

Abstract
The Serratia marcescens NucC protein is structurally and functionally homologous to the P2 Ogr family of eubacterial zinc finger transcription factors required for late gene expression in P2- and P4-related bacteriophages. These activators exhibit site-specific binding to a conserved DNA sequence, TGT-N3-R-N4-Y-N3-aCA, that is located upstream of NucC-dependent S. marcescens promoters and the late promoters of P2-related phages. In this report we describe the interactions of NucC with the P2 FETUD late operon promoter PF. NucC is shown to bind PF as a tetramer and to make 12 symmetrical contacts to the DNA phosphodiester backbone. The backbone contacts are centered on the TGT-N3-R-N4-Y-N3-aCA motif. Major groove base contacts can be seen at most positions within the ∼24-bp binding site. Minor groove contacts map to adjacent positions in the downstream half of the binding site, which corresponds to the area in which the DNA also appears to be bent by NucC binding. NucC binding provides a new example of protein-DNA interaction that is strikingly different from the DNA binding demonstrated for eukaryotic zinc-finger transcription factors.
The Serratia marcescens NucC protein was originally identified as a positive regulator of extracellular nuclease and bacteriocin 28b production (15, 23). NucC is a basic protein of 75 amino acids that is a member of the bacteriophage P2 Ogr family, a novel class of eubacterial zinc-binding transcription factors required for late gene expression in P2- and P4-related phages. All members of this family characterized thus far are at least somewhat functionally interchangeable and have significant amino acid sequence similarity in the N-terminal two-thirds of the protein. This similarity includes four essential Cys residues that are involved in coordination of zinc (17, 27, 30, 36), arranged in a Cys-X2-Cys-X22-Cys-X4-Cys motif. The function(s) of the nonconserved C-terminal residues is unclear. Viable C-terminal deletions of P2 ogr and phage 186 B suggest that this region is dispensable, although it likely contributes to full activity (18, 36). Genetic evidence is consistent with a mechanism for transcription activation that involves a direct interaction with the C-terminal domain of the α subunit(s) of RNA polymerase (1, 45, 50). Purified NucC, RNA polymerase holoenzyme, and template DNA have been shown to be the only necessary components for open complex formation and transcription from the P2 late promoter PF, and NucC induces a 90° bend in DNA containing the PF binding site (35).
Zinc fingers constitute a well-characterized motif for protein-DNA interaction, and structural data are available for a number of eukaryotic zinc finger transcription factors (reviewed in references 28, 29, and 32). Zinc finger proteins containing C2H2 zinc coordination motifs represent the largest group of transcription factors in eukaryotes (38, 47). These proteins typically contain several zinc fingers that wrap around the DNA in a spiral manner. The zinc coordinating motif in each finger contains an α-helix preceded by a β-hairpin. Amino acid residues on the surface of the α-helix make contacts to adjacent bases in the major groove, and in most cases the β-hairpin makes phosphate backbone contacts. Two other types of zinc coordination motifs are exemplified by the yeast Gal4 protein and the hormone receptor family of transcriptional activators. Gal4 binds as a homodimer to the sequence CCG-N11-CGG (4, 7, 19). The zinc coordinating motif is composed of a pair of α-helices that coordinate two zinc ions through six cysteine residues. The first helix contacts DNA bases in the major groove. The second helix contacts the phosphodiester backbone. Each zinc binding domain recognizes a CCG triplet (34). The hormone receptor family zinc-coordinating motif is characterized by two antiparallel α-helices capped by loops at their amino-terminal ends. Each helix-loop pair coordinates a single zinc ion. The first helix interacts with DNA bases in the major groove, while the loops and second helix make contacts to the phosphate backbone (41). The DNA-binding sites for the hormone receptors consist of two 6-bp consensus half sites (reviewed by Freedman and Luisi [16]). The orientation of the two half-sites and the spacing between them (3 to 6 bp) determines which subunits dimerize and the orientation between them (parallel or symmetrical).
In bacteria, transcription factors with zinc-binding motifs are uncommon. The Agrobacterium tumefaciens Ros protein contains a single Cys2His2 zinc finger domain and regulates expression of virulence genes and the plant oncogene ipt (8). Ros homologs have been identified primarily in other Rhizobiaceae. Although a plant homologue of Ros has not been found, phylogenetic analysis indicates that these proteins are probably of eukaryotic origin (3). The only other known example of a eubacterial zinc finger transcription factor is provided by the P2 Ogr family of proteins. Homologues can be identified by sequence similarity and by the presence of the conserved Cys-X2-Cys-X22-Cys-X4-Cys zinc coordination motif. Most are found in members of the Enterobacteriaceae (presumably in resident P2-related prophages) and in P2- and P4-related phages. Homologues have not been identified in other kingdoms. However, an internal region of the staphylococcal mec cassette recombinase CcrC (22) appears to contain a similar potential zinc-binding motif. Comparisons based on homology and secondary structure predictions do not reveal any significant similarities to other proteins.
The binding sites for the P2 Ogr family of activators contain a conserved element of hyphenated dyad symmetry, TGT-N3-R-N4-Y-N3-aCA (20, 24, 25, 48, 49). Replacement of either the TGT or ACA half-site almost completely inactivates promoter activity in vivo (10). In order to further elucidate the interactions between NucC and its binding site, we carried out high-resolution probing of NucC-DNA contacts. We describe here the interaction of NucC with PF DNA in vitro.
MATERIALS AND METHODS
Proteins and DNA.
NucC was purified as described previously (35). Three DNA templates were used for footprinting and are described in Fig. 1. The templates were made by PCR amplification, using 5′-end-labeled primers and Taq DNA polymerase. Primer end labeling was done with T4 kinase and [γ-32P]ATP. The primers are also shown in Fig. 1.
FIG. 1.

DNA templates used for footprinting analyses and the primers used for PCR and 5′-end labeling. The amplified region of pM37 is shown; the sense strand is the top strand of the double-stranded DNA sequence, and the template strand is the bottom strand. The TGT and cCA half-sites (centered at positions −65 and −50, respectively) are indicated with inverted arrows, and the transcription start site (+1) and direction is also indicated by an arrow. Template 1 was made by PCR using pM37 (35) and the primers 1204 M+n and M6. Template 2 was made by PCR using pM37 and the primers 1204 M+n and M6-3. Template 3 was made by PCR using pLYU2 and primers 1204 M+n and M6-3. PCR amplification adds two adenine residues (in primer M6-3) downstream of the PF sequence to templates 2 and 3. Plasmid pLYU2 is a derivative of pM37 with a T instead of a G at position −43 and a position C instead of an position A at −40 of the PF sequence. Upstream of PF nucleotide −69 is 37 bp of vector derived DNA, which is indicated by italics.
DNA footprinting and electrophoresis.
The DNA binding conditions for protection and interference footprinting consisted of ∼1 pmol of template DNA (300,000 to 500,000 cpm), 50 mM Tris-HCl, 50 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol, 100 μg of bovine serum albumin/ml, and 10% glycerol in a 10- to 20-μl volume, except where indicated otherwise. The amount of NucC used in the binding reactions (typically 50 to 200 ng) was adjusted for each experiment. Interference and protection products were isolated on polyacrylamide Tris-borate-EDTA gels. Free DNA and NucC-bound DNA were recovered from polyacrylamide gel slices as indicated for each experiment. The recovered DNA was further modified as described below. Footprinting reactions were resolved on 6.8% to 8% polyacrylamide-Tris-borate-EDTA-7.2 M urea gels.
Methylation protection and interference.
Methylation protection and interference assays were performed as described by Shaw and Stewart (42). For both assays, 20 μl of 0.01% dimethyl sulfoxide was added to 20 μl of template DNA for 2 min. In the protection assay, the modification was done in the presence of NucC. The reactions were stopped by the addition of 10 μl of 250 mM dithiothreitol. DNA modified in the absence of NucC was ethanol precipitated with 0.3 M sodium acetate twice before use in the interference assay. Reaction products were isolated on polyacrylamide gels, and the DNA was recovered from gel slices by electroelution and ethanol precipitated at −80°C. Piperidine cleavage was performed by resuspending the DNA in 50 μl of H2O, adding 50 μl of 2 M piperidine. and incubation at 90°C for 30 min. The DNA was precipitated with 120 μl of n-butanol, resuspended in 50 μl of 1% sodium dodecyl sulfate (SDS), and precipitated with 1 ml of n-butanol. For sodium hydroxide cleavage, the DNA was resuspended in 30 μl of sodium phosphate (pH 6.8)-1 mM EDTA and incubated at 92°C for 15 min. The samples were kept on ice before adding 3 μl of 1 M NaOH. The reaction was continued at 92°C for 30 min. The DNA was precipitated by adding 320 μl of 500 mM NaCl and 900 μl of ethanol, incubation on ice for 5 min, and centrifugation at 4°C for 10 min.
Depurination interference.
A total of 20 μl of 4% (vol/vol) formic acid was added to ∼2 μg of template DNA in 200 μl of H2O and transferred to a 37°C water bath for 20 min. The reaction was placed on ice and precipitated by adding 20 μl of 3 M sodium acetate (pH 7) and 600 μl of 95% ethanol. The DNA was pelleted by centrifugation for 10 min at room temperature. The pellets were washed with 75% ethanol, air dried, resuspended in H2O, and used in the gel shift assay. Reaction products were recovered from gel slices by electroelution into saturated ammonium acetate and ethanol precipitated overnight at −80°C. Piperidine strand scission was performed as described for the methylation assay.
Uracil substitution and missing thymine interference.
Uracil was randomly substituted for thymine by supplementing the PCR with 5 μM dUTP (200 μM concentrations of other deoxynucleoside triphosphates and 1.5 mM MgCl2). For the uracil substitution assay the DNA was recovered from the polyacrylamide gel used in the gel shift assay by passive diffusion, treated with uracil DNA glycosylase, phenol-chloroform extracted, and ethanol precipitated. Piperidene strand scission was performed as described for the methylation assay. For the missing thymine assay, the uracil substituted template was treated with uracil DNA glycosylase, phenol-chloroform extracted, and ethanol precipitated before the gel shift assay. The DNA was recovered from gel shift slices by electroelution and precipitated overnight at −80°C.
Ethylation interference.
Ethylation interference was performed as described by Manfield and Stockley (33). The DNA was resuspended in 100 μl of sodium cacodylate. After a brief incubation at 50°C, 100 μl of ethylnitrosourea-saturated ethanol was added, and incubation at 50°C was continued for 30 min. The ethylated DNA was precipitated by adding 4 μl of 4 M NaCl, 2 μg of Escherichia coli tRNA, and 150 μl of ethanol. The reactions were incubated on ice for 10 min, followed by centrifugation for 15 min at room temperature to recover the modified DNA, which was washed once with 70% ethanol, air dried, and dissolved in H2O for use in a gel shift assay. Bound and unbound DNA was recovered from gel slices by electroelution into saturated ammonium acetate and ethanol precipitated overnight at −80°C. The precipitated DNA was resuspended in 15 μl of 10 mM sodium phosphate (pH 6.8). For strand scission, 2.5 μl of 1 M NaOH was added, and the DNA was incubated at 90°C for 30 min. The reactions were placed on ice and then precipitated by adding 2.5 μl of 1 M acetic acid, 2 μl of 4 M NaCl, and 70 μl of ethanol. After a 10-min incubation on ice and a 15-min room temperature centrifugation, the pelleted DNA was washed with 70% ethanol and air dried.
Hydroxyl radical protection.
Hydroxyl radical cleavage reactions were performed as described by Schickor and Heumann (40). Aliquots (3.5 μl) of 20 mM sodium ascorbate, 0.6% hydrogen peroxide, and 2 mM iron(II) sulfate hexahydrate-4 mM EDTA were added to a 10-μl NucC binding reaction in the absence of glycerol. The cleavage reaction was stopped after 30 s by adding 1 volume of 20% glycerol, and the bound and free products were isolated by polyacrylamide gel electrophoresis (PAGE). The DNA was recovered from gel slices by passive diffusion at 4°C in H2O, ethanol precipitated, washed with 80% ethanol, and air dried.
Molecular mass estimation of protein-DNA complexes by native gel electrophoresis.
The molecular mass of the NucC-DNA complex was measured by using the method of Ferguson (14). Native gel electrophoresis was performed in a buffer containing 50 mM Tris base, 40 mM boric acid, and 10 mM EDTA. Oligonucleotide 5′-GTT GTG CTG TCG ATT AGA CAA C-3′ and its 22-bp complement were annealed to generate the NucC binding site. The migration of protein standards (Sigma), the DNA and the NucC-DNA complexes were analyzed on a series of gels containing different concentrations of polyacrylamide. The relative mobility (Rf) was calculated as the ratio of the mobility of a given protein to the mobility of bromophenol blue dye. For each protein, a derived value, 100[log(Rf × 100)], was plotted as a function of the acrylamide concentration (9, 21). The slopes of the lines generated for each standard protein were determined by linear regression analysis and plotted as a function of the logarithm molecular mass. This calibration curve, a Ferguson plot, was applied to determine the molecular mass of the NucC-DNA complex. The following polyacrylamide concentrations were used: 4, 5.5, 6, 7, 8, 9 and 10% for bovine serum albumin; 6, 7, 8, 9, 10, 11.5, and 12% for chicken egg albumin; 6, 7.5, 8.5, 9, 10, 11.5, and 13% for bovine erythrocyte carbonic anhydrase; 7, 11, 11.5, 12, and 13% for bovine milk α-lactobumin; and 6, 7.5, 8.5, 9, 10, 11.5, and 12% for the NucC-DNA complex. Migration of the unbound DNA was measured in the same gel as the NucC-DNA complexes. Protein standards were visualized by Coomassie blue staining. The NucC-DNA complexes were visualized by ethidium bromide staining.
SDS-PAGE analysis.
NucC was compared to protein molecular mass standards by SDS-PAGE with a Tricine buffer system and an 18% polyacrylamide gel. The two samples of NucC that were compared were stored at −80°C and at 4°C in 50 mM Tris-HCl (pH 7.0). Samples were prepared for electrophoresis by boiling for 5 min in 50 mM Tris-HCl (pH 6.8), 2% (wt/vol) SDS, 1% (wt/vol) β-mercaptoethanol, 10% glycerol, and 0.001% (wt/vol) bromophenol blue. Proteins were visualized by Coomassie blue staining.
RESULTS AND DISCUSSION
Base contacts. (i) Methylation protection and interference.
The alkylating reagent dimethyl sulfate (DMS) primarily methylates the N7 position of guanine in the major groove of DNA and the N3 position of adenine in the minor groove (43) and can be used to identify contacts in protection and premodification interference assays (42). Purine methylation results in the loss of a hydrogen bond acceptor and addition of a positive charge on the purine ring. These changes and steric blockage can affect protein binding. In addition to blocking the accessibility of the modified guanine and adenine atoms to DMS, protein binding can alter the methylation pattern of DNA by changing the conformation of the DNA and by creating areas of altered hydrophobicity. For the protection assay it was important to isolate the bound DNA before strand scission to avoid footprinting nonspecific NucC-DNA complexes.
NucC binding affected the methylation of eleven guanines and two adenines within the binding site (Fig. 2A). NucC binding decreased methylation at positions −68, −65, −60, and −57 on the sense strand and positions −62, −58, −50, and −47 on the template strand. NucC increased the methylation of guanines at positions −69 and −52 on the sense strand and position −51 on the template strand. Although methylated adenines are not usually resolved by piperidine strand scission, two bands corresponding to the adenines at positions −55 and −54 are apparent in the footprint. The adenine at position −54 was slightly hypermethylated in the presence of NucC, while the adenine at position −55 was slightly protected. NucC binding also affected the methylation of several bases that are outside of the area predicted to be contacted by the protein. These modifications are consistent with a NucC-induced structural alteration that changes the conformation of the major groove in these areas.
FIG. 2.

Major groove contacts to guanine N7 atoms and minor groove contacts to adenine N3 atoms probed by methylation protection and interference. (A) Methylation protection. Template 1 DNA was end labeled on either the sense strand or the template strand. DNA bound to NucC was isolated by gel shift to avoid footprinting of nonspecific complexes. Unbound or NucC-bound DNA, as indicated, was cleaved by using a reaction specific for methylated guanines prior to analysis on a polyacrylamide-urea sequencing gel (described in Materials and Methods). Asterisks designate two adenine residues that were also cleaved by DMS in this assay. Guanines protected from methylation by NucC binding are indicated by closed diamonds; open diamonds indicate guanines that are hypermethylated when NucC is bound. A G+A sequencing ladder of the same DNA was used as a marker. (B) Methylation interference. Template 1 DNA was end labeled on either the sense strand or the template strand and methylated prior to NucC binding. Bound and unbound fragments were isolated by gel shift and cleaved by using a reaction specific for methylated guanines and adenines (described in Materials and Methods) prior to analysis on a polyacrylamide-urea sequencing gel. Closed circles indicate nucleotides that decreased NucC binding when methylated; open circles indicate residues where methylation enhanced NucC binding. (C) Summary of the contacts mapped in the methylation protection and interference assays; symbols as described above. The conserved bases in the TGt-N12-cCA motif are boxed.
In the interference assay, a strand scission reaction that equally resolves guanine and adenine methylation was used. Many of the modifications detected in the interference assay were also detected in the protection assay. Methylation of ten guanines and two adenines affected NucC binding (Fig. 2B). Methylation of guanines at positions −68, −65, −60, and −52 on the sense strand and at positions −62, −50, and −47 on the template strand decreased the binding, and methylation of the guanines at positions −69 and −63 on the sense strand and at position −46 on the template strand increased binding. Methylation of adenines at position −53 on the sense strand and at position −55 on the template strand also increased NucC binding.
(ii) Missing purine interference.
Methylation protection and premodification interference assays provide very specific and limited information regarding the chemical features involved in DNA recognition. For instance, contacts to guanine 6-carbonyl and adenine N7 and 6-methyl groups are not expected to be detected in this assay. We addressed additional contributions of purine samples by using a less specific assay, depurination interference (5).
Depurination at most positions within the predicted binding site decreased NucC binding (Fig. 3). The two positions where binding was not affected were −69 and −54. Purine removal at position −55 increased binding. The results at positions −54 and −55 stand in contrast to the results obtained in the methylation interference assay, where methylation of the adenine at position −54 decreased binding while methylation of adenine at position −55 increased binding. The apparent discrepancy between the results of the depurination and methylation assays at positions −55 and −54 likely reflect a local distortion of the DNA helix that accompanies NucC-induced bending.
FIG. 3.
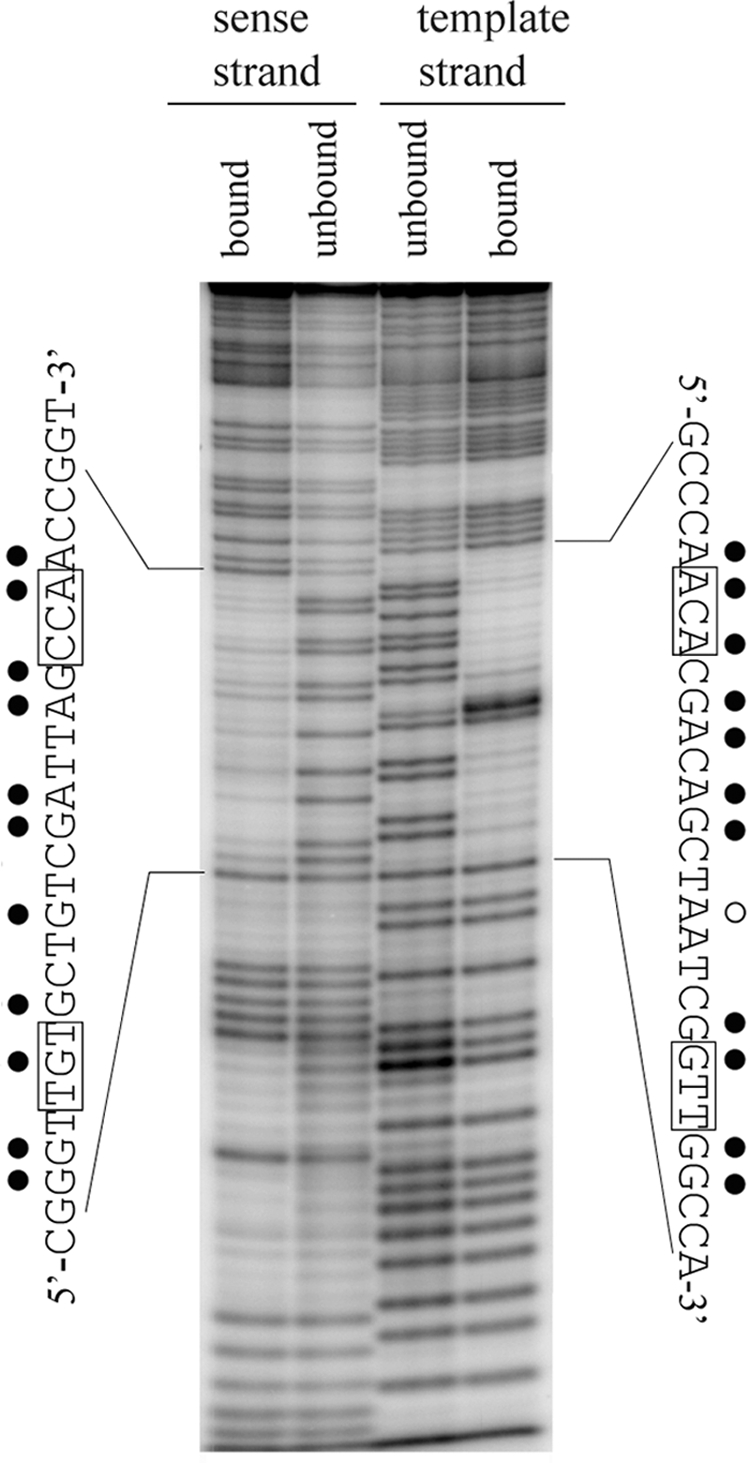
Missing purine interference. Template 3, 5′ end labeled on either the sense or template strand, as indicated, was randomly depurinated as described in Materials and Methods. Interference products were isolated by gel shift prior to strand cleavage of the NucC-bound and unbound fractions and analysis on a polyacrylamide-urea sequencing gel. The sequence of each strand of the binding site, indicating the positions at which purine removal affects NucC binding, is displayed along the sides of the gel. Symbols: •, purine removal decreases binding; ○, purine removal enhances binding. The conserved bases in the TGt-N12-cCA motif are boxed.
(iii) Uracil substitution and missing thymine interference.
Substitution of uracil for thymine in DNA results in the loss of the thymine 5-methyl group. The uracil substitution assay (37) was used to further probe areas of the major groove contacted by NucC. Uracil substitution at positions −67, −66, −64, and −59 on the sense strand and at positions −56, −53, −49, and −48 on the template strand decreased NucC binding (Fig. 4A). Uracil substitution in the template strand also interferes with NucC binding at what appears to be positions −96 and/or −99, which are well upstream of the region predicted to be contacted by NucC. We do not have an explanation for this result. This area of the probe does not correspond to PF DNA and was derived the from the plasmid template used to generate the probe.
FIG. 4.

Uracil substitution and thymine removal interference. (A) Uracil substitution interference. Uracil was randomly substituted for thymine in template 2 by PCR amplification with Taq polymerase and dUTP. The interference products (bound and unbound) for DNA end labeled on each strand were isolated by gel shift prior to strand scission at the positions of substituted uracil and analysis on the polyacrylamide-urea sequencing gel. Individual thymine residues that interfered with NucC binding when replaced with uracil are indicated. (B) Thymine removal interference. Uracil was randomly substituted for thymine in template 2 by PCR. The substituted uracil bases were removed by using uracil deglycosylase prior to isolating the interference products by gel shift, strand cleavage, and analysis on the polyacrylamide-urea sequencing gel. Individual thymine residues that affected NucC binding when absent are indicated. (C) Summary of the contacts mapped in the uracil substitution and thymine removal assays. Closed circles indicate residues where both uracil substitution and thymine removal decreased NucC binding. Open circles indicate residues where thymine removal increased binding. The conserved bases in the TGt-N12-cCA binding site motif are boxed.
The only positions within the NucC binding site that were not affected by uracil substitution are −61, −55, and −54. Uracil substitution does not provide information regarding contacts to the uracil 4-carbonyl group in the major groove or to the uracil 2- and 4-carbonyl groups in the minor and major grooves, respectively. To address the potential for these and other thymine contacts and to examine the effects of DNA flexibility on NucC binding, we used the missing thymine interference assay (11). The results (Fig. 4B) are identical to the results of the uracil substitution assay at every position except two; removal at positions −55 and −54 on the sense strand increased NucC binding. These are two of the three positions within the NucC binding site that were not detected as contacts in the uracil substitution assay.
Phosphodiester backbone contacts: ethylation interference.
Modification of DNA with the alkylating reagent ethylnitrosourea results in the addition of ethyl groups to the nonesterified oxygens in the phosphodiester backbone (43), which can be detected as contacts in an interference assay (see reference 33 and references therein). Interference with protein binding occurs when the addition of an ethyl group causes steric blockage or disrupts electrostatic interaction with the phosphodiester backbone.
NucC ethylation interference occurred at positions corresponding to phosphodiester groups between positions −69 and −68, −66 and −65, −61 to −59, and −53 to −51 on the sense strand and between positions −64 and −61, −56 and −54, −50 and −49, and −47 and −46 on the template strand (Fig. 5). Phosphodiester contacts at these positions were confirmed in multiple experiments. Other positions where ethylation appears to affect binding in the gel shown could not be reproduced on a consistent basis. These may result from base modifications that also affect binding but are poorly resolved under the experimental conditions.
FIG. 5.
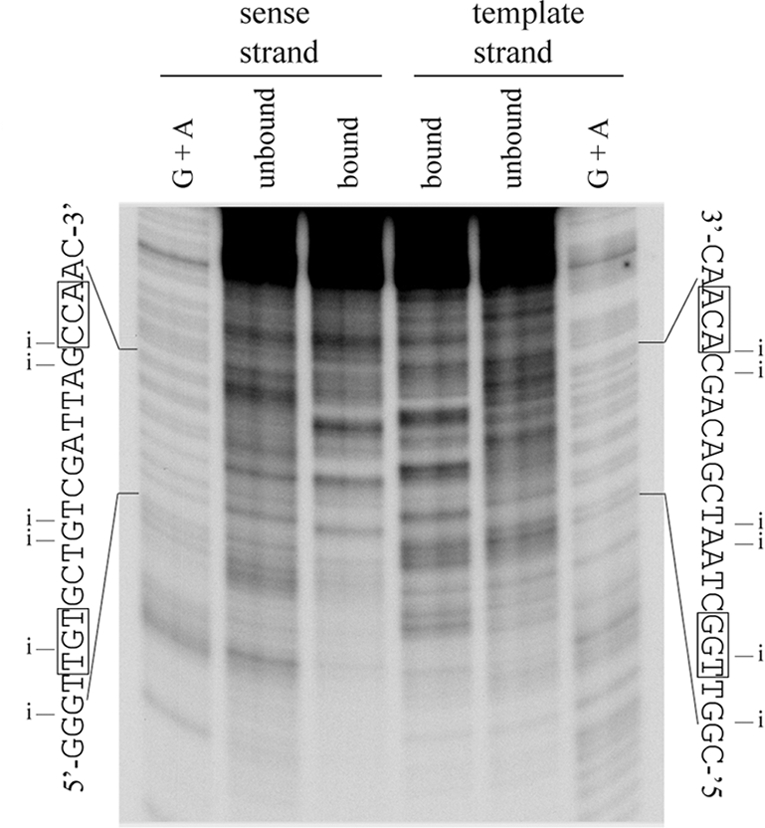
NucC phosphate backbone contacts. Template 2 was ethylated with ethylnitrosourea, and interference products were isolated by gel shift prior to strand cleavage at the ethylated positions and analysis of the bound and unbound fractions, as indicated, on a polyacrylamide-urea sequencing gel. Fragments were radiolabeled at one 5′ end, on the indicated strand. A G+A sequencing ladder of the same end-labeled DNA was used as a marker. The sequence of each strand of the binding site is shown alongside the gel. Positions where ethylation of the phosphate backbone interferes with NucC binding are indicated by an “i.” The conserved bases in the TGt-N12-cCA motif are boxed.
Minor groove interactions: hydroxyl radical protection.
Hydroxyl radical footprinting is a way to measure the solvent accessibility of sugar protons in DNA at high resolution (46). In canonical B-DNA, H-4′ and H-5″ are the sugar protons most accessible to solvent and are located on the minor groove face of the DNA. Changes in the reactivity of these protons to hydroxyl radicals can be attributed to protein contacts that block access or to protein-induced structural alterations to the DNA (6, 12).
There are variations in the pattern of DNA reactivity to hydroxyl radicals in the presence of NucC (Fig. 6). Areas of decreased reactivity were centered at approximately positions −66 and −54 on the sense strand and at positions −64 and −47 on the template strand. These results are consistent with NucC binding to this region, but it is unclear whether these changes are a direct result of close interactions of NucC within the minor groove. Changes in the pattern of reactivity are seen outside of the NucC binding site as well, and these changes as well as those within the binding site are consistent with NucC-induced structural alterations of the DNA.
FIG. 6.

Hydroxyl radical protection. Template 2, end labeled on either strand as indicated, was modified with hydroxyl radicals in the presence or absence of NucC. DNA was isolated by gel shift prior to analysis on a polyacrylamide sequencing gel to avoid footprinting nonspecific complexes. A G+A sequencing ladder of the same end-labeled DNA was used as a marker for each strand. The sequence of each strand of the binding site is shown alongside the gel. The approximate areas of decreased hydroxyl radical cleavage in the presence of NucC are indicated by brackets. The conserved bases in the TGt-N12-cCA motif are boxed.
Binding stoichiometry. (i) Molecular mass analyses of NucC-DNA complexes by native gel electrophoresis.
The degree to which the relative mobility of a macromolecule changes with the polyacrylamide concentration of gels used for native gel electrophoresis is determined by the mass and shape of the molecule or macromolecular complex. An unknown molecular mass can be measured by comparing the relative mobility to protein molecular mass standards in polyacrylamide gels of various concentrations under native conditions (14). We used this method to measure the mass of NucC-DNA complexes in order to determine binding stoichiometry.
A minimal 22-bp binding site was used in order to minimize the contribution of the shape of the DNA to electrophoretic mobility. A Ferguson plot of the mobility data for the protein standards, the DNA probe and the NucC-DNA complexes was used to measure the molecular mass of the NucC-DNA complex (Fig. 7). The measured molecular mass for the NucC-DNA complex was 42.1 kDa. NucC has a predicted molecular mass of 8.38 kDa, and the molecular mass of the DNA fragment is 13.47 kDa. Using these values, 3.42 molecules of NucC are calculated to bind per molecule of DNA.
FIG. 7.
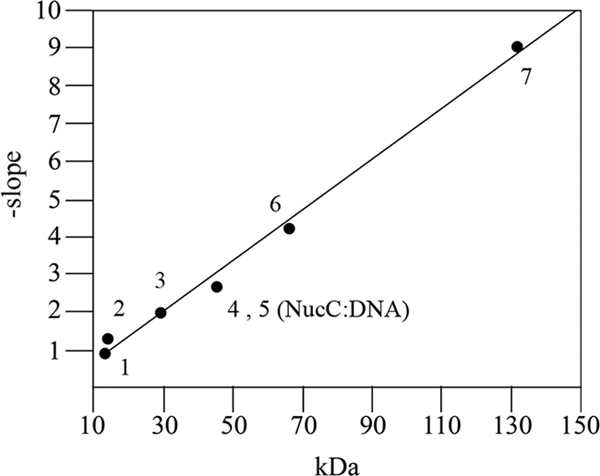
Molecular mass estimation of NucC-DNA complexes by native gel electrophoresis (Ferguson plot) analysis. Protein standards and NucC bound to a 22-bp DNA binding site were run on a series of gels with different polyacrylamide concentrations. The relative mobility (Rf) of each species was calculated as the ratio of mobility to the mobility of bromophenol blue and a derived value, 100[log(Rf × 100)], was plotted as a function of acrylamide concentration (data not shown). The slopes of the lines generated were determined by linear regression analysis and plotted as a function of the molecular mass to generate the calibration curve (Ferguson plot). Data points: 1, unbound DNA (13.47 kDa); 2, α-lactobumin (14.2 kDa); 3, carbonic anhydrase (29 kDa); 4, egg albumin (45 kDa); 5, NucC-DNA complex; 6, bovine albumin monomer (66 kDa); 7, bovine albumin dimer (132 kDa). The Rf of the NucC-DNA complex was intermediate to that of the egg albumin charge isomers over the range of polyacrylamide gel concentrations measured.
(ii) SDS-PAGE analysis of NucC.
As has been reported previously for the related PSP3 Pag and φR73 Delta proteins (24), NucC monomers migrate on SDS-polyacrylamide gels with an apparent molecular mass of ∼6.2-kDa (Fig. 8). The reason for this aberrant migration is not understood. Surprisingly, freshly purified NucC migrates as an apparent dimer on an SDS-polyacrylamide gel (Fig. 8). When the protein is stored at 4°C for several days, it assumes the electrophoretic mobility of monomeric NucC but loses its DNA-binding activity. The related Delta protein of satellite phage P4 contains two head-to-tail NucC-like domains in tandem, both of which are required for activity (26). These results, in conjunction with the dyad symmetry present in the binding site, strongly imply that the binding stoichiometry is constrained to an even number. Based on the molecular mass determined for the protein-DNA complexes, we conclude that four molecules of NucC are binding to the DNA.
FIG. 8.
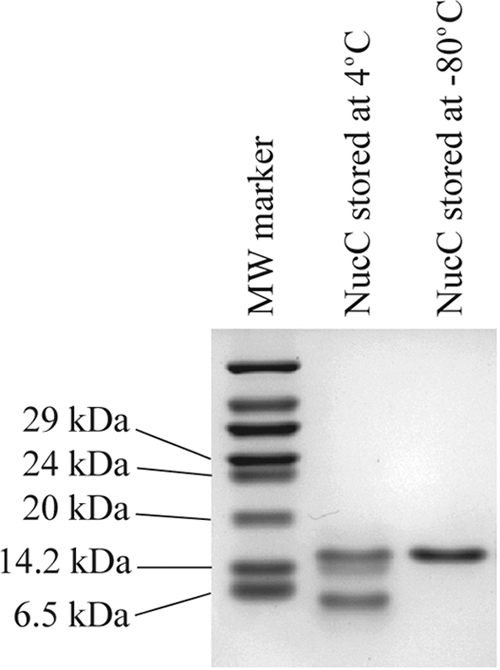
SDS-PAGE analysis of purified NucC. Samples, stored as indicated, were prepared for electrophoresis as described in Materials and Methods and run on an 18% polyacrylamide gel. NucC stored at 4°C progressively lost DNA-binding activity (data not shown).
There are several reported examples of proteins that form noncovalent oligomers or complexes resistant to SDS denaturation, even after boiling, including streptavidin (2), ubiquitinlike proteins (31), prionlike proteins (44), and membrane proteins (39). Such tight noncovalent associations are generally thought to be due to strong hydrophobic interactions between protein molecules. Further studies of NucC structure and function will be required to determine what part of the protein is involved in this unusual dimerization.
(iii) Compilation and structural modeling of DNA contacts.
Specific features of the DNA recognized by NucC are modeled on the B-form DNA and summarized in Fig. 9. The DNA is displayed as four 90° rotations of the molecule. For clarity, nitrogen modifications detected in the methylation assays are shown in blue on the model.
FIG. 9.

Compilation and molecular modeling of NucC contacts. The DNA is modeled as 90° right-hand rotations. In the molecular model, NucC contacts to the unesterified oxygens of the phosphate backbone are shown in yellow, thymine 5-methyl contacts are shown in red, and guanine N7 and adenine N3 contacts are shown in blue. In the sequence logo, the red and green bases indicate positions at which base removal decreased and increased binding, respectively. Labels: i, phosphate backbone contacts; 1, guanine N7 and adenine N3 atoms protected from methylation in the presence of NucC; 2, guanine N7 and adenine N3 atoms hypermethylated in the presence of NucC; 3, guanine N7 and adenine N3 atoms that interfered with binding when methylated; 4, guanine N7 and adenine N3 that increased binding when methylated; 5, contacts to thymine 5-methyl groups.
The 12 phosphodiester contacts form a symmetrical distribution. Major groove contacts are mapped at 18 positions within the binding site. Minor groove contacts are mapped to adenine N3 atoms at positions −55, −54, and −53. Removal of thymine bases at positions −55 and −54 increases binding, indicating that this area of the DNA is bent. A NucC-induced PF DNA bend of 90° has been measured by gel shift analysis (35). A distortion of the DNA helix could result in extrahelical base conformations or non-Watson-Crick base pairing at positions −55 and −54, with adenine N3 atoms facing the major groove. Another feature in the downstream half of the binding site is the degree of hypermethylation; methylation increases NucC binding or NucC binding increases methylation at positions −55 to −51. Outside of this cluster, hypermethylation is only detected at three positions: −69, −63, and −46. The interface between the protein and DNA appears to be more hydrophobic in the downstream half of the binding site.
The conserved trinucleotide inverted repeat in the NucC binding site is separated by 12 nonconserved bases. A contributing role for these nonconserved bases in the PF binding site has been demonstrated in previous genetic studies (10). In addition to showing that the conserved trinucleotide repeats in each half of the binding site are absolutely essential for binding and activity, this genetic analysis also indicated smaller effects for nucleotide substitutions in the intervening sequence. Furthermore, complementary base substitutions in the upstream half of the central bases of PF (positions −63 to −58) were shown to cause a ∼25% greater decrease in transcriptional activation by P4 Delta than complementary base substitutions at the corresponding positions in the downstream half (positions −57 to −52). The asymmetric effects of these substitutions can now be interpreted in light of the current data, in which we have shown that the area from positions −55 to −53 is characterized by putative minor groove contacts, increased binding upon base removal at positions −55 and −54 and a more hydrophobic protein-DNA interface. It appears likely that NucC is interacting with the DNA differently in this area of the binding site.
These studies demonstrate that DNA binding by NucC differs in important respects from the DNA binding demonstrated for eukaryotic zinc finger transcription factors. The yeast zinc finger transcriptional activator Gal4 binds as a dimer to the consensus binding site CCG-N11-CGG (4, 7, 13, 19), and the only direct base contacts in the Gal4 crystal structure are to the conserved CCG repeats (34). In contrast, NucC binds as a tetramer and major groove contacts have been mapped throughout the NucC binding site. Thus, NucC and the related proteins in the P2 Ogr family of eubacterial zinc finger transcription factors provide a new paradigm for protein-DNA interaction. Further comparisons to other transcriptional activators will require structural studies of the NucC-DNA complex. Of particular interest will be the positions of the individual NucC subunits and the distribution of the DNA bend.
Acknowledgments
This study was supported by National Science Foundation grant MCB9982525 (to G.E.C.) and by National Institutes of Health MARC Fellowship F31 GM17594 (to V.J.M.).
Plasmid pLY2 was constructed by a laboratory rotation student, Ying Liu.
Footnotes
Published ahead of print on 15 May 2009.
REFERENCES
- 1.Ayers, D. J., M. G. Sunshine, E. W. Six, and G. E. Christie. 1994. Mutations affecting two adjacent amino acid residues in the alpha subunit of RNA polymerase block transcriptional activation by the bacteriophage P2 Ogr protein. J. Bacteriol. 1767430-7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bayer, E. A., H. Ben-Hur, and M. Wilchek. 1990. Isolation and properties of streptavidin. Methods Enzymol. 18480-89. [DOI] [PubMed] [Google Scholar]
- 3.Bouhouche, N., M. Syvanen, and C. I. Kado. 2000. The origin of prokaryotic C2H2 zinc finger regulators. Trends Microbiol. 877-81. [DOI] [PubMed] [Google Scholar]
- 4.Bram, R. J., N. F. Lue, and R. D. Kornberg. 1986. A GAL family of upstream activating sequences in yeast: roles in both induction and repression of transcription. EMBO J. 5603-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunelle, A., and R. F. Schleif. 1987. Missing contact probing of DNA-protein interactions. Proc. Natl. Acad. Sci. USA 846673-6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burkhoff, A. M., and T. D. Tullius. 1987. The unusual conformation adopted by the adenine tracts in kinetoplast DNA. Cell 48935-943. [DOI] [PubMed] [Google Scholar]
- 7.Carey, M., H. Kakidani, J. Leatherwood, F. Mostashari, and M. Ptashne. 1989. An amino-terminal fragment of GAL4 binds DNA as a dimer. J. Mol. Biol. 209423-432. [DOI] [PubMed] [Google Scholar]
- 8.Chou, A. Y., J. Archdeacon, and C. I. Kado. 1998. Agrobacterium transcriptional regulator Ros is a prokaryotic zinc finger protein that regulates the plant oncogene ipt. Proc. Natl. Acad. Sci. USA 955293-5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chrambach, A., and D. Rodbard. 1971. Polyacrylamide gel electrophoresis. Science 172440-451. [DOI] [PubMed] [Google Scholar]
- 10.Christie, G. E., D. L. Anders, V. McAlister, T. S. Goodwin, B. Julien, and R. Calendar. 2003. Identification of upstream sequences essential for activation of a bacteriophage P2 late promoter. J. Bacteriol. 1854609-4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devchand, P. R., J. D. McGhee, and J. H. van de Sande. 1993. Uracil-DNA glycosylase as a probe for protein-DNA interactions. Nucleic Acids Res. 213437-3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dlakic, M., K. Park, J. D. Griffith, S. C. Harvey, and R. E. Harrington. 1996. The organic crystallizing agent 2-methyl-2,4-pentanediol reduces DNA curvature by means of structural changes in A-tracts. J. Biol. Chem. 27117911-17919. [DOI] [PubMed] [Google Scholar]
- 13.Fedor, M. J., N. F. Lue, and R. D. Kornberg. 1988. Statistical positioning of nucleosomes by specific protein-binding to an upstream activating sequence in yeast. J. Mol. Biol. 204109-127. [DOI] [PubMed] [Google Scholar]
- 14.Ferguson, K. A. 1964. Starch-gel electrophoresis: application to the classification of pituitary proteins and polypeptides. Metabolism 13SUPPL:985-1002. [DOI] [PubMed] [Google Scholar]
- 15.Ferrer, S., M. B. Viejo, J. F. Guasch, J. Enfedaque, and M. Regue. 1996. Genetic evidence for an activator required for induction of colicin-like bacteriocin 28b production in Serratia marcescens by DNA-damaging agents. J. Bacteriol. 178951-960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freedman, L. P., and B. F. Luisi. 1993. On the mechanism of DNA binding by nuclear hormone receptors: a structural and functional perspective. J. Cell. Biochem. 51140-150. [DOI] [PubMed] [Google Scholar]
- 17.Gebhardt, K. 1994. Structure and function analysis of the bacteriophage P2 Ogr protein. Dissertation/thesis. University of Oslo, Oslo, Norway.
- 18.Gebhardt, K., R. A. King, G. E. Christie, and B. H. Lindqvist. 1993. Mutational analysis of the bacteriophage P2 Ogr protein: truncation of the carboxy terminus. J. Bacteriol. 1757724-7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giniger, E., S. M. Varnum, and M. Ptashne. 1985. Specific DNA binding of GAL4, a positive regulatory protein of yeast. Cell 40767-774. [DOI] [PubMed] [Google Scholar]
- 20.Grambow, N. J., N. K. Birkeland, D. L. Anders, and G. E. Christie. 1990. Deletion analysis of a bacteriophage P2 late promoter. Gene 959-15. [DOI] [PubMed] [Google Scholar]
- 21.Hedrick, J. L., and A. J. Smith. 1968. Size and charge isomer separation and estimation of molecular weights of proteins by disc gel electrophoresis. Arch. Biochem. Biophys. 126155-164. [DOI] [PubMed] [Google Scholar]
- 22.Ito, T., X. X. Ma, F. Takeuchi, K. Okuma, H. Yuzawa, and K. Hiramatsu. 2004. Novel type V staphylococcal cassette chromosome mec driven by a novel cassette chromosome recombinase, ccrC. Antimicrob. Agents Chemother. 482637-2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin, S., Y. Chen, G. E. Christie, and M. J. Benedik. 1996. Regulation of the Serratia marcescens extracellular nuclease: positive control by a homolog of P2 Ogr encoded by a cryptic prophage. J. Mol. Biol. 256264-278. [DOI] [PubMed] [Google Scholar]
- 24.Julien, B., and R. Calendar. 1996. Bacteriophage PSP3 and φR73 activator proteins: analysis of promoter specificities. J. Bacteriol. 1785668-5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Julien, B., and R. Calendar. 1995. Purification and characterization of the bacteriophage P4 delta protein. J. Bacteriol. 1773743-3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Julien, B., P. Lefevre, and R. Calendar. 1997. The two P2 Ogr-like domains of the delta protein from bacteriophage P4 are required for activity. Virology 230292-299. [DOI] [PubMed] [Google Scholar]
- 27.Julien, B., D. Pountney, G. E. Christie, and R. Calendar. 1998. Mutational analysis of a satellite phage activator. Gene 223129-134. [DOI] [PubMed] [Google Scholar]
- 28.Klug, A., and J. W. Schwabe. 1995. Protein motifs. 5. Zinc fingers. FASEB J. 9597-604. [PubMed] [Google Scholar]
- 29.Krishna, S. S., I. Majumdar, and N. V. Grishin. 2003. Structural classification of zinc fingers: survey and summary. Nucleic Acids Res. 31532-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee, T. C., and G. E. Christie. 1990. Purification and properties of the bacteriophage P2 ogr gene product: a prokaryotic zinc-binding transcriptional activator. J. Biol. Chem. 2657472-7477. [PubMed] [Google Scholar]
- 31.Luders, J., G. Pyrowolakis, and S. Jentsch. 2003. The ubiquitin-like protein HUB1 forms SDS-resistant complexes with cellular proteins in the absence of ATP. EMBO Rep. 41169-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luscombe, N. M., S. E. Austin, H. M. Berman, and J. M. Thornton. 2000. An overview of the structures of protein-DNA complexes. Genome Biol. 1REVIEWS001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manfield, I., and P. G. Stockley. 1994. Ethylation interference. Methods Mol. Biol. 30125-139. [DOI] [PubMed] [Google Scholar]
- 34.Marmorstein, R., M. Carey, M. Ptashne, and S. C. Harrison. 1992. DNA recognition by GAL4: structure of a protein-DNA complex. Nature 356408-414. [DOI] [PubMed] [Google Scholar]
- 35.McAlister, V., C. Zou, R. H. Winslow, and G. E. Christie. 2003. Purification and in vitro characterization of the Serratia marcescens NucC protein, a zinc-binding transcription factor homologous to P2 Ogr. J. Bacteriol. 1851808-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pountney, D. L., R. P. Tiwari, and J. B. Egan. 1997. Metal- and DNA-binding properties and mutational analysis of the transcription activating factor, B, of coliphage 186: a prokaryotic C4 zinc-finger protein. Protein Sci. 6892-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pu, W. T., and K. Struhl. 1992. Uracil interference, a rapid and general method for defining protein-DNA interactions involving the 5-methyl group of thymines: the GCN4-DNA complex. Nucleic Acids Res. 20771-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riechmann, J. L., J. Heard, G. Martin, L. Reuber, C. Jiang, J. Keddie, L. Adam, O. Pineda, O. J. Ratcliffe, R. R. Samaha, R. Creelman, M. Pilgrim, P. Broun, J. Z. Zhang, D. Ghandehari, B. K. Sherman, and G. Yu. 2000. Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science 2902105-2110. [DOI] [PubMed] [Google Scholar]
- 39.Sagne, C., M. F. Isambert, J. P. Henry, and B. Gasnier. 1996. SDS-resistant aggregation of membrane proteins: application to the purification of the vesicular monoamine transporter. Biochem. J. 316(Pt. 3)825-831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schickor, P., and H. Heumann. 1994. Hydroxyl radical interference. Methods Mol. Biol. 3033-41. [DOI] [PubMed] [Google Scholar]
- 41.Schwabe, J. W., L. Chapman, J. T. Finch, and D. Rhodes. 1993. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell 75567-578. [DOI] [PubMed] [Google Scholar]
- 42.Shaw, P. E., and A. F. Stewart. 1994. Identification of protein-DNA contacts with dimethyl sulfate: methylation protection and methylation interference. Methods Mol. Biol. 8779-87. [DOI] [PubMed] [Google Scholar]
- 43.Siebenlist, U., and W. Gilbert. 1980. Contacts between Escherichia coli RNA polymerase and an early promoter of phage T7. Proc. Natl. Acad. Sci. USA 77122-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Speransky, V. V., K. L. Taylor, H. K. Edskes, R. B. Wickner, and A. C. Steven. 2001. Prion filament networks in [URE3] cells of Saccharomyces cerevisiae. J. Cell Biol. 1531327-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sunshine, M. G., and B. Sauer. 1975. A bacterial mutation blocking P2 phage late gene expression. Proc. Natl. Acad. Sci. USA 722770-2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tullius, T. D., and B. A. Dombroski. 1986. Hydroxyl radical “footprinting”: high-resolution information about DNA-protein contacts and application to lambda repressor and Cro protein. Proc. Natl. Acad. Sci. USA 835469-5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tupler, R., G. Perini, and M. R. Green. 2001. Expressing the human genome. Nature 409832-833. [DOI] [PubMed] [Google Scholar]
- 48.Van Bokkelen, G. B., E. C. Dale, C. Halling, and R. Calendar. 1991. Mutational analysis of a bacteriophage P4 late promoter. J. Bacteriol. 17337-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winslow, R. H., B. Julien, R. Calendar, and G. E. Christie. 1998. An upstream sequence element required for NucC-dependent expression of the Serratia marcescens extracellular nuclease. J. Bacteriol. 1806064-6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wood, L. F., N. Y. Tszine, and G. E. Christie. 1997. Activation of P2 late transcription by P2 Ogr protein requires a discrete contact site on the C terminus of the alpha subunit of Escherichia coli RNA polymerase. J. Mol. Biol. 2741-7. [DOI] [PubMed] [Google Scholar]