

Abstract
Pearl millet slurries, mixed with groundnuts or not, were chosen as a model to investigate the feasibility of obtaining a rapid overview of community structure and population dynamics of fermented foods using pyrosequencing of tagged 16S rRNA gene amplicons. From 14 fermented samples collected either in a traditional small-scale processing unit in Burkina Faso or at laboratory scale, 137,469 sequences of bacterial 16S rRNA gene amplicons were characterized. Except for a few Proteobacteria, almost all the bacterial sequences were attributed to cultivable bacteria. This approach enabled 80.7% of the sequences to be attributed to a family and 70% to a genus but did not enable identification to the species level. The bacterial sequences were assigned to four phyla, with Firmicutes representing the highest diversity, followed by Proteobacteria, Actinobacteria, and Bacteroidetes, which were found only in the slurries prepared in traditional production units. Most of the Firmicutes were lactic acid bacteria, mainly represented by members of the Lactobacillus, Pediococcus, Leuconostoc, and Weissella genera, whose ratio varied from the onset to the end of the fermentation. The other bacteria present at the beginning of fermentation were generally no longer detected at the end, which is consistent with already-known patterns in the microbial ecology of fermented foods. In conclusion, this method seems very promising for rapid and preliminary microbial characterization in many samples of an unknown food sample, by determining numerous nucleic sequences simultaneously without the need for cloning and cultivation-dependent methods.
Ben-saalga is a traditional gruel made in Burkina Faso by cooking a diluted lactic acid-fermented slurry of pearl millet (Pennisetum glaucum) (24). In Ouagadougou, the capital of Burkina Faso, this gruel is consumed by adults and by about 37% of children under 5 years old as a food complementary to breast feeding (J. P. Guyot, C. Mouquet-Rivier, E. H. Tou, E. Counil, A. S. Traoré, and S. Trèche, presented at the 2nd International Workshop “Food-Based Approaches for a Healthy Nutrition in West Africa,” Ouagadougou, Burkina Faso, November 2003). As the characteristics of ben-saalga do not meet the nutritional requirements of young children, its traditional processing has been studied to find ways to improve both its nutritional and sanitary qualities (21, 24). To improve the nutritional value of ben-saalga, millet and groundnut were fermented together using a modified processing method. This method included a precooking step in the traditional preparation of the slurry, followed by addition of malt and inoculation by back slopping (25, 26) (Fig. 1).
FIG. 1.

Description of the processing methods used to produce the fermented pearl millet slurries.
The study of ben-saalga provided an opportunity to investigate the microbiota responsible for the fermentation of the pearl millet slurry. Lactic acid bacteria (LAB) were shown to predominate throughout fermentation (25). Some of the LAB were responsible for one or more enzymatic activities of functional interest in nutrition such as those of α-amylases, phytases, or α-galactosidases (21).
Nevertheless, these studies were conducted using classical microbiological methods, and although molecular approaches can also introduce their own forms of bias mainly linked to the PCR method, they are currently the most powerful tools available for the characterization of microbial communities. In the field of food microbial ecology, pozol, a traditional fermented maize dough, was the first fermented food to which a culture-independent approach was used for community analysis based on PCR-denaturing gradient gel electrophoresis (DGGE) (1). Also, culture-independent approaches were extensively applied to analyze the microbiota of other cereal fermentations such as wheat and rye sourdough (16, 19). The PCR-DGGE method based on 16S rRNA gene amplification used in those studies is still one of the most popular for community analysis (7). PCR amplification, cloning, and sequencing of the 16S rRNA gene content of microbial samples are also of interest but are expensive and time-consuming (17). Recently developed methods based on microarray technology hold promise for large-scale studies, but they do not capture novel sequences (3).
Similarly to other groups working in different fields of microbial ecology of nonfood niches, we have developed a method based on 454-pyrosequencing to monitor microbial communities in fermented foods. Massive parallel pyrosequencing means that the compositions of more than 300,000 sequences can be determined simultaneously, and it does not require cloning of the samples, thus eliminating many of the problems associated with this step of metagenomic methods (6). A highly variable region of the 16S rRNA gene is amplified using primers that target adjacent conserved regions, followed by direct sequencing of individual PCR products (10, 18). This method has been used to describe deep-mine microbial ecology and deep-sea environments (5, 20).
In this study, we investigated for the first time the feasibility of the method to analyze the structure of the microbiomes of fermented foods, using as a case study different pearl millet slurries mixed with groundnuts or not. 454-pyrosequencing was applied to decipher bacterial 16S rRNA genes amplified from the total DNA or cDNA extracted from slurries prepared in a traditional processing unit following the traditional process (24) and from slurries prepared according to the modified process described above in a traditional production unit and in the laboratory. A third alternative consisted in extracting the metagenome of slurries prepared at lab scale and inoculated with a mix of pure strains of LAB.
MATERIALS AND METHODS
Samples of fermented slurries.
The traditional way of preparing ben-saalga has already been described in detail (24). It comprises the following main steps: washing and soaking the pearl millet grains and milling them in wet conditions; kneading and sieving the dough, leaving the diluted slurry to ferment naturally (fermentation step); and finally cooking the fermented slurry to obtain the ben-saalga gruel. We worked on fermented slurries made using the traditional or modified processes as described in Fig. 1. For all experiments, pearl millet grains were purchased at a local market in Ouagadougou (Burkina Faso).
The traditional fermented slurries were coded BT followed by numeral 1, 2, or 3, corresponding to each of the three traditional small-scale production units in Ouagadougou where the sampling was done. This number was followed by a subscript corresponding to the sampling time: “0” for samples at the beginning of the fermentation and “24” for samples taken after 24 h of fermentation. BT10 and BT124 were sampled in the same production unit in the same batch of pearl millet fermentation, and BT224 and BT324 were sampled in two other production units for purposes of comparison.
The slurries prepared following the modified processes were coded BI followed by numeral 1, 2, or 3 according to the fabrication process and followed by the duration of the fermentation (0 or 24 h). Samples BI10 and BI124 were collected in a traditional production unit that used the modified process involving cofermentation of pearl millet and groundnuts, precooking of the sieved slurry, and inoculation by back slopping (26). Samples BI2 and BI3 were produced in the laboratory by using a combination of precooking and inoculation either by back slopping (BI20 and BI224) or with a starter culture composed of a mix of bacteria (BI30 and BI324). This mix of bacteria was designed especially for this study to mimic fermentation due to back slopping and contained the LAB species usually found in cereal fermentations: Lactobacillus plantarum ATCC 14917T, Lactobacillus fermentum ATCC 14931, Leuconostoc mesenteroides ATCC 10880, P. pentosaceus ATCC 43200, and W. confusa ATCC 10881T (23). The LAB were grown in deMan, Rogosa, and Sharpe broth (Difco, Le Pont de Claix, France). Three percent of overnight cultures of each LAB were inoculated into the slurry. All the samples were kept at −20°C until DNA extraction and at −80°C until RNA extraction.
Nucleic acid extraction.
As the slurries were very sticky, they were diluted 10 times in 9‰ NaCl and centrifuged for 10 min at 1,000 × g twice to eliminate the starch and then for 10 min at 10,000 × g to pellet the bacteria.
DNA was extracted from the pellet using the Wizard genomic DNA purification kit (Promega, Charbonnières, France) with an additional lysis step using an amalgamator with zirconium beads (VWR, Fontenay-sous-Bois, France).
RNA was extracted from the samples obtained in the laboratory (BI20, BI224, BI30, and BI324) by using Trizol reagent (Fischer, Illkirch, France) following the manufacturer's instructions with an additional lysis step using an amalgamator with zirconium beads. The RNA was then converted into cDNA by using the avian myeloblastosis virus reverse transcriptase system with random primers (Promega) before the PCR. The results were named BI20 cDNA, BI224 cDNA, BI30 cDNA, and BI324 cDNA (Table 1).
TABLE 1.
Number of reads, unique sequences, and nonvalid sequences per sample of slurry
| Process and production | Sample | No. of sequences
|
||
|---|---|---|---|---|
| Total | Unique | Nonvalid | ||
| Traditional (traditional production unit) | BT10 | 3,895 | 1,227 | 694 |
| BT124 | 1,830 | 499 | 287 | |
| BT224 | 12,636 | 3,244 | 1,753 | |
| BT324 | 3,753 | 1,174 | 594 | |
| Modified | ||||
| Traditional production unit (back slopping) | BI10 | 13,380 | 2,429 | 1,934 |
| BI124 | 641 | 241 | 109 | |
| Laboratory scale | ||||
| Back slopping | BI20 | 7,847 | 2,023 | 1,142 |
| BI224 | 5,617 | 1,056 | 949 | |
| BI20 cDNA | 466 | 189 | 80 | |
| BI224 cDNA | 4,512 | 801 | 660 | |
| Mix of LAB | BI30 | 8,498 | 1,889 | 1,156 |
| BI324 | 16,410 | 3,156 | 1,937 | |
| BI30 cDNA | 8,009 | 1,617 | 1,309 | |
| BI324 cDNA | 3,918 | 939 | 717 | |
PCR.
In our setup, a 196-nucleotide sequence of the V3 region of the 16S rRNA gene (Escherichia coli positions 338 to 534) was amplified by PCR (14). For each sample (n = 14), the 16S rRNA gene was amplified from the extracted DNA and cDNA by using the composite forward primer 5′-GCCTCCCTCGCGCCATCAGNNACTCCTACGGGAGGCAGCAG-3′, where the underlined sequence is that of 454 Life Science primer A and the sequence in italics is the broad-range bacterial primer 338f. The reverse primer was 5′-GCCTTGCCAGCCCGCTCAGNNATTACCGCGGCTGCTGG-3′, where the underlined sequence is that of 454 Life Science primer B and the sequence in italics is the broad-range universal primer 518r. NN designates the unique two-base bar code used to tag each PCR product. For each sample, a 50-μl PCR mix was prepared containing 1× PCR buffer, 200 μM deoxynucleoside triphosphate mix (Promega), 0.5 μM of each primer (Eurogentec, Angers, France), and 1.25 U of GoTaq Hot Start polymerase (Promega). To each reaction mixture, 1 μl of the extracted template DNA was added. The PCR conditions used were 95°C for 4 min and 30 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 1 min, followed by one cycle at 72°C for 7 min.
Gel purification and pyrosequencing.
PCR products with an approximate length of 200 to 250 nucleotides were excised from the agarose gel and purified. The concentration and quality of the DNA were assessed on a Bioanalyser 2100 (Agilent, Massy, France) using the DNA 1000 lab chip (Agilent). Equal amounts of the samples with different sample-specific bar code sequences were pooled and used for pyrosequencing on an eight-lane Picotiter plate. The first pool contained the samples BT10, BT124, BI10, BI20, BI224, BI30, and BI324, and the second contained the remainder of the samples (Fig. 1). Pyrosequencing was performed at the 454 GS FLX Nextgenplatform (Cogenics, Grenoble, France).
Taxonomic assignment of sequence reads.
In order to reduce BLAST analysis time, for each sample, strictly identical sequences were counted and only one copy was conserved. The sequences were then compared with the Ribosomal Database Project (RDP) 16S database using BLASTN.
Analysis of BLAST output files was performed using MEGAN software version 2beta14 (8). This software reads the results of a BLAST comparison as input and attempts to place each read on a node in the NCBI taxonomy. RNA sequences that have no BLAST matches are assigned to the special node “no hits,” and those unassigned due to algorithmic reasons (e.g., below an applied threshold) are assigned to the special node “unassigned.” The result of the analysis is displayed as a tree representation of the NCBI taxonomy (as of September 2008). The results were manually inspected; in particular, the BLAST hit lists with poor taxonomic resolution (mainly the taxonomic levels “cellular organism” and “domain” and several prokaryotic “phyla”) were analyzed in detail. For example, a single Lactobacillus reference sequence wrongly assigned to another bacterial phylum would result in a drastic underrepresentation of Lactobacillus, because the algorithm of the MEGAN software would group the Lactobacillus 16S rRNA amplicons at the Bacteria domain level. In the analysis of a natural community, this would introduce a strong artificial community shift, biasing against Lactobacillus. Suspicious reference sequences were compared by BLAST against the NCBI nonredundant nucleotide database and removed from the hit lists. With the application of this iterative procedure, about 100 bacterial sequences with incorrect affiliations in the NCBI taxonomy were removed from the analysis. We used 10% of the BLASTN top hits as a relative cutoff.
RESULTS
Characteristics of the pyrosequencing data.
The average sequence length was 178 bases (range, 44 to 429). Among the 137,469 reads, 46,057 (34%) did not have a recognizable identifier, so that they could not be related to a specific sample. Table 1 summarizes the number of reads per sample. For each one, strictly identical sequences were counted and only one copy was conserved in order to reduce BLAST analysis time. Some of them were considered nonvalid because they did not match 5′ or 3′ primers (nonvalid sequences). All the unique sequences were then compared with the RDP 16S database by using BLASTN. Surprisingly, the number of total sequences varied from 16,410 to 466, depending on the sample.
Diversity of the microbiomes of the fermented pearl millet slurry.
Among the 20,482 unique sequences, the domain Bacteria was by far the most abundant with 83.2% of the unique sequences. Archaea had less than 1% of the unique sequences, and these were similar only to uncultured Crenarchaeota sequences. About 9% of the sequences were related to Eukarya and were of plant origin as they resembled maize or sorghum sequences.
The bacterial sequences were assigned to four phyla, namely, Actinobacteria, Proteobacteria, Firmicutes, and Bacteroidetes, with a higher representation of the Firmicutes and Proteobacteria. A few sequences were assigned to uncultured Proteobacteria. A total of 70.0% of the sequences could be attributed to a genus and 80.7% to a family. For the remaining sequences, we were unable to identify more than the domain, phylum, class, or order.
Microbiomes of traditional fermented pearl millet slurry.
We compared the microbiomes of traditional fermented pearl millet slurries collected in the same traditional small-scale production unit at the beginning (BT10) and at the end (BT124) of fermentation (Fig. 1). Figure 2 presents a MEGAN tree with the taxonomic affiliation of 16S rRNA gene amplicons identified by BLASTN. The fermentation of pearl millet resulted in simplification of the bacterial microbiota. Indeed, from the 11 families present at the beginning of the fermentation only two, Lactobacillaceae and Leuconostocaceae, including the genera Weissella, Pediococcus, and Lactobacillus, were found after 24 h of fermentation. At the beginning of fermentation, Bacteria and Archaea accounted for 67.2% and 4.8%, respectively, of the unique sequences. Maximum diversity was represented by Firmicutes, but a few sequences attributed to Proteobacteria, Bacteroidetes, and Actinobacteria were also found, although they were no longer detected at the end of fermentation. Firmicutes included only Lactobacillales and Lactobacillaceae, representing around 70% of the unique sequences, the Lactobacillus genus being the most common. The Weissella and Pediococcus genera represented around 20% of the unique sequences, while Streptococcus and Lactococcus represented less than 3%.
FIG. 2.

In-depth taxonomic community profiling of traditional fermented pearl millet slurries sampled at the beginning (BT10) and at the end (BT124) of fermentation. Each circle of the MEGAN tree represents a taxon in the NCBI taxonomy and is labeled by its name and the number of reads that were assigned to the taxon and not to a subtaxon. The size of the circle is scaled logarithmically to represent the number of reads.
In addition, we compared three samples collected in different traditional small-scale production units at the end of fermentation (Fig. 1, samples BT124, BT224, and BT324). The results showed that the microbiota differed considerably depending on the sample (Fig. 3). Indeed, we found three to seven LAB genera with different distributions. For example, sample BT224 was the only one that contained the seven LAB genera, and sample BT124 was the least complex, with only the Lactobacillus, Pediococcus, and Weissella genera. Furthermore, Proteobacteria were present only in samples BT224 and BT324.
FIG. 3.

In-depth taxonomic community profiling of traditional fermented pearl millet slurries collected in traditional small-scale production units at the end of fermentation (BT124, BT224, and BT324). MEGAN tree with the taxonomic affiliation of ribo-tags identified by BLASTN of all 16S rRNA sequences according to NCBI taxonomy. The numbers and sizes of the circles at the tree nodes refer to the affiliation with the respective taxon.
Microbial diversity of improved fermented pearl millet slurry prepared in a traditional small-scale production unit.
Analysis of the sequences obtained from cofermentation of pearl millet and groundnuts including precooking and back slopping (samples BI10 and BI124, Fig. 1) revealed that Bacteria represented up to 88% of the sequences. Archaea were found only at the beginning of fermentation and represented 1.7% of the sequences. All the Bacteria belonged to the Firmicutes phylum and were divided into four families, three belonging to the LAB group (Fig. 4) and one being Clostridiaceae. Enterococcaceae were represented by the Enterococcus genus and were found only at the end of fermentation, representing 0.2% of the unique sequences. In contrast, Streptococcaceae, Lactobacillaceae, and Leuconostocaceae were present from the beginning to the end of fermentation. While Streptococcaceae diversity remained constant, the diversity of the Lactobacillaceae family increased dramatically, whereas that of Leuconostocaceae decreased. Streptococcaceae diversity was mainly due to Streptococcus, as Lactococcus accounted only for 1.9 to 0.1% at the beginning and at the end of fermentation, respectively. The diversity of Lactobacillaceae was distributed between Lactobacillus and Pediococcus at the beginning of fermentation and was due to only Lactobacillus at the end of fermentation. All the Leuconostocaceae family was represented by the Weissella genus.
FIG. 4.
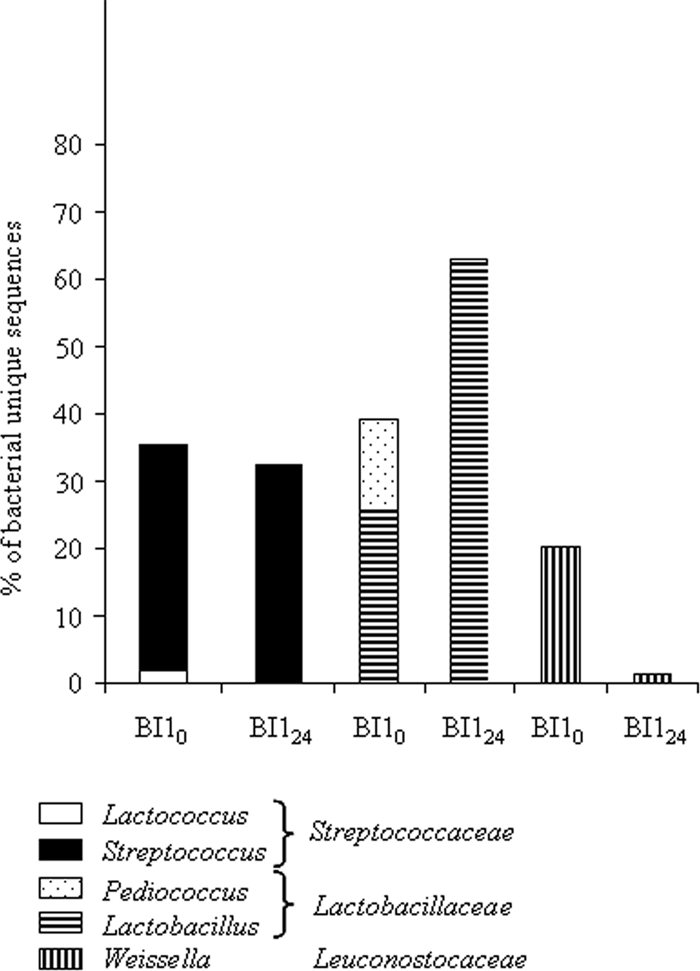
Structure of the LAB community of fermented slurries obtained after cofermentation of pearl millet and groundnuts and inoculation by back slopping.
Microbial diversity of fermented pearl millet slurry prepared in the laboratory.
When we considered the sequences obtained from the slurries prepared in the laboratory with precooking and inoculation by back slopping, at the beginning of fermentation (samples BI20 and BI224, Fig. 1), only 53% of the sequences were attributed to Bacteria, while at the end 92.61% were attributed to this domain. A few Archaea (1.7%) were present only at the beginning of fermentation. The Bacteria were mostly Firmicutes, representing 84.9% to 93.3% of the unique sequences at the beginning and at the end of fermentation, respectively; the other Bacteria were Proteobacteria. The number of phyla decreased during the course of fermentation, especially the non-LAB. Indeed, Proteobacteria were represented only by Acetobacteraceae at the end of fermentation, while they were divided between Acetobacteraceae, Enterobacteriaceae, and Pseudomonadaceae at the beginning. As in the sequences obtained with the traditional process, Firmicutes were divided into four families (Fig. 5). Enterococcaceae remained constant throughout fermentation, accounting for around 14% of diversity. Enterococcaceae were represented only by the Enterococcus genus. Streptococcaceae and Lactobacillaceae diversity increased simultaneously from 22.7% to 42.4% and from 0.8% to 16.5% of the unique sequences, respectively. At the same time, the proportion of unique sequences attributed to Leuconostocaceae was halved. Streptococcaceae were represented only by the Lactococcus genus. The proportion of unique sequences attributed to Lactobacillaceae increased dramatically during the course of fermentation, and the increase was due to Lactobacillus, as the proportion of Pediococcus remained constant. Leuconostocaceae were mainly represented by the Weissella genus, but the Leuconostoc genus was also present.
FIG. 5.
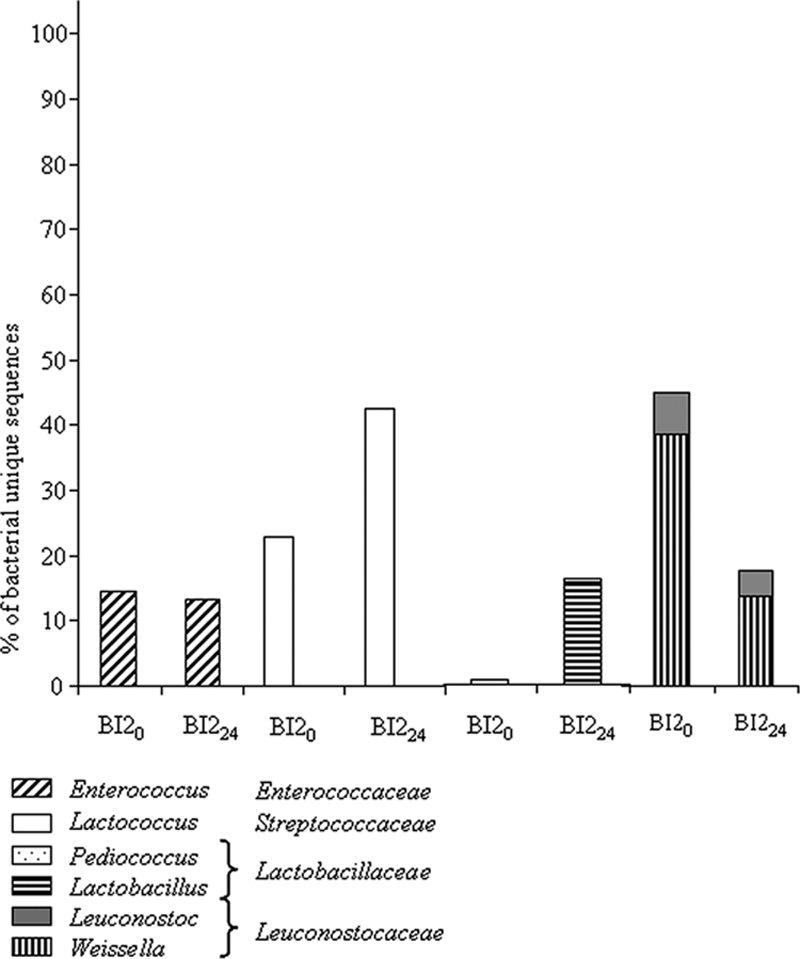
Structure of the LAB community of fermented slurries obtained after fermentation of pearl millet and inoculation by back slopping.
For the positive control, we made a slurry that was precooked and then inoculated with a mix of pure LAB species instead of using back slopping. The LAB mixture was composed of species frequently found in cereal fermentations: L. plantarum ATCC 14917T, L. fermentum ATCC 14931, Leuconostoc mesenteroides ATCC 10880, P. pentosaceus ATCC 43200, and W. confusa ATCC 10881T. The analysis of sequences from these slurries showed that Bacteria represented 52.7% to 93.6% of the unique sequences at the beginning and end of fermentation, respectively. The Bacteria were exclusively represented by Firmicutes, except at the beginning of fermentation, when 0.2% of the sequences were Proteobacteria represented by Acetobacteraceae. Among the Firmicutes, Lactobacillaceae and Leuconostocaceae accounted for almost all the diversity (Fig. 6). At the beginning of fermentation, 0.2% of the sequences were assigned to Streptococcaceae, represented by the genus Streptococcus, whereas no Streptococcaceae were detected at the end of fermentation. Lactobacillaceae were almost equally distributed between Lactobacillus and Pediococcus. Leuconostocaceae were divided into Weissella and Leuconostoc, with higher diversity in Weissella.
FIG. 6.
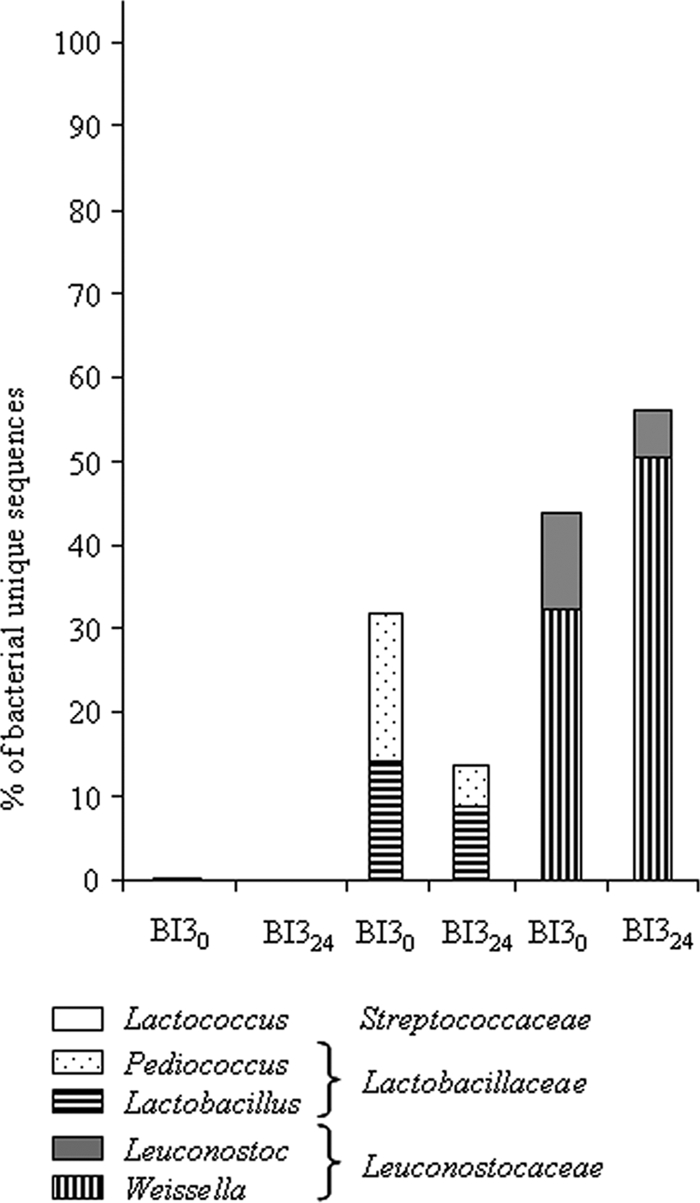
Structure of the LAB community of fermented slurries obtained after fermentation of pearl millet and inoculation with a mix of bacteria.
As these slurries were made in the laboratory, we were able to collect samples under good conditions allowing RNA extraction and reverse transcription. All in all, the data obtained with cDNA followed the same dynamics as those obtained with DNA analysis, but the proportion of unique sequences was different (Table 1). For example, in sample BI20, 84.9% of the unique sequences were attributed to Firmicutes with DNA amplicons, while only 67% of cDNA sequences were assigned to the same phylum. In contrast, 59.0% of the unique sequences were attributed to Enterococcaceae with cDNA analysis and only 14.8% with DNA analysis (data not shown).
DISCUSSION
In this study we used for the first time pyrosequencing of tagged 16S rRNA gene amplicons to decipher the microbiome of a fermented food. This work provides a detailed picture of the structures of the microbiomes of 10 samples of fermented pearl millet slurries made with different processes either in a traditional small-scale processing unit or at laboratory scale.
Nevertheless, two main difficulties were encountered. First, as only 200 bp of the 16S rRNA gene was amplified, and the bacterial species involved in the fermentation process are phylogenetically close, taxonomic assignment was possible only up to the genus level. But this problem was also encountered in other widely used methods such as PCR-DGGE followed by the sequencing of the bands, as the same region of the gene is amplified (1, 7). Furthermore, Liu et al. (10) showed that the sequencing effort is best focused on gathering more short sequences than fewer longer ones, provided that the primers are chosen wisely (10). Sundquist et al. showed that with a wide diversity of bacteria there is no need for read lengths of much more than 100 bp, even if the sample profile may be more relevant with longer reads (22). These authors also suggested that the errors of the RDP phylogeny limit the ability of the method to unambiguously classify reads down to the lowest levels of the phylogeny. A more accurate database classification will help to identify the genera and species of the reads (22, 27).
Second, we noticed that the ribo-tag used for sample allocation, even though it allowed us to pool the samples and thus greatly reduced the cost of analysis, could lead to dramatic underrepresentation of the bacterial diversity of the sample. For example, sample BI10 contained 641 sequences attributed to three families while sample BI124 contained 13,380 sequences attributed to four families. In the first case, it can be supposed that the rare sequences were lost. This bias could be due to the emulsion-based clonal amplification step that enables single molecule sequencing without cloning the target sequences into bacteria. It could also be because quantification of the PCR products was not precise enough and led to strong deviation during pooling of the samples. This problem has not been reported before, when many samples are pooled as we did. For example, Andersson et al. (2) pooled three samples to analyze the human gut microbiota, while we pooled seven. In their study of the macaque gut microbiome, McKenna et al. (11) showed that all bar codes were well populated, with an average of 1,404 sequences per community tested, and only 0.01% of sequences were likely to be miscataloged due to errors in parsing the bar codes. These authors used four base sequences instead of two as in our case (11). Dowd et al. also showed good representation of all samples of cattle feces with 1,732 to 3,224 sequences (4). However, indications are lacking on how many samples were pooled in these two studies. It appears from the literature and from the global approach allowed by pyrosequencing that microbial diversity in fermented foods is far lower than that of other niches such as soils, sediments, human gut, or cattle feces, which in our case justified pooling a larger number of samples. Notwithstanding, since it is the first time that this approach has been applied to fermented foods, some further refinements will be required for this type of ecological niche.
Depending on the pearl millet sample that we analyzed, 53 to 94% of the sequences were attributed to the Bacteria domain. This is quite surprising, since one of the primers was designed to amplify the bacterial 16S rRNA gene. The nonbacterial sequences were attributed to Archaea (0 to 4.8%) or Eukarya (0 to 43%), with Archaea sequences related to uncultured Crenarchaeota and Eukarya sequences of plant origin. This is in agreement with a previous study in which the authors sequenced bands of PCR-DGGE profiles and found Zea mays chloroplasts by using the same primers (1). Within the Bacteria domain, we found four phyla, with Firmicutes having the highest diversity, followed by Proteobacteria, Actinobacteria, and Bacteroidetes, which were found only in the traditional slurries. Given the processing conditions in traditional units, i.e., fermentation in the open air without any safety precautions, it is not surprising to find bacteria known to belong to gut microbiota or to the processing unit environment (workers' hands, soil, water, etc.) (12). Proteobacteria were found in 6 out of 10 samples of slurries. This phylum was represented by Acetobacteraceae, which are mainly environmental Bacteria; Enterobacteriaceae, which are present in the human gut and contain many opportunistic pathogens; Pseudomonadaceae and Comamonadaceae, which are widely distributed in nature and contain many pathogens; and Moraxellaceae, which, together with Acetobacteraceae, are occasionally the cause of human infections (9). Proteobacteria may represent up to 15.13% of the unique sequences, so they could be responsible for diseases after consumption. Nevertheless, at the end of the fermentation step most were no longer detected, which is consistent with the reported effect of lactic acid fermentation, which inhibits the growth and survival of food-borne pathogens (15). Furthermore, cooking the slurries after fermentation to produce gruels is a further step that helps reduce the level of contamination by potentially pathogenic bacteria.
As expected, sequences attributed to LAB were the most frequent. Indeed, fermentation of ben-saalga is lactic acid fermentation (13, 25). In addition, the results obtained from the cDNA analysis revealed the same microbiome pattern, suggesting that the assignments made from the DNA analysis corresponded to living bacteria. Analysis of samples from fermentations in a traditional small-scale producing unit and in the laboratory showed that the use of DNA bar coding and pyrosequencing not only allowed determination of the structure of the community, taking into account representative Bacteria phyla, but could also be a powerful tool to depict population dynamics during fermentation. Whereas considerable diversity was found at the beginning of fermentation, at the end of fermentation only representatives of Lactobacillaceae (Pediococcus and Lactobacillus), Leuconostocaceae, and Enterococcaceae were found. In all samples of slurries fermented either spontaneously or by inoculation by back slopping (samples BT, BI1, and BI2), a common pattern was observed for Lactobacillaceae, which increased, whereas Leuconostocaceae decreased. Surprisingly, a completely different pattern was observed in the slurries fermented by the mix of pure LAB cultures. This mix was composed only of Lactobacillaceae (L. plantarum ATCC 14917T, L. fermentum ATCC 14931, and P. pentosaceus ATCC 43200) and of Leuconostocaceae (Leuconostoc mesenteroides ATCC 10880 and W. confusa ATCC 10881T). Consistently, the pyrosequencing approach allowed detection of these families and of the corresponding genus at the beginning and at the end of fermentation. However, in contrast to the samples fermented by the natural LAB populations, at the end of fermentation the number of sequences corresponding to Lactobacillaceae decreased whereas the number of those corresponding to Leuconostocaceae increased. One possible explanation is that the bacterial strains chosen for the control were purchased from a culture collection and were not adapted to this food environment.
In the field of food fermentation, studies of population structure and dynamics are rather difficult to perform by means of culture-dependent methods due to the problems involved in processing large numbers of samples. Culture-independent methods enable processing of numerous samples. In the field of cereal fermentations, the microbiota of wheat and rye sourdoughs have been thoroughly investigated using such methods. For instance, in a very interesting study, LAB community structures in 39 samples of different traditional Belgium sourdoughs were characterized by PCR-DGGE (19). Thereafter, the use of transversal cluster analysis of the composite data set from culture-dependent and -independent approaches enabled analysis of the overall stability of LAB communities between two sampling campaigns (19). Our work focused mainly on the feasibility of using pyrosequencing for food microbiome analysis on samples prepared in different ways and collected at the beginning and end of fermentation, which showed that several samples (10 in this study) can be analyzed at the same time. These results suggest that this method would allow analysis of enough food samples taken at regular times, generating in the same run an overall view of community structure and population dynamics without the need for cultivation techniques.
In conclusion, this method seems very promising for rapid preliminary microbial characterization of an unknown food sample. For a more detailed view up to the species level, DNA bar coding and pyrosequencing could allow further refinement of the targeting of species with appropriate molecular primers in combination with other techniques such as real-time PCR.
Acknowledgments
This work was supported by the European Commission (Project Cerefer, contract no. ICA4-CT-2002-10047).
We thank Diana Fernandez (IRD) for the interesting discussions about this project and Claire Mouquet-Rivier (IRD) for the field samplings.
Footnotes
Published ahead of print on 1 May 2009.
REFERENCES
- 1.Ampe, F., N. ben Omar, C. Moizan, C. Wacher, and J. P. Guyot. 1999. Polyphasic study of the spatial distribution of microorganisms in Mexican pozol, a fermented maize dough, demonstrates the need for cultivation-independent methods to investigate traditional fermentations. Appl. Environ. Microbiol. 65:5464-5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersson, A. F., M. Lindberg, H. Jakobsson, F. Backhed, P. Nyren, and L. Engstrand. 2008. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 3:e2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bae, J. W., S. K. Rhee, J. R. Park, W. H. Chung, Y. D. Nam, I. Lee, H. Kim, and Y. H. Park. 2005. Development and evaluation of genome-probing microarrays for monitoring lactic acid bacteria. Appl. Environ. Microbiol. 71:8825-8835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dowd, S. E., T. R. Callaway, R. D. Wolcott, Y. Sun, T. McKeehan, R. G. Hagevoort, and T. S. Edrington. 2008. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol. 8:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edwards, R. A., B. Rodriguez-Brito, L. Wegley, M. Haynes, M. Breitbart, D. M. Peterson, M. O. Saar, S. Alexander, E. C. Alexander, Jr., and F. Rohwer. 2006. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 7:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Edwards, R. A., and F. Rohwer. 2005. Viral metagenomics. Nat. Rev. Microbiol. 3:504-510. [DOI] [PubMed] [Google Scholar]
- 7.Ercolini, D. 2004. PCR-DGGE fingerprinting: novel strategies for detection of microbes in food. J. Microbiol. Methods 56:297-314. [DOI] [PubMed] [Google Scholar]
- 8.Huson, D. H., A. F. Auch, J. Qi, and S. C. Schuster. 2007. MEGAN analysis of metagenomic data. Genome Res. 17:377-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laskin, A. I., and H. A. Lechevalier. 1977. Handbook of microbiology, vol. 1. CRC Press, Boca Raton, FL.
- 10.Liu, Z., C. Lozupone, M. Hamady, F. D. Bushman, and R. Knight. 2007. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 35:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenna, P., C. Hoffmann, N. Minkah, P. P. Aye, A. Lackner, Z. Liu, C. A. Lozupone, M. Hamady, R. Knight, and F. D. Bushman. 2008. The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS Pathog. 4:e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Motarjemi, Y. 2002. Impact of small scale fermentation technology on food safety in developing countries. Int. J. Food Microbiol. 75:213-229. [DOI] [PubMed] [Google Scholar]
- 13.Mouquet-Rivier, C., C. Icard-Vernière, J. P. Guyot, E. H. Tou, I. Rochette, and S. Trèche. 2008. Consumption pattern, biochemical composition and nutritional value of fermented pearl millet gruels in Burkina Faso. Int. J. Food Sci. Nutr. 59:716-729. [DOI] [PubMed] [Google Scholar]
- 14.Muyzer, G., E. C. de Waal, and A. G. Uitterlinden. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59:695-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nout, M. J. R., and Y. Motarjemi. 1997. Assessment of fermentation as a household technology for improving food safety: a joint FAO/W.H.O. workshop. Food Control 8:221-226. [PMC free article] [PubMed] [Google Scholar]
- 16.Randazzo, C. L., H. Heilig, C. Restuccia, P. Giudici, and C. Caggia. 2005. Bacterial population in traditional sourdough evaluated by molecular methods. J. Appl. Microbiol. 99:251-258. [DOI] [PubMed] [Google Scholar]
- 17.Randazzo, C. L., S. Torriani, A. D. Akkermans, W. M. de Vos, and E. E. Vaughan. 2002. Diversity, dynamics, and activity of bacterial communities during production of an artisanal Sicilian cheese as evaluated by 16S rRNA analysis. Appl. Environ. Microbiol. 68:1882-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ronaghi, M., and E. Elahi. 2002. Pyrosequencing for microbial typing. J. Chromatogr. B 782:67-72. [DOI] [PubMed] [Google Scholar]
- 19.Scheirlinck, I., R. Van der Meulen, A. Van Schoor, M. Vancanneyt, L. De Vuyst, P. Vandamme, and G. Huys. 2008. Taxonomic structure and stability of the bacterial community in Belgian sourdough ecosystems as assessed by culture and population fingerprinting. Appl. Environ. Microbiol. 74:2414-2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sogin, M. L., H. G. Morrison, J. A. Huber, D. M. Welch, S. M. Huse, P. R. Neal, J. M. Arrieta, and G. J. Herndl. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc. Natl. Acad. Sci. USA 103:12115-12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Songré-Ouattara, L. T., C. Mouquet-Rivier, C. Vernière, C. Humblot, B. Diawara, and J. P. Guyot. 2008. Enzyme activities of lactic acid bacteria from a pearl millet fermented gruel (ben-saalga) of functional interest in nutrition. Int. J. Food Microbiol. 128:395-400. [DOI] [PubMed] [Google Scholar]
- 22.Sundquist, A., S. Bigdeli, R. Jalili, M. L. Druzin, S. Waller, K. M. Pullen, Y. Y. El-Sayed, M. M. Taslimi, S. Batzoglou, and M. Ronaghi. 2007. Bacterial flora typing with deep, targeted, chip-based pyrosequencing. BMC Microbiol. 7:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tou, E. 2007. Caractérisation et amélioration du prcédé traditionnel de préparation de la bouillie de mil fermenté, ben-saalga, utilisée comme aliment de complément au Burkina-Faso. Université de Ouagadougou, Ouagadougou, Burkina Faso.
- 24.Tou, E. H., J. P. Guyot, C. Mouquet-Rivier, I. Rochette, E. Counil, A. S. Traore, and S. Treche. 2006. Study through surveys and fermentation kinetics of the traditional processing of pearl millet (Pennisetum glaucum) into ben-saalga, a fermented gruel from Burkina Faso. Int. J. Food Microbiol. 106:52-60. [DOI] [PubMed] [Google Scholar]
- 25.Tou, E. H., C. Mouquet-Rivier, C. Picq, A. S. Traoré, S. Trèche, and J. P. Guyot. 2007. Improving the nutritional quality of ben-saalga, a traditional fermented millet-based gruel, by co-fermenting millet with groundnut and modifying the processing method. LWT-Food Sci. Technol. 40:1561-1569. [Google Scholar]
- 26.Tou, E. H., C. Mouquet-Rivier, I. Rochette, A. S. Traoré, S. Trèche, and J. P. Guyot. 2007. Effect of different process combinations on the fermentation kinetics, microflora and energy density of ben-saalga, a fermented gruel from Burkina Faso. Food Chem. 100:935-943. [Google Scholar]
- 27.Urich, T., A. Lanzen, J. Qi, D. H. Huson, C. Schleper, and S. C. Schuster. 2008. Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS ONE 3:e2527. [DOI] [PMC free article] [PubMed] [Google Scholar]