

Abstract
The aim of this work was to elucidate the molecular mechanisms of flucytosine (5FC) resistance and 5FC/fluconazole (FLC) cross-resistance in 11 genetically and epidemiologically unrelated clinical isolates of Candida lusitaniae. We first showed that the levels of transcription of the FCY2 gene encoding purine-cytosine permease (PCP) in the isolates were similar to that in the wild-type strain, 6936. Nucleotide sequencing of the FCY2 alleles revealed that 5FC and 5FC/FLC resistance could be correlated with a cytosine-to-thymine substitution at nucleotide 505 in the fcy2 genes of seven clinical isolates, resulting in a nonsense mutation and in a putative nonfunctional truncated PCP of 168 amino acids. Reintroducing a FCY2 wild-type allele at the fcy2 locus of a ura3 auxotrophic strain derived from the clinical isolate CL38 fcy2(C505T) restored levels of susceptibility to antifungals comparable to those of the wild-type strains. In the remaining four isolates, a polymorphic nucleotide was found in FCY1 where the nucleotide substitution T26C resulted in the amino acid replacement M9T in cytosine deaminase. Introducing this mutated allele into a 5FC- and 5FC/FLC-resistant fcy1Δ strain failed to restore antifungal susceptibility, while susceptibility was obtained by introducing a wild-type FCY1 allele. We thus found a correlation between the fcy1 T26C mutation and both 5FC and 5FC/FLC resistances. We demonstrated that only two genetic events occurred in 11 unrelated clinical isolates of C. lusitaniae to support 5FC and 5FC/FLC resistance: either the nonsense mutation C505T in the fcy2 gene or the missense mutation T26C in the fcy1 gene.
Treatment of fungal infections is challenged by a limited number of available antifungal agents and by the emergence of antifungal resistance. Therefore, during the last few years, research has focused on elucidating the molecular mechanisms of antifungal resistance (4, 28). The haploid opportunistic yeast Candida lusitaniae (teleomorph, Clavispora lusitaniae) is a good model for studying antifungal resistance. Although much less common than other Candida species, this yeast is characterized by its propensity to develop resistance to antifungal agents during treatment, mainly to amphotericin B, but also to azole drugs and to flucytosine (5FC) (11, 16, 17, 22, 26).
5FC is a fluorinated pyrimidine that is used in combination with other antifungals to treat human cryptococcosis and sometimes candidiasis (3, 33). 5FC is actively transported into the cell through the action of purine-cytosine permease (PCP), encoded by the FCY2 gene. Once inside the cell, 5FC is rapidly converted to 5-fluorouracil (5FU) by cytosine deaminase (encoded by the FCY1 gene) and to 5-fluorouridine monophosphate (5FUMP) by uracil phosphoribosyltransferase (UPRTase; encoded by the FUR1 gene). 5FUMP is then converted to 5-fluorouridine triphosphate, which, when incorporated into fungal RNA instead of uridylic acid, disrupts protein synthesis. 5FUMP is also converted to 5-fluorodeoxyuridine monophosphate, which is a potent inhibitor of thymidylate synthase, an enzyme involved in DNA synthesis.
The mechanisms of 5FC resistance are based on mutations that result in deficiencies in the enzymes involved in antifungal uptake or metabolism (33). In Cryptococcus neoformans, it was shown that mutations in either the FCY1 or the FCY2 gene could confer 5FC resistance (34). In Candida albicans, Dodgson et al. (10) were able to correlate the primary 5FC resistance specific to clade I with a defective UPRTase due to a single nucleotide change in the FUR1 gene at residue 101. In a previous work, we demonstrated that 5FC resistance in four C. lusitaniae isolates was monogenic and was due to a defect in PCP encoded by the FCY2 gene, which was unable to transport 5FC (23). In addition, these strains were specifically cross-resistant to fluconazole (FLC) when both antifungals, 5FC and FLC, were used in combination. It was then hypothesized that extracellular 5FC would behave as a competitive inhibitor of FLC uptake (23). We recently provided molecular evidence that inactivation of the C. lusitaniae FCY2 and FCY1 genes promoted FLC resistance in the presence of subinhibitory 5FC concentrations (7, 24).
The present work explores the molecular events supporting 5FC resistance and 5FC/FLC cross-resistance in 11 genetically unrelated C. lusitaniae clinical isolates from our collection, including the 4 previously described and characterized at the biochemical level (23). In particular, we analyzed the sequences of the FCY2 and FCY1 genes in order to identify point mutations potentially involved in antifungal resistance. On the basis of gene replacement experiments, we first demonstrated that in seven clinical isolates, 5FC resistance and 5FC/FLC cross-resistance were correlated with a single cytosine-to-thymine substitution at nucleotide 505 in the fcy2 gene, resulting in a nonsense mutation and then in a putative nonfunctional truncated PCP. Second, in the remaining four isolates, we showed that the molecular events supporting resistance could be correlated with a single thymine-to-cytosine substitution at nucleotide 26 in the fcy1 gene, resulting in the amino acid substitution M9T at the protein level.
MATERIALS AND METHODS
C. lusitaniae strains and growth conditions.
The yeast strains used in this study are listed in Table 1. Strains 6936 (Centraalbureau voor Schimmelcultures, Utrecht, The Netherlands) and 42720 (American Type Culture Collection, Manassas, VA) were used as reference strains for FCY2 and FCY1 sequence analysis. Strain 6936 was also used for the cloning of FCY2, FCY21, and FCY1 wild-type genes and as a susceptible reference strain for antifungal agent susceptibility tests.
TABLE 1.
C. lusitaniae strains and isolates used in this study
| Strain | Genotypea | MIC (μg/ml)b
|
Origin/referencec | ||
|---|---|---|---|---|---|
| 5FC | FLC | 5FC + FLC | |||
| 42720 | MATα URA3 FCY2 | ≤0.5 | 2 | NO | ATCC |
| 6936 | MATaURA3 FCY2 | ≤0.5 | 2 | NO | CBS |
| fcy1Δ | MATaura3(D95V) fcy1::REP | 128 | 2 | 8-32 | 24 |
| fcy1ΔFCY1 | MATaura3(D95V) fcy1::(REP URA3 FCY1) | ≤0.5 | 2 | NO | This study |
| fcy1Δfcy1-26 | MATaura3(D95V) fcy1::(REP URA3 fcy1-26) | 128 | 2 | 8-32 | This study |
| fcy1Δfcy1-119 | MATaura3(D95V) fcy1::(REP URA3 fcy1-119) | 128 | 2 | 8-32 | This study |
| CL38-5 | MATα ura3-Δ360 fcy2 | NO | NO | NO. | This study |
| CL38-5URA3 | MATα ura3-Δ360::URA3 fcy2 | 32 | 2 | 8-16 | This study |
| CL38-5 FCY2 | MATα ura3-Δ360 fcy2::(URA3 FCY2) | ≤0.5 | 2 | NO | This study |
| CL38-5 FCY21 | MATα ura3-Δ360 fcy2 FCY21::(URA3 FCY21) | 32 | 2 | 8-16 | This study |
| CL26 | MATaURA3 FCY2 | 64 | ≤0.25 | 8-16 | Clin., blood/15, 23, this study |
| CL29 | MATα URA3 fcy2 | 128 | 2 | 8-32 | Clin., urine/15, 23 |
| CL31 | MATα URA3 fcy2 | 32 | 2 | 4-16 | Clin., feces/15, 23 |
| CL32 | MATaURA3 fcy2 | 128 | ≤0.25 | 8-16 | Clin., bedsore/15, 23, this study |
| CL38 | MATα URA3 fcy2 | 32 | 2 | 8-16 | Clin., nose/15, 23 |
| CL42 | MATaURA3 fcy2 | 64 | 4 | 8-32 | Clin., blood/15, 23 |
| CL48 | MATα URA3 fcy2 | 32 | 2 | 4-16 | Clin., catheter/this study |
| CL119 | MATα URA3 FCY2 | 32 | 1 | 8-16 | Clin., urine/this study |
| CL128 | MATα URA3 fcy2 | 32 | 1 | 8-16 | Clin., pharynx/this study |
| CL174 | MATα URA3 FCY2 | 64 | 1 | 8-16 | Clin., cutaneous sample/this study |
| CL216 | MATaURA3 FCY2 | 32 | 1 | 8-32 | Clin., urine/this study |
The mating types of C. lusitaniae clinical isolates were assigned as described previously (15).
Testing for the occurrence of a 5FC/FLC cross-resistance was performed using a 5FC concentration gradient from 256 to 0.5 μg/ml associated with a constant concentration of 16 μg/ml FLC, except for clinical isolates CL26, CL32, and CL174, for which 5FC/FLC cross-resistance was observed at a constant concentration of 8 μg/ml FLC. The values correspond to ranges of 5FC concentrations allowing growth of strains. NO, not observed.
CBS, Centraalbureau voor Schimmelcultures (Utrecht, The Netherlands). Clin., clinical isolate.
A collection of C. lusitaniae isolates currently containing 120 clinical isolates was screened for clinical isolates with a 5FC/FLC cross-resistance phenotype. Some of these isolates have been previously described (11, 21, 23, 25). Each of the 11 isolates selected for this study owing to their antifungal resistance phenotypes was recovered from an individual patient hospitalized in an intensive care unit in a French medical center between 1991 and 2003. Clinical isolates CL26, CL29, CL31, CL32, CL38, CL48, CL128, CL174, and CL216 were obtained from St. Joseph's Hospital or North Hospital (Marseille, France), whereas clinical isolates CL42 and CL119 were isolated at the Pitié-Salpêtrière Hospital (Paris, France). Clinical isolates CL26, CL29, CL31, CL32, CL38, and CL42 were previously screened for a 5FC-resistant phenotype (15, 23). It had been further demonstrated that 5FC resistance was due to a defect in PCP in four clinical isolates, CL29, CL31, CL38, and CL42 (23).
Yeast strains were routinely cultivated in liquid YPD medium (1% yeast extract, 2% peptone, 2% dextrose) at 35°C under agitation (250 rpm). Solid media were produced with 2% agar (Sigma).
Antifungal agents and susceptibility testing.
Stock solutions of 3.2 mg/ml FLC (ICN Biomedicals Inc.) and 12.8 mg/ml 5FC (Sigma) were prepared in sterile distilled water.
Each strain was initially tested for antifungal susceptibility by using microdilution assays according to CLSI standards (9). Yeast suspensions were diluted with RPMI 1640 medium (Sigma) to a final cell count of approximately 103 cells/ml, as previously described (7). The 96-well microtiter plates were incubated for 48 h at 35°C. Endpoint readings were recorded with an automated microtiter reader (Molecular Devices) and were calculated as MICs corresponding to the lowest concentration of drug that inhibited growth by 50% compared to the drug-free growth control. Final antifungal concentrations ranged from 0.5 to 256 μg/ml for 5FC and 0.25 to 128 μg/ml for FLC. Testing for the occurrence of 5FC/FLC cross-resistance was performed using a 5FC concentration gradient associated with a constant concentration of FLC of 8 or 16 μg/ml.
DNA and RNA extractions.
Genomic DNA was extracted by following the protocol described by Scherer and Stevens (29), except that zymolyase was replaced by lyticase (60 U/ml) for production of spheroplasts. Total RNAs were extracted using the RNeasy Mini Kit (Qiagen) associated with the RNase-free DNase set (Qiagen) according to the manufacturer's instructions.
PCR amplifications of FCY2, FCY21, and FCY1 genes; semiquantitative reverse transcription (RT)-PCR; and sequencing.
PCRs from genomic DNA were performed with Hot-Start Taq DNA polymerase (Qiagen). All the primers were synthesized by Invitrogen and are listed in Table 2. PCR conditions for amplification were those indicated by the supplier. The annealing temperatures were 60°C for the primer set FC21A/FC21S and 58°C for the primer pairs FC5/FC6, FC1N/FC1Sp, and FC1ex5/FC1ex3. PCR products were purified using the QIAquick PCR purification kit or the MinElute gel extraction kit (Qiagen) according to the manufacturer's instructions.
TABLE 2.
Oligonucleotides
| Primer | Sequence (5′ to 3′)a | PCR amplification | Reference |
|---|---|---|---|
| FC1 | AGCGAAGGGTTGAGTTGC | FCY2 gene (5′ primer) | 7 |
| FC2 | ATGTCGAGTTCCGTTCTC | FCY2 gene (3′ primer) | 7 |
| FC3 | CTGGTAGAATACAACACCG | FCY21 gene (5′ primer) | 7 |
| FC4 | CGGAACGTACGAGAGAGAC | FCY21 gene (3′ primer) | 7 |
| FC21A | TCGAGAGACGTCTTAGGTGGAACTGTACTG | FCY21 gene (5′ primer, AatII site) | This study |
| FC21S | CGAGCTCCGCGGCGGAACGTACGAGAGAGAC | FCY21 gene (3′primer, SacII site) | This study |
| FC5 | ACGGTTCGAGCGCAACTGG | FCY1 gene (5′ primer) | This study |
| FC6 | TTCTGCTACCAGCAAACGG | FCY1 gene (3′ primer) | This study |
| FC1N | GAGTCATGCATATAGTGGAAAGCCCCAATTAC | FCY1 gene (5′ primer, NsiI site) | This study |
| FC1Sp | GAGCCACTAGTCAATCGCTTACAATACAAGAC | FCY1 gene (3′ primer, SpeI site) | This study |
| FC1ex5 | TTCTGCTACTCACTCCAGGTGC | FCY1 gene (5′ primer) | This study |
| FC1ex3 | CTACAAGTGTCTCAAGTCATATTGG | FCY1 gene (3′ primer) | This study |
| FC1RT1 | TGATGACAAATTGGGGATGC | FCY1 mRNA (5′ primer) | This study |
| FC1RT2 | ATCTCGCCGTGCAAAGTCGC | FCY1 mRNA (3′ primer) | This study |
| ACT1 | AGCTCTGAATCTCTCGTTACC | ACT1 mRNA (5′ primer) | 8 |
| ACT2 | GTCGGTGACGAAGCTCAGTCC | ACT1 mRNA (3′ primer) | 8 |
Restriction sites are underlined.
RNA (0.5 μg) was reverse transcribed by using the RevertAid H Minus First Strand cDNA synthesis kit (Fermentas) and oligo(dT)18 primers. PCR was performed with Hot Star Taq DNA polymerase (Qiagen). One microliter of cDNA was used as a template for PCR. Oligonucleotides FC1RT1 and FC1RT2 (Table 2) were designed to allow amplification of a 165-bp 3′-end cDNA fragment from the respective mRNA. The conditions for amplification were 15 min at 95°C, followed by 30 cycles of 30 s at 94°C, 30 s at 58°C, 30 s at 72°C, and a final extension of 7 min at 72°C. The PCR products were visualized by electrophoresis in 2% agarose gels and quantified by using a DC290 camera coupled with the Kodak 1D 3.5.3 software. Expression of the C. lusitaniae actin-encoding gene (ACT1) was used as an internal control, as described previously (8).
All the amplified DNA or cDNA fragments were sequenced in both directions by Millegen (Labège, France).
Sequence analysis.
The open reading frames (ORFs) of FCY2 and FCY1 genes from strain 42720 were retrieved from the C. lusitaniae genome database available on the Broad Institute Fungal Genome website (http://www.broad.mit.edu/annotation/fungi/candida_lusitaniae/index.html). Similarity searches in the database were performed with the BLAST algorithm (2) using FCY2 (GenBank accession no. 506866) or FCY1 (GenBank accession no. DQ372926) of C. lusitaniae strain 6936.
The ORFs were identified from the sequenced nucleotide fragments using the Multiple Translation Program available on the Biosupport website (http://bioinfo.hku.hk/services/).
Consensus multiple alignments for nucleotide and amino acid sequences were done with the ClustalW program (32) available on the Pôle Bioinformatique Lyonnais website (http://pbil.univ-lyon1.fr/).
Plasmid constructions.
The plasmid pG-ura3[Δ360], which harbors a 360-bp deletion (nucleotides 213 to 572) located in the core of the ura3 gene, was previously described (8). This plasmid was used to transform the clinical isolate CL38 in order to obtain an auxotroph isolate for uracil. The pUFCY2 complementation plasmid (6.6 kb) was previously described (7). It contains the full-length FCY2 gene (2,117 bp) subcloned into the SpeI-NsiI restriction sites of the pGEM-U plasmid containing the C. lusitaniae URA3 gene (14).
To construct the pUFCY21 complementation plasmid, a 2,234-bp DNA fragment, overlapping the full-length FCY21 gene, flanked with the AatII and SacII restriction sites, was obtained by PCR amplification with the primer set FC21A/FC21S (Table 2). The resulting PCR product was then subcloned into the AatII-SacII restriction sites of the pGEM-U plasmid to generate the complementation plasmid pUFCY21 (6.7 kb).
To construct the pUFCY1 complementation plasmid, an 840-bp DNA fragment, overlapping the ORF of the FCY1 gene and flanked by SpeI and NsiI restriction sites, was obtained by PCR amplification with the primer set FC1N and FC1Sp (Table 2). The resulting PCR product was then subcloned into the SpeI-NsiI restriction sites of the pGEM-U plasmid to generate the complementation plasmid pUFCY1 (5.3 kb). In the same way, the ORFs of the fcy1 alleles from the clinical isolates CL26 (allele fcy1-26) and CL119 (allele fcy1-119) were amplified by the same primer pair and cloned into the pGEM-U plasmid to generate pUFCY1-26 and pUFCY1-119 plasmids.
Yeast transformation.
Yeast transformation was performed by the electroporation procedure, as previously described (14). To obtain the auxotrophic ura3-Δ360 fcy2 MATα CL38-5 strain, the clinical isolate CL38 was transformed with the circular plasmid pG-ura3[Δ360]. The yeast cells were then plated onto YNB solid medium (0.67% yeast nitrogen base without amino acids [Difco Laboratories], 2% glucose, 2% agar) supplemented with 1 M sorbitol and 25 μg/ml uracil (Fermentas) and incubated for 24 h at 35°C. Then, the yeast cells were washed and the ura3-Δ360 genotype was selected by plating the cells onto YNB solid medium supplemented with 1 mg/ml 5-fluoroorotic acid (Fermentas) and 25 μg/ml uracil.
For complementation experiments with the fcy2 mutation, the CL38-5 strain was transformed with the plasmid pUFCY2, pUFCY21, or pGEM-U, previously linearized within FCY2 (BamHI restriction site), FCY21 (BamHI restriction site), or URA3 (PstI restriction site), respectively, in order to obtain a greater efficiency of plasmid integration at the relevant homologous locus. For complementation of the fcy1 deletion, the fcy1Δ strain was transformed with plasmid pUFCY1, pUFCY1-26, or pUFCY1-119, previously linearized within FCY1 (BclI restriction site).
Ura+ transformants were selected on YNB medium supplemented with 1 M sorbitol after 3 to 4 days of incubation at 35°C.
A control experiment was performed by transforming the wild-type strain 6936 with a 1,026-bp PCR amplification overlapping the ORF of an FCY1 wild-type allele and of the mutated alleles fcy1-26 and fcy1-119, obtained with the primer pair FC1Ex5/FC1Ex3 (Table 2). Transformants were then selected on RPMI medium supplemented with 16 μg/ml 5FC.
Southern and Northern hybridizations.
For Southern analysis, approximately 10 μg of C. lusitaniae DNA was digested with appropriate restriction enzymes, separated by electrophoresis in a 0.8% agarose gel, and transferred onto a nylon membrane (Hybond N+; Roche Molecular Biochemicals). The membranes were hybridized with digoxigenin-labeled DNA probes synthesized with a PCR DIG probe synthesis kit (Roche Molecular Biochemicals) as recommended by the supplier. The FCY21 DNA probe (homologous to FCY21) and the FCY1 DNA probe (homologous to FCY1) were generated by PCR amplification, using primer pairs FC3/FC4 and FC1N/FC1Sp (Table 2), respectively, as described above. The Fi DNA probe, homologous to the core sequence of FCY2; the URA3 DNA probe, homologous to the C. lusitaniae URA3 locus; and the BLA DNA probe, specific to the plasmid sequence, were also generated with specific primers as previously described (7).
For Northern analysis, approximately 2 μg of RNA was separated in a 1% agarose gel containing formaldehyde, as described by Brown and Mackey (6). RNA was transferred onto a nylon membrane (Hybond N+; Roche Molecular Biochemicals) and hybridized with digoxigenin-labeled FCY2 and FCY21 RNA probes, antisense to FCY2 and FCY21 transcripts, synthesized with a DIG RNA labeling kit (Roche Molecular Biochemicals). For a loading control, membranes were stripped and rehybridized with an RNA probe complementary to a 460-bp fragment derived from the 18S rRNA gene of C. lusitaniae, as previously described (7).
For each experiment, probe-target hybrids were visualized by a chemiluminescent assay with the DIG luminescent detection kit (Roche Molecular Biochemicals), according to the manufacturer's instructions, and exposure of the blot to X-ray film for approximately 3 h.
RESULTS
In vitro susceptibility testing.
A collection of 120 C. lusitaniae clinical isolates was screened for 5FC/FLC cross-resistance phenotypes. The growth of each strain was thus measured by the twofold-microdilution method in the presence of FLC alone (a concentration gradient from 0.25 to 128 μg/ml), 5FC alone (a concentration gradient from 0.5 to 256 μg/ml), and 5FC in combination with 8 or 16 μg of FLC per ml. Eleven epidemiologically unrelated clinical isolates of C. lusitaniae were retrieved from the collection based on their 5FC/FLC cross-resistance phenotypes. They were isolated from different patients in geographically distinct hospitals and over a period of 13 years between the first and the last isolate. These strains were specifically cross-resistant to FLC when both antifungals, 5FC and FLC, were used in association, but they were FLC susceptible when FLC was used alone (Table 1). In the presence of 16 μg of FLC per ml, cross-resistance developed progressively in the presence of ranges of 5FC concentrations between 4 and 16 μg/ml (CL31 and CL48), 8 and 16 μg/ml (CL38, CL119, and CL128), and 8 and 32 μg/ml (CL29, CL42, and CL216). In clinical isolates CL26, CL32, and CL174, 5FC/FLC cross-resistance could be observed only in the presence of 8 μg/ml of FLC. Such an unusual interaction between 5FC and FLC had been previously characterized in the four isolates CL29, CL31, CL38, and CL42 and was shown to be derived from a defect in PCP encoded by FCY2 (23). All the isolates selected in this study were susceptible to 5FU, indicating that the genetic defect supporting both 5FC and 5FC/FLC resistance affected the genes working upstream of FUR1 in the pyrimidine salvage pathway, that is, either FCY2 or FCY1 (data not shown).
Expression analysis of FCY2 and FCY21 genes encoding PCP.
In a previous work, we showed that the PCP family was putatively encoded by two paralogs in C. lusitaniae, the FCY2 and FCY21 genes (7). In order to elucidate the molecular mechanism of 5FC/FLC cross-resistance in clinical isolates, we first compared the expression of the FCY2 and FCY21 genes in the clinical isolates CL29, CL31, CL38, and CL42 and in the wild-type strain 6936 by Northern blot analysis. We showed that the FCY2 gene was expressed at similar levels in all the strains analyzed and that, under our experimental conditions, the FCY21 mRNA could not be detected in the wild-type strain or in clinical isolates (Fig. 1). This analysis showed that the mechanism of resistance in these clinical isolates was unlikely to be correlated with a defect in FCY2 transcription. Comparable results were obtained when we analyzed the expression of FCY2 in the clinical isolates CL26, CL32, CL48, CL119, CL128, CL174, and CL216 (results not shown). These results also confirmed that FCY21 behaved as a pseudogene under our experimental conditions.
FIG. 1.
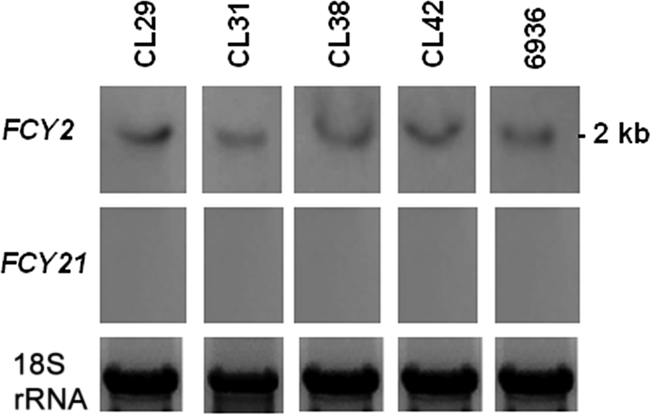
Expression analysis of C. lusitaniae FCY2 and FCY21 genes in clinical isolates and in the wild-type strain 6936. Shown is Northern blot analysis of total RNA from the clinical isolates CL29, CL31, CL38, and CL42 and from strain 6936 hybridized with FCY2 and FCY21 RNA probes. Membranes were stripped and hybridized with the 18S rRNA probe for RNA loading control.
Nucleotide sequencing of FCY2.
PCR products spanning the entire ORFs of the FCY2 gene were sequenced in the 11 clinical isolates. These sequences were aligned with those from the reference strains 6936 (GenBank accession no. 506866) and 42720 (retrieved from the C. lusitaniae genome database and referred to as CLUG 04172.1). Alignment of these 13 sequences first revealed that the percentage of variable codons in the ORF was approximately 1.5% and that they were randomly distributed along the coding regions of the different strains. However, the majority of the polymorphic nucleotide changes resulted in no change (synonymous mutation) in the respective amino acid residues (not shown).
The positions containing polymorphic nucleotides involved in an amino acid change are listed in Table 3. The FCY2 alleles showed 15 nonsynonymous polymorphic nucleotide sites dispersed throughout the gene. Among these 15 polymorphic sites, only 6 (at nucleotides 505, 922, 976, 1057, 1300, and 1450) could be potentially involved in point mutations linked to 5FC resistance, because the others were also found in the 5FC-susceptible 6936 or 42720 reference strain.
TABLE 3.
Polymorphisms in the FCY2 genes of 5FC/FLC cross-resistant C. lusitaniae clinical isolates
| Reference strain or isolatea | Codon and amino acid at positionb:
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 283 | 289 | 442 | 505 | 703 | 832 | 922 | 928 | 949 | 976 | 1057 | 1291 | 1300 | 1450 | 1477 | |
| 6936 | GTG | GCC | CTT | CAG | CAC | TAC | AAC | CAC | GAG | TAC | AAC | AGT | TCT | GGT | ATT |
| Val | Ala | Leu | Gln | His | Tyr | Asn | His | Glu | Tyr | Asn | Ser | Ser | Gly | Ile | |
| 42720 | TTG | GGC | TTT | CAG | ACC | TTT | ACC | GAC | AAG | TAC | AAC | AAC | ACT | CGT | GTT |
| Leu | Gly | Phe | Gln | Thr | Phe | Thr | Asp | Lys | Tyr | Asn | Asn | Thr | Gly | Val | |
| CL26 | TTG | GGC | TTT | CAG | ACC | TAC | ACC | GAC | AAG | TAC | AAC | AAC | ACT | CGT | GTT |
| Leu | Gly | Phe | Gln | Thr | Tyr | Thr | Asp | Lys | Tyr | Asn | Asn | Thr | Gly | Val | |
| CL29 | GTG | GCC | TTT | TAG | ACC | TAC | ACC | CAC | GAG | TGC | AAC | AGT | TCT | CGT | GTT |
| Val | Ala | Phe | Stop | Thr | Tyr | Thr | His | Glu | Cys | Asn | Ser | Ser | Gly | Val | |
| CL31 | TTG | GCC | TTT | TAG | ACC | TTC | ACC | GAC | AAG | TAC | AGC | AAC | ACT | CGT | GTT |
| Leu | Ala | Phe | Stop | Thr | Phe | Thr | Asp | LyS | Tyr | Ser | Asn | Thr | Gly | Val | |
| CL32 | GTG | GCC | TTT | TAG | ACC | TAC | ACC | CAC | GAG | TAC | AAC | AGT | TCT | CGT | GTT |
| Val | Ala | Phe | Stop | Thr | Tyr | Thr | His | Glu | Tyr | Asn | Ser | Ser | Gly | Val | |
| CL38 | GTG | GCC | TTT | TAG | ACC | TAC | ACC | CAC | GAG | TAC | AAC | AGT | TCT | CGT | GTT |
| Val | Ala | Phe | Stop | Thr | Tyr | Thr | His | Glu | Tyr | Asn | Ser | Ser | Gly | Val | |
| CL42 | GTG | GCC | TTT | TAG | ACC | TAC | ACC | CAC | GAG | TAC | AAC | AGT | TCT | AGT | GTT |
| Val | Ala | Phe | Stop | Thr | Tyr | Thr | His | Glu | Tyr | Asn | Ser | Ser | Ser | Val | |
| CL48 | GTG | GCC | TTT | TAG | ACC | TAC | AAC | CAC | GAG | TAC | AAC | AGT | TTT | CGT | GTT |
| Val | Ala | Phe | Stop | Thr | Tyr | Asn | His | Glu | Tyr | Asn | Ser | Phe | Gly | Val | |
| CL119 | TTG | GGC | TTT | CAG | ACC | TAC | ACC | CAC | GAG | TAC | AAC | AGT | TCT | CGT | GTT |
| Leu | Gly | Phe | Gln | Thr | Tyr | Thr | His | Glu | Tyr | Asn | Ser | Ser | Gly | Val | |
| CL128 | TTG | GCC | TTT | TAG | ACC | TAC | ATC | CAC | GAG | TAC | AAC | AGT | TTT | CGT | GTT |
| Val | Ala | Phe | Stop | Thr | Tyr | Ile | His | Glu | Tyr | Asn | Ser | Phe | Gly | Val | |
| CL174 | TTG | GCC | TTT | CAG | ACC | TTC | AAC | CAC | GAG | TAC | AAC | AGT | TCT | CGT | GTT |
| Val | Ala | Phe | Gln | Thr | Phe | Asn | His | Glu | Tyr | Asn | Ser | Ser | Gly | Val | |
| CL216 | TTG | GCC | TTT | CAG | ACC | TAC | AAC | CAC | GAG | TAC | AAC | AGT | TCT | CGT | GTT |
| Val | Ala | Phe | Gln | Thr | Tyr | Asn | His | Glu | Tyr | Asn | Ser | Ser | Gly | Val | |
FCY2 genes of strain 6936 (GenBank accession number 506866) and of strain 42720 (retrieved from the C. lusitaniae genome database; contig CLUG 04172.1) are the reference sequences.
Codon positions are given as the number of the first nucleotide in the ORF of the gene. At position 505, the nucleotide substitution from a cytosine to a thymidine resulted in a change from a glutamine codon (CAG) to a stop codon (TAG), which is shown by boldface type. Elsewhere, specific amino acid changes (and the corresponding codons) that occurred only in some clinical isolates but not in reference strains are in italics.
In seven clinical isolates (CL29, CL31, CL32, CL38, CL42, CL48, and CL128), the FCY2 gene was found to have a cytosine-to-thymidine substitution (underlined) at nucleotide 505, resulting in a change from a glutamine (CAG) codon to a stop (TAG) codon. This nonsense mutation could result in a nonfunctional truncated PCP of 168 amino acids, whereas the PCP in a wild-type strain corresponds to a protein of 513 amino acids. This result strongly suggests that 5FC resistance could be linked to the stop mutation at position 505 in these clinical isolates, four of which were already known to have a PCP defect (23). For this reason, the genotype fcy2 was attributed to these isolates. The remaining five point mutations were all found in clinical isolates harboring the nonsense mutation downstream from position 505, and it is therefore very unlikely that they could play a role in 5FC resistance. For the other clinical isolates, CL26, CL119, CL174, and CL216, all amino acid changes that occurred in the FCY2 sequence were also present either in strain 6936 or in strain 42720.
Complementation of the fcy2 mutation.
The preliminary step for complementation experimentation was the construction of the CL38-5 strain derived from the clinical isolate CL38 fcy2, which was genetically engineered to become an auxotroph for uracil. For this purpose, the clinical isolate CL38 was transformed with the circular plasmid pG-ura3[Δ360], and the ura3-Δ360 genotype was counterselected on YNB solid medium supplemented with 5-fluoroorotic acid and uracil. The genetic organizations of 10 5FOA-resistant Ura− clones were analyzed by Southern blotting (not shown). All of the 5FOA-resistant clones analyzed displayed the hybridization fragments expected from gene replacement at the ura3 locus. The genotype ura3-Δ360 fcy2 was assigned to the 5FOA-resistant transformants. One of them was named CL38-5 and was used in the subsequent complementation experiments.
The linear plasmid pUFCY2, which harbored both URA3 and FCY2 wild-type alleles, was used to transform the strain CL38-5 ura3-Δ360 fcy2 to prototrophy. We screened by Southern blotting for homologous integration of the whole plasmid pUFCY2 that occurred at the fcy2(C505T) locus, resulting in the relevant genotype ura3-Δ360 fcy2::(URA3 FCY2) (strain CL38-5 FCY2). Genomic DNAs were digested with BamHI and hybridized with an Fi probe, homologous to part of the FCY2 gene, along with DNA of the parental strain, CL38-5. All fragments of the expected size are shown in Fig. 2. The molecular events were confirmed by hybridization with the URA3 and BLA probes. In the same way, the linear plasmids pUFCY21 and pGEM-U were each used to transform the strain CL38-5. We detected by Southern blotting homologous integration of the whole plasmids pUFCY21 and pGEM-U that occurred at the FCY21 and ura3 loci, respectively, resulting in the relevant genotypes ura3-Δ360 fcy2 FCY21::(URA3 FCY21) (strain CL38-5 FCY21) and ura3-Δ360::URA3 fcy2 (strain CL38-5 URA3). Genomic DNAs were digested with EcoRV and hybridized with FCY21, URA3, and BLA probes, according to the experiment (results not shown).
FIG. 2.

Southern blot hybridization (B) and schematic representation (A) of a resident fcy2 locus and of molecular events that occurred in transformants. (A) The signals revealed by the labeled probes (each marked with an asterisk) correspond to those expected from the genomic restriction map. The hybridization patterns were visualized with the Fi, URA3, and BLA probes of BamHI-digested genomic DNA from CL38-5 (a) and from the reintegrant strain CL38-5 FCY2 (b). (B) Hybridization of genomic DNA with the URA3 probe revealed an additional fragment of 19.4 kb in both strains, which corresponds to the resident locus URA3. DNA fragment sizes are indicated in kilobases.
Screening for 5FC resistance and 5FC/FLC cross-resistance phenotypes revealed that only strain CL38-5 FCY2 recovered antifungal susceptibility levels identical to those of the susceptible reference strain 6936 or 42720 (Table 1). The CL38-5 FCY21 and CL38-5 URA3 strains were still cross-resistant to these antifungals, as was the clinical isolate CL38. Reintroducing a FCY2 allele from a wild-type strain into a 5FC/FLC-resistant fcy2 isolate was sufficient to restore an antifungal susceptibility pattern, whereas reintroducing an FCY21 allele was not.
Expression analysis of the FCY1 gene encoding cytosine deaminase and sequencing of cDNAs.
As 5FC resistance in clinical isolates CL26, CL119, CL174, and CL216 could not be correlated with point mutation in the FCY2 gene, we compared the expression levels of FCY1 in these isolates and in the wild-type strain 6936 by semiquantitative RT-PCR analysis (Fig. 3). We showed that the FCY1 gene was expressed at similar levels in all the strains analyzed. As BLAST analysis of the C. lusitaniae database revealed the presence of a predicted intron in the FCY1 gene, we completed this study by sequencing the amplified cDNAs. We confirmed the occurrence of an intron in the C. lusitaniae FCY1 gene located at nucleotides 56 to 105 from the ATG codon. This intron seemed to be correctly spliced in the wild-type strain 6936 and in the four clinical isolates.
FIG. 3.
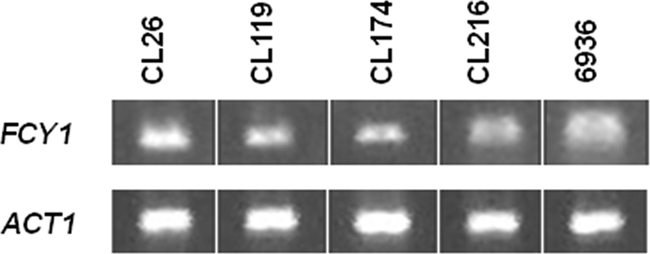
Expression levels of the FCY1 gene in the wild-type strain 6936 and in the clinical isolates CL26, CL119, CL174, and CL216. A representative semiquantitative RT-PCR analysis is shown (from three independent experiments). For each target gene, the amount of transcription was compared with that of the actin 1 (ACT1) gene.
The mechanism of resistance in these isolates was thus unlikely to be correlated with a defect in FCY1 transcription or with inappropriate splicing of the intron.
Nucleotide sequencing of FCY1.
PCR products spanning the entire ORFs of the FCY1 gene were then sequenced in isolates CL26, CL119, CL174, and CL216, as well as in all the fcy2 clinical isolates. These sequences were aligned with those from the reference strains 6936 (GenBank accession no. DQ372926) and 42720 (retrieved from the C. lusitaniae genome database and referred to as CLUG 05740.1). Alignment of these sequences revealed that the FCY1 gene has a level of polymorphism similar to that of FCY2 (1.5%), with only one polymorphism site, located at nucleotide 26, involved in an amino acid change. The reference strains have an ATG codon, which results in a methionine at position 9, whereas the isolates CL26, CL119, CL174, and CL216 have an ACG codon, which results in a threonine at the same position. It is worth noting that fcy2 clinical isolates do not have this mutation. These sequences were also aligned with those from Saccharomyces cerevisiae (GenBank accession no. NC001148), C. albicans (GenBank accession no. AJ616007), and Candida glabrata (GenBank accession no. CR380950). The amino acid residue methionine was also conserved in the cytosine deaminases of S. cerevisiae and C. glabrata, whereas in Fcy1p of C. albicans (GenBank accession no. AJ616007), a leucine was found at this position (Fig. 4).
FIG. 4.

Amino acid sequence alignment of cytosine deaminases encoded by the FCY1 gene from the C. lusitaniae wild-type strain 6936 (GenBank accession no. DQ372926), C. albicans (GenBank accession no. AJ616007), S. cerevisiae (GenBank accession no. NC001148), and C. glabrata (GenBank accession no. CR380950). The polymorphic amino acid residue that has been identified in Fcy1p sequences of C. lusitaniae at position 9 is framed. The regions involved in metal coordination and substrate protonation, described as signature sequences of the deaminase, family are shaded in gray. The amino acid residues that are thought to be essential for the binding of cytosine and 5FC are in boldface. Absolutely conserved residues are marked with asterisks, highly conserved residues are marked with colons, and weakly conserved residues are marked with periods.
Thus, the amino acid replacement M9T in cytosine deaminase occurred only in the four 5FC-resistant clinical isolates CL26, CL119, CL174, and CL216, and this point mutation was not present in the two susceptible reference strains and in the fcy2 clinical isolates.
Complementation of the fcy1 deletion.
In a previous work, a null mutant of the FCY1 gene was constructed by using an integrative transformation system based upon the URA3 blaster strategy (24). Antifungal susceptibility testing showed that the fcy1Δ mutant was resistant to 5FC and to 5FC/FLC association (Table 1).
The linear plasmids pUFCY1, pUFCY1-26, and pUFCY1-119, harboring both URA3 and a wild-type FCY1 gene, an fcy1-26 allele from isolate CL26, or an fcy1-119 allele from isolate CL119, respectively, were each used to transform the mutant fcy1Δ to prototrophy. We screened by Southern blotting homologous integration of the whole plasmids pUFCY1, pUFCY1-26, and pUFCY1-119 that occurred at the fcy1::REP locus, resulting in the relevant genotypes ura3(D95V) fcy1::(REP URA3 FCY1) (fcy1ΔFCY1 strain), ura3(D95V) fcy1::(REP URA3 fcy1-26) (fcy1Δfcy1-26 strain), and ura3(D95V) fcy1::(REP URA3 fcy1-119) (fcy1Δfcy1-119 strain). Genomic DNAs were digested with BamHI and hybridized with the FCY1 probe, homologous to a part of the FCY1 gene, along with DNA of the parental fcy1Δ strain. All fragments were the expected size (results not shown). The molecular events were confirmed by hybridization with the URA3 and BLA probes.
Screening for 5FC resistance and 5FC/FLC cross-resistance phenotypes revealed that only the fcy1ΔFCY1 strain recovered antifungal susceptibility levels identical to those of the susceptible reference strains 6936 and 42720 (Table 1). The fcy1Δfcy1-26 and fcy1Δfcy1 strains were still cross-resistant to these antifungals, as was the parental fcy1Δ mutant. Reintroducing an FCY1 allele from a wild-type strain into a 5FC/FLC-resistant fcy1Δ strain was sufficient to restore an antifungal susceptibility pattern, whereas reintroducing an fcy1-26 or fcy1-119 allele did not. A control experiment was performed by transforming the 5FC-susceptible wild-type strain 6936 with PCR products derived from amplification of an FCY1 wild-type allele and of the mutated alleles fcy1-26 and fcy1-119 and selecting transformants on medium supplemented with 16 μg/ml of 5FC. Fewer than 10 5FC-resistant transformants were obtained with the wild-type FCY1 allele, whereas several hundred transformants were obtained with the mutated alleles. Nucleotide sequencing of the FCY1 loci of 10 transformants obtained with the mutated alleles revealed that all of them had replaced the wild-type allele by integrating the M9T mutant allele. On the other hand, no change in the nucleotide sequence of the FCY1 loci of two transformants obtained with the wild-type allele was detected; the resistant phenotype was probably derived from mutations in other genes of the pyrimidine salvage pathway. Overall, these data strongly suggest that the M9T mutation supports 5FC resistance.
DISCUSSION
The aim of this work was to elucidate the molecular mechanisms of 5FC resistance and 5FC/FLC cross-resistance in 11 unrelated C. lusitaniae clinical isolates from our collection. We showed in previous works that the occurrence of 5FC/FLC cross-resistance in C. lusitaniae could be linked to inactivation either of FCY2 or of FCY1, whereas inactivation of the FUR1 gene led only to strong 5FC resistance (7, 24). Screening of our collection of 120 clinical isolates allowed us to select 12 5FC-resistant clinical isolates. Among them, only one isolate, CL60, was highly resistant to both 5FC and 5FU (MIC ≥ 256 μg/ml) and susceptible to 5FC/FLC association (result not shown), as previously described (23). Thus, this isolate could have a defective UPRTase encoded by the FUR1 gene. All 11 of the other 5FC-resistant isolates were also 5FC/FLC cross-resistant, suggesting that they could be affected in either the FCY2 or FCY1 gene. This indicates that the molecular mechanisms underlying 5FC resistance in C. lusitaniae are preferentially linked to a dysfunctioning of PCP or cytosine deaminase rather than of UPRTase, unlike what was described in C. albicans (18), where mutations in the FUR1 gene occurred more frequently (10, 18). Among the 5FC/FLC cross-resistant clinical isolates, four (CL29, CL31, CL38, and CL42) have been previously described as being defective in PCP (23). Thus, further characterization of the 11 clinical isolates was focused on the two paralogous genes that can code for a PCP in C. lusitaniae, FCY2 and FCY21. By detecting full-length mRNA specific to the FCY2 gene, we showed unequivocally that the expression level of the FCY2 gene in the clinical isolates was similar to that in the wild-type strain 6936 and, therefore, that a defect in PCP function was not linked to a decreased level of FCY2 transcription. We also demonstrated that, under our experimental conditions, FCY21 was not expressed in C. lusitaniae. It was further shown in this study that an FCY21 allele from a wild-type 5FC-susceptible strain could not functionally complement an fcy2-defective allele when introduced into a 5FC-resistant strain. Thus, it can be concluded that FCY21 has no role to play with regard to 5FC transport in C. lusitaniae.
The previously described nucleotide sequencing of the FCY2 alleles in clinical isolates CL29, CL31, CL38, and CL42 (23), as well as that of isolates CL32, CL48, and CL128 newly described in this study, revealed a nonsense C-T mutation at nucleotide 505, interrupting the ORF to give a putative truncated PCP of 168 amino acids (mRNAs are produced full length). In S. cerevisiae, functional analysis of Fcy2p revealed that two regions were crucial for PCP activity: (i) a transmembrane domain at positions 257 to 276 which could be part of a hydrophilic pore involved in the base-H+ translocation process and (ii) a segment at positions 371 to 377 located in a hydrophilic loop that was essential for uptake activity and specificity (12, 13). Sequence alignment of the C. lusitaniae PCP with S. cerevisiae Fcy2p showed that these two regions, relatively well conserved between the two organisms, were located at positions 236 to 255 and 349 to 355, respectively, in C. lusitaniae. As these crucial regions are located downstream from the point mutation in clinical isolates, if a truncated PCP is produced, it is probably not functional. For these reasons, and because a PCP defect had already been demonstrated by kinetic transport studies for four clinical isolates, the genotype fcy2 was attributed to these isolates. This genotype was then confirmed by complementation studies. Indeed, we demonstrated that reintegration of a wild-type FCY2 allele (derived from strain 6936) into the genome of a ura3Δ strain, fcy2(C505T) (derived from the clinical isolate CL38), was sufficient to restore 5FC and 5FC/FLC susceptibility.
In the four other clinical isolates, CL26, CL119, CL174, and CL216, we failed to correlate 5FC resistance and 5FC/FLC cross-resistance with point mutation in the FCY2 gene. As we showed in a previous work that disruption of the FCY1 gene, encoding cytosine deaminase, could also be responsible for 5FC resistance and 5FC/FLC cross-resistance in C. lusitaniae (24), a putative dysfunction of the cytosine deaminase gene in these four isolates was investigated. By semiquantitative RT-PCR analysis, we first showed that the expression levels of the FCY1 genes in the four clinical isolates were similar to that in the wild-type strain 6936 and therefore that a defect in cytosine deaminase function was not linked to a decreased level of FCY1 transcription. Nucleotide sequencing of the FCY1 alleles in these isolates allowed us to detect a nucleotide substitution, T26C, resulting in the amino acid replacement M9T in cytosine deaminase in the four 5FC-resistant clinical isolates CL26, CL119, CL174, and CL216. This point mutation was not present in the two susceptible reference strains and in the fcy2 clinical isolates. Multiple-sequence alignment of Fcy1p of Candida sp. and S. cerevisiae revealed that the active-site architecture involved in metal coordination and substrate protonation (HXE and CXXC), which constitutes the signature sequence of the deaminase family (20), was strongly conserved (HE and C[H/S/D]MC). These two regions were located at positions 55 to 57 and 84 to 87, respectively, in C. lusitaniae (Fig. 4). Furthermore, 4 residues of the S. cerevisiae cytosine deaminase (F114, W152, D155, and I156), especially important for the binding of cytosine and of the alternate substrate 5FC (19), were strictly conserved in an Fcy1p Candida sp. (Fig. 4). In C. albicans, it has been shown that a single 5FC-resistant isolate had a homozygous polymorphism in FCA1 that resulted in a glycine-to-aspartate substitution at position 28 in cytosine deaminase (18). This point mutation was thus located upstream of these noteworthy regions, like the methionine-to-threonine substitution at position 9 in the four isolates of C. lusitaniae, indicating that other regions in the N-terminal part of the protein are important for enzyme activity.
We then demonstrated that reintegration of an FCY1 allele from a wild-type strain into a 5FC/FLC-resistant fcy1Δ strain was sufficient to restore an antifungal susceptibility pattern, whereas reintroducing an fcy1-26 (from isolate CL26) or fcy1-119 (from isolate CL119) allele was not. For these reasons, the genotype fcy1 was attributed to isolates CL26, CL119, CL174, and CL216. Moreover, as a mutation at the same point occurred in four different isolates, these strains could also constitute a specific clade in C. lusitaniae, like the fcy2 clinical isolates.
The occurrence of the same single point mutation in the fcy2 genes of seven different clinical isolates and of the same single point mutation in the fcy1 genes of four clinical isolates raises the question of the origins of these mutations and why they were selected. These isolates, which have been isolated over a period of 13 years in different French medical centers, appeared to be genetically distant. For example, CL32 and CL42 were MATa, whereas the others were MATα, and some polymorphic nucleotide changes (mainly synonymous mutations) were equitably distributed in the fcy2 gene sequence. Thus, two hypotheses could be invoked. First, these mutations may have evolved under 5FC selection pressure as the sole possibilities to efficiently inactivate 5FC uptake or deamination. Such a hypothesis suggests that all patients have undergone 5FC antifungal therapy, which is not the case. Additionally, in the FCY2 gene, there are 48 possibilities for other putative mutations that would have resulted in a nonsense mutation before nucleotide 505. Alternatively, these strains could derive from a remote common ancestor in which inactivation of the pyrimidine salvage pathway by a mutation in FCY2 or FCY1 could have conferred a selective advantage over wild-type strains. In S. cerevisiae, it has been shown that the FUR4-encoded uracil permease, which catalyzes the first step of the pyrimidine salvage pathway, is downregulated by uracil, which is toxic to cells with high permease activity (5, 30). Thus, inactivation of the pyrimidine salvage pathway in clinical isolates could efficiently prevent a high level of pyrimidine nucleotides that could be toxic for the cell. These strains could constitute a specific clade in C. lusitaniae, like clade I of C. albicans (27), in which primary resistance to 5FC was linked to a single nucleotide change in the FUR1 gene (10). The occurrence of a specific 5FC-resistant clade had been also described in Candida dubliniensis (1) and in Candida tropicalis (31). It would be interesting to determine if the inactivation of the pyrimidine salvage pathway in these yeasts is selected for during evolution, notably in the context of better adaptation to the human host.
Overall, molecular characterization of 5FC resistance associated with 5FC/FLC cross-resistance in 11 C. lusitaniae clinical isolates allowed us to elucidate the genetic event occurring in the fcy2 genes of 7 isolates and in the fcy1 genes of 4 isolates. However, further investigations are necessary to decipher the origins of these mutations and the reason they were selected and maintained during the evolution of genetically and epidemiologically unrelated C. lusitaniae isolates.
Footnotes
Published ahead of print on 4 May 2009.
REFERENCES
- 1.Al Mosaid, A., D. J. Sullivan, I. Polacheck, F. A. Shaheen, O. Soliman, S. Al Hedaithy, S. Al Thawad, M. Kabadaya, and D. C. Coleman. 2005. Novel 5-flucytosine-resistant clade of Candida dubliniensis from Saudi Arabia and Egypt identified by Cd25 fingerprinting. J. Clin. Microbiol. 43:4026-4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balkis, M. M., S. D. Leidich, P. K. Mukherjee, and M. A. Ghannoum. 2002. Mechanisms of fungal resistance: an overview. Drugs 62:1025-1040. [DOI] [PubMed] [Google Scholar]
- 4.Barker, K. S., and P. D. Rogers. 2006. Recent insights into the mechanisms of antifungal resistance. Curr. Infect. Dis. Rep. 8:449-456. [DOI] [PubMed] [Google Scholar]
- 5.Blondel, M. O., J. Morvan, S. Dupre, D. Urban-Grimal, R. Haguenauer-Tsapis, and C. Volland. 2004. Direct sorting of the yeast uracil permease to the endosomal system is controlled by uracil binding and Rsp5p-dependent ubiquitylation. Mol. Biol. Cell 15:883-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown, T., and K. Mackey. 1997. Analysis of RNA by Northern and slot blot hybridization, p. 4.9.1-4.9.16. In F. M. Ausubel, R. Brendt, R. E. Kingston, et al. (ed.), Current protocols in molecular biology, vol. 1. John Wiley & Sons, New York, NY. [DOI] [PubMed] [Google Scholar]
- 7.Chapeland-Leclerc, F., J. Bouchoux, A. Goumar, C. Chastin, J. Villard, and T. Noel. 2005. Inactivation of the FCY2 gene encoding purine-cytosine permease promotes cross-resistance to flucytosine and fluconazole in Candida lusitaniae. Antimicrob. Agents Chemother. 49:3101-3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapeland-Leclerc, F., P. Paccallet, G. Ruprich-Robert, D. Reboutier, C. Chastin, and N. Papon. 2007. Differential involvement of histidine kinase receptors in pseudohyphal development, stress adaptation, and drug sensitivity of the opportunistic yeast Candida lusitaniae. Eukaryot. Cell 6:1782-1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.CLSI. 2007. Development of in vitro susceptibility testing criteria and quality ontrol parameters; approved standard, 3rd ed. CLSI document M23-A3. CLSI, Wayne, PA.
- 10.Dodgson, A. R., K. J. Dodgson, C. Pujol, M. A. Pfaller, and D. R. Soll. 2004. Clade-specific flucytosine resistance is due to a single nucleotide change in the FUR1 gene of Candida albicans. Antimicrob. Agents Chemother. 48:2223-2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Favel, A., A. Michel-Nguyen, F. Peyron, C. Martin, L. Thomachot, A. Datry, J. P. Bouchara, S. Challier, T. Noel, C. Chastin, and P. Regli. 2003. Colony morphology switching of Candida lusitaniae and acquisition of multidrug resistance during treatment of a renal infection in a newborn: case report and review of the literature. Diagn. Microbiol. Infect. Dis. 47:331-339. [DOI] [PubMed] [Google Scholar]
- 12.Ferreira, T., J. Chevallier, P. Paumard, C. Napias, and D. Brethes. 1999. Screening of an intragenic second-site suppressor of purine-cytosine permease from Saccharomyces cerevisiae. Possible role of Ser272 in the base translocation process. Eur. J. Biochem. 260:22-30. [DOI] [PubMed] [Google Scholar]
- 13.Ferreira, T., C. Napias, J. Chevallier, and D. Brethes. 1999. Evidence for a dynamic role for proline376 in the purine-cytosine permease of Saccharomyces cerevisiae. Eur. J. Biochem. 263:57-64. [DOI] [PubMed] [Google Scholar]
- 14.Francois, F., F. Chapeland-Leclerc, J. Villard, and T. Noel. 2004. Development of an integrative transformation system for the opportunistic pathogenic yeast Candida lusitaniae using URA3 as a selection marker. Yeast 21:95-106. [DOI] [PubMed] [Google Scholar]
- 15.Francois, F., T. Noel, R. Pepin, A. Brulfert, C. Chastin, A. Favel, and J. Villard. 2001. Alternative identification test relying upon sexual reproductive abilities of Candida lusitaniae strains isolated from hospitalized patients. J. Clin. Microbiol. 39:3906-3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guinet, R., J. Chanas, A. Goullier, G. Bonnefoy, and P. Ambroise-Thomas. 1983. Fatal septicemia due to amphotericin B-resistant Candida lusitaniae. J. Clin. Microbiol. 18:443-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hawkins, J. L., and L. M. Baddour. 2003. Candida lusitaniae infections in the era of fluconazole availability. Clin. Infect. Dis. 36:e14-e18. [DOI] [PubMed] [Google Scholar]
- 18.Hope, W. W., L. Tabernero, D. W. Denning, and M. J. Anderson. 2004. Molecular mechanisms of primary resistance to flucytosine in Candida albicans. Antimicrob. Agents Chemother. 48:4377-4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ireton, G. C., M. E. Black, and B. L. Stoddard. 2003. The 1.14 Å crystal structure of yeast cytosine deaminase: evolution of nucleotide salvage enzymes and implications for genetic chemotherapy. Structure 11:961-972. [DOI] [PubMed] [Google Scholar]
- 20.Ko, T. P., J. J. Lin, C. Y. Hu, Y. H. Hsu, A. H. Wang, and S. H. Liaw. 2003. Crystal structure of yeast cytosine deaminase. Insights into enzyme mechanism and evolution. J. Biol. Chem. 278:19111-19117. [DOI] [PubMed] [Google Scholar]
- 21.Michel-Nguyen, A., A. Favel, C. Chastin, M. Selva, and P. Regli. 2000. Comparative evaluation of a commercial system for identification of Candida lusitaniae. Eur. J. Clin. Microbiol. Infect. Dis. 19:393-395. [DOI] [PubMed] [Google Scholar]
- 22.Minari, A., R. Hachem, and I. Raad. 2001. Candida lusitaniae: a cause of breakthrough fungemia in cancer patients. Clin. Infect. Dis. 32:186-190. [DOI] [PubMed] [Google Scholar]
- 23.Noel, T., F. Francois, P. Paumard, C. Chastin, D. Brethes, and J. Villard. 2003. Flucytosine-fluconazole cross-resistance in purine-cytosine permease-deficient Candida lusitaniae clinical isolates: indirect evidence of a fluconazole uptake transporter. Antimicrob. Agents Chemother. 47:1275-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papon, N., T. Noel, M. Florent, S. Gibot-Leclerc, D. Jean, C. Chastin, J. Villard, and F. Chapeland-Leclerc. 2007. Molecular mechanism of flucytosine resistance in Candida lusitaniae: contribution of the FCY2, FCY1, and FUR1 genes to 5-fluorouracil and fluconazole cross-resistance. Antimicrob. Agents Chemother. 51:369-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peyron, F., A. Favel, H. Guiraud-Dauriac, M. El Mzibri, C. Chastin, G. Dumenil, and P. Regli. 1997. Evaluation of a flow cytofluorometric method for rapid determination of amphotericin B susceptibility of yeast isolates. Antimicrob. Agents Chemother. 41:1537-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pfaller, M. A., S. A. Messer, and R. J. Hollis. 1994. Strain delineation and antifungal susceptibilities of epidemiologically related and unrelated isolates of Candida lusitaniae. Diagn. Microbiol. Infect. Dis. 20:127-133. [DOI] [PubMed] [Google Scholar]
- 27.Pujol, C., M. A. Pfaller, and D. R. Soll. 2004. Flucytosine resistance is restricted to a single genetic clade of Candida albicans. Antimicrob. Agents Chemother. 48:262-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanglard, D., and F. C. Odds. 2002. Resistance of Candida species to antifungal agents: molecular mechanisms and clinical consequences. Lancet Infect. Dis. 2:73-85. [DOI] [PubMed] [Google Scholar]
- 29.Scherer, S., and D. A. Stevens. 1987. Application of DNA typing methods to epidemiology and taxonomy of Candida species. J. Clin. Microbiol. 25:675-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seron, K., M. O. Blondel, R. Haguenauer-Tsapis, and C. Volland. 1999. Uracil-induced down-regulation of the yeast uracil permease. J. Bacteriol. 181:1793-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tavanti, A., A. D. Davidson, E. M. Johnson, M. C. Maiden, D. J. Shaw, N. A. Gow, and F. C. Odds. 2005. Multilocus sequence typing for differentiation of strains of Candida tropicalis. J. Clin. Microbiol. 43:5593-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vermes, A., H. J. Guchelaar, and J. Dankert. 2000. Flucytosine: a review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J. Antimicrob. Chemother. 46:171-179. [DOI] [PubMed] [Google Scholar]
- 34.Whelan, W. L. 1987. The genetic basis of resistance to 5-fluorocytosine in Candida species and Cryptococcus neoformans. Crit. Rev. Microbiol. 15:45-56. [DOI] [PubMed] [Google Scholar]