

Abstract
Fanconi anemia (FA) is a genetic disease featuring genomic instability and cancer predisposition1. Nine FA genes have been identified, and their products participate in a DNA damage response network involving BRCA1 and BRCA22,3. We have previously purified a FA core complex containing the FANCL ubiquitin ligase and 6 other FA proteins4–6. Each protein in this complex is essential for monoubiquitination of FANCD2, a key reaction in the FA DNA damage response pathway2,7. Here we show that another component of this complex, FAAP250, is mutated in FA patients of a new complementation group (FA-M). FAAP250, renamed FANCM, has sequence similarity to known DNA repair proteins, including archaeal Hef, yeast Mph1 and human ERCC4/XPF. FANCM can dissociate DNA triplex, possibly due to its ability to translocate on duplex DNA. FANCM is essential for FANCD2 monoubiquitination and becomes hyperphosphorylated in response to DNA damage. Our data suggest an evolutionary link between FA proteins and DNA repair; FANCM may act as an engine that translocates the FA core complex along DNA.
Keywords: Fanconi anemia, FANCM, Hef, MPH1, XPF/ERCC4, FANCD2
We have previously shown that 7 out of 9 components of the FA core complex are FA proteins (FANC-A, B, C, E, F, G, and L)4–6. Using mass spectrometry, we identified another component, FAAP250 (Fig. 1a), as KIAA1596, a hypothetical protein with unknown function. Antibodies raised against KIAA1596 specifically recognized the 250 kD polypeptide of the FA core complex immunopurified by a FANCA antibody, supporting the identity of KIAA1596 as FAAP250 (Fig. 1b).
Figure 1. FAAP250 is an integral component of the FA core complex.
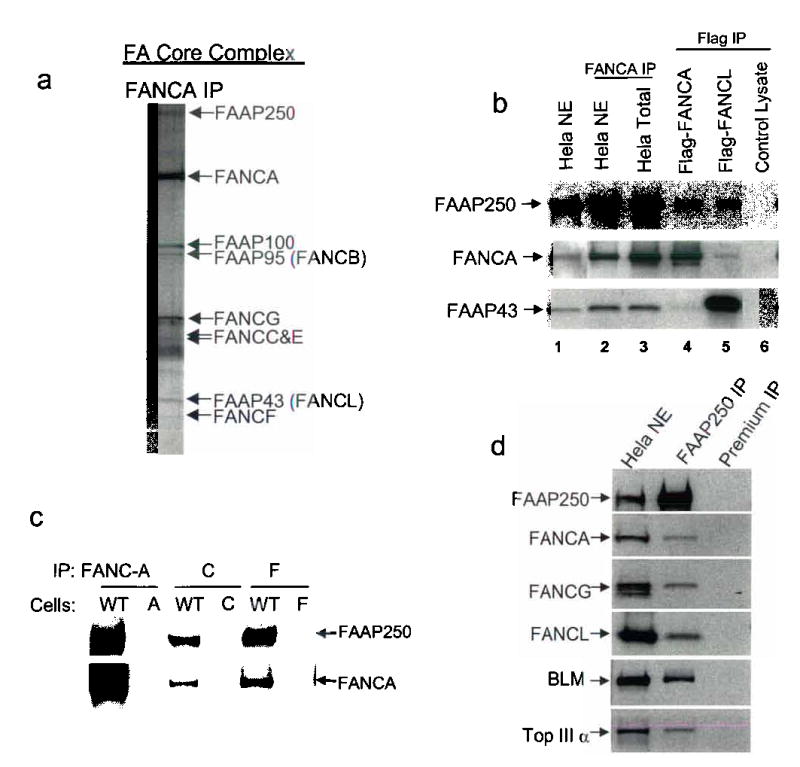
(a) A silver stained SDS gel showing the immunopurified FA core complex5. We have previously shown that this complex containing 5 known FA proteins (FANCA, -C, -E, -F and -G) and four novel components, which are termed FAAPs for FANCA -associated polypeptides4,5. We demonstrated that two FAAPs are also FA proteins: FAAP95 is FANCB6, and FAAP43 is FANCL, a novel E3 ubiquitin ligase5. (b) Immunoblotting to show the presence of FAAP250 in polypeptides obtained from immunoprecipitation (IP) using antibodies against FANCA from HeLa nuclear extract and whole cell extract. FAAP250 was also co-immunoprecipitated by Flag antibody from HEK293 cells stably expressing Flag-FANCA and Flag-FANCL. As a control, FAAP250 was not co-immunoprecipitated by a mock immunoprecipitation using HEK293 cells that do not express the Flag-tagged FA proteins (lane 6). (c) Immunoblotting to show co-IP of FAAP250 with FANCA, FANCC and FANCF from cell lysate of a normal individual (WT), but not from those of FA patients defective in the corresponding FA genes, as indicated. (d) Immunoblotting shows that multiple FA core complex proteins were co-immunoprecipited by a FAAP250 antibody. A preimmune serum was used in a control immunoprecipitation. The presence of BLM and Topo IIIa, which stably associate with the FA core proteins in the BRAFT complex4, was also shown.
Several lines of evidence suggest that FAAP250 is an integral component of the FA core complex. First, FAAP250 was detected in the FA core complex immunoisolated by an anti-Flag antibody from cells expressing either Flag-tagged FANCA, or Flag-tagged FANCL (Fig. 1b). Second, FAAP250 was coimmunoprecipitated by antibodies against multiple FA core components components (FANCA, C, and F) from lymphoblastoid cells of a normal individual, but not from patient cells deficient in the corresponding FA proteins (Fig. 1c). Third, reciprocal immunoprecipitation in HeLa cells using the FAAP250 antibody showed co-precipitation of multiple FA core complex components, such as FANCL, FANCA, and FANCG (Fig. 1d and data not shown).
Importantly, depletion of FAAP250 in HeLa and HEK293 cells by siRNA drastically reduced the levels of monoubiquitinated FANCD2 under both basal and mitomycin C (MMC)-induced conditions (Fig. 2a and data not shown). This is similar to observations following depletion of other core complex components5,6,8. FAAP250-depleted cells also showed increased MMC-sensitivity (Supplementary Fig. 1), a feature that resembles FA cells. We infer that FAAP250, like the other core complex components, has an essential function for FANCD2 monoubiquitination, and its gene likely underlies a distinct FA complementation group.
Fig. 2. FAAP250 is required for FANCD2 monoubiquitination and is hyperphosphorylated in response to DNA damage.
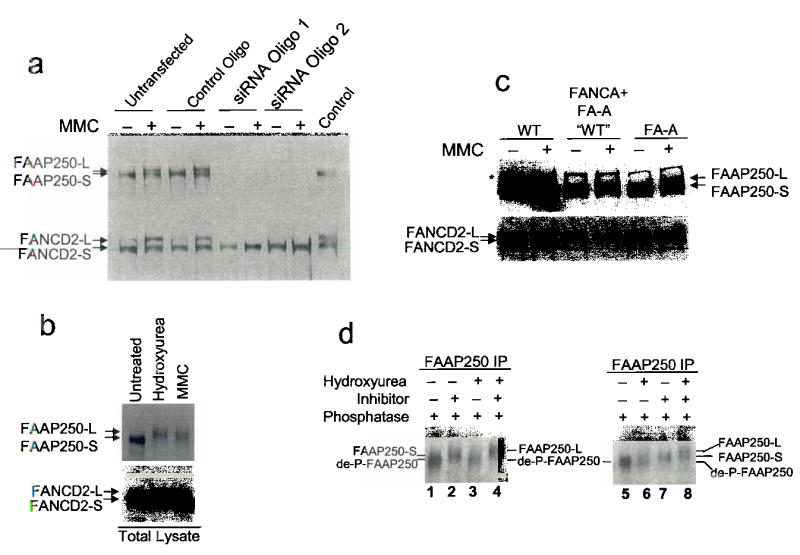
(a) Immunoblotting shows that HeLa cells depleted of FAAP250 by two different siRNA oligos have defective FANCD2 monoubiquitination. HeLa nuclear extract was used as a control. We noticed that a new form of FAAP250 with slower mobility, marked as FAAP250-L (long form), was induced in cells treated with MMC or other genotoxic agents. FAAP250 from unstressed cells was marked as FAAP250-S (short form). (b) Immunoblotting shows that FAAP250 alters its gel mobility when HEK293 cells were treated with hydroxyurea and MMC. FANCD2 monoubiquitination was used as a control. (c) Immunoblotting shows induction of FAAP250-L in lymphoblastoid cells from a normal individual (WT), an FA-A patient and the same patient cell line complemented by expression of exogenous FANCA. (d) Immunoblotting shows that both FAAP250-S and FAAP250-L increased their mobility upon treatment by lamda protein phosphatase. Notably, the de-phosphorylated FAAP250 (de-P-FAAP250) has a faster mobility than either FAAP250-S or FAAP250-L, suggesting both forms of FAAP250 are phosphorylated. We found that our immunoisolated FAAP250 may contain some contaminating phosphatases, which, in the presence of the phosphatase buffer, can dephosphorylate FAAP250 to some degree even without the addition of the exogenous phosphatase. To circumvent this problem, we used the phosphatase inhibitor-treated FAAP250 fraction as the un-dephosphorylated control.
Supplementary Fig. 1.
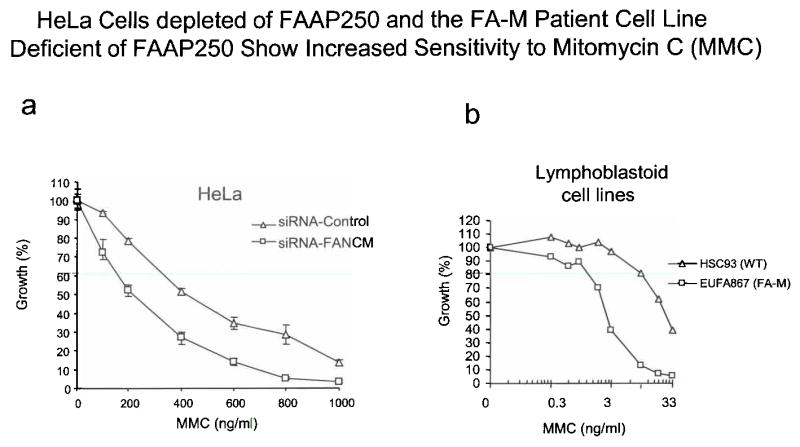
Legend: (a) A graph showing that HeLa cells depleted of FAAP250 (FANCM) by siRNA have increased MMC sensitivity compared to cells treated with a control siRNA oligo. (b) A graph showing that the FA-M patient-derived lymphoblastoid cell line (EUFA867) deficient of FAAP250(FANCM) has increased sensitivity to MMC compared to a cell line derived from a normal individual (WT). It was noticed that the increased MMC sensitivity is about 2-fold for HeLa cells, lower than that of FA-M cells (about 10-fold). This difference could be due to the limitation of siRNA technique that fails to completely deplete FAAP250. There is a difference in the two procedures: in experiment (a), cells were treated with a high dose of MMC for 2 hrs, whereas in experiment (b), cells were continuously exposed to a low dose of MMC for a period of 3 doubling times (between 3–14 days).
We noticed that a long form of FAAP250, termed FAAP250-L, was induced when cells were treated with MMC, hydroxyurea, and cisplatin (Fig. 2a, 2b, and data not shown), implying that FAAP250 is post-translationally modified in response to DNA damage or replication block. FAAP250-L was also detected when FA cells deficient in FANCA, FANCB, FANCE and FANCL were treated with MMC (Fig. 2c and data not shown), suggesting that this modification occurs independently of the ubiquitin ligase activity of the FA core complex. When treated with a phosphatase, both short (FAAP250-S) and long forms (FAAP250-L) showed increased mobility on SDS-gel (Fig. 2d), suggesting that FAAP250 is a phosphoprotein that becomes hyperphosphorylated in response to genotoxic stress.
FAAP250 has a single homologue in other vertebrate species. It contains two potential enzymatic domains: one homologous to a superfamily of DEAH-box helicases or DNA-stimulated ATPases; the other homologous to the endonuclease domain of ERCC4/XPF (Fig. 3). Interestingly, FAAP250 is closely related to archaeal protein Hef (E value 10−43), which possesses both helicase and endonuclease activities, and can resolve stalled replication forks 9,10. Hef has also been proposed to be the archaeal homolog of ERCC4/XPF11,12, an endonuclease essential for nucleotide excision repair in eukaryotes13,14. FAAP250 and ERCC4/XPF are the only proteins in the human genome with a helicase/ATPase domain at the N-terminus and an endonuclease domain at the C-terminus, suggesting that they may have a common ancestor.
Fig. 3. FAAP250 (FANCM) contains Potential DNA-Metabolizing Domains and is homologous to DNA repair proteins in Archea and yeast.
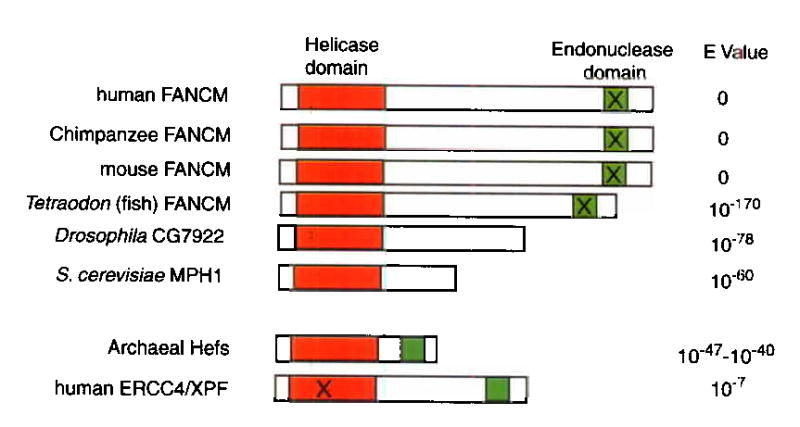
A schematic presentation of DNA-metabolizing domains in FAAP250 (named FANCM here; see text) and its sequence homologues. The helicase and endonuclease domains are marked by red and green, respectively, and their sequence alignments are shown in Supplementary Fig. 2 and 3. The domains marked by the “X” are predicted to be non-functional based on degeneracy of their sequences compared to the consensus. The helicase domains of ERCC4/XPF proteins have been suggested to be degenerate and perhaps nonfunctional 11,12. The E values were obtained by searching the NCBI database with the BLASTP program and the human FANCM as the query sequence. The homology between MPH1 and a partial EST sequence of FAAP250 has been described17.
While the helicase/ATPase domains of FAAP250 proteins are highly conserved in the seven motifs present in the SF2 family of helicases (Supplementary Fig. 2), their endonuclease domains are degenerate at several residues that are absolutely conserved in the ERCC4/XPF and the Mus81 family of endonucleases15 (Supplementary Fig. 3). Mutation of the corresponding residues in ERCC4/XPF either inactivates or strongly reduces its endonuclease activity15. It seems likely that during evolution, the nuclease domains of FAAP250 proteins may have diverged, becoming dispensable and non-functional. Indeed, proteins with high homology to the helicase domains of FAAP250 are present in lower eukaryotic species such as Drosophila (CG7922) and yeast (MPH1) (E values: 10−78 and 10−60, respectively). These proteins may have the same antecedents as FAAP250, but along with the loss of nuclease domains during evolution. MPH1 protein has a DNA helicase activity16 and participates in an error-free DNA damage bypass pathway to protect the genome from spontaneous and chemically-induced damage17,18. The similarity between FAAP250 and MPH1 suggests that they may be part of common mechanisms that guard genome stability.
Supplementary Fig. 2.

Legend: Sequence alignment of predicted helicase domains of FANCM and its sequence homologs. The seven motifs present in all superfamily II helicases are underlined, and the conserved residues are highlighted. The arrow indicates the conserved lysine (K117) in motif I that has been replaced by arginine in Flag-FANCIvl ATPase mutant. ERCC4/XPF and Mei9 (the Drosophila homolog of ERCC4/XPF) are included in the alignment.
Supplementary Fig. 3.

Legend: The alignment of the C-terminal endonuclease domains of FANCM (FAAP250) proteins with those in ERCC4/XPF and Hefs. The residues that are absolutely conserved in ERCC4/XPF, Mus81 and Hef families of proteins are highlighted, and those that are degenerate in FANCM protein are underlined. A summary of the endonuclease activity of various ERCC4/XPF point mutants15 is shown at the top. The different signs are: “++” for wildtype activity; “+” for no detectable activity in the presence of Mg++, but residue activity in the presence of Mn++; “(+)”, little or no activity; “−”, no activity. A conserved motif found in many nucleases is indicated by a bracket.
The helicase domains of ERCC4/XPF proteins are also degenerate and perhaps nonfunctional 11,12 (Supplementary Fig. 2). Thus, FAAP250 and ERCC4/XPF may have derived from a common ancestor that, like archaeal Hef, has both functional helicase and nuclease domains. They may have subsequently diverged, with ERCC4/XPF retaining its endonuclease activity and FAAP250 maintaining its helicase/ATPase activity.
To assess possible biochemical activity of FAAP250, we generated cell lines that stably express FAAP250 tagged with a Flag-epitope and immunoisolated the tagged protein by using the flag antibody (Fig. 4a). As a control, we made a point mutant that substitutes arginine for a conserved lysine within the helicase motif I (Supplementary Fig. 2; Fig. 4a). This substitution in other helicase domains completely inactivates ATPase and helicase activities19,20. Consistent with previous instances, the wildtype FAAP250 protein, but not its mutant, exhibited single- and double- stranded DNA-stimulated ATPase activity (Fig. 4b). The fact that the mutant protein has no detectable ATPase activity indicates that this activity is derived from the helicase domain of FAAP250.
Fig. 4. FAAP250 has an ATP-dependent DNA translocase activity.

(a) A silver-stained SDS-gel showing the immunoisolated Flag-FAAP250 of wildtype and K117R mutant from HEK293-derived cells stably expressing these proteins. Polypeptides immunoisolated from HEK293 cells that do not express the Flag-tagged proteins are shown as a control (Mock). (b) A histogram shows that Flag-FAAP250 has DNA-stimulated ATPase activity, whereas a point mutation within the conserved helicase motif I (K117R) inactivates such activity. The double-stranded (ds) and single-stranded (ss) DNA are indicated. (c) An autoradiograph showing that Flag-FAAP250 exihibits no detectable helicase activity in a double-strand displacement assay. BLM helicase-associated complexes (BLM)4 were used as a positive control. The DNA substrate has 13-nucleotide single-stranded tails on both sides of a 20-bp double-stranded region, so that it should be able to detect activity of helicases of either 5′ to 3′ or 3′ to 5′ directions. (d) Autoradiograph showing that Flag-FAAP250 displays a triple-helix unwinding activity, whereas its ATPase mutant (K117R) lacks such activity. A chromatin-remodeling complex, ATRX complex30, was used as a positive control. The substrate consists of a 40-base homopyrimidine oligonucleotide located centrally on a 190 bp double-stranded DNA23. (e) Autoradiograph showing that Flag-FAAP250 does not displace a blunt triple helix. The substrate is the same as in (d), except that the duplex DNA is the same size (40 bp) as the homopyrimidine oligonucleotide, to create the blunt triplex. The band with faster mobility is from the designed blunt triplex substrate. The band with slower mobility (marked with an arrowhead) is from the precursor of the substrate.
We found that the FAAP250 protein displayed no detectable helicase activity in strand-displacement assays using substrates consisting of double-stranded DNA, 4-way junction, or replication fork, which can be resolved by either MPH1 or Hef10,16 (Fig. 4c and Supplementary Fig. 4). The absence of detectable helicase activity in FAAP250 argues that it may function differently from MPH1 and Hef. However, we cannot rule out the possibility that FANCM may be a structure-specific helicase, and the absence of a detectable helicase activity can be due to lack of normal physiological environment in our assays.
Supplementary Fig. 4.
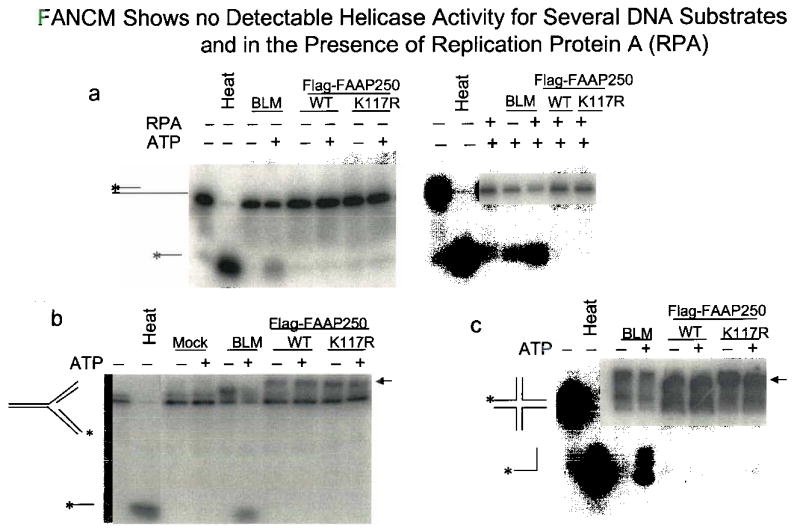
Legend: (a) An audioradiograph shows that the presence of a 40-nucleotide 3′ single-stranded DNA tail in the substrate and the addition of RPA (100 nM) fail to activate the potential helicase activity of Flag-FANCM. These conditions can activate the helicase activity of MPH116, the yeast orthologue of FANCM. The presence of a 40-nucleotide 5′ single-stranded DNA tail in the substrate also fails to activate the helicase activity of FANCM (data not shown), (b) and (c) Autoradiographs show that FANCM cannot unwind two structured DNA substrates that can be resolved by Hef10, the archaeal ortholgue of FANCM. The arrow indicates a possible complex between structured DNA and Flag-FAAP250.
Many proteins with SF2 helicase domains can use the energy of ATP-hydrolysis to translocate on DNA21–23. One way to demonstrate translocase activity is the triple-helix displacement assay. In this assay, a triple helix (H-DNA) is formed in which each nucleotide of the third strand forms Hoogsteen base pairs with Watson-Crick base pairs of the duplex. When the translocase proceeds through the triplex, it displaces the third strand. Wildtype FAAP250, but not its mutant, displayed triple-helix displacement activity (Fig. 4d). To exclude the possibility that FAAP250 may displace the triplex through direct binding and dissociation, we used a blunt triplex in this assay, and found that FAAP250 failed to displace this substrate (Fig. 4e). Together, these results suggest that FAAP250 can dissociate DNA triplex, possibly due to an ability of the protein to translocate on duplex DNA.
We screened FA patient derived cells for FAAP250 mutations by immunoblotting. FAAP250 was detected in cells from all known complementation groups (Fig. 5a and data not shown). This result implied that if FAAP250 is mutated in FA patients, it should be in a new group. FAAP250 was undetectable in a cell line from patient EUFA867 that had been excluded from many known groups (A, B, C, E, F, G) by linkage analysis, cell fusion and cDNA transfection (data not shown). Sequencing of the patient’s cDNA revealed a nonsense mutation (2171C>A; S724X) in exon 13, and the absence of the sequence encoded by exon 15, which was caused by a 2554 bp deletion spanning a part of intron 14 and almost the entire exon 15 (Fig. 5b; Supplementary Fig. 5). In theory, these mutations could lead to truncated products of 80 kD and 160 kD, respectively. However, neither product was detectable (data not shown), suggesting that they may be unstable.
Fig. 5. FAAP250 is mutated in Fanconi anemia patients and is essential for FA core complex assembly and function.

(a) Immunoblotting shows that FAAP250 is absent in a FA patient of a new complementation group, EUFA867. The phenotype of this patient is typical of FA, with increased chromosomal breakage on treatment of cells with MMC and DEB, progressive bone marrow failure, and characteristic bilateral radial anomalies. A nonspecific polypeptide crossreactive with FAAP250 antibody (marked with an asterisk) could be used as a loading control. (b) A schematic representation of the genomic structure of FAAP250 and the mutations (S724X and exon 15 deletion) detected EUFA867 and her sibling, who also has FA. The nonsense mutation was also detected in the mother of the two patients. (c) Immunoblotting shows that in EUFA867 cell extract, the levels of FANCA and FANCG are strongly reduced, whereas that of FANCL is moderately reduced, when compared to the extract from a normal individual (WT). In addition, monoubiquitinated FANCD2 (FANCD2-L) was not detectable in the extract of EUFA867 cells. (d) Immunoblotting shows defective nuclear localization of FANCA and FANCL in EUFA867 cells. HeLa nuclear extract (NE) was included as a control. BLM and caspase 3 were used as markers for nuclear (NE) and cytoplasmic (cyt) extracts, respectively. (e) Immunoprecipitation-coupled immunoblotting shows that FANCM failed to co-immunoprecipitate with FANCA from extracts of FA patient cells lacking various core complex proteins, as indicated on the top. (f) Immunoprecipitation coupled-immunoblotting shows reduced co-immunoprecipitation of FANCA and FANCL by an FANCM antibody in FA-A and FA-B cell extracts.
Supplementary Fig. 5.

Legend: (a) Sequence of the F.4AO/genomic DNA surrounding the deleted region (underlined). Exons are marked with red. The two PCR primers used amplify the region deleted in the patient in (B) are indicated in blue.
(b) BD0232 and BD0233 are lymphoblastoid cell lines from the father and mother of EUFA867. The PCR product from the wild type allele is non-detectable, due to the large size of this region which cannot be efficiently amplified under the current condition. Only the PCR product from the deleted allelle is detected (marked with an arrow). The PCR on exon 13 was performed as a control and shows the integrity of the genomic DNA.
Another FA patient in the same family, a sibling of EUFA867, was found to carry the identical mutations in his blood DNA, providing additional evidence for FAAP250 as the gene responsible for FA in these patients. The nonsense mutation was detected in DNA from blood and from a lymphoblastoid cell line of the patients’ healthy mother, consistent with maternal inheritance of this allele. The genomic deletion was not detected in DNA from blood or a lymphoblastoid cell line of the healthy father, but linkage analysis with tightly linked flanking markers (D14S259 and D14S1027) showed that both siblings had inherited the same paternal allele, and correct paternity was established with multiple markers (data not shown). Because it is unlikely that the same deletion has occurred independently twice in EUFA867 and her sibling, the most likely explanation is that the father is mosaic for this deletion: his germ cells, but not blood cells, carry the mutation, which was thus transmitted to his children. In summary, our results show that FAAP250 is mutated in patients of a new FA complementation group (FA-M), and is therefore termed FANCM.
Numerous attempts to use FANCM cDNA to complement FA-M (EUFA867) cells for MMC-sensitivity (Supplementary Fig. 1) and defective FANCD2 monoubiquitination (see below) have failed. The main reason is that we were unable to ectopically express FANCM in FA-M (EUFA867) cells using both plasmid and viral vectors with either constitutive or inducible promoters. As an alternative, we used other approaches to evaluate the importance of FANCM in the FA core complex and pathway. First, FA-M cells display drastically reduced levels of FANCA and FANCG, and modestly reduced level of FANCL (Fig. 5c). This feature resembles FA cells lacking FANCA or FANCB, in which the levels of FANCG and FANCL are correspondingly reduced. The fact that FANCA and FANCG are unstable in FA-M cells underscores the importance of FANCM for the integrity of the core complex. Second, FA-M cells exhibit deficient nuclear localization of FANCA and FANCL (Fig. 5d), a characteristic also observed in FA-B cells. Third, the interaction between FANCM and FANCA is defective in FA cells lacking other core complex components (Fig. 5e, 5f), and the interaction between FANCM and FANCL is absent in FA-A and FA-B cells (Fig. 5f), providing additional evidence for FANCM as part of the core complex.
Notably, we found that the FA-M cells are defective in FANCD2 monoubiquitination (Fig. 5c), which is a common feature of all FA patient cells with a mutated core complex component. This result is also consistent with siRNA data in HeLa and HEK293 cells where depletion of FANCM drastically reduced levels of monoubiquitinated FANCD2 (Fig. 2a and data not shown). Together, these data indicate that FANCM, like the other 7 FA core complex proteins, is necessary for FA core complex assembly and FANCD2 monoubiquitination.
Although FA proteins have been implicated in DNA repair, the interactions between FA proteins and DNA are poorly understood, because the known FA proteins lack DNA-related enzymatic domains or activities. Here we demonstrated that FANCM has a conserved helicase domain and a DNA-translocase activity. A companion paper identified another FA protein, FANCJ, as BACH1/BRIP124, a known DNA helicase25. The discovery of two FA proteins with helicase domains or activities suggests a mechanism of direct participation in DNA repair by the FA proteins.
We propose that FANCM may have at least three roles in this process. First, it may have a structural role to allow assembly of the FA core complex; because in its absence, the nuclear localization and stability of several FA proteins are defective (Fig. 5c and d). Second, FANCM may act as an engine that translocates the core complex along DNA. Speculatively, this translocation may allow the core complex to sense and locate to the damaged DNA, which could be an important step either before or after FANCD2 monoubiquitination. Third, FANCM is hyperphosphorylated in response to DNA damage, suggesting that it may serve as a signal transducer through which the activity of the core complex is regulated. A DNA damage checkpoint kinase, ATR, has been shown to act upstream of the FA pathway26,27, but its substrate has not been defined. FANCM contains multiple predicted ATR phosphorylation sites (data not shown), and may serve as a substrate through which ATR regulates FANCD2 monoubiquitination.
In contrast to FANCM which acts upstream of the FA pathway (in terms of FANCD2 monoubiquitination) and displays no helicase activity, FANCJ functions downstream28 and possesses a 5′ to 3′ helicase activity25. Possibly, FANCJ may directly repair damaged DNA with this activity. It remains to be determined how the function of FANCJ is connected to the FA pathway. Notably, BLM, a helicase with 3′ to 5′ polarity (opposite of FANCJ), associates with FANCM (Fig. 1d) and the rest of the FA core complex4. The presence of three proteins with distinct helicase properties in the FA-related complexes suggests a coordinated action of FA proteins in DNA repair.
Materials and Methods
Cell lines
Epstein Barr virus immortalized lymphoblastoid cell lines were established from blood samples of FA patient EUFA867 and her parents who had been referred to the EUFAR cell repository for genetic subtyping. The research was carried out after approval by the Guy’s Hospital Research Ethics Committee (London) and the Institutional Review Board (VU University Medical Center, Amsterdam). We have obtained informed consent on all human subjects involved in our work. Cell lines were cultured in RPMI with 10% fetal calf serum.
Protein purification and analysis
We immunopurified FA core complex with a FANCA antibody from HeLa cells as described4. We identified FAAP250 by liquid chromatography-mass spectrometry (LC/MS) analysis. The mass spectrometry data are not shown but available upon request. We made two polyclonal rabbit antibodies (FAM-1Ab and FAM-2Ab) against FAAP250 (FANCM) by using two chimeric proteins containing regions of FAAP250 (aa. 1190–1273 and aa. 1643 to 2048 respectively) fused to maltose-binding protein (MBP) as immunogen. These fusion proteins were expressed and purified from E.coli according to manufacturer’s protocols (New England Biolabs), and used for immunization as well as for purification of the antibody. The immunoprecipitation condition has been described4. We prepared nuclear and cytoplasmic extracts as described4.
Immunoprecipitation and de-phosphorylation of FANCM
HEK293 cells were either treated with hydroxyurea for 24hrs or untreated. They were then extracted with lysis buffer (50mM Tris-HCl, pH 7.5, 300mM NaCl, 1% Triton X-100 and 1mM DTT) supplemented with a protease inhibitor cocktail (Roche) and phosphatase inhibitors(50mM NaF and 2mM Na3V04). FAAP250 was immunoprecipitated using FAM-1Ab. The immunoprecipitate was washed twice with λ phosphatase buffer (New England Biolabs), and resuspended in 50 μl of λ phosphatase buffer either in the presence or absence of phosphatase inhibitors. Four hundred units of λ phosphatase (New England Biolabs) was added to each sample, followed by incubation at 30°C for 1 hr. Samples were separated by 6% SDS-PAGE and immunoblotted for FAAP250 using FAM-1Ab antibody.
Cloning of FANCM and construction of vectors
We purchased a KIAA1596 cDNA clone from Origen Inc. Sequencing revealed that this clone contains an open reading frame of 2048 aa., longer than the predicted sequence of XP_048128 (2014 aa.) at NCBI. The cDNA was subcloned by standard molecular biology techniques into mammalian expression vectors, pCEP4 and pMEP4 (InVitrogen). The cDNA was also linked with the Flag-epitope at the N-terminus, and cloned into pIRESneo3 vector (Clonetech). The Flag-tagged FAAP250 cDNA has also been cloned into the pCEP4 vector and the retroviral vector using the Gateway recombinant DNA system (InVitrogen). These constructs have been used for complementation and expression studies.
Transfection and complementation
Transfection of HeLa cells using siRNA oligonucleotides was performed as described5. The FAAP250 siRNA oligo used two different 19-nucleotide target sequences (CUAUGCUUAUUGCCAGGUU and AGGCUGUGCAACAAGUUAU) and was purchased from Dharmacon Research. HEK293 cells were transfected with Flag-FANCM cDNA in a pIRESneo3 vector, and were selected for G418-resistant clones. These clones were screened for expression of Flag-FANCM by immunoblotting. Those expressing high levels of Flag-FANCM were grown in large scale for immunopurification of the protein. Patient cell line EUFA867 has been transfected with pIRESneo3, pCEP4 and pMEP4 vectors containing FANCM cDNA and selected on medium containing either G418 (0.4 mg/ml) or hygromycin (0.1 mg/ml). For inducible expression of FANCM in EUFA867 cells, the GeneSwitch system (InVitrogen) was used. Cross-linker sensitivity for EUFA867 cells was determined using a MMC-induced growth inhibition assay, as described6. The MMC-sensitivity assay for FAAP250-depleted HeLa cells was done as described29, with the following modifications. HeLa cell were transfected with either a FAAP250 siRNA oligo or a control oligo, and grown for 60 hrs. They were then plated at a density of 500 cells per 10 cm dish. After 8 hrs, cells were treated MMC at the indicated concentration (Supplementary Fig. 1) for 2 hrs. The drug was then washed away and fresh media were added. After 9–12 days, the colonies were fixed, stained and counted. FANCM expression was determined by either direct immunoblotting or by immunoprecipitation-coupled immunoblotting.
Biochemical analysis of FANCM
Flag-FANCM wildtype and mutant proteins were immunoisolated from HEK293 cell clones that have been selected for their high expression. The protocols were essentially as described 5, but with an important modification: the nuclear extract was depleted with a BLM antibody prior to immunoprecipitation with the Flag antibody, since FANCM, like the other FA core complex components, stably associates with the BLM helicase and Topo IIIα, so that if Flag-FANCM is immunoisolated directly from the extract, the intrinsic helicase activity of BLM is high enough to interfere with our analysis of the FANCM activity. We therefore thoroughly depleted BLM until the Flag-FANCM ATPase mutant protein (K117R) displays no detectable ATPase activity. The Flag-FAAP250 fraction obtained contains low levels of FA core complex, as revealed by presence of other FA core complex components by Western (data not shown). However, the majority flag-FAAP250 did not appear to be assembled into the core complex, possibly due to over-expression of the exogenous protein (which is at least 10-fold higher than that of the endogenous protein; data not shown). The Flag-FANCM proteins obtained were then analyzed for ATPase, helicase, and triple-helix unwinding activities as described4,23,30. About 50 ng of Flag-FANCM was used in the different assays. The final concentration of ATP in the ATPase assay is 1 mM. The reaction was incubated for 30 min at 30 °C. Two DNA duplex substrates (Supplementary Fig. 4) containing 40-nucleotide single stranded DNA tails at either 5′- or 3′- end of the duplex region used the exact sequences as described16.
Amplification of FANCM sequences
Total RNA was isolated from lymphoblastoid cell line EUFA867 using a Qiagen RNeasy kit (Qiagen, Venlo, The Netherlands). cDNA was prepared using 10 pmol oligo-dT20 primers and Superscript™ II RT polymerase (Invitrogen, Carlsbad, California, USA). The PCR reactions for amplification of FANCM were performed on cDNA using Platinum Taq polymerase (Invitrogen) and primers that covered the whole cDNA sequence. Genomic DNA (gDNA) was isolated from whole blood and lymphoblastoid cell lines of patients and family members using a Qiagen Blood mini kit (Qiagen). The PCR reactions for amplification of exon 13 of FANCM were performed on 200 ng gDNA using Platinum Taq polymerase (Invitrogen) and primers in intron 12 and 13. The deletion-containing region of the genomic DNA was amplified using Pfx polymerase (Invitrogen) and primers in intron 14 and 16.
Sequencing of FANCM
PCR products were purified using a SAP/EXO treatment (Amersham Biosciences, Uppsala, Sweden) according to manufactures instructions. Sequencing reactions were prepared using 10 pmol primer and Big Dye terminator cycle sequencing kit (Applied Biosystems, Foster City, CA, USA) and performed with the GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA, USA). Samples were analysed on ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA).
Accession numbers
The accession number for human FANCM in this study is DQ140356, which is longer than the predicted KIAA1596 sequence in the database (XP_048128.5). The numbers for FANCM orthologues in other species are: chimpanzee (XP_509928.1), mouse (NP_849243.2), fish (tetraodon nigroviridis; CAG10454.1), rat (XP_234239.2), dog (XP_537429.1); Drosophila (CG7922), yeast (MPH1), and Archaeal Hef proteins (AAB89786, AAB99518, and AAB85892),
Acknowledgments
We thank the FA patients and families for contributing to this study, N. Sherman for mass spectrometry analysis, S. Brill for providing Mus81, R. Wood for XPF, R. Brosh and M. Kenny for RPA, R.Pattni for genotyping, M. Rooimans for technical assistance, and the National Cell Culture Center for providing cells, D. Schlessinger for critical reading of the manuscript. Financial support was from the FA Research Fund, Eugene, Oregon, U.S.A., Ellison medical Foundation (WW), NIH grant CA112775 (MEH), Daniel Ayling Fanconi Anaemia Trust (CGM) and the intramural program of National Institute of Health, National Institute on Aging (WW). JPW and ALM are supported by Dutch Cancer Society and the Netherlands Organization for Health Research and Development.
Footnotes
Competing Interests Statement The authors declare that they have no competing financial interests.
References
- 1.Joenje H, Patel KJ. The emerging genetic and molecular basis of fanconi anaemia. Nat Rev Genet. 2001;2:446–59. doi: 10.1038/35076590. [DOI] [PubMed] [Google Scholar]
- 2.Garcia-Higuera I, et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–62. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 3.Howlett NG, et al. Biallelic Inactivation of BRCA2 in Fanconi Anemia. Science. 2002;13:13. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 4.Meetei AR, et al. A multiprotein nuclear complex connects Fanconi anemia and bloom syndrome. Mol Cell Biol. 2003;23:3417–3426. doi: 10.1128/MCB.23.10.3417-3426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meetei AR, et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet. 2003;35:165–70. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- 6.Meetei AR, et al. X-linked inheritance of Fanconi anemia complementation group B. Nat Genet. 2004;36:1219–24. doi: 10.1038/ng1458. [DOI] [PubMed] [Google Scholar]
- 7.Taniguchi T, D’Andrea AD. The Fanconi anemia protein, FANCE, promotes the nuclear accumulation of FANCC. Blood. 2002;100:2457–62. doi: 10.1182/blood-2002-03-0860. [DOI] [PubMed] [Google Scholar]
- 8.Bruun D, et al. siRNA depletion of BRCA1, but not BRCA2, causes increased genome instability in Fanconi anemia cells. DNA Repair (Amst) 2003;2:1007–13. doi: 10.1016/s1568-7864(03)00112-5. [DOI] [PubMed] [Google Scholar]
- 9.Komori K, Fujikane R, Shinagawa H, Ishino Y. Novel endonuclease in Archaea cleaving DNA with various branched structure. Genes Genet Syst. 2002;77:227–41. doi: 10.1266/ggs.77.227. [DOI] [PubMed] [Google Scholar]
- 10.Komori K, et al. Cooperation of the N-terminal Helicase and C-terminal endonuclease activities of Archaeal Hef protein in processing stalled replication forks. J Biol Chem. 2004;279:53175–85. doi: 10.1074/jbc.M409243200. [DOI] [PubMed] [Google Scholar]
- 11.Sgouros J, Gaillard PH, Wood RD. A relationship betweena DNA-repair/recombination nuclease family and archaeal helicases. Trends Biochem Sci. 1999;24:95–7. doi: 10.1016/s0968-0004(99)01355-9. [DOI] [PubMed] [Google Scholar]
- 12.Aravind L, Walker DR, Koonin EV. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 1999;27:1223–42. doi: 10.1093/nar/27.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sijbers AM, et al. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell. 1996;86:811–22. doi: 10.1016/s0092-8674(00)80155-5. [DOI] [PubMed] [Google Scholar]
- 14.Mu D, Hsu DS, Sancar A. Reaction mechanism of human DNA repair excision nuclease. J Biol Chem. 1996;271:8285–94. doi: 10.1074/jbc.271.14.8285. [DOI] [PubMed] [Google Scholar]
- 15.Enzlin JH, Scharer OD. The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif. Embo J. 2002;21:2045–53. doi: 10.1093/emboj/21.8.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prakash, R. et al. Saccharomyces cerevisiae MPH1 gene, required for homologous recombination-mediated mutation avoidance, encodes a 3′ to 5′ DNA helicase. J Biol Chem (2005). [DOI] [PubMed]
- 17.Scheller J, Schurer A, Rudolph C, Hettwer S, Kramer W. MPH1, a yeast gene encoding a DEAH protein, plays a role in protection of the genome from spontaneous and chemically induced damage. Genetics. 2000;155:1069–81. doi: 10.1093/genetics/155.3.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schurer KA, Rudolph C, Ulrich HD, Kramer W. Yeast MPH1 gene functions in an error-free DNA damage bypass pathway that requires genes from Homologous recombination, but not from postreplicative repair. Genetics. 2004;166:1673–86. doi: 10.1534/genetics.166.4.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zavitz KH, Marians KJ. ATPase-deficient mutants of the Escherichia coli DNA replication protein PriA are capable of catalyzing the assembly of active primosomes. J Biol Chem. 1992;267:6933–40. [PubMed] [Google Scholar]
- 20.Sung P, Higgins D, Prakash L, Prakash S. Mutation of lysine-48 to arginine in the yeast RAD3 protein abolishes its ATPase and DNA helicase activities but not the ability to bind ATP. Embo J. 1988;7:3263–9. doi: 10.1002/j.1460-2075.1988.tb03193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Komen S, Petukhova G, Sigurdsson S, Stratton S, Sung P. Superhelicity-driven homologous DNA pairing by yeast recombination factors Rad51 and Rad54. Mol Cell. 2000;6:563–72. doi: 10.1016/s1097-2765(00)00055-1. [DOI] [PubMed] [Google Scholar]
- 22.Havas K, et al. Generation of superhelical torsion by ATP-dependent chromatin remodeling activities. Cell. 2000;103:1133–42. doi: 10.1016/s0092-8674(00)00215-4. [DOI] [PubMed] [Google Scholar]
- 23.Saha A, Wittmeyer J, Cairns BR. Chromatin remodeling by RSC involves ATP-dependent DNA translocation. Genes Dev. 2002;16:2120–34. doi: 10.1101/gad.995002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.M. Levitus, Q. W., B.C. Godthelp, Y. de Vries, S. Hussain, W.W., Wiegant, E. E.-M., J. Steltenpool, M.A. Rooimans, G. Pals, F. & Arwert, C. G. M., M.Z. Zdzienicka, K. Hiom, J.P. De Winter, H. Joenje. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat GenetThis issue (2005). [DOI] [PubMed]
- 25.Cantor S, et al. The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci U S A. 2004;101:2357–62. doi: 10.1073/pnas.0308717101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pichierri P, Rosselli F. The DNA crosslink-induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways. Embo J. 2004;23:1178–87. doi: 10.1038/sj.emboj.7600113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andreassen PR, D’Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004;18:1958–63. doi: 10.1101/gad.1196104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levitus M, et al. Heterogeneity in Fanconi anemia: evidence for 2 new genetic subtypes. Blood. 2004;103:2498–503. doi: 10.1182/blood-2003-08-2915. [DOI] [PubMed] [Google Scholar]
- 29.Demuth I, Digweed M, Concannon P. Human SNM1B is required for normal cellular response to both DNA interstrand crosslink-inducing agents and ionizing radiation. Oncogene. 2004;23:8611–8. doi: 10.1038/sj.onc.1207895. [DOI] [PubMed] [Google Scholar]
- 30.Xue Y, et al. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci U S A. 2003;100:10635–40. doi: 10.1073/pnas.1937626100. [DOI] [PMC free article] [PubMed] [Google Scholar]