

Abstract
In the eubacterial cell, the peptidoglycan is perpetually hydrolyzed throughout the cell cycle by different enzymes such as lytic transglycosylases, endopeptidases, and amidases. In Escherichia coli, four N-acetylmuramoyl-l-alanine amidases, AmiA, -B, -C, and -D, are present in the periplasm. AmiA, -B, and -C are soluble enzymes, whereas AmiD is a lipoprotein anchored in the outer membrane. To determine more precisely the specificity and the kinetic parameters of AmiD, we overproduced and purified the native His-tagged AmiD in the presence of detergent and a soluble truncated form of this enzyme by removing its signal peptide and the cysteine residue responsible for its lipidic anchorage. AmiD is a zinc metalloenzyme and is inactivated by a metal chelator such as EDTA. Native His-tagged and truncated AmiD hydrolyzes peptidoglycan fragments that have at least three amino acids in their peptide chains, and the presence of an anhydro function on the N-acetylmuramic acid is not essential for its activity. The soluble truncated AmiD exhibits a biphasic kinetic time course that can be explained by the inactivation of the enzyme by the substrate. This behavior highlights a new strategy to inhibit this class of enzymes.
The peptidoglycan or murein is a semirigid macromolecule surrounding the cytoplasmic membrane of eubacteria. It is an essential component of the cell wall that confers their shape to bacterial cells and protects them against high internal osmotic pressures. The peptidoglycan is composed of glycan chains of alternating units of N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) that are interconnected by covalent cross-links between short peptides (29, 31). The peptide is bound to the carboxyl of the lactyl moiety of MurNAc; in Escherichia coli, its primary structure is l-Ala-γ-d-Glu-mA2pm-d-Ala (with mA2pm as an abbreviation for meso-diaminopimelic acid). The peptidoglycan cross-link is achieved by an amide bond between the carboxyl group of the C-terminal d-Ala of one peptide and the free amino group of the mA2pm side chain of another peptide borne by an adjacent glycan chain (29, 31). Far from behaving as a static structure, the peptidoglycan is continuously hydrolyzed throughout the cell cycle by different enzymes such as lytic transglycosylases, endopeptidases, and amidases (22, 32). These different hydrolytic enzymatic activities involved in peptidoglycan turnover are essential both to allow the growth of the cell and the incorporation of new peptidoglycan chains by cell wall synthetases (transglycosylases and d,d-transpeptidases) and to separate daughter cells during the division (18).
In E. coli, the peptidoglycan fragments generated by peptidoglycan turnover, mainly the GlcNAc-1,6-anhydro-N-acetylmuramic acid (GlcNAc-anhydroMurNAc)-tripeptides and tetrapeptides, are transported into the cytoplasm to be recycled (24). Among the different cytoplasmic enzymes involved in the degradation of the recycled products, the anhydroMurNAc-l-alanine amidase AmpD liberates the peptides from the sugar moieties and the free tripeptides are reintroduced in the biosynthesis of the cytoplasmic cell wall precursor (19, 20).
Cytoplasmic AmpD is not the only peptidoglycan l-alanine amidase present in E. coli; four other extracellular amidases are also present, namely, AmiA, AmiB, AmiC (15, 16, 18), and YbjR, now renamed AmiD (11, 28) (S. Petrella, R. Herman, C. Généreux, A. Pennartz, E. Sauvage, B. Joris, and P. Charlier, crystal structure of native AmiD at 1.8 angström; RSCB Protein Data Bank, 2006) (Fig. 1). Based on their amino acid sequence similarities, these proteins can be subdivided into two families. The first one groups AmiA, -B, and -C enzymes, which are members of the amidase_3 superfamily (PF01520) of the Pfam database of protein families (http://pfam.sanger.ac.uk/). The second one contains AmiD and AmpD amidases, which belong to the amidase_2 superfamily (PF01510 in the Pfam database).
FIG. 1.

Schematic structure of the E. coli peptidoglycan. Cleavage sites of N-acetylmuramoyl-l-alanine amidases are indicated by the arrows. Tetrapeptides, tripeptides, and cross-linked tetrapeptides (octapeptides) are boxed.
During the E. coli cell cycle, AmiC amidase is localized to the septal ring and is involved in the splitting of the peptidoglycan septum during cell division (32). AmiA appears to be dispersed throughout the periplasm (32). However, it seems that AmiA and B can take over the role of AmiC in the divisome complex (26). The AmpD three-dimensional structure shows a high resemblance to the T7 bacteriophage lysozyme structure, which is also a peptidoglycan l-alanine amidase, and to those of the eukaryotic peptidoglycan recognition proteins (7) involved in the innate response against bacteria (8). AmpD exhibits a strict specificity for the 1,6-anhydro-muropeptides (12) and, as the T7 bacteriophage lysozyme, has a zinc ion in its catalytic site (3, 21, 23). The peptidoglycan recognition proteins possessing catalytic activity also have a zinc ion in their catalytic sites. In contrast, those proteins without enzymatic activity do not chelate a zinc ion but act as receptors (8).
The AmiD primary structure exhibits 40% identity to the AmpD sequence. Although AmiD is described in the literature as an anhydroMurNAc-l-alanine amidase, it seems that its enzymatic specificity would be more extended and that AmiD could hydrolyze muropeptides with or without an anhydro function on the MurNAc residue (28).
AmiD is synthesized with a precursor signal peptide (residues 1 to 16), which is cleaved by signal peptidase II. The peptidase cuts upstream of the Cys17 residue to which a glyceride fatty acid lipid is attached (14). AmiD thus becomes a lipoprotein anchored in the outer membrane of E. coli (28). To determine the specificity and the kinetic parameters of AmiD, we overproduced and purified the membrane-bound AmiD and a truncated form of this enzyme (sAmiD). In this work, we report the preliminary substrate specificity analysis of the membrane-bound AmiD and a detailed kinetic characterization of the truncated protein. Unexpectedly, the hydrolysis of short peptidoglycan fragments by sAmiD shows that they can act as sAmiD inhibitors.
MATERIALS AND METHODS
Bacterial strains, plasmids, and DNA manipulations.
E. coli strains C43 (Novagen, San Diego, CA) and DH5α (Invitrogen, Merelbeke, Belgium) were used as hosts for the plasmids as well as for the production of the His-tagged AmiD lipoprotein (AmiD-His6). E. coli LMG194 was used to overproduce sAmiD. E. coli cells were made competent and transformed with plasmid DNA by the method of Dagert and Ehrlich (4). Chromosomal E. coli JC7623 (30) and DH5α DNAs were prepared by using a Wizard genomics DNA purification kit (Promega Corporation, Madison, WI) and used as templates for amiD amplifications. PCRs were carried out by using an Expand High Fidelity PCR system (Roche Applied Science, Vilvoorde, Belgium). Amplified DNA fragments and plasmids were purified using the Wizard purification system and a Nucleospin plasmid extraction system (Macherey-Nagel, Düren, Germany), respectively. Restriction endonucleases and sequencing enzymes were purchased from Amersham Pharmacia and Gibco BRL. Oligonucleotides and primers for DNA sequencing were obtained from Eurogentec (Seraing, Belgium). Standard molecular biology experiments were performed according to standard published procedures (27).
The amiD-His6 gene was obtained by PCR by using the following primers: 5′-AAAACGCGTTCATGAGAAGATTCTT-3′ and 5′-AAAACTGCCGGATCCATCCTGCCCG-3′ The forward primer was designed to incorporate a BspHI site (in italics) into the amiD initiation codon (underlined), whereas the reverse primer introduced a BamHI site (in italics) and removed the amiD stop codon. The amplified fragment was digested by BspHI and BamHI and inserted between the compatible NcoI and BglII sites of pTrcHis60 vector (25) to generate a pR60 plasmid. In the resulting plasmid, amiD is fused in frame at its 3′ end with a short sequence coding for a Gly-Ser-His6 tag and is under the control of the trc promoter.
The pBAD/Myc-HisAKr plasmid, a pBAD/Myc-His derivative in which the ampicillin resistance gene was replaced by a kanamycin resistance gene (5), was used as a vector for the overexpression of the amiD gene encoding sAmiD (samiD). The latter gene was amplified by PCR using purified genomic DNA as the template. The amplimers used were as follows: forward, 5′-CCATGGGAGCAGGCGAAAAAGGCATTGTCGAG-3′; reverse, 5′-CGAATTCCTAATCCTGCCCGTATTTCTCCAGC-3′. The first amplimer was designed to introduce a NcoI site (in italics) behind the Cys17 AmiD codon, whereas the second introduced an EcoRI site (in italics) behind the amiD stop codon.
The amplified fragment was purified and cloned into pGEM-T-Easy vector (Promega, Corporation, Madison, WI) to generate pCIP701. After its purification, the pCIP701 plasmid was digested with NcoI and EcoRI, and the fragment corresponding to samiD was purified by agarose gel electrophoresis and ligated into pBAD/Myc-HisAKr vector digested with the same restriction endonucleases to generate pCIP702, in which samiD is under the control of the arabinose pBAD promoter. In all cases, the sequence of the amplified fragment was confirmed by sequencing.
Purification of the AmiD-His6 protein.
E. coli C43/pR60 cells were grown at 37°C in 1 liter of 2× yeast tryptone medium containing ampicillin (100 μg/ml). When the absorbance at 600 nm (A600) reached 0.7, isopropyl-β-d-thiogalactopyranoside (IPTG) was added to achieve a final concentration of 0.5 mM and incubation was continued overnight at 20°C. The cells were harvested by centrifugation at 4°C, and the pellet was washed with 50 ml of cold 25 mM phosphate buffer (pH 7.5) supplemented with 1 mM 1,4-dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 0.5 μg/ml leupeptin, and 0.7 μg/ml pepstatin. After centrifugation, the bacteria were resuspended in 25 mM Tris-HCl (pH 7.8) containing 150 mM NaCl, 15% glycerol, 1 mM MgCl2, and 1 mM 2-mercaptoethanol (buffer A) (15 ml) and disrupted by sonication at 0°C using a Bioblock Vibracell 72412 sonicator (Bioblock Fisher, Tournai, Belgium). The resulting suspension was centrifuged at 4°C for 30 min at 200,000 × g with a Beckman TL100 centrifuge (Beckman, Villepinte, France). The pellet was treated at 4°C for 2 h with n-β-dodecyl-maltoside (0.9%) (DDM) in 25 mM sodium phosphate buffer (pH 7.5) containing 300 mM NaCl and 10% glycerol (buffer B) (6 ml). After this treatment, the solubilized AmiD-His6 was separated from the insoluble material by centrifugation at 200,000 × g for 30 min to obtain the soluble fraction (DDM1). The solubilization step was repeated twice for the resulting pellet, and two new soluble extracts (DDM2 and DDM3) (6 ml each) were obtained by using 0.9 and 1.3% DDM, respectively.
The AmiD-His6 protein was purified on Ni2+-nitrilotriacetate-agarose at 4°C according to the recommendations of the manufacturer (Qiagen, Venlo, The Netherlands). The DDM fractions (4 ml) were mixed with 1 ml of gel previously washed with buffer A supplemented with 0.2% DDM (buffer C). The suspension was gently stirred for 2 h using a test tube rotator. A step gradient of imidazole (10, 20, 50, 75, 100, 150, and 200 mM in 2-ml fractions) in buffer C was used to eluate the bound proteins. Under these conditions, solubilized AmiD-His6 was eluted between at 50 and 75 mM imidazole. The fractions of interest were concentrated to 1 ml on Centricon YC filters (Millipore, Billerica, MA). Imidazole was eliminated by dialysis against 25 mM sodium phosphate (pH 7.4) containing 150 mM NaCl, 10% glycerol, and 0.2% DDM (buffer D).
Purification of sAmiD.
Three liters of minimal RM medium (Invitrogen, Merelbeke, Belgium) supplemented with glucose (2 mg/ml) and kanamycin (50 μg/ml) was inoculated with 60 ml of an overnight culture of E. coli LMG194/pCIP702 and incubated at 37°C under conditions of agitation. When the A600 reached 0.7, expression of samiD was induced by addition of arabinose (2 g/liter) and the culture was further incubated for 4 h. Cells were collected by centrifugation, washed with 150 mM NaCl, resuspended in 20 mM sodium citrate buffer (pH 5.6) containing 1 mM DTT, and disrupted (Constant system, Daventry, United Kingdom). After the addition of Benzonase (Merck, Calbiochem, San Diego, CA) (50 U/liter of culture), the soluble cell extract obtained by centrifugation (13,000 × g for 25 min at 4°C) was dialyzed against the buffer (20 mM sodium citrate; pH 5.6) used to equilibrate an SP-Sepharose Fast Flow column (Amersham Pharmacia Biotech, Uppsala, Sweden) (2.6 by 30 cm; 150 ml). After adsorption on a cation exchanger, the proteins were eluted by a linear NaCl gradient (0 to 0.7 M; total volume, 750 ml). The sAmiD-containing fractions detected by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis were pooled, concentrated by ultrafiltration (Amicon; Vivascience, Sartorius, Germany) (cutoff, 10,000), and loaded onto a Sephacryl S100 HR column (Amersham Pharmacia Biotech, Uppsala, Sweden) (2.6 by 95 cm; 500 ml). After SDS-PAGE analysis, the sAmiD-containing fractions were pooled, dialyzed against 20 mM Tris-HCl buffer (pH 8.5), and subjected to anion-exchange chromatography on a Q Sepharose Fast Flow column (Amersham Pharmacia Biotech, Uppsala, Sweden) (1.6 by 13.5 cm; 27 ml) equilibrated in the dialysis buffer. Elution was performed by a linear NaCl gradient (0 to 0.2 M; total volume, 150 ml). The fractions containing sAmiD were pooled and dialyzed against 20 mM sodium citrate buffer (pH 5.6) and stored in this buffer at 4°C.
Determination of the sAmiD zinc content.
The purified enzyme was dialyzed against HEPES buffer (10 mM; pH 7) and analyzed by inductive coupled plasma mass spectroscopy using a spectrometer (Fison-VG-Plasmaquad-PQII+; Fison, United Kingdom) as previously described (23).
Kinetic studies and sources of substrates.
The different muropeptides used in this study are listed in Table 1. Radiolabeled UDP-MurNAc-tripeptides were prepared by enzymatic synthesis using the purified Mur synthetases MurC, MurD, and MurE from E. coli as previously described (1, 6). MurNAc peptides were obtained by mild hydrolysis of the UDP-MurNAc peptide precursors as previously described (17). Dansylated lipids I and II (Nɛ-dansyl-A2pm) and radioactive lipid I (l-[14C]Ala) were enzymatically synthesized by using purified MraY and MurG enzymes (9). Isolation of peptidoglycan from E. coli DH5α cells was performed as already described (6, 13).
TABLE 1.
Muropeptides and HPLC internal standards used in this study
| Muropeptide | Structure | Reference(s) or source | Internal standardb (source) |
|---|---|---|---|
| Dansylated lipid I or lipid I (l-[14C]Ala) | Undecaprenyl-pyrophospho-MurNAc-l-Ala-γ-d-Glu-mA2pm(ɛ-Dansyl)-d-Ala-d-Ala | 9 | |
| Dansylated lipid II | Undecaprenyl-pyrophospho-MurNAc-(l-Ala-γ-d-Glu-mA2pm(ɛ-Dansyl)-d-Ala-d-Ala)-GlcNAc | 9 | |
| UDP-MurNAc-tripeptide | UDP-MurNAc-l-Ala-γ-d-Glu-mA2pm | 1,6 | |
| GlcNAc-anhydroMurNAc-tetrapeptidea | GlcNAc-anhydroMurNAc-l-Ala-γ-d-Glu-mA2pm-d-Ala | 17 | Gly-Phe (Sigma) |
| MurNAc-pentapeptide | MurNAc-l-Ala-γ-d-Glu-mA2pm-d-Ala-d-Ala | 17 | Phe-Phe (Sigma) |
| MurNAc-tripeptide | MurNAc-l-Ala-γ-d-Glu-mA2pm | 17 | Gly-Phe (Sigma) |
| MurNAc-tripeptide (Lys) | MurNAc-l-Ala-γ-d-Glu-l-Lys | 17 | |
| MurNAc-dipeptide | MurNAc-l-Ala-d-Glu | 17 | Phe-Phe (Sigma) |
| G1050 | MurNAc-d-Ala-d-Glu | Bachem | Ala-Ala-Ala-Ala (Sigma) |
| G1060 | MurNAc-l-Ala-l-Glu | Bachem | Ala-Ala-Ala-Ala (Sigma) |
Also referred to in the literature as tracheal cytotoxin (2).
Internal standards used to improve the precision of the estimation of muropeptides hydrolysis followed by HPLC.
The AmiD-His6 amidase activity was measured by monitoring the release of radioactive tripeptide in mixtures (final volume, 50 μl) containing 25 mM sodium phosphate (pH 7.4), 1 mM MurNAc-[14C]tripeptide (500 Bq), and enzyme (15 μl of an appropriate dilution in buffer B containing 0.4% DDM). After 30 min at 37°C, the reaction was stopped by the addition of 10 μl of glacial acetic acid. Before high-performance liquid chromatography (HPLC) analysis, samples were reduced and lyophilized. The reduction was performed using 0.25 M sodium borate (pH 9) by the addition of 2 to 3 mg of sodium borohydride for 30 min at room temperature. The lyophilized residue was dissolved in 100 μl of 50 mM ammonium formate (pH 3.5), and 80 μl was injected into a Nucleosil 100C18 5-μm column (Alltech, France) (4.6 by 150 mm), using the same buffer (at a flow rate of 0.6 ml/min) as that used for the mobile phase. Detection was performed by radioactive flow detection (model LB 506-C1; Berthold, Bad Wildbad, Germany) using a Quicksafe Flow 2 scintillator (Zinsser Analytic, Frankfurt, Germany) at 0.6 ml/min. Quantitation was carried out with Winflow software (Berthold, Bad Wildbad, Germany).
Native AmiD-His6 activity on a UDP-MurNAc tripeptide (50 nanomoles) was measured under the same conditions as those used for MurNAc peptides. The products of the reaction were separated by paper electrophoresis in 50 mM formic acid at 800 V for 2 h and quantified with a radioactivity scanner (model Multi-Tracemaster LB285; EG&G Wallac/Berthold).
Dansylated lipids I and II (2 μl) or radioactive lipid I (4 μl [500 Bq]) was desiccated before the addition of 2 μl of pure native AmiD-His6 (1.5 μg) in buffer D and 2 μl of buffer E (4% Triton X-100, 400 mM Tris-HCl, 600 mM NaCl). The reaction mixture was incubated for 5 min at room temperature and analyzed by thin-layer chromatography (9) developed with 2-propanol/ammonium hydroxide/water (6/3/1 [vol/vol]).
Th activity of native AmiD-His6 on peptidoglycan was assayed by overnight incubation at 37°C (final volume of 200 μl) of partially purified native AmiD-His6 (DDM2 fraction; 100 μg of total protein) with insoluble purified peptidoglycan equivalent to 200 nmol of MurNAc in 50 mM sodium phosphate buffer (pH 7.4) containing 0.2% DDM. The reaction mixture was centrifuged (Eppendorf, Hamburg, Germany) at 9,000 × g for 10 min to eliminate insoluble residual material, and the supernatant was reduced and lyophilized (13). This material was dissolved in 0.5 ml of 0.05% trifluoroacetic acid and then applied to a Nucleosil 100C18 5-μm column (4.6 by 150 mm). Elution was performed with 0.05% trifluoroacetic acid for 10 min, followed by a linear gradient of acetonitrile from 0 to 10% during the next 50 min, at a flow rate of 0.6 ml/min. Peaks were detected at 215 nm. Under these conditions, the main peptides released from the macromolecule, namely, the free tripeptide and tetrapeptide, and their cross-linked forms, the heptapeptide (tri-tetra) and octapeptide (tetra-tetra), were eluted at 11 and 17 min and at 37 and 41 min, respectively (see Fig. 2). The identity of these compounds was confirmed by amino acid analysis and mass spectrometry analysis (data not shown).
FIG. 2.

Release of peptides from the peptidoglycan polymer by the purified AmiD-His6. Purified peptidoglycan from E. coli was digested overnight by AmiD-His6, and the reaction mixture was analyzed using HPLC and a Nucleosil 100C18 column as described in Materials and Methods. Peaks were detected at 215 nm. The main hydrolysis products identified were the free tripeptide (l-Ala-γ-d-Glu-mA2pm; peak 1) and tetrapeptide (l-Ala-γ-d-Glu-mA2pm-d-Ala; peak 2) and their corresponding cross-linked forms, the heptapeptide (tri-tetra; peak 3) and octapeptide (tetra-tetra; peak 4). No release of peptides was observed following incubation of peptidoglycan in the absence of AmiD-His6 (lower profile).
For time course kinetic experiments, purified sAmiD was diluted in 50 mM phosphate buffer (pH 7) containing 10 nM ZnCl2 and incubated at 30°C with the different muropeptides listed in Table 1. Prior to addition of the enzyme and depending on the tested substrate, a different peptide was added as an internal standard to normalize peak integration after HPLC (Table 1). Time course samples (100 μl) were withdrawn from the reaction mixture, and the reaction was stopped by the addition of 10 μl of 1% (wt/vol) H3PO4. The hydrolysis of the different substrates was followed by reverse-phase HPLC (Nucleosil 100-5 C18; Agilent Technologies, Diegem, Belgium) (4 by 250 mm; 5 μm) by using an Alliance 9680 chromatographer (Waters, Zellik, Belgium). The column was equilibrated with 50 mM sodium phosphate (pH 4.5) (buffer C) and eluted with a linear gradient (0 to 100%) of buffer D (buffer C containing 25% of methanol [vol/vol]) at a flow rate of 0.6 ml/min. The eluate was monitored by measuring the UV absorbance at 205 nm with a Waters 996 diode array detector (photodiode array detector; Waters, Zellik, Belgium). The peak areas of the substrates were calculated using Empower software (Waters, Zellik, Belgium), and Grafit software was used to analyze the results (Erithacus Software, United Kingdom).
Inhibition of the enzyme by EDTA.
Purified sAmiD (1 μM) was incubated for 30 min at 30°C with increasing concentrations of EDTA (5, 10, 20, 50, 100, and 200 μM and 1 mM). The remaining activity was measured by incubation with 180 μM GlcNAc-anhydroMurNAc tetrapeptide substrate (Table 1) for 30 min at 30°C.
RESULTS
Expression and purification of the membrane AmiD-His6.
Plasmid pR60, carrying the wild-type amiD gene fused at its 3′ end to a sequence encoding a His6 tag, was introduced into the two E. coli DH5α and C43 strains. After induction assays, AmiD-His6 production was better when E. coli C43 was used as the host. However, in both cases, the AmiD-His6 production at 37°C led to cell lysis after IPTG induction. To overcome this lysis, the cultures were grown overnight at 20°C after addition of the inducer. After sonication of induced bacteria and centrifugation, the supernatant and the resuspended pellet were assayed for N-acetylmuramoyl-l-alanine amidase activity by using radioactive MurNAc tripeptide as the substrate (Table 1). The amidase activity was found associated with the insoluble fraction, which confirmed that the recombinant protein was correctly processed and associated with the outer membrane. The solubilization of AmiD-His6 by DDM was undertaken as described in Materials and Methods, and its purification was carried out by affinity chromatography on Ni2+-nitrilotriacetate-agarose. A protein with a molecular mass of 32 kDa was observed by the use of SDS-PAGE in the fractions eluted between 50 and 75 mM imidazole in the presence of 0.2% DDM. After this purification step, AmiD-His6 was 75% pure (data not shown).
However, the lytic effect of the AmiD-His6 accumulation in the recombinant E. coli cells, combined with the difficulty of purifying this AmiD-His6 lipoprotein in the presence of detergent, resulted in very low AmiD-His6 production (about 0.3 to 0.5 mg of purified protein per liter of culture). To overcome this problem, a truncated form of AmiD that could be overproduced in the E. coli cytoplasm was constructed.
Cloning, production, and purification of sAmiD.
The E. coli samiD gene was amplified from the E. coli DH5α genomic DNA as described in Materials and Methods. In the final construct, the gene fragment encoding the first 17 amino acid residues (MRRFFWLVAAALLLAGC) of the AmiD preprotein was replaced by an MG dipeptide to remove the signal peptide and the C17 cysteine residue whose modification was responsible for the lipid anchorage of the mature AmiD into the outer membrane. As expected, arabinose-induced E. coli LMG194/pCIP702 cells overproduced a soluble cytoplasmic protein of 30 kDa corresponding to the soluble sAmiD. Starting with 3 liters of minimal RM medium inoculated with E. coli/pCIP702, three successive chromatographic steps were necessary to obtain 34 mg of pure sAmiD (3.4 mg/ml; final purity, 98%).
sAmiD is a zinc metalloenzyme.
The zinc content of sAmiD (0.03 mM in 10 mM HEPES [pH 7]) determined by inductive coupled plasma mass spectroscopy showed that 1 mol of sAmiD contained 0.7 ± 0.2 mol of Zn, indicating that the zinc ion was bound to sAmiD. The purified enzyme (100 nM) was inhibited by an excess of the zinc chelator EDTA (5, 10, 20, 50, 100, and 200 μM and 1 mM). The inhibition in the presence of 5 μM of EDTA was already complete after 30 min. In similarity to AmpD and the majority of peptidoglycan recognition proteins (7, 34), AmiD is a metalloenzyme containing a zinc ion in its active site.
Substrate specificity of AmiD-His6.
Tests of the amidase specificity of AmiD-His6 were performed using the DDM2 and DDM3 fractions as enzyme preparations (see Materials and Methods) in comparison with the same fractions obtained from the control strain DH5α/pTrcHis60 without an amiD gene inserted in the plasmid. After overnight incubation, MurNAc-tripeptides containing either mA2pm or l-lysine at the third position of the peptide (Table 1) were hydrolyzed by AmiD-His6. However, neither the nucleotide precursor UDP-MurNAc-tripeptide nor the lipid I and II intermediates were hydrolyzed by the purified protein. HPLC analysis of peptidoglycan incubated with AmiD-His6 showed that the macromolecule was solubilized by the action of the enzyme. The main soluble degradation products released were identified as the free tripeptide and tetrapeptide and the corresponding cross-linked forms, the heptapeptide (tri-tetra) and octapeptide (tetra-tetra) (Fig. 2).
Substrate specificity and kinetic characterization of sAmiD.
Kinetic analyses of the N-acetylmuramoyl-l-alanine amidase sAmiD were carried out by using a discontinuous time course assay and reverse-phase HPLC to determine the remaining concentrations of the different substrates listed in Table 1. The decrease in the substrate concentration observed after different periods of incubation was computed by integrating the area of the peak and by using an internal standard (for details, see Materials and Methods and Table 1). For the analysis of the data and curve fittings, the product concentrations at different times were obtained as follows: P = S0 − St (where S0 and St represent the substrate concentrations at times 0 and t, respectively).
The hydrolysis of GlcNAc-anhydroMurNAc-l-Ala-γ-d-Glu-mA2pm-d-Ala (27 μM), which is usually reported in the literature as the tracheal cytotoxin (2), by the enzyme (106 nM) exhibited biphasic behavior characterized by a rapid exponential phase preceding a linear phase, a typical burst reaction (10) (Fig. 3). The concentration of the product formed in the rapid exponential phase was much greater than the concentration of the enzyme, suggesting the presence of a branched pathway (33). For this type of kinetic scheme, the equation describing the progress curve of the product as a function of time is as follows:
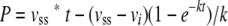 |
(1) |
where P represents the concentration of the product at time t and vi and vss represent the initial and the stationary rates, respectively (33). The rate constant, k, characterizes the rate decrease and is referred to here as the burst constant (Fig. 3). The vi/vss ratio is also characteristic of the burst and corresponds to the value of its amplitude. This kinetic curve signals the likelihood of a branched pathway that includes the presence of two enzyme isomers, E and E′ (Fig. 4). In the absence of a substrate, the enzyme is principally present in its fully active form (E) whereas the poorly active form (E′) is induced by the substrate.
FIG. 3.

Time course of product formation with the GlcNAc-anhydroMurNAc-tetrapeptide as the substrate. The substrate (27 μM) was incubated in the presence of 106 nM sAmiD. The graph highlights biphasic behavior characteristic of a burst kinetic. The slopes of the dotted and dashed lines represent the initial and stationary state rates, respectively, describing the burst phenomenon (see the text for details). The curve fitting (solid line) was obtained by using equation 1.
FIG. 4.

Kinetic schemes for a substrate-induced inactivation mechanism. (A) General scheme; (B) simplified scheme. E, native enzyme; E′, inactive or poorly active form of the enzyme induced by the substrate; S, substrate; P, product. E·S and E′·S represent substrate-enzyme complexes. K1 to K4 represent dissociation constants corresponding to the different equilibria. kp and k′p represent the rate constants characterizing the hydrolytic decay of E·S and E′·S, respectively. The kinetic scheme shown in panel A can be simplified to the scheme shown in panel B when the conversion of E′ into E is fast and the equilibrium E ↔ E′ is completely displaced toward E.
The MurNAc-tripeptide and MurNAc-pentapeptide were substrates of the amidase and also exhibited burst kinetics (Table 2). The MurNAc-dipeptide was hydrolyzed neither by sAmiD nor inhibitor when GlcNAc-anhydroMurNAc-tetrapeptide was used as the reporter substrate. Two other muramyl-dipeptides, G1050 and G1060, which are diastereoisomer variants of MurNAc-dipeptide (Table 1), were tested and did not interact with sAmiD.
TABLE 2.
Initial (vi) and steady state (vss) rates for the hydrolysis by sAmiD of the different substrates and the burst constants (k) characterizing the inactivation rates and the vi/vss ratiosa
| Muropeptide | Muropeptide concn (μM) | Enzyme concn (nM) | vi (μM/min) | k (1/min) | vss (μM/min) | vi/vss |
|---|---|---|---|---|---|---|
| GlcNAc-anhydroMurNAc-tetrapeptide | 27 | 106 | 4.7 ± 0.8 | 0.6 ± 0.1 | 0.178 ± 0.001 | 26 ± 1 |
| MurNAc-pentapeptide | 27 | 500 | 0.33 ± 0.04 | 0.028 ± 0.005 | 0.051 ± 0.005 | 6.5 ± 0.2 |
| MurNAc-tripeptide | 16 | 34 | 1 ± 0.2 | 0.17 ± 0.04 | 0.13 ± 0.03 | 7.7 ± 0.6 |
| MurNAc-dipeptide | 16 | 20 × 103 | No hydrolysis | No hydrolysis | No hydrolysis | |
| G1050 | 50 | 106 | No hydrolysis | No hydrolysis | No hydrolysis | |
| G1060 | 50 | 106 | No hydrolysis | No hydrolysis | No hydrolysis |
The parameters were obtained by curve fitting using equation 1.
The GlcNAc-anhydroMurNAc-tetrapeptide was the best substrate tested; compared to the nonanhydro MurNAc-peptides, its vi value was 5- to 10-fold higher (Table 2). To be a substrate of AmiD, the peptide bound to the MurNAc residue should apparently contain at least three residues (l-Ala-γ-d-Glu-mA2pm), and its optimal length seems to be three or four residues. Indeed, in the MurNAc-peptide series, the vi value reached a maximum at a residue length of three, and the presence of an mA2pm residue seemed to be critical for activity. The vi/vss ratios were very similar for the two substrates MurNAc-pentapeptide and MurNAc-tripeptide (6.5 and 7.7, respectively) and appeared to be maximal for the GlcNAc-anhydroMurNAc-tetrapeptide (vi/vss = 26), a finding highlighting the importance of the sugar moiety for the substrate-induced inactivation.
Reversibility of the substrate-induced inactivation.
GlcNAc-anhydroMurNAc-tetrapeptide (14 μM) was hydrolyzed; after completion of the burst (85% of substrate consumption), a fresh sample of the same substrate was added. A second burst was observed, and its amplitude was similar to that of the first burst (Fig. 5A). This result was in agreement with a reversible inactivation of sAmiD by the substrate. In the general kinetic A scheme in Fig. 4, the E·S substrate-enzyme complex is reversibly converted into an E′·S complex that could be unproductive or could slowly decompose to enzyme and product. This transient substrate-induced inactivation of the enzyme is interpreted in terms of conformational isomerization of the enzyme. In the case of sAmiD, the conversion between the inactive E′ and active E forms was fast. Indeed, after the depletion of the substrate, the addition of a fresh sample of substrate gave rise to a similar burst, which can be explained by the occurrence of a fast return to the active E form. So, the general branched pathway can be simplified in the B scheme (Fig. 4), in which the E·S intermediate is reversibly converted into a dead-end complex. This result also showed that the product of the reaction did not interact with the enzyme and consequently did not inhibit its activity.
FIG. 5.

Evidence for reversible substrate inactivation. (A and B) Time courses of product formation during the hydrolysis of GlcNAc-anhydroMurNAc-tetrapeptide by sAmiD. In both cases, the initial enzyme concentration was fixed to 106 nM, whereas the initial substrate concentrations were 14 μM and 27 μM, respectively. (A) Fresh substrate (14 μM) was added when 85% of the initial substrate was hydrolyzed; (B) a fresh enzyme solution (106 nM) was added after the burst. The arrows indicate the times of addition of fresh substrate (A) or enzyme (B). Each value represents the mean of the results of three determinations. The dotted and dashed lines highlight the two steps of the experiment. See the text for details.
The addition of a fresh sample of sAmiD after the exponential phase (Fig. 5B) resulted in a second burst, confirming the proposed kinetic B scheme. Indeed, in this scheme the addition of fresh enzyme in the presence of the equilibrated E, E·S, and E′·S species displaces the equilibrium to the formation of E·S and E·S′.
It is somewhat difficult to give a quantitative interpretation of the results represented in Fig. 5A, because the Vmax and Km values could not be measured for each phase on the basis of our results. Nevertheless, simple simulations based on both models showed that after 85% of the substrate was hydrolyzed, a new burst could be obtained upon the addition of fresh substrate.
DISCUSSION
The sAmiD N-acetylmuramoyl-l-alanine amidase is a zinc metalloenzyme that probably binds a single divalent zinc ion in its active site and can be inactivated by a metal chelator such as EDTA. We recently determined the three-dimensional structure of sAmiD (2BGX and 2BH7 in the Protein Data Bank; S. Petrella, unpublished data) and confirmed the presence of a zinc ion in the active site of the enzyme. In the crystal, sAmiD is present as a dimer formed by domain swapping, whereas in solution, sAmiD behaves as a monomer (Petrella, unpublished).
Concerning the AmiD specificity, our results show that AmiD hydrolyzes muropeptides with or without an anhydro function on MurNAc (see Results and Table 2), as suspected by Uehara and Park (28). In the latter study, the authors tested two muropeptides (GlcNAc-anhydroMurNAc-tripeptide and anhydroMurNAc-tripeptide) and showed that the latter was much (40×) more slowly hydrolyzed by AmiD. They concluded that a GlcNAc residue is required for AmiD activity. Here, we demonstrate that the MurNAc tri- and pentapeptides are also good substrates for sAmiD (Table 2). The latter was hydrolyzed five times more efficiently than the GlcNAc-anhydroMurNAc-tetrapeptide which was used as a reference substrate by Uehara and Park (28) and us. In addition, AmiD is active on the native peptidoglycan, and its overexpression is toxic for the cell (our study and reference 28). All these results suggest that AmiD acts as an autolysin, with the peptidoglycan being its substrate in in vivo situations.
The most striking feature of our results is the AmiD kinetic behavior. Indeed, for all the tested substrates, AmiD exhibited a biphasic kinetic curve that can be explained by the presence of a branched pathway involving the inactivation of the enzyme by the substrate. It is the first time that this type of behavior has been reported for an enzyme interacting with peptidoglycan derivatives. It is difficult to decide whether this kinetic behavior has a physiological role or is a curiosity highlighted by the in vitro study of AmiD amidase. If AmiD is inhibited in growing cells by muropeptides liberated by the autolytic machinery, it should be inactivated when its substrate accumulates in the periplasm. This situation prevails in the presence of β-lactam antibiotics or other compounds stimulating the autolytic enzymes. In consequence, this AmiD substrate-inactivation phenomenon should facilitate the entry of muropeptides into the cytoplasm for recycling or to signal the presence of cell wall stress that requires a cellular adaptation. Peptidoglycan fragments are not usually considered to be possible inhibitors of cell wall hydrolases. Nevertheless, the inactivation of this class of enzymes results in an increase in the susceptibility of the E. coli cell to β-lactam antibiotics (15). The use of muropeptides could be a new way to inhibit cell wall hydrolases. To explore this new approach, original chemical strategies must be established to obtain large quantities of synthetic muropeptides. This represents a new challenge for organic chemists.
Acknowledgments
This research was supported in part by the Belgian Program on Interuniversity Poles of Attraction initiated by the Belgian state (grants P5/33 and P6/19), the Actions de Recherche Concertées (grant 03/08-297), the Fonds de la Recherche Fondamentale Collective (grants 2.4511.06 and 2.4524.03), the Fonds de la Recherche en Sciences Médicales (grant 3.4586.05), Eur-Intafar (grant LSHM-CT-2004-512138), and the Centre National de la Recherche Scientifique (France). Anne Pennartz is a recipient of a fellowship from the Fonds pour la Recherche dans l'Industrie et l'Agriculture. Bernard Joris is a research associate of the Fonds National de la Recherche Scientifique.
We thank Mireille Hervé for providing substrates and advice for HPLC analysis, Georges Dive for reading the paper, and Jean Marie Frère for fruitful discussions.
Footnotes
Published ahead of print on 23 February 2009.
REFERENCES
- 1.Bouhss, A., S. Dementin, J. van Heijenoort, C. Parquet, and D. Blanot. 2002. MurC and MurD synthetases of peptidoglycan biosynthesis: borohydride trapping of acyl-phosphate intermediates. Methods Enzymol. 354:189-196. [DOI] [PubMed] [Google Scholar]
- 2.Chang, C. I., Y. Chelliah, D. Borek, D. Mengin-Lecreulx, and J. Deisenhofer. 2006. Structure of tracheal cytotoxin in complex with a heterodimeric pattern-recognition receptor. Science 311:1761-1764. [DOI] [PubMed] [Google Scholar]
- 3.Cheng, X., X. Zhang, J. W. Pflugrath, and F. W. Studier. 1994. The structure of bacteriophage T7 lysozyme, a zinc amidase and an inhibitor of T7 RNA polymerase. Proc. Natl. Acad. Sci. USA 91:4034-4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dagert, M., and S. D. Ehrlich. 1979. Prolonged incubation in calcium chloride improves the competence of Escherichia coli cells. Gene 6:23-28. [DOI] [PubMed] [Google Scholar]
- 5.Duez, C., M. Vanhove, X. Gallet, F. Bouillenne, J. Docquier, A. Brans, and J. M. Frère. 2001. Purification and characterization of PBP4a, a new low-molecular-weight penicillin-binding protein from Bacillus subtilis. J. Bacteriol. 183:1595-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duncan, K., J. van Heijenoort, and C. T. Walsh. 1990. Purification and characterization of the d-alanyl-d-alanine-adding enzyme from Escherichia coli. Biochemistry 29:2379-2386. [DOI] [PubMed] [Google Scholar]
- 7.Dziarski, R., and D. Gupta. 2006. Mammalian PGRPs: novel antibacterial proteins. Cell Microbiol. 8:1059-1069. [DOI] [PubMed] [Google Scholar]
- 8.Dziarski, R., and D. Gupta. 2006. The peptidoglycan recognition proteins (PGRPs). Genome Biol. 7:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El Ghachi, M., A. Bouhss, H. Barreteau, T. Touzé, G. Auger, D. Blanot, and D. Mengin-Lecreulx. 2006. Colicin M exerts its bacteriolytic effect via enzymatic degradation of undecaprenyl phosphate-linked peptidoglycan precursors. J. Biol. Chem. 281:22761-22772. [DOI] [PubMed] [Google Scholar]
- 10.Frieden, C., and J. Fernandez-Sousa. 1975. Kinetic studies on pig heart cytoplasmic malate dehydrogenase. J. Biol. Chem. 250:2106-2113. [PubMed] [Google Scholar]
- 11.Généreux, C. 2003. Etude de l'anhydro-N-acétylmuramyl-l-alanine amidase d'Amp D de Citrobacter freundii. Ph.D. thesis. Université de Liège, Liège, Belgium.
- 12.Généreux, C., D. Dehareng, B. Devreese, J. Van Beeumen, J. M. Frère, and B. Joris. 2004. Mutational analysis of the catalytic centre of the Citrobacter freundii AmpD N-acetylmuramyl-l-alanine amidase. Biochem. J. 377:111-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Girardin, S. E., L. H. Travassos, M. Hervé, D. Blanot, I. G. Boneca, D. J. Philpott, P. J. Sansonetti, and D. Mengin-Lecreulx. 2003. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J. Biol. Chem. 278:41702-41708. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi, S., and H. C. Wu. 1990. Lipoproteins in bacteria. J. Bioenerg. Biomembr. 22:451-471. [DOI] [PubMed] [Google Scholar]
- 15.Heidrich, C., M. F. Templin, A. Ursinus, M. Merdanovic, J. Berger, H. Schwarz, M. A. de Pedro, and J. V. Höltje. 2001. Involvement of N-acetylmuramyl-l-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol. Microbiol. 41:167-178. [DOI] [PubMed] [Google Scholar]
- 16.Heidrich, C., A. Ursinus, J. Berger, H. Schwarz, and J. V. Höltje. 2002. Effects of multiple deletions of murein hydrolases on viability, septum cleavage, and sensitivity to large toxic molecules in Escherichia coli. J. Bacteriol. 184:6093-6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hervé, M., A. Boniface, S. Gobec, D. Blanot, and D. Mengin-Lecreulx. 2007. Biochemical characterization and physiological properties of Escherichia coli UDP-N-acetylmuramate:l-alanyl-γ-d-glutamyl-meso-diaminopimelate ligase. J. Bacteriol. 189:3987-3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Höltje, J. V., and C. Heidrich. 2001. Enzymology of elongation and constriction of the murein sacculus of Escherichia coli. Biochimie 83:103-108. [DOI] [PubMed] [Google Scholar]
- 19.Höltje, J. V., U. Kopp, A. Ursinus, and B. Wiedemann. 1994. The negative regulator of β-lactamase induction AmpD is a N-acetyl-anhydromuramyl-l-alanine amidase. FEMS Microbiol. Lett. 122:159-164. [DOI] [PubMed] [Google Scholar]
- 20.Jacobs, C., L. J. Huang, E. Bartowsky, S. Normark, and J. T. Park. 1994. Bacterial cell wall recycling provides cytosolic muropeptides as effectors for β-lactamase induction. EMBO J. 13:4684-4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeruzalmi, D., and T. A. Steitz. 1998. Structure of T7 RNA polymerase complexed to the transcriptional inhibitor T7 lysozyme. EMBO J. 17:4101-4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitano, K., E. Tuomanen, and A. Tomasz. 1986. Transglycosylase and endopeptidase participate in the degradation of murein during autolysis of Escherichia coli. J. Bacteriol. 167:759-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liepinsh, E., C. Généreux, D. Dehareng, B. Joris, and G. Otting. 2003. NMR structure of Citrobacter freundii AmpD, comparison with bacteriophage T7 lysozyme and homology with PGRP domains. J. Mol. Biol. 327:833-842. [DOI] [PubMed] [Google Scholar]
- 24.Park, J. T. 1993. Turnover and recycling of the murein sacculus in oligopeptide permease-negative strains of Escherichia coli: indirect evidence for an alternative permease system and for a monolayered sacculus. J. Bacteriol. 175:7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pompeo, F., J. van Heijenoort, and D. Mengin-Lecreulx. 1998. Probing the role of cysteine residues in glucosamine-1-phosphate acetyltransferase activity of the bifunctional GlmU protein from Escherichia coli: site-directed mutagenesis and characterization of the mutant enzymes. J. Bacteriol. 180:4799-4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Priyadarshini, R., D. L. Popham, and K. D. Young. 2006. Daughter cell separation by penicillin-binding proteins and peptidoglycan amidases in Escherichia coli. J. Bacteriol. 188:5345-5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 28.Uehara, T., and J. T. Park. 2007. An anhydro-N-acetylmuramyl-l-alanine amidase with broad specificity tethered to the outer membrane of Escherichia coli. J. Bacteriol. 189:5634-5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Heijenoort, J. 1998. Assembly of the monomer unit of bacterial peptidoglycan. Cell. Mol. Life Sci. 54:300-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Visick, J. E., and S. Clarke. 1997. RpoS- and OxyR-independent induction of HPI catalase at stationary phase in Escherichia coli and identification of rpoS mutations in common laboratory strains. J. Bacteriol. 179:4158-4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vollmer, W., D. Blanot, and M. A. de Pedro. 2008. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32:149-167. [DOI] [PubMed] [Google Scholar]
- 32.Vollmer, W., B. Joris, P. Charlier, and S. Foster. 2008. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 32:259-286. [DOI] [PubMed] [Google Scholar]
- 33.Waley, S. G. 1991. The kinetics of substrate-induced inactivation. Biochem. J. 279:87-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang, Z. M., X. Li, R. R. Cocklin, M. Wang, K. Fukase, S. Inamura, S. Kusumoto, D. Gupta, and R. Dziarski. 2003. Human peptidoglycan recognition protein-L is an N-acetylmuramoyl-l-alanine amidase. J. Biol. Chem. 278:49044-49052. [DOI] [PubMed] [Google Scholar]