treated mice showed increased guarding behavior compared to control-treated mice. Sarcoma-treated mice with morphine infusion showed more guarding behavior compared to sarcoma-treated mice with saline infusion. (c) Mice injected with control media receiving morphine infusion demonstrated lower paw withdrawal thresholds on test days 10 and 12 (3 and 5 days into morphine infusion) indicating tactile hypersensitivity. All sarcoma-treated mice showed lower paw withdrawal thresholds on test days 10 and 12, with mice receiving morphine infusions showing lower paw withdrawal thresholds compared to sarcoma-treated mice with saline infusions. (d) Sarcomatreated mice showed limping behaviors (score of 3), with sarcoma-treated mice with morphine infusion developing limping behaviors prior to saline-infused mice (days 10 vs. 12, respectively). All graphs show means ± SEM. *Indicates significant difference from control saline group, #indicates significant difference between saline- and morphine-treated mice within the sarcoma or the control groups.
3.3. Sustained morphine enhances sarcoma-induced evoked pain
To assess the effects of sustained morphine (20 mg/kg/day) on sarcoma-induced tactile hypersensitivity, the response thresholds of sarcoma or control media-treated mice receiving sustained morphine or saline infusion were measured with von Frey filaments (Fig. 1c). A slight decrease in response thresholds was observed in all mice 6 days following surgery, likely due to the inflammatory consequences of the surgery. Consistent with previous studies, mice injected with osteolytic sarcoma cells developed enhanced responses to tactile stimuli and movement-evoked pain by days 10 and 12 post-femoral injections (Honore et al., 2000a) and morphine administration induced tactile hypersensitivity in control mice within 3 days that was maintained through 5 days of morphine infusion (days 10 and 12 post-control media injection, respectively) (Vanderah et al., 2001). Sarcoma-treated mice receiving sustained morphine exposure showed significantly lower paw withdrawal thresholds than sarcoma-treated mice receiving saline infusion 10 and 12 days following sarcoma injection. To assess whether sustained morphine exposure alters sarcoma-induced movement-evoked pain, limb use was rated in mice as previously described (Honore et al., 2000a). Sarcoma-treated mice with sustained morphine administration were more likely to be rated for limping behavior 10 days post-sarcoma injection compared to sarcoma-treated mice with saline infusion (Fig. 1d). Sarcoma- treated mice with saline infusion did not show significant limping behavior compared to mice treated with control media at the 10 day time-point. Both the saline-and the morphine-treated mice showed significantly more limping behavior 12 days post-sarcoma injection with no significant differences in limb use between saline- and morphine-treated mice at this time-point. These data indicate that sustained morphine administration enhances sarcoma-induced evoked pain behaviors.
3.4. Sustained morphine enhances sarcoma-induced spontaneous and evoked pain in a dose-related manner
To examine whether the sustained morphine-induced enhancement of sarcoma-induced pain is dose-dependent, we measured spontaneous and evoked pain behaviors in the presence of 1, 3, 10 and 20 mg/kg/day morphine. On day 12 after the surgery and 5 days into morphine infusion, there was a dose-dependent increase in sarcoma-induced spontaneous flinching (Fig. 2a) and guarding behavior (Fig. 2b), indicating that morphine elicited a dose-related increase in sarcoma-induced spontaneous pain behaviors. Of note, no flinching or guarding behaviors were observed in mice treated with control media indicating that morphine fails to induce spontaneous pain behaviors at any of the doses tested (data not shown). Sarcoma-induced evoked pain was measured at the same time-point, and sustained morphine infusion enhanced sarcoma-induced tactile hypersensitivity (Fig. 2c) and decreased limb use (Fig. 2d) in a dose-dependent fashion, indicating that infusion of morphine increases sarcoma-induced evoked pain behaviors in a dose-dependent manner. Sustained morphine infusion did not alter limb use of mice treated with control media at any of the doses tested (data not shown) but decreased evoked paw withdrawal thresholds in the control mice (R2 = .99, p < 0.05). Collectively, these data indicate that the observed effects of sustained morphine administration on spontaneous and evoked behaviors are dose related.
Fig. 2.

Sarcoma cells were injected into the intramedullary space of the femur. Saline or morphine osmotic minipumps delivering morphine sulfate at 1, 3, 10, or 20 mg/kg/days were implanted (s.c.) 7 days after injection and were tested 12 days after injection (5 days after pump implantation). (a) Sarcoma-treated mice with morphine infusion showed increased flinching in a dose-dependent manner. (b) Sarcoma-treated mice with morphine infusion showed increased guarding behavior in a dose-dependent manner. (c) Sarcoma-treated mice with morphine infusion showed increased tactile hypersensitivity in a dose-dependent manner. (d) Sarcoma-treated mice with morphine infusion showed decreased limb use. All graphs show means ± SEM with linear regression lines. For all analyses, the slopes of the regression lines were significantly different from zero. Separate groups of six mice were used for each data point.
3.5. Sustained morphine-induced effects in DRG cells
To determine possible mechanisms underlying sustained morphine-induced increase in sarcoma-induced pain, neural markers were evaluated in cell bodies of primary afferent sensory fibers located within the dorsal root ganglion (DRG) 12 days following femoral injections of cancer cells (Fig. 3). Consistent with previous studies, sarcoma, but not control media, elicited ATF3 expression in DRG cells (Peters et al., 2005; Sevcik et al., 2005). ATF3 expression was not produced by morphine treatment in control mice. However, ATF3 expression was exacerbated by morphine treatment in sarcoma mice. Morphine infusion doubled the percentage of total DRG cells showing ATF3 expression, from 11 ± 1%of total DRG cells in sarcoma–saline-treated animals to 22 ± 2% of total DRG cells in sarcoma–morphine-treated mice. Control media-treated mice showed very low levels of ATF3 expression in both saline- and morphine-treated animals, suggesting that morphine treatment alone was not producing cell damage. Consistent with previous reports (Honore et al., 2000b), sarcoma–saline-treated mice did not show an increased number of SP or CGRP positive DRG cells compared to saline-control animals cells (Honore et al., 2000b). However, in both control media and sarcoma injected mice, sustained morphine exposure significantly increased the percent of SP and CGRP positive DRG cells (Fig. 3).
Fig. 3.

Immunofluorescent staining for ATF3, SP, or CGRP within the ipsilateral DRG (L4) on day 12. Graphs indicate % total DRG cells that are ATF3-ir, SP-ir, or CGRP-ir positive. Mice injected with control media show very low levels of ATF3-ir, with less than 5% ATF3-ir positive cell bodies within the DRG, irrespective of saline or morphine infusion. Sarcoma treatment produced a significant increase in ATF3-ir positive cells, with morphine infusion doubling the percentage of ATF3-ir positive cell bodies. Morphine infusion approximately doubles the percentage of SP-ir positive cell bodies compared to saline-treated mice equally sarcoma and control media-treated mice. Sarcoma treatment did not increase the percentage of SP-ir positive cell bodies compared to control media treatment. Morphine treatment approximately doubles the percentage of CGRP-ir positive cell bodies equally in sarcoma and control media-treated mice, and sarcoma treatment does not increase the percentage of CGRPP-ir positive cell bodies compared to control media treatment. Graphs show means ± SEM. *Indicates significant difference from control saline group, #indicates significant difference between saline- and morphine-treated mice within the sarcoma or the control groups.
3.6. Sustained morphine accelerates sarcoma-induced bone loss and fracture
To assess possible effects of sustained morphine exposure on sarcoma-induced bone loss, radiographic images were taken following behavioral testing. Radiographs of bones on day 12 (5 days into morphine or saline infusion) show that sustained morphine administration increased sarcoma-induced bone loss in a dose-dependent fashion (Fig. 4). In the sarcoma-treated mice, the most severe bone loss was observed in the distal head of the bone, with bone loss extending along the femur to the proximal head (Fig. 4a). In sarcoma-treated mice with morphine infusion, there was a significant increase in bone destruction in both the proximal and distal heads of the femur, with fractures indicated by full-cortical bone loss as indicated by the arrows (Fig. 4a and b). Consistent with previous reports (Luger et al., 2001), visual ratings of the radiographs by an observer blinded to the experimental conditions show that bone loss is observed by 6 days following femoral injection of sarcoma cells (Fig. 4c). Pre-morphine bone ratings were equivalent between groups, indicating no baseline group differences of bone loss prior to morphine and saline infusion. Sarcoma-induced bone loss increased in both morphine and saline-treated mice in a time-dependent manner. Sustained morphine exposure enhanced sarcoma-induced bone loss compared to sarcoma-treated mice receiving saline infusion in a time-dependent manner, with a significant increase in sarcoma-induced bone loss by 12 days following femoral injection, 5 days into morphine infusion (Fig. 4c). Moreover, the morphine induced increase in bone loss 5 days into morphine infusion was dose-dependent (Fig. 4d). Sustained administration of morphine across 3 and 5 days doubled the rate of sarcoma-induced spontaneous fracture rate, indicated by bone loss across the full thickness of the cortical bone (Fig. 4e). Morphine treatment also increased the rate of sarcoma-induced spontaneous fracture in a dose-dependent fashion, with higher doses (10 and 20 mg/kg/day) of morphine doubling the incidence of sarcoma-induced fracture by 5 days into infusion, 12 days following sarcoma injection into the femur (Fig. 4e). Of note, in our experiments neither saline nor morphine-infused control mice developed bone loss (Fig. 4a).
Fig. 4.

(a) Radiograph images of the injected femur on day 12. Sarcoma-induced bone loss is greatest at the distal end of the femur, progressing along the femur to the proximal head of the femur. Sarcoma-induced bone loss is more extensive in mice that received morphine infusion across 5 days compared to saline-treated mice. Fractures, indicated by arrows, were defined as full-thickness cortical loss. (b) Enlarged radiograph images of the distal and proximal ends of the femur showing sustained morphine infusion across 5 days increases sarcoma-induced bone loss and increases fractures (indicated by arrows). (c) Bone loss ratings of sarcoma-treated mice with saline or morphine infusions 6, 10, and 12 days following sarcoma injection show that some sarcoma-induced bone loss is observed 6 days following, with no difference between morphine- and saline-treated mice. Unicortical fractures begin to develop 10 days following sarcoma injection. Mice receiving morphine infusion across 5 days demonstrated more sarcoma-induced bone destruction on day 12 compared with saline-treated mice, with more mice showing bicortical fractures. Graphs show means ± SEM. *Indicates significant difference from control saline group. #Indicates significant difference between saline- and morphine-treated mice within the sarcoma or the control groups. (d) Sustained morphine enhanced sarcoma-induced bone loss in a dose-dependent manner. Graphs shows mean ± SEM with linear regression lines. The slope of the regression line was significantly non-zero. Separate groups of six mice were used for each data point. (e) Morphine infusion doubled the percentage of mice with sarcoma-induced spontaneous fractures compared to saline-infused animals. (f) Morphine infusion increased the incidence of sarcoma-induced spontaneous fractures in a dose-dependent manner. Graphs shows means ± SEM with linear regression lines. The slope of the regression line was significantly non-zero. Separate groups of six mice were used for each data point.
3.7. Naloxone antagonizes morphine-induced enhanced pain and bone loss
To examine whether the observed effects of morphine are opioid receptor mediated we administered naloxone (10 mg/kg/day, s.c.) alone or together with the highest dose of morphine used in our study (20 mg/kg, s.c.). Spontaneous and evoked pain behaviors were measured 5 days into infusion, 12 days following injection of sarcoma cells into the femur. Co-administration of naloxone prevented the development of sustained morphine induced increase in sarcoma-induced spontaneous and evoked pain behaviors (Fig. 5). Naloxone prevented morphine-induced enhancement of spontaneous pain behaviors, including enhanced flinching behavior (Fig. 5a) and guarding behavior (Fig. 5b). Naloxone also prevented development of sustained morphine-induced enhanced evoked pain behaviors, tactile hypersensitivity (Fig. 5c) and decreased limb use (Fig. 5d). Naloxone administration (without morphine) did not alter sarcoma-induced spontaneous or evoked pain when compared to sarcoma-treated mice receiving saline.
Fig. 5.

Sarcoma cells were injected into the intramedullary space of the femur. Naloxone (10 mg/kg/day, s.c.) was administered alone and in parallel, through separate minipumps, with the highest dose of morphine (20 mg/kg/day, s.c.). Minipumps were implanted (s.c.) 7 days after injection and were tested 12 days after injection (5 days after pump implantation). (a) Administration of naloxone with morphine prevented the sustained morphine-induced increase in sarcoma-induced flinching. (b) Administration of naloxone with morphine prevented the sustained morphine-induced increase in sarcoma-induced guarding. (c) Administration of naloxone with morphine prevented the sustained morphine-induced increase in tactile hypersensitivity. (d) Administration of naloxone with morphine prevented sustained morphine-induced increase in limping behavior. (e) Administration of naloxone with morphine prevented sustained morphine-induced increase in bone loss. (f) Administration of naloxone with morphine prevented the doubling in the incidence of spontaneous fracture. Naloxone alone had no effect on sarcoma-induced behaviors. Graphs (a–e) represent means ± SEM. *Indicates significant difference from control saline group.
Co-administration of naloxone with morphine also blocked sustained morphine-induced enhanced sarcoma- induced bone loss and increased spontaneous fracture (Fig. 5e and f). Bone rating scores of sarcoma-treated mice receiving both naloxone and morphine infusion as well as naloxone alone did not differ (p > 0.05) from sarcoma-treated mice receiving saline infusion indicating full reversal of sustained morphine-induced increase in sarcoma-induced bone loss (Fig. 5e). Co-administration of naloxone with morphine also completely blocked the increased rate of spontaneous fracture, with 100% of the mice treated with morphine showing spontaneous sarcoma-induced fracture, and 50% of the mice treated with naloxone/morphine infusion showing spontaneous sarcoma-induced fracture; this value was the same as that seen for sarcoma-treated mice treated with saline infusion or naloxone infusion (Fig. 5f). Administration of naloxone alone did not alter sarcoma-induced effects on bone loss.
3.8. Sustained morphine fails to affect tumor burden but enhances osteoclastogenesis
To determine the possible underlying mechanisms of morphine-induced enhanced bone loss we treated fibrosarcoma cells with different doses of morphine (−4, −5, −6, −7, −8 M) for 2, 4, or 6 days. Addition of morphine to the fibrosarcoma cells failed to alter tumor growth in vitro, indicating that morphine does not directly alter the growth of these cells (Fig. 6a). To determine whether sustained morphine altered tumor burden in vivo, femurs were collected 10 days following femoral injection, 3 days into infusion, and were stained with hematoxylin and eosin (H&E) to visualize normal marrow elements and cancer cells. Stained sections demonstrated equivalent distribution of sarcoma cells, with sarcoma observed in 100% of the metaphysis of the distal head of the femur in both saline- and morphine-treated mice (Fig. 6b). Analysis of tumor burden within the intra-medullary space of the bone showed that sustained morphine failed to alter tumor burden, with sarcoma observed in 75% of the bone in saline and morphine-infused mice. These data indicate that sustained morphine exposure fails to alter tumor growth or tumor burden within the bone.
Fig. 6.
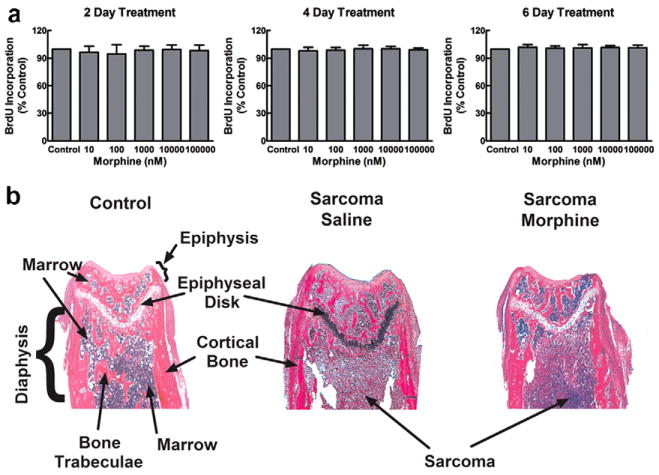
(a) Sarcoma cell growth was assessed in vitro by measuring BrdU incorporated into the sarcoma cells by ELISA. Sarcoma cells were untreated (control), or treated with various doses of morphine for 2, 4, or 6 days prior to assay. All assays were carried out in 96-well plates. Morphine treatment was timed such that the designated treatment period ended 4 days after plating, when the cells reached 50–70% confluency in the culture wells. The optical density at 450 nm in the morphine-treated samples is expressed as % of that in the untreated control cells done in parallel, and data are means ± SEM from three independent experiments. Morphine had no effect on the BrdU immunoreactivity incorporated into the cultured cells compared to control cells that were not treated with morphine. (b) Femur sections were stained with hematoxylin and eosin (H&E) on day 10. The distal ends of femurs from control mice show normal bone marrow, trabecular bone and cortical bone. The distal ends of sarcoma-treated femurs show tumor cells throughout the metaphysis of the distal head of the femur in both the saline- and morphine-treated mice.
Previous studies have shown that osteolytic cancers, such as the sarcoma cell line used in these studies, upregulate osteoclasts within the bone (Honore et al., 2000a; Sabino et al., 2002b). To determine whether morphine infusion enhanced sarcoma-induced bone loss by altering in bone resorption, osteoclasts were stained and counted within the metaphysis of the distal head of the femur (Fig. 7a and b). Osteoclast staining was significantly increased in sarcoma-treated animals compared to control animals. Morphine infusion did not alter osteoclast staining in control animals, suggesting that sustained morphine itself did not alter osteoclastogenesis. However, sustained morphine infusion increased sarcoma-induced upregulation of osteoclasts, indicating that sustained morphine increases sarcoma-induced upregulation of osteoclastogenesis.
Fig. 7.
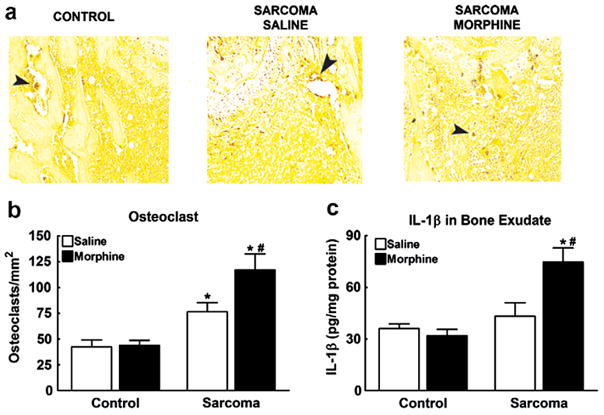
(a) Femur sections were stained with tartrate-resistant acid phosphatase (TRAP) on day 10 to visualize osteoclasts that stained dark red (indicated by arrows). Osteoclast staining was increased in sarcoma-treated mice compared to control mice. (b) Sarcoma increased the number of osteoclasts within the metaphysis of the femur. Morphine infusion further increased osteoclasts compared to saline-infused mice. Osteoclast counts did not differ between morphine- and saline-infused mice treated with control media. (c) IL-1β protein content was measured in exudate from the intramedullary space of the femurs on day 12. Sustained morphine infusion increased IL-1β protein content in sarcoma-treated mice. Sustained morphine infusion had no effect on IL-1β protein levels in control media-treated mice. Graphs show means ± SEM. *Indicates significant difference from control saline group, #indicates significant difference between saline- and morphine-treated mice within the sarcoma or the control groups.
3.9. Sustained morphine upregulates osteoclastogenic mediators in sarcoma-treated femurs
To determine potential underlying mechanisms of sustained morphine-induced increase in sarcomainduced upregulation of osteoclasts, we examined IL- 1β protein content in exudate from the intramedullary space of the femurs as IL-1 has been shown to play an important role in osteoclastogenesis (Blair et al., 2005; Wei et al., 2005). Sarcoma-treated mice receiving morphine infusion across 5 days showed enhanced IL-1β levels compared to all other groups (Fig. 7c).
4. Discussion
Many bone cancer patients receive treatment with opioids administered through oral administration of controlled release tablets or transdermal patches designed to keep steady blood plasma levels of the drug (Heiskanen and Kalso, 1997; Warfield, 1998; Mercadante, 1999b; Allan et al., 2001; Hanks et al., 2001). Using a mouse model in which osteolytic sarcoma cells are injected and sealed into the bone, we found that sustained morphine administration through osmostic minipumps that maintain consistent blood levels of morphine across the 7-day administration period: (a) enhanced, rather than alleviated, sarcoma-induced spontaneous and evoked pain in a dose-dependent and naloxone-sensitive manner; (b) increased expression of “pronociceptive” neural markers in the DRG not seen with sarcoma treatment alone; (c) increased neural markers of cell damage in sarcoma, but not control, mice; (d) increased osteoclastogenesis, accelerated sarcoma- induced bone loss and increased incidence of spontaneous fracture in a dose-dependent and naloxone-sensitive manner. The present data with morphine raise the possibility that maintaining cancer patients on sustained morphine might worsen, rather than alleviate, cancer-induced pain, and might accelerate cancer-induced bone loss in osteolytic cancers. It should be noted, however, that many factors are likely to influence the effects of prolonged opioid exposure on cancer-induced pain including: the specific opioid administered, the type of metastasis, whether osteolytic vs. osteoblastic lesions are observed, and the route, dose and/or schedule of opioid administration (Sacerdote et al., 2003; Tegeder et al., 2003; Ossipov et al., 2004; Urch et al., 2005).
Clinical reports have demonstrated that opioids administered through different routes of administration (e.g. spinal, transdermal, oral) can unexpectedly produce hyperalgesia in some patients, and may be associated with rapid opioid escalation (De Conno et al., 1991; Sjogren et al., 1993, 1994; Jacobsen et al., 1995; Mercadante et al., 2003; Chu et al., 2006). Increasing opioid dose and opioid rotation are used to achieve full-pain relief in this group of patients (Mercadante et al., 2003, 2005). A significant genetic basis for opioid- induced hyperalgesia may underlie individual variability in opioid response (Kest et al., 2002; Klepstad et al., 2005; Liang et al., 2005; Mogil et al., 2005). Numerous reports indicate that prolonged opiate administration can elicit hyperalgesia in rodents (Laulin et al., 1999; Vanderah et al., 2000; Gardell et al., 2002, 2006; Johnston et al., 2004; Juni et al., 2006). In the present study, morphine was delivered in a manner designed to produce stable blood morphine levels through minipumps (Feng et al., 2006) and was found to elicit a dose-dependent enhancement, rather than alleviation, of sarcoma-induced spontaneous and evoked pain behaviors. Moreover, while naloxone treatment alone did not alter sarcoma-induced pain, the antagonist prevented morphine-induced enhancement of sarcoma-induced pain. These data, and the observed agonist dose–response relationship, indicate that sustained morphine enhancement of sarcoma-induced pain occurs via morphine interaction with opiate receptors. While many cancer patients respond well to opioids for pain management (Zech and Lehmann, 1995; Mercadante, 1999a), these data raise the possibility that prolonged opioid administration might exacerbate bone cancer-induced pain in some patients and thus require supplemental pain medication to overcome both opioid- and cancer-induced hyperalgesia and pain.
Sarcoma-induced changes in neural markers of pain were similar to previous reports, including increased expression of activating transcription factor 3 (ATF3), a marker for neuronal damage (Peters et al., 2005; Sevcik et al., 2005), and no change in SP and CGRP within cell bodies of primary afferent fibers (Honore et al., 2000b). Sustained morphine administration increased the excitatory neurotransmitters, SP and CGRP, within primary afferent fibers in mice treated with control media as previously reported (Ma et al., 2000; Gardell et al., 2002; King et al., 2005a) as well as in mice treated with sarcoma. The enhanced expression of SP and CGRP in sarcoma-treated mice receiving morphine could represent an “add-on” mechanism of pain not normally participating in sarcoma-induced pain. In addition, morphine administration doubled the number of cells positive for ATF3 in sarcoma-treated mice, but not control, mice. These results indicate that while sustained morphine exposure does not produce cell damage alone, it increases sarcoma-induced neuronal damage. The morphine-induced pronociceptive neuroplastic changes within the primary afferent fibers in combination with the sarcoma-induced neuroplastic changes within the pain pathways likely play an important role in the observed morphine-induced increase in sarcoma-induced pain behaviors. Additionally, morphine treatment accelerated sarcoma-induced bone loss, and doubled the incidence of sarcoma-induced fracture 10 and 12 days following sarcoma injection. Such increased bone loss and fracture rate is likely to contribute to the morphine-induced pain in these animals.
One potential contributor to the enhanced sarcoma-induced bone loss in morphine-treated mice is a change in sarcoma growth and tumor burden in these animals. There are mixed reports on the effects of morphine on growth of cancer cells which are dependent upon the specific cell line examined, and the dose/concentration of morphine used in the studies. Several studies report that morphine inhibits cancer cell growth (Maneckjee and Minna, 1990, 1994; Sueoka et al., 1996; Sueoka et al., 1998; Tegeder et al., 2003; Zagon and McLaughlin, 2005) while others report that morphine increases cancer growth and instance of metastasis (Simon and Arbo, 1986; Ishikawa et al., 1993; Gupta et al., 2002) or no effect on cancer cell growth (Zagon and McLaughlin, 1984; Tegeder et al., 2003; Zagon and McLaughlin, 2005). In the murine sarcoma cell line used in these studies, sustained morphine exposure failed to alter tumor growth or tumor burden, suggesting that the enhanced sarcoma-induced bone loss observed in the morphine-infused mice may be due to alterations in bone resorption. Bone resorption and rebuilding take place continuously for bone maintenance and repair and are mediated by osteoclast (resorption) and osteoblast (building) activity (Boyce et al., 1999, 2003). Previous studies have shown that osteolytic cancers, such as the sarcoma cell line used in these studies, upregulate osteoclasts within the bone resulting in bone loss (Honore et al., 2000a; Luger et al., 2001; Sabino et al., 2002a). Consistent with these reports, osteoclast staining was increased in sarcoma-treated mice. Additionally, however, morphine infusion significantly increased sarcoma-induced upregulation of osteoclasts.
IL-1 plays an important role in osteoclastogenesis through direct mechanisms such as stimulating differentiation of osteoclast precursors and indirect mechanisms such as increasing RANKL expression, which plays a critical role in osteoclast maturation, activity and survival (Clohisy et al., 2003; Blair et al., 2005; Dougall and Chaisson, 2006). Previous studies demonstrated that sustained morphine administration increases IL-1 protein within the spinal cord and the lumbosacral CSF (Johnston et al., 2004). In our studies, morphine treatment increased IL-1β levels within the bone in sarcoma- treated mice, an effect that is likely to contribute to the observed osteoclastogenesis. IL-1β has also been implicated in peripheral sensitization after injury leading to inflammatory pain (Sommer and Kress, 2004). Therefore, morphine-induced increase in IL-1β within the bone is also likely to play a role in the sustained morphine- induced increase in sarcoma-induced pain.
An alternate explanation for the observed enhanced bone loss in morphine-treated sarcoma mice is that sustained morphine infusion produces pain relief leading to increased limb use which in turn results in enhanced bone destruction. This possibility seems unlikely as our data show that morphine exposure increased limping and guarding behavior, indicating more pain resulting in less weight applied to the limb and the likelihood of less limb use. It is well established that reduced loading on the skeleton leads to bone loss (Giangregorio and Blimkie, 2002; Damrongrungruang et al., 2004; Kondo et al., 2005). Indeed, limb disuse (rather than increased use) is associated with increased bone resorption and decreased bone formation, resulting in bone atrophy and fragility (Giangregorio and Blimkie, 2002). Thus, enhanced pain resulting in reduced bone use and diminished mechanical load on the femur may have contributed to the observed enhanced bone loss in the sarcoma-treated animals receiving morphine infusion.
In humans, pain resulting from bone metastases is primarily, and appropriately, treated with opioids. Bolus opioid administration has been shown to effectively alleviate bone cancer pain in humans as well as in animal models (Mercadante, 1991, 1998; Urch et al., 2005). However, sustained opioid delivery through patches (i.e. fentanyl) or extended release formulations (i.e. controlled-release morphine or oxycodone tablets) is becoming more commonly used for pain management in these patients (Heiskanen and Kalso, 1997; Warfield, 1998; Allan et al., 2001; Hanks et al., 2001). Our experiments demonstrate that prolonged morphine treatment may have negative effects on pain and bone mass. Increased understanding of opioid-induced changes in this controlled model will allow examination of other variables such as other opioids, drug interaction with other treatments such as bisphosphonates, NSAIDs, or chemotherapeutic treatments, and opioid effects on other cancers that metastasize to the bone, such as human prostate and breast cancers. The present data may lay foundations for studies that will further our basic understanding of opioid-induced actions in cancer pain, disease progression, and effects within the bone microenvironment and allow for the development of strategies which may improve the use of opioids for pain management.
Acknowledgments
Histological data on the bones were generated by the Tissue Acquisition and Cellular/Molecular Analysis Shared Service core at the Arizona Cancer Center, supported by NIH Grant CA23074. The authors would like to thank Kathy McDaniel, Erika Dexter and Wendy Molina for the technical expertise on histological analysis of the bones. The work in this manuscript was supported in part by NIH Grants DA11823, DA12656, DA1643, CA56666, and by the Arizona Biomedical Research Commission #9007.
References
- Allan L, Hays H, Jensen N-H, de Waroux BLP, Bolt M, Donald R, et al. Randomised crossover trial of transdermal fentanyl and sustained release oral morphine for treating chronic non-cancer pain. BMJ. 2001;322:1154–60. doi: 10.1136/bmj.322.7295.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baamonde A, Lastra A, Juarez L, Garcia V, Hidalgo A, Menendez L. Effects of the local administration of selective mu-, delta- and kappa-opioid receptor agonists on osteosarcoma-induced hyperalgesia. Naunyn Schmiedebergs Arch Pharmacol. 2005;372:213–9. doi: 10.1007/s00210-005-0013-6. [DOI] [PubMed] [Google Scholar]
- Beeton CA, Bord S, Ireland D, Compston JE. Osteoclast formation and bone resorption are inhibited by megakaryocytes. Bone. 2006;39:985–90. doi: 10.1016/j.bone.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Blair HC, Robinson LJ, Zaidi M. Osteoclast signalling pathways. Biochem Biophys Res Commun. 2005;328:728–38. doi: 10.1016/j.bbrc.2004.11.077. [DOI] [PubMed] [Google Scholar]
- Blum RH, Novetsky D, Shasha D, Fleishman S. The multidisciplinary approach to bone metastases. Oncology (Huntington) 2003;17:845–57. discussion 862–3, p. 867. [PubMed] [Google Scholar]
- Boyce BF, Hughes DE, Wright KR, Xing L, Dai A. Recent advances in bone biology provide insight into the pathogenesis of bone diseases. Lab Invest. 1999;79:83–94. [PubMed] [Google Scholar]
- Boyce BF, Xing L, Shakespeare W, Wang Y, Dalgarno D, Iuliucci J, et al. Regulation of bone remodeling and emerging breakthrough drugs for osteoporosis and osteolytic bone metastases. Kidney Int Suppl. 2003:S2–5. doi: 10.1046/j.1523-1755.63.s85.2.x. [DOI] [PubMed] [Google Scholar]
- Celerier E, Gonzalez JR, Maldonado R, Cabanero D, Puig MM. Opioid-induced hyperalgesia in a murine model of postoperative pain: role of nitric oxide generated from the inducible nitric oxide synthase. Anesthesiology. 2006;104:546–55. doi: 10.1097/00000542-200603000-00023. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chu LF, Clark DJ, Angst MS. Opioid tolerance and hyperalgesia in chronic pain patients after one month of oral morphine therapy: a preliminary prospective study. J Pain. 2006;7:43–8. doi: 10.1016/j.jpain.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Clohisy DR, Mantyh PW. Bone cancer pain. Cancer. 2003;97:866–73. doi: 10.1002/cncr.11144. [DOI] [PubMed] [Google Scholar]
- Clohisy DR, Ogilvie CM, Carpenter RJ, Ramnaraine ML. Localized, tumor-associated osteolysis involves the recruitment and activation of osteoclasts. J Orthop Res. 1996;14:2–6. doi: 10.1002/jor.1100140103. [DOI] [PubMed] [Google Scholar]
- Clohisy DR, Ogilvie CM, Ramnaraine ML. Tumor osteolysis in osteopetrotic mice. J Orthop Res. 1995;13:892–7. doi: 10.1002/jor.1100130613. [DOI] [PubMed] [Google Scholar]
- Clohisy JC, Frazier E, Hirayama T, Abu-Amer Y. RANKL is an essential cytokine mediator of polymethylmethacrylate particleinduced osteoclastogenesis. J Orthop Res. 2003;21:202–12. doi: 10.1016/S0736-0266(02)00133-X. [DOI] [PubMed] [Google Scholar]
- Coleman RE. Skeletal complications of malignancy. Cancer. 1997;80:1588–94. doi: 10.1002/(sici)1097-0142(19971015)80:8+<1588::aid-cncr9>3.3.co;2-z. [DOI] [PubMed] [Google Scholar]
- Coleman RE. Metastatic bone disease: clinical features, pathophysiology and treatment strategies. Cancer Treat Rev. 2001;27:165–76. doi: 10.1053/ctrv.2000.0210. [DOI] [PubMed] [Google Scholar]
- Damrongrungruang T, Kuroda S, Kondo H, Aoki K, Ohya K, Kasugai S. A simple murine model for immobilization osteopenia. Clin Orthop Relat Res. 2004:244–51. doi: 10.1097/00003086-200408000-00035. [DOI] [PubMed] [Google Scholar]
- De Conno F, Caraceni A, Zecca E, Spoldi E, Ventafridda V. Continuous subcutaneous infusion of hyoscine butylbromide reduces secretions in patients with gastrointestinal obstruction. J Pain Symptom Manage. 1991;6:484–6. doi: 10.1016/0885-3924(91)90005-o. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–62. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Dougall WC, Chaisson M. The RANK/RANKL/OPG triad in cancerinduced bone diseases. Cancer Metastasis Rev. 2006;25:541–9. doi: 10.1007/s10555-006-9021-3. [DOI] [PubMed] [Google Scholar]
- Feng P, Rahim RT, Cowan A, Liu-Chen L-Y, Peng X, Gaughan J, et al. Effects of mu, kappa or delta opioids administered by pellet or pump on oral Salmonella infection and gastrointestinal transit. Eur J Pharmacol. 2006;534:250–7. doi: 10.1016/j.ejphar.2006.01.048. [DOI] [PubMed] [Google Scholar]
- Gardell LR, King T, Ossipov MH, Rice KC, Lai J, Vanderah TW, et al. Opioid receptor-mediated hyperalgesia and antinociceptive tolerance induced by sustained opiate delivery. Neurosci Lett. 2006;396:44–9. doi: 10.1016/j.neulet.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Gardell LR, Wang R, Burgess SE, Ossipov MH, Vanderah TW, Malan TP, Jr, et al. Sustained morphine exposure induces a spinal dynorphin-dependent enhancement of excitatory transmitter release from primary afferent fibers. J Neurosci. 2002;22:6747–55. doi: 10.1523/JNEUROSCI.22-15-06747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giangregorio L, Blimkie CJ. Skeletal adaptations to alterations in weight-bearing activity: a comparison of models of disuse osteoporosis. Sports Med. 2002;32:459–76. doi: 10.2165/00007256-200232070-00005. [DOI] [PubMed] [Google Scholar]
- Gupta K, Kshirsagar S, Chang L, Schwartz R, Law P-Y, Yee D, et al. Morphine stimulates angiogenesis by activating proangiogenic and survival-promoting signaling and promotes breast tumor growth. Cancer Res. 2002;62:4491–8. [PubMed] [Google Scholar]
- Halleen JM, Karp M, Viloma S, Laaksonen P, Hellman J, Kakonen SM, et al. Two-site immunoassays for osteoclastic tartrate-resistant acid phosphatase based on characterization of six monoclonal antibodies. J Bone Miner Res. 1999;14:464–9. doi: 10.1359/jbmr.1999.14.3.464. [DOI] [PubMed] [Google Scholar]
- Hanks GW, Conno F, Cherny N, Hanna M, Kalso E, McQuay HJ, et al. Morphine and alternative opioids in cancer pain: the EAPC recommendations. Br J Cancer. 2001;84:587–93. doi: 10.1054/bjoc.2001.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiskanen T, Kalso E. Controlled-release oxycodone and morphine in cancer related pain. Pain. 1997;73:37–45. doi: 10.1016/s0304-3959(97)00072-9. [DOI] [PubMed] [Google Scholar]
- Honore P, Luger NM, Sabino MA, Schwei MJ, Rogers SD, Mach DB, et al. Osteoprotegerin blocks bone cancer-induced skeletal destruction, skeletal pain and pain-related neurochemical reorganization of the spinal cord. Nat Med. 2000a;6:521–8. doi: 10.1038/74999. [DOI] [PubMed] [Google Scholar]
- Honore P, Rogers SD, Schwei MJ, Salak-Johnson JL, Luger NM, Sabino MC, et al. Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience. 2000b;98:585–98. doi: 10.1016/s0306-4522(00)00110-x. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Tanno K, Kamo A, Takayanagi Y, Sasaki K. Enhancement of tumor growth by morphine and its possible mechanism in mice. Biol Pharm Bull. 1993;16:762–6. doi: 10.1248/bpb.16.762. [DOI] [PubMed] [Google Scholar]
- Itoh N, Kasai H, Ariyoshi W, Harada E, Yokota M, Nishihara T. Mechanisms involved in the enhancement of osteoclast formation by enamel matrix derivative. J Periodontal Res. 2006;41:273–9. doi: 10.1111/j.1600-0765.2005.00868.x. [DOI] [PubMed] [Google Scholar]
- Jacobsen LS, Olsen AK, Sjogren P, Jensen NH. Morphine-induced hyperalgesia, allodynia and myoclonus – new side-effects of morphine? Ugeskr Laeger. 1995;157:3307–10. [PubMed] [Google Scholar]
- Johnston IN, Milligan ED, Wieseler-Frank J, Frank MG, Zapata V, Campisi J, et al. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci. 2004;24:7353–65. doi: 10.1523/JNEUROSCI.1850-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juni A, Klein G, Kest B. Morphine hyperalgesia in mice is unrelated to opioid activity, analgesia, or tolerance: evidence for multiple diverse hyperalgesic systems. Brain Res. 2006;1070:35–44. doi: 10.1016/j.brainres.2005.11.054. [DOI] [PubMed] [Google Scholar]
- Kest B, Hopkins E, Palmese CA, Adler M, Mogil JS. Genetic variation in morphine analgesic tolerance: a survey of 11 inbred mouse strains. Pharmacol Biochem Behav. 2002;73:821–8. doi: 10.1016/s0091-3057(02)00908-5. [DOI] [PubMed] [Google Scholar]
- King T, Gardell LR, Wang R, Vardanyan A, Ossipov MH, Philip Malan TP, Jr, et al. Role of NK-1 neurotransmission in opioidinduced hyperalgesia. Pain. 2005a;116:276–88. doi: 10.1016/j.pain.2005.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T, Ossipov MH, Vanderah TW, Porreca F, Lai J. Is paradoxical pain induced by sustained opioid exposure an underlying mechanism of opioid antinociceptive tolerance? Neurosignals. 2005b;14:194–205. doi: 10.1159/000087658. [DOI] [PubMed] [Google Scholar]
- Klepstad P, Dale O, Borchgrevink PC, Kaasa S, Skorpen F. Genetic variation – important for the clinical effect of opioids? Tidsskr Nor Laegeforen. 2005;125:2655–8. [PubMed] [Google Scholar]
- Kondo H, Nifuji A, Takeda S, Ezura Y, Rittling SR, Denhardt DT, et al. Unloading induces osteoblastic cell suppression and osteoclastic cell activation to lead to bone loss via sympathetic nervous system. J Biol Chem. 2005;280:30192–200. doi: 10.1074/jbc.M504179200. [DOI] [PubMed] [Google Scholar]
- Laulin JP, Celerier E, Larcher A, Le Moal M, Simonnet G. Opiate tolerance to daily heroin administration: an apparent phenomenon associated with enhanced pain sensitivity. Neuroscience. 1999;89:631–6. doi: 10.1016/s0306-4522(98)00652-6. [DOI] [PubMed] [Google Scholar]
- Laulin JP, Larcher A, Celerier E, Le Moal M, Simonnet G. Longlasting increased pain sensitivity in rat following exposure to heroin for the first time. Eur J Neurosci. 1998;10:782–5. doi: 10.1046/j.1460-9568.1998.00083.x. [DOI] [PubMed] [Google Scholar]
- Liang D, Liao G, Peltz G, Clark DJ. IASP 11th World Congress on Pain. 2005 [Google Scholar]
- Luger NM, Honore P, Sabino MA, Schwei MJ, Rogers SD, Mach DB, et al. Osteoprotegerin diminishes advanced bone cancer pain. Cancer Res. 2001;61:4038–47. [PubMed] [Google Scholar]
- Luger NM, Sabino MAC, Schwei MJ, Mach DB, Pomonis JD, Keyser CP, et al. Efficacy of systemic morphine suggests a fundamental difference in the mechanisms that generate bone cancer vs. inflammatory pain Pain. 2002;99:397–406. doi: 10.1016/S0304-3959(02)00102-1. [DOI] [PubMed] [Google Scholar]
- Ma W, Zheng WH, Kar S, Quirion R. Morphine treatment induced calcitonin gene-related peptide and substance P increases in cultured dorsal root ganglion neurons. Neuroscience. 2000;99:529–39. doi: 10.1016/s0306-4522(00)00226-8. [DOI] [PubMed] [Google Scholar]
- Maneckjee R, Minna JD. Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc Natl Acad Sci USA. 1990;87:3294–8. doi: 10.1073/pnas.87.9.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maneckjee R, Minna JD. Opioids induce while nicotine suppresses apoptosis in human lung cancer cells. Cell Growth Differ. 1994;5:1033–40. [PubMed] [Google Scholar]
- Menendez L, Lastra A, Fresno MF, Llames S, Meana A, Hidalgo A, et al. Initial thermal heat hypoalgesia and delayed hyperalgesia in a murine model of bone cancer pain. Brain Res. 2003a;969:102–9. doi: 10.1016/s0006-8993(03)02284-4. [DOI] [PubMed] [Google Scholar]
- Menendez L, Lastra A, Hidalgo A, Meana A, Garcia E, Baamonde A. Peripheral opioids act as analgesics in bone cancer pain in mice. Neuroreport. 2003b;14:867–9. doi: 10.1097/00001756-200305060-00018. [DOI] [PubMed] [Google Scholar]
- Menendez L, Lastra A, Meana A, Hidalgo A, Baamonde A. Analgesic effects of loperamide in bone cancer pain in mice. Pharmacol Biochem Behav. 2005;81:114–21. doi: 10.1016/j.pbb.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Mercadante S. What is the definition of breakthrough pain? Pain. 1991;45:107–8. doi: 10.1016/0304-3959(91)90172-T. [DOI] [PubMed] [Google Scholar]
- Mercadante S. Predictive factors and opioid responsiveness in cancer pain. Eur J Cancer. 1998;34:627–31. doi: 10.1016/s0959-8049(97)10053-3. [DOI] [PubMed] [Google Scholar]
- Mercadante S. Opioid rotation for cancer pain: rationale and clinical aspects. Cancer. 1999a;86:1856–66. doi: 10.1002/(sici)1097-0142(19991101)86:9<1856::aid-cncr30>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Mercadante S. Problems of long-term spinal opioid treatment in advanced cancer patients. Pain. 1999b;79:1–13. doi: 10.1016/S0304-3959(98)00118-3. [DOI] [PubMed] [Google Scholar]
- Mercadante S, Arcuri E, Ferrera P, Villari P, Mangione S. Alternative treatments of breakthrough pain in patients receiving spinal analgesics for cancer pain. J Pain Symptom Manage. 2005;30:485–91. doi: 10.1016/j.jpainsymman.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Mercadante S, Ferrera P, Villari P, Arcuri E. Hyperalgesia: an emerging iatrogenic syndrome. J Pain Symptom Manage. 2003;26:769–75. doi: 10.1016/s0885-3924(03)00258-6. [DOI] [PubMed] [Google Scholar]
- Mercadante S, Fulfaro F. World Health Organization guidelines for cancer pain: a reappraisal. Ann Oncol. 2005;16:132–5. doi: 10.1093/annonc/mdi922. [DOI] [PubMed] [Google Scholar]
- Mercadante S, Portenoy RK. Opioid poorly-responsive cancer pain. part 2: basic mechanisms that could shift dose response for analgesia. J Pain Symptom Manage. 2001;21:255–64. doi: 10.1016/s0885-3924(00)00236-0. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Ritchie J, Smith SB, Strasburg K, Kaplan L, Wallace MR, et al. Melanocortin-1 receptor gene variants affect pain and muopioid analgesia in mice and humans. J Med Genet. 2005;42:583–7. doi: 10.1136/jmg.2004.027698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Malan TP, Jr, Hruby VJ, et al. Antinociceptive and nociceptive actions of opioids. J Neurobiol. 2004;61:126–48. doi: 10.1002/neu.20091. [DOI] [PubMed] [Google Scholar]
- Peters CM, Ghilardi JR, Keyser CP, Kubota K, Lindsay TH, Luger NM, et al. Tumor-induced injury of primary afferent sensory nerve fibers in bone cancer pain. Exp Neurol. 2005;193:85–100. doi: 10.1016/j.expneurol.2004.11.028. [DOI] [PubMed] [Google Scholar]
- Pud D, Cohen D, Lawental E, Eisenberg E. Opioids and abnormal pain perception: new evidence from a study of chronic opioid addicts and healthy subjects. Drug Alcohol Depend. 2006;82:218–23. doi: 10.1016/j.drugalcdep.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Ren WP, Markel DC, Zhang R, Peng X, Wu B, Monica H, et al. Association between UHMWPE particle-induced inflammatory osteoclastogenesis and expression of RANKL, VEGF, and Flt-1 in vivo. Biomaterials. 2006;27:5161–9. doi: 10.1016/j.biomaterials.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Ripamonti C, Dickerson ED. Strategies for the treatment of cancer pain in the new millennium. Drugs. 2001;61:955–77. doi: 10.2165/00003495-200161070-00005. [DOI] [PubMed] [Google Scholar]
- Sabino MA, Ghilardi JR, Jongen JL, Keyser CP, Luger NM, Mach DB, et al. Simultaneous reduction in cancer pain, bone destruction, and tumor growth by selective inhibition of cyclooxygenase-2. Cancer Res. 2002a;62:7343–9. [PubMed] [Google Scholar]
- Sabino MC, Ghilardi JR, Feia KJ, Jongen JL, Keyser CP, Luger NM, et al. The involvement of prostaglandins in tumorigenesis, tumorinduced osteolysis and bone cancer pain. J Musculoskelet Neuronal Interact. 2002b;2:561–2. [PubMed] [Google Scholar]
- Sacerdote P, Limiroli E, Gaspani L. Experimental evidence for immunomodulatory effects of opioids. Adv ExpMed Biol. 2003;521:106–16. [PubMed] [Google Scholar]
- Schwei MJ, Honore P, Rogers SD, Salak-Johnson JL, Finke MP, Ramnaraine ML, et al. Neurochemical and cellular reorganization of the spinal cord in a murine model of bone cancer pain. J Neurosci. 1999;19:10886–97. doi: 10.1523/JNEUROSCI.19-24-10886.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevcik MA, Ghilardi JR, Peters CM, Lindsay TH, Halvorson KG, Jonas BM, et al. Anti-NGF therapy profoundly reduces bone cancer pain and the accompanying increase in markers of peripheral and central sensitization. Pain. 2005;115:128–41. doi: 10.1016/j.pain.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Simon RH, Arbo TE. Morphine increases metastatic tumor growth. Brain Res Bull. 1986;16:363–7. doi: 10.1016/0361-9230(86)90057-2. [DOI] [PubMed] [Google Scholar]
- Sjogren P, Jensen NH, Jensen TS. Disappearance of morphine-induced hyperalgesia after discontinuing or substituting morphine with other opioid agonists. Pain. 1994;59:313–6. doi: 10.1016/0304-3959(94)90084-1. [DOI] [PubMed] [Google Scholar]
- Sjogren P, Jonsson T, Jensen NH, Drenck NE, Jensen TS. Hyperalgesia and myoclonus in terminal cancer patients treated with continuous intravenous morphine. Pain. 1993;55:93–7. doi: 10.1016/0304-3959(93)90188-U. [DOI] [PubMed] [Google Scholar]
- Sommer C, Kress M. Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett. 2004;361:184–7. doi: 10.1016/j.neulet.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Sueoka E, Sueoka N, Kai Y, Okabe S, Suganuma M, Kanematsu K, et al. Anticancer activity of morphine and its synthetic derivative, KT-90, mediated through apoptosis and inhibition of NF-kappaB activation. Biochem Biophys Res Commun. 1998;252:566–70. doi: 10.1006/bbrc.1998.9695. [DOI] [PubMed] [Google Scholar]
- Sueoka N, Sueoka E, Okabe S, Fujiki H. Anti-cancer effects of morphine through inhibition of tumour necrosis factor-alpha release and mRNA expression. Carcinogenesis. 1996;17:2337–41. doi: 10.1093/carcin/17.11.2337. [DOI] [PubMed] [Google Scholar]
- Takano H, Ariyoshi W, Kanno T, Fukuhara E, Ichimiya H, Matayoshi T, et al. Induction of osteoclast-like cells derived from the synovial lavage fluids of patients with temporomandibular joint disorders. Osteoarthritis Cartilage. 2006;15:291–9. doi: 10.1016/j.joca.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Takano H, Tomita T, Toyosaki-Maeda T, Maeda-Tanimura M, Tsuboi H, Takeuchi E, et al. Comparison of the activities of multinucleated bone-resorbing giant cells derived from CD14- positive cells in the synovial fluids of rheumatoid arthritis and osteoarthritis patients. Rheumatology (Oxford) 2004;43:435–41. doi: 10.1093/rheumatology/keh077. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Grosch S, Schmidtko A, Haussler A, Schmidt H, Niederberger E, et al. G protein-independent G1 cell cycle block and apoptosis with morphine in adenocarcinoma cells: involvement of p53 phosphorylation. Cancer Res. 2003;63:1846–52. [PubMed] [Google Scholar]
- Urch CE, Donovan-Rodriguez T, Gordon-Williams R, Bee LA, Dickenson AH. Efficacy of chronic morphine in a rat model of cancer-induced bone pain: behavior and in dorsal horn pathophysiology. J Pain. 2005;6:837–45. doi: 10.1016/j.jpain.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Gardell LR, Burgess SE, Ibrahim M, Dogrul A, Zhong CM, et al. Dynorphin promotes abnormal pain and spinal opioid antinociceptive tolerance. J Neurosci. 2000;20:7074–9. doi: 10.1523/JNEUROSCI.20-18-07074.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP, Jr, Lai J, Porreca F. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J Neurosci. 2001;21:279–86. doi: 10.1523/JNEUROSCI.21-01-00279.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C, He Q, Li G. Osteoclastogenesis in the nonadherent cell population of human bone marrow is inhibited by rhBMP-2 alone or together with rhVEGF. J Orthop Res. 2006;24:29–36. doi: 10.1002/jor.20010. [DOI] [PubMed] [Google Scholar]
- Warfield C. Controlled-release morphine tablets in patients with chronic cancer pain. Cancer. 1998;82:2299–306. doi: 10.1002/(sici)1097-0142(19980615)82:12<2299::aid-cncr1>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL. IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest. 2005;115:282–90. doi: 10.1172/JCI23394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. Cancer pain relief and palliative care: technical report series. Geneva, Switzerland: World Health Organization; 1986. [PubMed] [Google Scholar]
- Zagon IS, McLaughlin PJ. Opioids alter tumor cell growth and differentiation in vitro. NIDA Res Monogr. 1984;49:344–50. [PubMed] [Google Scholar]
- Zagon IS, McLaughlin PJ. Opioids and differentiation in human cancer cells. Neuropeptides. 2005;39:495–505. doi: 10.1016/j.npep.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Zech DF, Lehmann KA. Transdermal fentanyl in combination with initial intravenous dose titration by patient-controlled analgesia. Anticancer Drugs. 1995;6:44–9. doi: 10.1097/00001813-199504003-00008. [DOI] [PubMed] [Google Scholar]