

Abstract
Rotavirus NSP1 has been shown to function as an E3 ubiquitin ligase that mediates proteasome-dependent degradation of interferon (IFN) regulatory factors (IRF), including IRF3, -5, and -7, and suppresses the cellular type I IFN response. However, the effect of rotavirus NSP1 on viral replication is not well defined. Prior studies used genetic analysis of selected reassortants to link NSP1 with host range restriction in the mouse, suggesting that homologous and heterologous rotaviruses might use their different abilities to antagonize the IFN response as the basis of their host tropisms. Using a mouse embryonic fibroblast (MEF) model, we demonstrate that heterologous bovine (UK and NCDV) and porcine (OSU) rotaviruses fail to effectively degrade cellular IRF3, resulting in IRF3 activation and beta IFN (IFN-β) secretion. As a consequence of this failure, replication of these viruses is severely restricted in IFN-competent wild-type, but not in IFN-deficient (IFN-α/β/γ receptor- or STAT1-deficient) MEFs. On the other hand, homologous murine rotaviruses (ETD or EHP) or the heterologous simian rotavirus (rhesus rotavirus [RRV]) efficiently degrade cellular IRF3, diminish IRF3 activation and IFN-β secretion and are not replication restricted in wild-type MEFs. Genetic reassortant analysis between UK and RRV maps the distinctive phenotypes of IFN antagonism and growth restriction in wild-type MEFs to NSP1. Therefore, there is a direct relationship between the replication efficiencies of different rotavirus strains in MEFs and strain-related variations in NSP1-mediated antagonism of the type I IFN response.
Group A rotaviruses are segmented double-stranded RNA viruses that cause severe dehydrating diarrhea in infants and young children worldwide (28). The viruses replicate primarily in mature enterocytes of the small intestine, but viremia and systemic infections are well documented in both humans and animals (6). The role of adaptive immunity in rotavirus infection has been extensively studied in the mouse model. Effectors, such as CD4+ and CD8+ T cells, play a critical role in the timely clearance of primary infection, while B cells are more important in resistance to reinfection (14, 27). The role of innate immunity in rotavirus infection, however, remains poorly understood.
Early studies showed that levels of type I and II interferon (IFN) are elevated in rotavirus-infected children and animals (2, 9, 22, 31, 35). Alpha IFN (IFN-α) was used successfully to treat rotavirus diarrhea in bovine and porcine models (23, 34). Exogenous type I or II IFN inhibited rotavirus infection in human intestinal HT-29 and CaCo-2 cells when the cells were treated 24 h or more prior to infection (5). However, homologous murine rotavirus-induced diarrhea and virus shedding in feces were not significantly altered in type I or II receptor knockout (KO) mice or suckling mice treated with exogenous IFN (1). In these studies, combined type I and II receptor KO mice were not studied. Vancott et al. showed that rotavirus shedding in feces was enhanced in adult, but not in suckling, signal transducer and activator of transcription 1 (STAT1) KO mice compared to wild-type mice, even though the shedding duration was not changed (33). More recently, we reported that enteric and systemic replication of a rhesus rotavirus (RRV) is significantly increased in suckling mice deficient in both type I and II IFN signaling (IFN-α/β/γ receptor [IFNR] KO or STAT1 KO), resulting in lethal pancreatitis, hepatitis, and biliary atresia, while replication of a murine rotavirus (EC), either enteric or systemic, was virtually identical between healthy and IFN-deficient mice (13). The diverse effects of IFN on rotavirus infection suggest that the virus may possess anti-host IFN mechanisms that have variable efficacy depending on the host, cell type, and virus species origin.
The rotavirus nonstructural protein NSP1, the product of gene segment 5, has been recently proposed to function as an E3 ubiquitin ligase and to promote the proteasome-mediated degradation of cellular IFN regulatory factor 3 (IRF3), IRF5, and IRF7 and to concomitantly suppress early IFN responses (3, 4, 16). While NSP1 deletion mutant viruses exhibit small plaque size in infected cells, NSP1 is not required for viral replication (32). The simian rotavirus strain SA11 with NSP1 truncation exhibits cell-type-specific reduction in replication and can grow to higher titers in IRF3/IRF7 knockdown experiments. However, differences in the replication capacities of nonmutant rotavirus strains encoding full-length NSP1 proteins due to variations in the ability of NSP1 to interfere with the host IFN response have not been directly demonstrated (4). Interestingly, in a mouse study using classic genetic analysis of reassortants between a highly enterovirulent homologous murine rotavirus (EW) and a significantly less enterovirulent heterologous simian strain (RRV), NSP1 was associated with host range restriction, defined as the ability of a strain to spread efficiently among a litter of suckling mice (7). Presently, it is not clear whether rotavirus host range restrictions are determined in part or completely by the differential effects of NSP1 on degrading IRFs and antagonizing host IFN among homologous or heterologous virus strains.
In this study, we used primary mouse embryonic fibroblasts (MEFs) from wild-type or IFN-deficient (IFNR KO or STAT1 KO) mice to investigate the different roles of the IFN system in regulating the replication of selected heterologous and homologous rotavirus strains. We then used viral genetics to determine the viral gene associated with differential replication. We found that replication of heterologous bovine (UK and NCDV) or porcine (OSU) rotaviruses was significantly restricted in wild-type MEFs compared to MEFs lacking an intact IFN signaling response. In contrast, the replication of heterologous RRV or tissue culture-adapted murine rotaviruses (ETD and EHP) was identical in wild-type and IFN-deficient MEFs. Using a library of reassortants derived from an IFN-sensitive (bovine [UK]) and a resistant (simian [RRV]) strain, we demonstrated that the ability of RRV to replicate efficiently in wild-type MEFs cosegregated with gene segment 5, which encodes NSP1. In addition, we found that infection of mouse fibroblasts with RRV or ETD, but not UK, resulted in a significant loss of endogenous IRF3, prevented its phosphorylation on serine 396, suppressed IRF3 nuclear translocation, and inhibited IFN-β secretion in MEFs. These properties were also linked to the NSP1 gene. These data are the first to establish a direct association of the efficiency of NSP1-mediated IRF3 degradation and IFN suppression with levels of rotavirus replication in primary cells.
MATERIALS AND METHODS
Mice, cells, and viruses.
IFN-α/β-IFN-γ receptor double-KO (IFNR KO) mice and wild-type control 129SV mice were generous gifts from H. Virgin (24). STAT1 KO mice, which are also on the 129SV background, were purchased from Taconic (Germantown, NY). All mice were maintained at the Veterinary Medical Unit of the Palo Alto VA Health Care System. All animal studies were approved by the Stanford Institutional Animal Care Committee.
Isolation of MEFs from wild-type 129SV, IFNR KO, and STAT1 KO mice was performed following the protocol from Millipore (Billerica, MA). Briefly, embryos from 14-day-gestation mice were digested in 5% trypsin with EDTA (Sigma) for 20 min at 37°C. After digestion, cells were collected, washed, and passed through a 40-μm cell strainer (BD Bioscience, Bedford, MA) to obtain single-cell suspensions. The isolated cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) at 37°C in a humidified 5% CO2 incubator. MEFs used in this study were passaged less than five times.
The tissue culture-adapted simian strains RRV and SA11 (Baylor clone 3), the bovine strains UK and NCDV, the porcine strain OSU, the murine strains ETD (a cell culture-adapted version of the original wild-type EDIM strain) and EHP, and reassortant viruses derived from coinfection by UK and RRV were propagated in MA104 cells. The MA104 cells were maintained in M199 medium supplemented with 10% FCS. Titers of stock rotavirus were determined by plaque assay in MA104 cells and expressed as PFU per milliliter as described previously (20).
Rotavirus infections and determination of virus yields.
Rotaviruses were activated by trypsin (5 μg/ml; Sigma-Aldrich, St. Louis, MO), diluted in DMEM lacking FCS (iDMEM), and used to infect fully confluent MEFs from wild-type, IFNR KO, or STAT1 KO mice in 96-well plates at multiplicities of infection (MOIs) of 10, 1, or 0.1 in quadruplicate. The virus was allowed to absorb for 1 h at 37°C and then was removed, and the plates were washed with iDMEM. Serum-free DMEM containing 0.5 μg/ml trypsin was then added to the cells, which were maintained for an additional 23 h (total, 24 h) at 37°C and 5% CO2. Following infection, the MEFs were freeze-thawed to release the virus. Virus yields were determined by focus-forming assay in MA104 cells as described previously (21). Briefly, MEF lysates were serially diluted in M199 without FCS and added to MA104 cells in 96-well plates. Following 1 h of absorption, the cells were washed and cultured for 15 h in M199 without FCS or trypsin prior to fixation with 10% formalin and permeabilization with 1% Triton X-100 (Sigma-Aldrich). After being washed, the cells were stained with polyclonal guinea pig anti-rotavirus antibody and peroxidase-conjugated goat anti-guinea pig immunoglobulin G antibody (Chemicon, Temecula, CA). 3-Amino-9-ethylcarbazole was used for color development (Vector Laboratories, Inc., Burlingame, CA). The virus titers were expressed as focus-forming units (FFU) per milliliter of lysate. To measure the relative magnitudes of virus yields between wild-type and IFN-deficient MEFs, a virus yield ratio was calculated as follows: yield from IFNR or STAT1 KO MEF (FFU/ml)/yield from wild-type MEF (FFU/ml).
Measurement of IFN-β by ELISA.
MEFs were plated in 48-well plates until they were confluent and were infected with different rotaviruses at an MOI of 10 or 1, as indicated, in quadruplicate as described above. Supernatants (SN) were collected 16 h postinfection (p.i.), clarified by centrifugation (20,000 × g for 10 min), and stored at −80°C prior to being tested. IFN-β in SN was determined by enzyme-linked immunosorbent assay (ELISA) with a kit from R&D Systems, Inc. (Minneapolis, MN), according to the manufacturer's instructions. The amount of IFN-β was expressed as pg/ml of SN.
Fluorescence microscopy detection of IRF3 in rotavirus-infected MEFs.
Wild-type MEFs were grown on human fibronectin-coated cover glasses (BD Bioscience, San Jose, CA) in six-well plates. The cells were infected at an MOI of less than 0.5. Following 1 h of virus absorption, the cells were washed and cultured for an additional 5 h in the presence of iDMEM lacking trypsin. The cells were fixed with cold methanol and acetone (1:1) for 30 min and permeabilized with 1% Triton X-100 for 2 min. The presence of rotavirus was detected with Texas red-labeled mouse monoclonal anti-rotavirus VP6 antibody, clone 1E11, which reacts with all of the rotavirus strains used. IRF3 was detected with a polyclonal goat anti-IRF3 antibody (R&D Systems, Inc.) and Alexa 488-labeled rabbit anti-goat immunoglobulin G (Invitrogen, Carlsbad, CA). Cell nuclei were stained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Invitrogen). Immunofluorescent staining was observed under a Nikon Eclipse TE300 inverted fluorescence microscope equipped with a QImaging Retica 200R charge-coupled-device camera (QImaging, Surrey, BC, Canada). Images were acquired and analyzed with the QCapture Pro program (QImaging).
Immunoblotting.
Wild-type MEFs were infected with UK, RRV, ETD, and selected UK- or RRV-like reassortant viruses (4-1-1 and 24-1-1) at an MOI of 30. After 1 h of absorption, the virus was aspirated and the cells were washed and maintained in fresh iDMEM without trypsin. At 16 h p.i., the cells were washed twice in phosphate-buffered saline and lysed in 2× Laemmli buffer containing 2% sodium dodecyl sulfate (SDS) at room temperature for 20 min. The lysates were passed through a 25-gauge needle six to eight times and denatured by being boiled for 5 min. The proteins were separated by electrophoresis on 12% SDS-polyacrylamide gel electrophoresis gels and transferred to nitrocellulose membranes prior to being immunoblotted. The Western blots were probed for endogenous levels of IRF3 with polyclonal rabbit anti-IRF3 antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA), for IRF3 phosphorylated at S396 (pS396 IRF3) with rabbit monoclonal antibody (MAb) (clone 4D4G; Cell Signaling Technology, Canvers, MA), for phosphorylated STAT1 with rabbit MAb (clone Y701; Cell signaling), for rotavirus VP6 with mouse MAb (clone 2B4; Abcam Inc., Cambridge, MA), or for actin with mouse anti-β-actin MAb (clone AC15; Sigma). Horseradish peroxidase-labeled anti-rabbit or anti-mouse secondary antibodies were purchased from GE Healthcare Bio-Science Corp. (Piscataway, NJ), and signals were developed using enhanced chemiluminescence (GE Healthcare Bio-Science Corp.).
Analysis of the IFN response to inactivated rotavirus.
A titered stock of UK virus was divided into aliquots for comparison of live and inactivated virus. Virus was inactivated with psoralen (Sigma; 40 μg/ml) and by exposure to a UV light source (GBL-100C utilizing 100-W mercury reflector lamps; G. B. Gate and Co., Franklin Square, NY) for 40 min as previously described (18). Residual infectivity in the inactivated virus preparation could not be detected by plaque assay (data not shown). NIH 3T3 fibroblasts were grown in DMEM supplemented with 10% FCS and infected at the indicated MOIs, as described above. Six hours p.i., the cells were washed twice in Dulbecco's PBS and lysed in 2× Laemmli buffer containing 2% SDS for 20 min at room temperature. The lysates were boiled for 5 min and analyzed by SDS-polyacrylamide gel electrophoresis and immunoblotting as described above.
Statistical analysis.
Differences in virus yields and IFN-β production in MEFs were analyzed by analysis of variance (ANOVA) using Statview (SAS Institute, Cary, NC). Comparison of wild-type and IFN-deficient MEFs was performed using a post hoc Scheffe test. Analysis of rotavirus genes cosegregating with virus growth phenotypes in MEFs was analyzed by the permutation method of Brown and Fears (8).
RESULTS
Differential replication of rotaviruses in wild-type and IFN-deficient MEFs.
In order to study the role of IFN response in the regulation of selected heterologous and homologous rotavirus strains in mouse cells, wild-type and IFN-signaling-deficient MEFs were infected with different strains of rotaviruses, and the virus yields were measured at 24 h p.i. (Fig. 1). The virus yields of bovine (UK and NCDV) and porcine (OSU) rotaviruses from wild-type MEFs were more than 2 log10 units lower than that from IFN-deficient (IFNR KO or STAT1 KO) MEFs (P < 0.01) (Fig. 1), indicating the IFN sensitivity of these strains in wild-type MEFs. In contrast, the replication rates of simian RRV and homologous murine ETD and EHP strains were similar in both wild-type and IFN-deficient MEFs (P > 0.05) (Fig. 1). The growth of another simian strain, SA11, was reduced 8- to 10-fold in wild-type compared to IFN-deficient MEFs (P < 0.01) (Fig. 1). Collectively, these experiments revealed that the heterologous bovine and porcine rotavirus strains are highly sensitive to the antiviral effects of the mouse IFN system, while the two homologous murine rotavirus strains, as well as the simian RRV, replicate optimally despite the presence of an intact IFN signaling system. The SA11 strain appears to have an intermediate phenotype.
FIG. 1.

Virus yield in wild-type or IFN-deficient MEFs following infection with the indicated strains of rotavirus. Wild-type 129SV, IFNR KO, or STAT1 KO MEFs were infected with different rotavirus strains at an MOI of 1 for 24 h. The virus yield after infection was determined by focus-forming assay in MA104 cells and expressed as FFU/ml (see Materials and Methods). Each bar represents the average from quadruple data points plus the standard deviation. *, significant difference in the virus yield compared to IFNR KO and STAT1 KO MEFs (P < 0.01) by ANOVA and Scheffe test.
Genetic basis for host IFN restriction of heterologous rotaviruses.
The data in Fig. 1 indicate that some strains of rotavirus (UK, NCDV, and OSU) exhibit markedly restricted growth in wild-type MEFs compared to IFN-deficient MEFs, while other virus strains (heterologous RRV and the homologous ETD and EHP) display no such restriction. In order to identify the genetic basis for this striking difference in IFN-dependent growth restriction in different rotavirus strains, we used a series of reassortant viruses derived from cells coinfected with UK (IFN restricted) and RRV (nonrestricted). Wild-type or IFN-deficient MEFs were infected with UK × RRV reassortant viruses, and the virus yield ratio between IFN-deficient MEFs and wild-type MEFs for each reassortant virus was calculated (Table 1). Based on the yield ratios, the reassortants could be divided into UK-like (ratio greater than 100) and RRV-like (ratio less than 20) groups. We found that the UK growth phenotype always cosegregated with UK gene 5, while the RRV phenotype always cosegregated with RRV gene 5 (Table 1); this segregation was significantly different from that expected by chance (P < 0.001). Notably, no other rotavirus genes showed statistically significant correlation with the growth phenotype in the two MEF types (P > 0.05).
TABLE 1.
Genetic analysis of deferential growth in wild type and IFN-deficient MEFs using UK × RRV reassortant virusesa
| Reassortant | Gene
|
Virus growth ratio
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5b | 6 | 7 | 8 | 9 | 10 | 11 | IFNR KO/wild type | STAT1 KO/wild type | |
| 25-1-1 | R | U | U | R | R | U | R | U | R | R | U | 1.4 | 0.9 |
| 24-1-1 | R | U | U | R | R | U | R | U | R | U | U | 2.0 | 3.0 |
| 7-1-1 | R | R | R | R | R | R | R | R | R | R | U | 2.8 | 3.6 |
| 19-1-1 | R | R | R | R | R | R | R | U | R | R | R | 3.7 | 1.0 |
| 14-1-1 | U | R | U | R | R | R | R | R | R | R | R | 4.0 | 5.0 |
| 11-2-1-1 | R | U | R | R | R | U | R | R | R | R | U | 4.1 | 5.9 |
| 27-3-1 | U | R | R | R | R | R | R | R | R | R | R | 4.7 | 3.6 |
| 25-2-1 | R | R | R | R | R | U | R | R | R | U | R | 7.4 | 5.9 |
| 13-1-1 | R | R | R | R | R | U | R | R | R | R | R | 7.7 | 7.5 |
| 22-1-1 | R | R | R | R | R | R | R | U | R | R | R | 7.9 | 8.6 |
| 8-1-1 | R | R | U | R | R | R | R | R | R | R | U | 8.8 | 16.4 |
| 1-2-1 | R | U | R | R | U | R | R | R | R | R | U | 133.3 | 104.9 |
| 36-2-1 | U | U | U | U | U | U | U | U | R | U | R | 141.0 | 125.4 |
| 6-1-1 | R | R | R | R | U | U | R | U | R | R | R | 142.4 | 135.6 |
| 27-2-1 | R | R | R | R | U | U | R | R | R | R | R | 160.0 | 135.8 |
| 4-1-1 | R | R | R | R | U | U | R | R | R | R | U | 178.2 | 214.3 |
| 36-1-1 | R | U | R | R | U | R | R | U | U | R | R | 221.8 | 141.8 |
| 21-1-1 | U | U | U | U | U | R | R | U | R | R | R | 230.0 | 151.8 |
| 9-8-1 | U | U | U | U | U | U | R | U | U | U | U | 271.4 | 256.7 |
| 27-1-1-1 | U | U | R | U | U | R | U | R | U | U | U | 287.6 | 139.8 |
| 32-2-1-1 | U | U | R | U | U | U | R | U | U | U | R | 393.8 | 358.1 |
| 20-1-1 | U | R | U | U | U | R | U | U | U | R | R | 582.1 | 410.3 |
| UK | U | U | U | U | U | U | U | U | U | U | U | 232.1 | 338.5 |
| RRV | R | R | R | R | R | R | R | R | R | R | R | 4.8 | 4.2 |
R represents gene from RRV, and U represents gene from UK. Wild-type, IFNR KO, and STAT1 KO MEFs were infected with UK, RRV, or the indicated UK × RRV reassortant viruses at an MOI of 1 for 24 h. Virus yields were determined by focus-forming assay on MA104 cells. The virus growth ratio was calculated as follows: yield of IFNR KO or STAT1 KO MEF (FFU/ml)/yield of wild-type MEF (FFU/ml). The ratios presented are the means of quadruple data points. Boldface type highlights gene 5 segregation.
Cosegregation of gene 5 with a distinct UK or RRV growth phenotype in MEFs is significantly different from random distribution (P < 0.001 by the permutation method of Brown and Fears [8]).
IFN-β secretion in rotavirus-infected MEFs.
Since rotavirus NSP1, which is encoded by gene 5, has been shown to degrade cellular IRF3 and -7, resulting in downregulation of the host IFN response (3, 4), we measured IFN-β secretion in wild-type MEFs following infection with UK, RRV, ETD, and selected UK- or RRV-like reassortant viruses by ELISA. As shown in Fig. 2, UK and a UK-like reassortant, 4-1-1, which has UK gene 5 but most other genes from RRV, induced a robust IFN-β response in wild-type MEFs at an MOI of 10 after 16 h of infection (Fig. 2). A reduced, but clear, IFN-β response was also observed at 6 h p.i. or at a lower MOI of 1 (data not shown). In contrast, RRV; an RRV-like reassortant, 24-1-1, which has RRV gene 5 but most other genes from UK; and the murine ETD strain significantly suppressed IFN-β secretion in wild-type MEFs at an MOI of 10 (Fig. 2). These results demonstrated that the presence of the RRV NSP1-encoding gene was correlated with the absence of IFN-β secretion in MEFs and is in agreement with our genetic analysis correlating the growth phenotype in wild-type versus IFN-deficient MEFs with RRV NSP1.
FIG. 2.
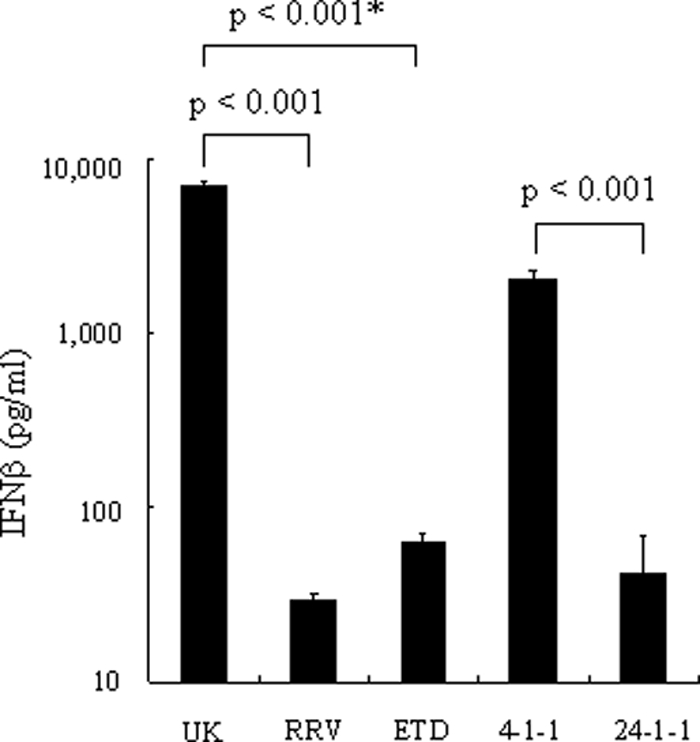
IFN-β secretion from wild-type MEFs following infection with different strains of rotavirus. Wild-type MEFs were infected with UK, RRV, ETD, UK-like reassortant 4-1-1, and RRV-like reassortant 24-1-1 at an MOI of 10 for 16 h. IFN-β secretion in the SN was determined by ELISA. Each bar represents quadruple data points plus the standard deviation. *, statistically significant difference between groups by ANOVA and Scheffe test.
Rotavirus-induced IRF3 nuclear translocation in MEFs.
It has been shown that activation of IRF3 results in its phosphorylation and nuclear translocation (26). We found that infection with UK and the reassortant 4-1-1 induced IRF3 nuclear translocation in wild-type MEFs as early as 6 h p.i. (Fig. 3A to D). Interestingly, IRF3 nuclear translocation was observed in cells both positive and negative for VP6 staining, presumably due to a secondary response to secreted IFN-β from neighboring infected cells. This conclusion was supported by the observation that IRF3 nuclear translocation occurred only in VP6 positively stained cells from IFNR-deficient MEFs (Fig. 3K and L); however, the possibility that some of the antigen-negative cells represented infection at levels too low to be detected by immunostaining cannot be excluded. Similar IRF3 nuclear translocation was also observed in MEFs infected with the bovine NCDV or porcine OSU strain (data not shown). In contrast, IRF3 nuclear translocation was not detectable in MEFs infected with either RRV, reassortant 24-1-1 (encoding RRV NSP1), or ETD (Fig. 3E to J) These results suggest that the growth restriction of heterologous bovine or porcine rotavirus strains in the presence of an intact murine fibroblast IFN response is likely related to the inability of their NSP1 proteins to inhibit IRF3 function.
FIG. 3.

IRF3 nuclear translocation in wild-type MEFs following infection with the indicated strains of rotavirus. Wild-type MEFs were infected with the UK (A and B), RRV (C and D), ETD (E and F), UK-like reassortant 4-1-1 (G and H), or RRV-like reassortant 24-1-1 (I and J) strain, and IFNR KO MEFs were infected with UK (K and L) at an MOI of 0.5 for 6 h. The cells were stained for IRF3 (green), rotavirus VP6 (red), and nuclei (blue). The panels on the left show all three colors, except panel K, which shows only red and green, and the panels on the right show only IRF3 (green). The small arrows indicate antigen-positive cells with or without IRF3 nuclear translocation. The large arrows indicate antigen-negative cells with IRF3 nuclear translocation. Magnification, ×600.
IRF3 degradation in wild-type MEFs following rotavirus infection.
The microscopy data suggested that cellular IRF3 function was inhibited by RRV and the homologous ETD strain but enhanced by UK infection. In order to examine this possibility further, we measured levels of endogenous total IRF3 and pS396 IRF3 in MEFs by immunoblotting following virus infection (Fig. 4). At 16 h p.i., levels of total IRF3 in RRV-, 24-1-1-, and ETD-infected wild-type MEFs were significantly reduced compared to those in uninfected or UK- or reassortant 4-1-1-infected cells. Interestingly, pS396 IRF3, a marker for virus-induced IRF3 activation (25), could be detected only in cells infected with UK or reassortant 4-1-1, but not RRV, reassortant 24-1-1, or ETD. Thus, rotavirus strains possessing the RRV NSP1 gene could efficiently direct IRF3 degradation, and no pS396 IRF3 accumulated in their presence. Conversely, a lack of IRF3 degradation and accumulation of pS396 IRF3 isoforms was observed in the presence of viruses encoding a UK NSP1 protein.
FIG. 4.

Immune blot analysis of total IRF3 and pS396 IRF3 in wild-type MEFs infected with the indicated strains of rotavirus. Wild-type MEFs were infected with selected rotaviruses at an MOI of 30 for 16 h or mock infected. Proteins in the cell lysates were analyzed for total IRF3, pS396 IRF3 (pIRF3), and β-actin by immune blotting.
Effects of inactivated rotavirus on IFN-β secretion and IRF3 activation.
We investigated whether the ability of UK rotavirus to activate IRF3 and stimulate IFN-β was dependent on viral replication in NIH 3T3 cells. NIH 3T3 cells were chosen for this experiment because they have higher levels of endogenous IRF3 than MEFs and secrete comparable amounts of IFN-β in response to UK virus infection (data not shown). NIH 3T3 cells treated with psoralen-inactivated UK rotavirus (MOI = 1) secreted little or no IFN-β, while infectious UK stimulated approximately 100-pg/ml secretion after 16 h of infection (data not shown). In addition, NIH 3T3 cells treated with live, but not psoralen-inactivated, UK virus demonstrated IRF3 phosphorylation, consistent with the conclusion that the IFN response in these cells is dependent on viral replication (Fig. 5). Similarly, only replication-competent UK virus induced significant phosphorylation of STAT1 (Fig. 5), an event that was correlated with the ability of UK to activate IRF3 and induce IFN-β secretion.
FIG. 5.

Role of viral replication in UK virus-mediated activation of the IFN response. NIH 3T3 cells were infected with equal amounts of live (UK) or psoralen-inactivated (i-UK) virus as indicated, and total protein lysates were examined by immunoblotting at 6 h p.i.
DISCUSSION
Using a model of rotavirus infection in primary MEFs, we demonstrated that the type I IFN host response was efficiently stimulated by several wild-type cell culture-adapted heterologous strains of rotavirus, including bovine UK and NCDV or porcine OSU, leading to IFN-β secretion, IRF3 nuclear translocation, and phosphorylation of S396 on IRF3. Consequently, replication of these viruses was significantly restricted in IFN-competent wild-type MEFs compared to IFN-deficient MEFs. In contrast, wild-type cell culture-adapted homologous murine strains, such as ETD and EHP, efficiently suppressed the host type I IFN response in wild-type cells, and removal of functional IFN signaling components in IFNR KO or STAT1 KO MEFs provided no significant growth advantage to these murine strains. Interestingly, the heterologous simian rotavirus RRV behaved similarly to murine viruses both in suppression of host IFN responses and in replication ratios in wild-type and IFN-deficient MEFs.
Using classic rotavirus gene reassortment analysis between UK and RRV parental strains, we observed that rotavirus gene segment 5, encoding NSP1, is a significant determinant of the distinctive growth phenotypes of these two viruses in wild-type and IFN-deficient MEFs. While other genes undoubtedly play roles in determining cell tropism under various conditions, in this study of primary wild-type MEFs, only gene 5 was significantly associated with a differential growth advantage in the presence of an intact IFN signaling system. It has been shown for several virus strains and cell systems that rotavirus NSP1 inhibits the host cell type I IFN response by mediating proteasomal degradation of IRF3, -5, and -7 (3, 4, 16). In this study, we confirmed that in MEFs, ETD or RRV infection induced IRF3 degradation, reduced IRF3 activation as measured by nuclear translocation assay and S396 phosphorylation, and inhibited IFN-β secretion in MEFs. We mapped the IRF3 degradation activity of RRV to the NSP1 protein using UK × RRV reassortant viruses. ETD NSP1 presumably behaves similarly to RRV NSP1 in MEFs, since it antagonized cellular IRF3 and IFN-β secretion in a similar way and exhibited a lack of enhanced replication in IFN-deficient MEFs that was identical to that of RRV.
In contrast to the rotavirus-mediated inhibition of IFN responses, a second phenotype uncovered in these experiments was rotavirus-induced activation of the murine IFN response. Specifically, we found that UK-infected MEFs responded with high levels of IFN-β secretion, translocation of IRF3 to the nuclei of infected fibroblasts, phosphorylation of S396, and absence of IRF3 degradation. These properties of UK also mapped to the NSP1 protein by reassortment analysis. The restricted growth of the NCDV and OSU strains in wild-type MEFs may also be mediated by NSP1, since these viruses induced IRF3 nuclear translocation in infected wild-type MEFs similarly to UK virus (data not shown). Of note, NCDV has been previously shown to effectively degrade IRF3 in MA104 cells (15). In addition, the NSP1-encoding genes of NCDV and UK viruses used in these experiments are able to efficiently degrade IRF3 when transiently overexpressed in certain nonmurine cell types (unpublished observation). This indicates that the NCDV and UK viruses used here are not intrinsically defective in IRF3 degradation. They simply do not degrade murine IRF3 in mouse fibroblasts. Of note, inactivated UK rotavirus did not efficiently activate IRF3 or induce IFN secretion in MEFs, indicating that in these cells, at a minimum, viral transcription is required to initiate the IFN response. This finding is compatible with prior studies that linked IFN-β secretion with RIG I-mediated signaling in HT29 intestinal epithelial cells (19). Future studies are needed to determine whether this transcription-dependent IFN signaling mechanism is utilized by murine cells of other lineages. Taken together, our results provide new evidence that strain-specific differences in NSP1-directed IRF3 inhibition and suppression of IFN secretion in primary MEFs underlie the differences in virus tropism for these cells. Whether these differences in growth capacity are also present in other primary cell types, such as epithelial or lymphoid cells, either in vitro or in vivo and whether they are also associated with differences in NSP1 remain to be tested.
Barro and Patton demonstrated that a mutant SA11 lacking the full-length NSP1 exhibits cell-type-dependent defects in virus replication that can be partially restored with small interfering RNA to IRF3 or -7 (4). However, the effects of the IFN response on the replication of different rotavirus strains encoding full-length NSP1 proteins has not been directly examined within a particular cell type. In this study, we have used strains of rotavirus with wild-type NSP1s and a genetic reassortment approach to clearly demonstrate that the differential viral replication ability of UK versus RRV rotaviruses seen in wild-type and IFN-deficient MEFs is directly related to their NSP1 proteins and their ability to degrade IRF3. Since the results in this study were obtained using viruses with full-length wild-type NSP1, our results are likely to be relevant to future investigations into the molecular mechanism of rotavirus strain-specific IFN antagonism and virus host range restriction in vivo.
A recent study by Graff et al. indicates that in addition to IRF3 degradation, NSP1 can also mediate proteasome-dependent degradation of β-TrCP, resulting in reduction of NF-κB activation and the magnitude of the IFN response (15). While we did not measure β-TrCP degradation in this study, it will be interesting to investigate the status of β-TrCP in MEFs following infection with different strains of rotavirus. However, as we have shown, UK virus fails to degrade cellular IRF3 and stimulates a robust IFN-β response, while RRV efficiently degrades IRF3 and suppresses the host IFN-β response in MEFs. Therefore, at least for these two strains of virus replicating in murine fibroblasts, NSP1-mediated inhibition of β-TrCP is unlikely to be relevant for IFN induction.
A species- or cell-type-specific viral antagonism of host IFN responses has been described in other virus systems, such as simian virus 5 (SV5), a member of the Paramyxovirus family. SV5 is permissive in human, but not murine, cells, and the differential growth is correlated with different levels of the IFN response stimulated in human or murine cells after infection (10). SV5 suppresses host IFN antiviral activity through V protein-mediated degradation of STAT1; however, the species specificity of IFN antagonism is determined by viral interaction with the STAT2 protein (29).
NSP1 is the most genetically diverse rotavirus protein, although phylogenetic analysis reveals that sequences from viruses isolated from the same species tend to cluster, suggesting that rotavirus NSP1 may have coevolved with the cognate host species (12). It is interesting that the NSP1 proteins of the simian rotaviruses RRV and SA11 are closer to those of murine viruses than to those of either bovine or porcine strains. Whether this phylogeny in sequence plays any significant role in the similar behavior observed between simian and murine rotavirus strains in MEFs in our experiments is presently unknown and an area of our current research. Graff et al. have reported that the ineffective degradation of IRF3 in MA104 cells by OSU NSP1 is related to its weak binding to the target protein (16). The NSP1 N-terminal zinc-binding domain, which is proposed to function as an E3 ligase in directing IRF3 degradation, is highly conserved. The C terminus, which is said to be related to NSP1 binding to IRF3 (17), is significantly more variable among different viral strains. At this time, no specific sequence features of NSP1 have been identified that can unambiguously explain the strain-specific IRF3 degradation in MEFs or other cells. We are currently investigating the molecular mechanism of this rotavirus species- or cell-type-specific IFN antagonism.
We have previously shown that RRV replication is significantly enhanced in IFNR-deficient mice, suggesting that RRV is sensitive to an intact host IFN system in vivo, especially in its replicative ability in systemic organs, such as the biliary tract and pancreas (13). Therefore, it is somewhat paradoxical that our current data demonstrate that RRV replication is not restricted in IFN-competent wild-type MEFs. Of note, Douagi et al. (11) reported that, despite the efficient inhibition of type I IFN secretion by RRV in MEFs, RRV infection efficiently elicits type I IFN secretion by murine dendritic cells (DC). Additionally, we have found that purified primary human DCs readily respond to exposure to RRV with a brisk type I IFN response, and this response is independent of viral transcription (unpublished data). These findings suggest that, in vivo, DCs could provide a source of type I IFN that is not subject to viral NSP1-mediated antagonism. RRV is not intrinsically resistant to type I IFN, since its replication is inhibited in cells pretreated with type I IFN (5). Hence, we can speculate that in vivo RRV antagonism of the local host IFN response facilitates its replication in targets such as intestinal epithelial cells while IFN produced by DCs (and possibly other cell types) would eventually control the RRV infection. This hypothesis is supported by our observation that initial levels of RRV in the gut and systemically are similar in wild-type and IFN-deficient mice and that a significant increase in systemic virus loads occurs at later times p.i. in IFN KO mice (13). RRV infection is cleared after 5 days p.i. in wild-type mice, but replication persists much longer in IFN-deficient mice, especially in the biliary tract and pancreas, and induces severe inflammatory responses. In addition, RRV-induced biliary atresia in newborn mice can also be treated with exogenous type I IFN (30), a finding also consistent with our hypothesis.
The response of the mouse IFN system to murine rotaviruses is apparently distinct from that to simian RRV. Similar to RRV, murine rotaviruses efficiently suppressed a cellular IFN response, and their replication was not restricted in wild-type MEFs. However, unlike that of RRV, murine rotavirus replication in vivo is not markedly restricted in the presence of an intact IFN system (13). This suggests that murine rotaviruses are likely to successfully negate or avoid IFN responses in target cells, such as primary human DCs, that detect RRV infection. Clearly, further studies are needed to determine whether this is the case. Interestingly, we observed that murine rotavirus replication was not significantly affected in MEFs pretreated with type I IFN (data not shown). This finding is consistent with a previous study showing the ineffectiveness of exogenous IFN treatment in murine rotavirus-induced enteric infection (1) and suggests that in the mouse, murine rotaviruses may be more generally resistant to IFN antiviral activity than RRV. A possibility suggested by these findings is that murine rotaviruses may encode additional mechanisms to negate IFN-induced antiviral responses. Moreover, RRV and murine rotavirus clearly have other distinct cell tropisms in mice, since murine rotavirus is not found in the pancreas in either wild-type or IFN-deficient mice (13) and does not efficiently infect biliary epithelial cells in mice (reference 13 and data not shown). Thus, combined differences in IFN sensitivity and cell tropism between RRV and murine rotavirus may explain the observed discrepancy between in vitro and in vivo behaviors. Nevertheless, our studies clearly indicate that cell-type-specific inhibition of the early innate immune response mediated by NSP1 determines the abilities of different heterologous and homologous strains of rotavirus to replicate in murine fibroblasts and is likely to form at least part of the basis for rotavirus host and/or cell tropism in vivo.
Acknowledgments
We sincerely thank Herbert W. Virgin IV for his kind gift of IFNR KO and wild-type mice. We also thank Tyson H. Holmes for his assistance in statistical analysis.
This study was supported in part by a VA Merit Award and NIH grants R01 AI021362-24 and P30DK56339.
Footnotes
Published ahead of print on 6 May 2009.
REFERENCES
- 1.Angel, J., M. A. Franco, H. B. Greenberg, and D. Bass. 1999. Lack of a role for type I and type II interferons in the resolution of rotavirus-induced diarrhea and infection in mice. J. Interferon Cytokine Res. 19655-659. [DOI] [PubMed] [Google Scholar]
- 2.Azim, T., M. H. Zaki, G. Podder, N. Sultana, M. A. Salam, S. M. Rahman, K. Sefate, and D. A. Sack. 2003. Rotavirus-specific subclass antibody and cytokine responses in Bangladeshi children with rotavirus diarrhoea. J. Med. Virol. 69286-295. [DOI] [PubMed] [Google Scholar]
- 3.Barro, M., and J. T. Patton. 2005. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc. Natl. Acad. Sci. USA 1024114-4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barro, M., and J. T. Patton. 2007. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J. Virol. 814473-4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bass, D. M. 1997. Interferon gamma and interleukin 1, but not interferon alfa, inhibit rotavirus entry into human intestinal cell lines. Gastroenterology 11381-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blutt, S. E., and M. E. Conner. 2007. Rotavirus: to the gut and beyond! Curr. Opin. Gastroenterol. 2339-43. [DOI] [PubMed] [Google Scholar]
- 7.Broome, R. L., P. T. Vo, R. L. Ward, H. F. Clark, and H. B. Greenberg. 1993. Murine rotavirus genes encoding outer capsid proteins VP4 and VP7 are not major determinants of host range restriction and virulence. J. Virol. 672448-2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown, C. C., and T. R. Fears. 1981. Exact significance levels for multiple binomial testing with application to carcinogenicity screens. Biometrics 37763-774. [PubMed] [Google Scholar]
- 9.De Boissieu, D., P. Lebon, J. Badoual, Y. Bompard, and C. Dupont. 1993. Rotavirus induces alpha-interferon release in children with gastroenteritis. J. Pediatr. Gastroenterol. Nutr. 1629-32. [DOI] [PubMed] [Google Scholar]
- 10.Didcock, L., D. F. Young, S. Goodbourn, and R. E. Randall. 1999. Sendai virus and simian virus 5 block activation of interferon-responsive genes: importance for virus pathogenesis. J. Virol. 733125-3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Douagi, I., G. M. McInerney, A. S. Hidmark, V. Miriallis, K. Johansen, L. Svensson, and G. B. Karlsson Hedestam. 2007. Role of interferon regulatory factor 3 in type I interferon responses in rotavirus-infected dendritic cells and fibroblasts. J. Virol. 812758-2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunn, S. J., T. L. Cross, and H. B. Greenberg. 1994. Comparison of the rotavirus nonstructural protein NSP1 (NS53) from different species by sequence analysis and northern blot hybridization. Virology 203178-183. [DOI] [PubMed] [Google Scholar]
- 13.Feng, N., B. Kim, M. Fenaux, H. Nguyen, P. Vo, M. B. Omary, and H. B. Greenberg. 2008. Role of interferon in homologous and heterologous rotavirus infection in the intestines and extraintestinal organs of suckling mice. J. Virol. 827578-7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franco, M. A., N. Feng, and H. B. Greenberg. 1996. Rotavirus immunity in the mouse. Arch. Virol. Suppl. 12141-152. [DOI] [PubMed] [Google Scholar]
- 15.Graff, J. W., K. Ettayebi, and M. E. Hardy. 2009. Rotavirus NSP1 inhibits NFκB activation by inducing proteasome-dependent degradation of beta-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog. 5e1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graff, J. W., J. Ewen, K. Ettayebi, and M. E. Hardy. 2007. Zinc-binding domain of rotavirus NSP1 is required for proteasome-dependent degradation of IRF3 and autoregulatory NSP1 stability. J. Gen. Virol. 88613-620. [DOI] [PubMed] [Google Scholar]
- 17.Graff, J. W., D. N. Mitzel, C. M. Weisend, M. L. Flenniken, and M. E. Hardy. 2002. Interferon regulatory factor 3 is a cellular partner of rotavirus NSP1. J. Virol. 769545-9550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Groene, W. S., and R. D. Shaw. 1992. Psoralen preparation of antigenically intact noninfectious rotavirus particles. J. Virol. Methods 3893-102. [DOI] [PubMed] [Google Scholar]
- 19.Hirata, Y., A. H. Broquet, L. Menchen, and M. F. Kagnoff. 2007. Activation of innate immune defense mechanisms by signaling through RIG-I/IPS-1 in intestinal epithelial cells. J. Immunol. 1795425-5432. [DOI] [PubMed] [Google Scholar]
- 20.Hoshino, Y., R. G. Wyatt, H. B. Greenberg, J. Flores, and A. Z. Kapikian. 1984. Serotypic similarity and diversity of rotaviruses of mammalian and avian origin as studied by plaque-reduction neutralization. J. Infect. Dis. 149694-702. [DOI] [PubMed] [Google Scholar]
- 21.Ishida, S., N. Feng, B. Tang, J. M. Gilbert, and H. B. Greenberg. 1996. Quantification of systemic and local immune responses to individual rotavirus proteins during rotavirus infection in mice. J. Clin. Microbiol. 341694-1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang, B., L. Snipes-Magaldi, P. Dennehy, H. Keyserling, R. C. Holman, J. Bresee, J. Gentsch, and R. I. Glass. 2003. Cytokines as mediators for or effectors against rotavirus disease in children. Clin. Diagn. Lab. Immunol. 10995-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lecce, J. G., J. M. Cummins, and A. B. Richards. 1990. Treatment of rotavirus infection in neonate and weanling pigs using natural human interferon alpha. Mol. Biother. 2211-216. [PubMed] [Google Scholar]
- 24.Leib, D. A., T. E. Harrison, K. M. Laslo, M. A. Machalek, N. J. Moorman, and H. W. Virgin. 1999. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J. Exp. Med. 189663-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin, R., C. Heylbroeck, P. M. Pitha, and J. Hiscott. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 182986-2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin, R., Y. Mamane, and J. Hiscott. 1999. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol. Cell. Biol. 192465-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McNeal, M. M., J. L. VanCott, A. H. Choi, M. Basu, J. A. Flint, S. C. Stone, J. D. Clements, and R. L. Ward. 2002. CD4 T cells are the only lymphocytes needed to protect mice against rotavirus shedding after intranasal immunization with a chimeric VP6 protein and the adjuvant LT(R192G). J. Virol. 76560-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parashar, U. D., C. J. Gibson, J. S. Bresse, and R. I. Glass. 2006. Rotavirus and severe childhood diarrhea. Emerg. Infect. Dis. 12304-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parisien, J. P., J. F. Lau, and C. M. Horvath. 2002. STAT2 acts as a host range determinant for species-specific paramyxovirus interferon antagonism and simian virus 5 replication. J. Virol. 766435-6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petersen, C., E. Bruns, M. Kuske, and P. von Wussow. 1997. Treatment of extrahepatic biliary atresia with interferon-alpha in a murine infectious model. Pediatr. Res. 42623-628. [DOI] [PubMed] [Google Scholar]
- 31.Qi, T., L. Xie, Y. Wang, J. Wang, H. Chen, and L. Zhou. 2002. Dynamic variation of serum and stool level of interleukin-2, interleukin-6 and interferon-alpha in children with rotavirus enteritis and its relation to clinical manifestations. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi 16270-273. [PubMed] [Google Scholar]
- 32.Taniguchi, K., K. Kojima, and S. Urasawa. 1996. Nondefective rotavirus mutants with an NSP1 gene which has a deletion of 500 nucleotides, including a cysteine-rich zinc finger motif-encoding region (nucleotides 156 to 248), or which has a nonsense codon at nucleotides 153 to 155. J. Virol. 704125-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vancott, J. L., M. M. McNeal, A. H. Choi, and R. L. Ward. 2003. The role of interferons in rotavirus infections and protection. J. Interferon Cytokine Res. 23163-170. [DOI] [PubMed] [Google Scholar]
- 34.Vanden Broecke, C., A. Schwers, L. Dagenais, A. Goossens, M. Maenhoudt, P. P. Pastoret, and J. Werenne. 1984. Interferon response in colostrum-deprived newborn calves infected with bovine rotavirus: its possible role in the control of the pathogenicity. Ann. Rech. Vet. 1529-34. [PubMed] [Google Scholar]
- 35.Vargues, F., A. Samb, S. M'Boup, A. Gaye, M. Sene, M. P. David, and F. Cisse. 1984. Parasitologic, bacteriologic and virological studies of feces in diarrhea in 212 non-hospitalized children in Dakar. Application of the concept of mass opportunistic infections. Bull. Soc. Pathol. Exot. Filiales 7797-103. [PubMed] [Google Scholar]