

Summary
Pathogenic Vibrio cholerae cycle between the nutrient rich human intestinal tract and nutrient poor aquatic environments and currently few bacterial factors are known that aid in the transition between these disparate environments. We hypothesized that the ability to store carbon as glycogen would facilitate both bacterial fitness in the aquatic environment and transmission of V. cholerae to new hosts. To investigate the role of glycogen in V. cholerae transmission, we constructed mutants that cannot store or degrade glycogen. Here, we provide the first report of glycogen metabolism in V. cholerae and demonstrate that glycogen prolongs survival in nutrient poor environments that are known ecological niches of V. cholerae, including pond water and rice-water stool. Additionally, glycogen contributes to the pathogenesis of V. cholerae in a transmission model of cholera. A role for glycogen in the transmission of V. cholerae is further supported by the presence of glycogen granules in rice-water stool vibrios from cholera patients, indicating that glycogen is stored during human infection. Collectively, our findings indicate that glycogen metabolism is critical for V. cholerae to transition between host and aquatic environments.
Keywords: Vibrio cholerae, glycogen, virulence, aquatic environment
Introduction
Cholera is an acute intestinal infection caused by toxigenic strains of the gram-negative bacterium Vibrio cholerae. Cholera remains a major global burden as outbreaks frequently occur in many developing regions, including Southeast Asia, Africa and South America (Sack et al., 2006). V. cholerae is a native member of aquatic environments and pathogenic strains are found in freshwater and estuaries in cholera endemic areas (Colwell, 1996, Reidl & Klose, 2002). V. cholerae is acquired by ingestion of contaminated food or water, and once acquired it colonizes and multiplies in the human small intestine. Induction of cholera toxin expression after colonization of the small intestine results in the production of large quantities of a secretory diarrhea, or rice-water stool (Lee et al., 1999, Reidl & Klose, 2002). Shedding of rice-water stool from cholera patients disseminates large numbers of V. cholerae back into aquatic environments, facilitating the transmission of cholera during outbreaks.
During its life-cycle, pathogenic V. cholerae survives in disparate environments as it passes between the human small intestine and aquatic environment. V. cholerae exits the host in rice-water stool, which is nutrient poor, alkaline and incapable of supporting growth (Freter et al., 1961). Once shed, V. cholerae faces dramatic changes in its environment, including downward shifts in osmolarity, pH and temperature. Additionally, essential nutrients, including phosphate and fixed nitrogen, are limiting (Nelson et al., 2008, Schild et al., 2007). Currently, we lack a comprehensive understanding of the physiological changes V. cholerae makes to successfully transition between the host and aquatic environments. However, factors important for this transition are beginning to be identified. Recently, V. cholerae was shown to induce expression of a number of genes specifically during late stages of infection in the infant mouse model of infection. Many of these ‘late-genes’ are dispensable for infection but are necessary for environmental survival, suggesting V. cholerae prepares for entry into the aquatic environment prior to exiting the host (Schild et al., 2007). Once in the environment, it has been suggested that V. cholerae survives by utilizing chitin as its primary carbon and nitrogen source and may associate with phytoplankton, zooplankton, and insect egg masses (Lipp et al., 2002, Halpern et al., 2004). Additionally, the ability to form biofilms (Watnick & Kolter, 2000) and to produce large amounts of polyphosphate likely aid in environmental survival (Jahid et al., 2006, Ogawa et al., 2000). Here, we report an additional strategy V. cholerae employs to persist in aquatic environments and enhance its transmission to new hosts: accumulation of internal stores of carbon in the form of glycogen.
Glycogen is a highly branched polysaccharide that consists of glucose units linked in a linear chain by α-1,4- bonds with α-1,6- branches. Glycogen has been shown to accumulate in bacteria upon entry into stationary phase or when growth is inhibited due to limitation of required nutrients (e.g. nitrogen or phosphate) in the presence of excess carbon (Preiss & Romeo, 1989). While the precise role of glycogen storage in bacteria is not understood, glycogen stores may serve as a reservoir of carbon and energy that can be mobilized for survival during periods of carbon starvation (Preiss & Romeo, 1994).
The genetic basis of glycogen synthesis and degradation has been extensively characterized in Escherichia coli. In E. coli, glgC, glgA and glgB encode glycogen synthesis enzymes and glgP and glgX encode enzymes for glycogen degradation. GlgC encodes an ADP-glucose pyrophosphorylase that catalyzes the first step of glycogen biosynthesis: the synthesis of ADP-glucose and pyrophosphate from ATP and glucose-1-phosphate. In subsequent steps, the glucosyl moieties of ADP-glucose are linked in α-1,4 and α-1,6 linkages by the action of a glycogen synthase (GlgA) and glycogen branching enzyme (GlgB), respectively. Deletion of glgC has been reported to prevent glycogen synthesis (Preiss & Romeo, 1994), although recent reports suggested that some glycogen can be synthesized in glgC mutants during growth in certain conditions (Moran-Zorzano et al., 2007, Eydallin et al., 2007a). The role of glgS in glycogen synthesis is unclear as it was shown to be important for glycogen accumulation in a certain strain of E. coli but is dispensable in another (Hengge-Aronis & Fischer, 1992, Eydallin et al., 2007b). To degrade glycogen, glycogen phosphorylase, encoded by glgP, sequentially removes glucose units from the non-reducing ends of glycogen (Alonso-Casajus et al., 2006). The α-1,6 linkages of glycogen are hydrolyzed by GlgX, a glycogen debranching enzyme (Dauvillee et al., 2005). Deletion of either glgP or glgX prevents degradation of internal stores of glycogen (Alonso-Casajus et al., 2006, Dauvillee et al., 2005).
V. cholerae O1 El Tor biotype strain N16961 contains most of the genes involved in glycogen metabolism in E. coli, although it lacks an annotated glgS or glgP. Interestingly, V. cholerae contains two GlgC homologs, GlgC1 and GlgC2, which are 58% identical in amino acid sequence and are located on each of the two chromosomes of V. cholerae. This unique genetic arrangement is conserved in other Vibrio species, including V. parahaemolyticus and V. vulnificus (http://cmr.jcvi.org/tigrscripts/CMR/CmrHomePage.cgi).
While a number of genes in V. cholerae have been annotated as playing roles in glycogen metabolism based on homology, the role of glycogen metabolism in the lifecycle of V. cholerae has not been experimentally investigated. A role for glycogen in cholera infection has been suggested in gene expression studies that showed glgB and glgX are induced during human infection whereas glgX is repressed when V. cholerae is shed in rice-water stool (Nelson et al., 2008, Lombardo et al., 2007, Merrell et al., 2002). Here, we report V. cholerae stores glycogen during human infection and that glycogen stores prolong survival in niches encountered during the lifecycle of V. cholerae, including in rice-water stool and pond water. Our data are consistent with glycogen playing an important role in the fitness and transmission of V. cholerae.
Results
Nitrogen limitation induces glycogen accumulation in V. cholerae
The role of glycogen on V. cholerae survival and pathogenesis was investigated by constructing strains that harbor in-frame deletions of genes implicated in glycogen synthesis (glgC1 and glgC2) or degradation (glgX). Glycogen accumulation has been reported to occur when bacterial growth is limited due to depletion of an essential nutrient for growth, with nitrogen limitation serving as strongest inducer (Preiss & Romeo, 1989, Preiss & Romeo, 1994). To identify conditions under which V. cholerae stores glycogen, wild-type (WT) and each of the glgC or glgX mutant strains were patched on M9-glucose minimal media with decreasing concentrations of nitrogen and glycogen accumulation was qualitatively assayed by potassium iodine staining (Damotte et al., 1968). The data (not shown) revealed that WT V. cholerae stores glycogen during growth under nitrogen limitation.
Because the potassium iodine staining method is qualitative, we quantified internal glycogen stores by enzymatically hydrolyzing glycogen with amyloglucosidase (Parrou & Francois, 1997) and measured liberated glucose using a tetrazolium blue reducing sugar assay (Jue & Lipke, 1985). After generating standard curves using purified glycogen, we applied this assay to cells grown under high nitrogen conditions. We detected very low levels of glycogen in WT and in glgC2, and none in glgC1 or a glgC1 glgC2 double mutant (Fig 1A). Deletion of glgX resulted in 10-fold more glycogen accumulation compared to WT. The presence of glycogen in a glgX mutant under conditions in which WT stores little glycogen suggests that V. cholerae continually synthesizes and degrades glycogen under high nitrogen conditions. This finding is consistent with studies in E. coli and Mycobacterium smegmatis that showed that glycogen is continuously synthesized and degraded (Alonso-Casajus et al., 2006, Belanger & Hatfull, 1999, Dauvillee et al., 2005), and in Corynebacterium glutamicum where inactivation of glgX results in aberrant glycogen accumulation compared to WT (Seibold & Eikmanns, 2007).
Figure 1. Nitrogen limitation induces glycogen storage in WT and glgC2.
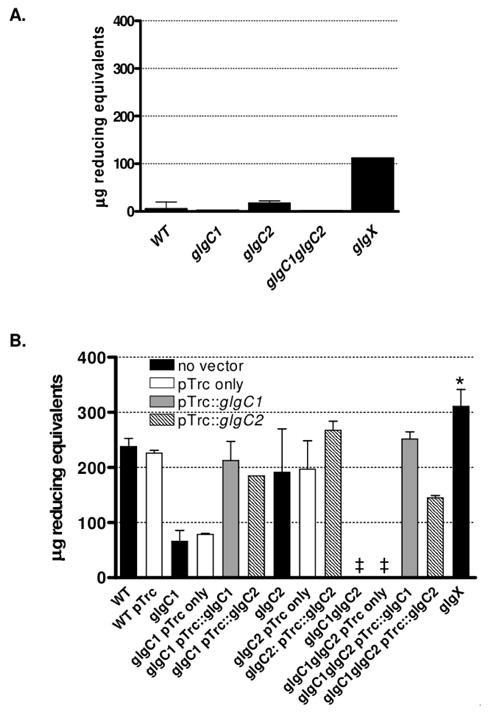
(A–B) Glycogen in WT, mutant and complemented strains was quantified after growth on high (A) or low (B) nitrogen M9-glucose agar by enzymatic hydrolysis of glycogen into glucose monomers. Liberated glucose was subsequently measured using the tetrazolium blue reducing sugar assay and expressed as μg reducing equivalents in 109 V. cholerae/mL. The median and interquartile range of two independent experiments each performed in triplicate are shown. The glgX strain accumulated significantly more glycogen than WT by a two-tailed student’s t-test (*p<0.005). The cross symbol indicates the amount of glycogen was below the limit of detection.
Glycogen accumulation was found to increase dramatically under low nitrogen conditions. WT accumulated ~15-fold more glycogen under low nitrogen conditions than under high nitrogen conditions (Fig 1A vs 1B). The glgC1 mutant accumulated 3.5-fold less glycogen than WT whereas the glgC2 mutant accumulated glycogen to similar levels as WT (Fig 1B). In contrast, no glycogen stores were detected in a glgC1 glgC2 double mutant under the conditions tested. Deletion of glgX resulted in an over-accumulation of glycogen compared to WT, consistent with findings in E. coli (Dauvillee et al., 2005).
The defect in glycogen storage in glgC1 and glgC1 glgC2 was fully complemented by expression of glgC1 in trans. Expression of glgC2 in trans partially rescued the glycogen accumulation defect in both glgC1 and glgC1 glgC2. The accumulation of glycogen after expression of GlgC2 in trans suggests glgC2 does encode an active ADP-glucose pyrophosphorylase.
Glycogen granules were visualized in each strain by lead citrate staining and transmission electron microscopy (TEM). A small number of electron dense granules were detected in WT, glgC1 and glgC2 during growth under high nitrogen conditions (Fig 2A–C) consistent with the biochemical assay results presented above. The glgX mutant contained more glycogen granules than WT under high nitrogen conditions (2E). In contrast, under low nitrogen conditions, thin sections of WT, glgC2 and glgX are saturated with electron dense granules, indicating that a large volume of the cell is occupied by glycogen (Fig 2F, H, J). The glgC1 mutant contained fewer glycogen granules than WT (Fig 2G) and <5% of the glgC1 glgC2 double mutant cells contained any detectable granules (Fig 2I). These observations are consistent with the results of the biochemical assay used to quantify glycogen and support our findings that both glgC1 and glgC1 glgC2 have profound defects in glycogen accumulation.
Figure 2. Visualization of glycogen granules by transmission electron microscopy.
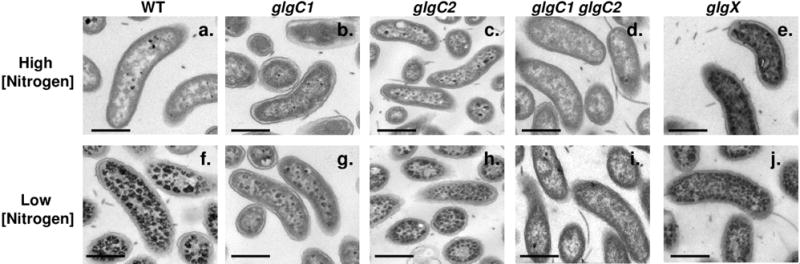
WT, glgC1, glgC2, glgC1 glgC2 and glgX were grown on M9-glucose agar with high or low nitrogen and grown at 37°C. Fixed, embedded sections of cells were stained with lead citrate. Glycogen appears as electron dense granules. Scale bar = 0.5 μm.
glgC1 and glgC2 are differentially expressed
Deletion of glgC1 results in a 3.5-fold reduction in glycogen accumulation compared to WT while deletion of glgC2 is inconsequent. The differences in glycogen storage between glgC1 and glgC2 could be due to differential regulation of the two glgC homologs. To address this possibility, the steady-state expression levels of glgC1 and glgC2 were analyzed by qRT-PCR. We found that both glgC1 and glgC2 are expressed during growth under low nitrogen conditions (Fig 3). However, the glgC1 transcript level was 9-fold higher relative to glgC2 during mid- and late-exponential growth under nitrogen limitation (Fig 3). In each mutant strain, the transcript levels of the remaining glgC homolog and other glycogen metabolism genes were equivalent to WT levels (data not shown). These results collectively suggest that the difference in glycogen accumulation between the glgC1 and glgC2 strains is not due to altered expression levels of downstream biosynthesis or degradation genes but rather is dependent on the elevated transcript level of glgC1 relative to glgC2. Furthermore, the finding that ectopic expression of glgC2 from a multi-copy plasmid resulted in glycogen accumulation in the glgC1 and glgC1 glgC2 strains (Fig 1B), suggests that when expressed, GlgC2 can stimulate glycogen accumulation. Thus, glgC1 and glgC2 are differentially regulated at the level of transcription and glgC1 is the dominant message expressed during nitrogen-limited growth on glucose.
Figure 3. The glgC1 transcript is expressed to higher levels than glgC2 in low nitrogen conditions.
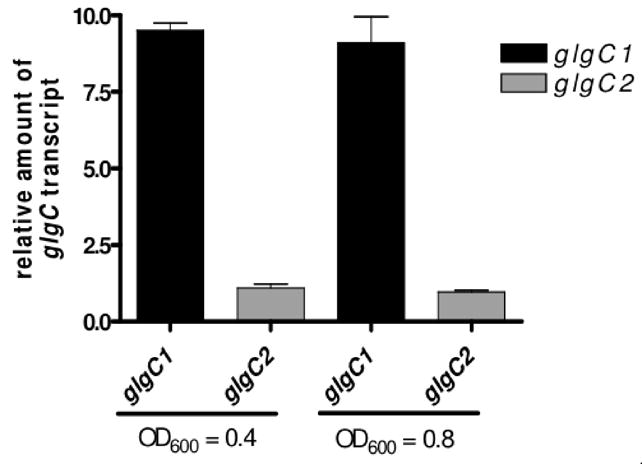
Quantitative RT-PCR (qRT-PCR) analysis of glgC1 and glgC2 expression during growth on M9-glucose under nitrogen limitation at 37°C. Samples were collected during mid- and late-exponential growth (OD600 = 0.4 and OD600 = 0.8). All samples were prepared in triplicate and expression levels were normalized to rpoB expression in each sample and expressed relative to glgC2 expression in WT. The mean and standard deviation of a representative experiment is shown.
Glycogen synthesis mutants are impaired for growth during the glycogen accumulation phase
The glgC1 and glgC1 glgC2 mutants exhibit growth defects in high and low nitrogen conditions compared to WT, glgC2 or glgX (Fig 4A, 4C). During growth in high nitrogen conditions, both glgC1 and glgC1 glgC2 have a moderate growth defect upon entry into stationary phase (Fig 4A). The observed growth defects of these strains occur during entry into stationary phase and seem to correspond to the period during the growth curve when a spike in glycogen accumulation occurs in WT (Fig 4B), consistent with the kinetics of glycogen accumulation in E. coli (Preiss & Romeo, 1989). Growth of glgC1 and glgC1 glgC2 is more drastically reduced during growth under nitrogen limitation (Fig 4C). Specifically, growth halts in late-exponential phase, corresponding to the point when significant stores of glycogen accumulate in WT (Fig 4D). These data suggest that growth of V. cholerae halts under glycogen inducing conditions when glycogen cannot accumulate. The observed growth defects in each strain can be complemented by expression of glgC1 or glgC2 in trans (data not shown).
Figure 4. Glycogen storage mutants exhibit growth defects during growth under high and low nitrogen conditions, corresponding to the period in the growth curve when glycogen accumulates.
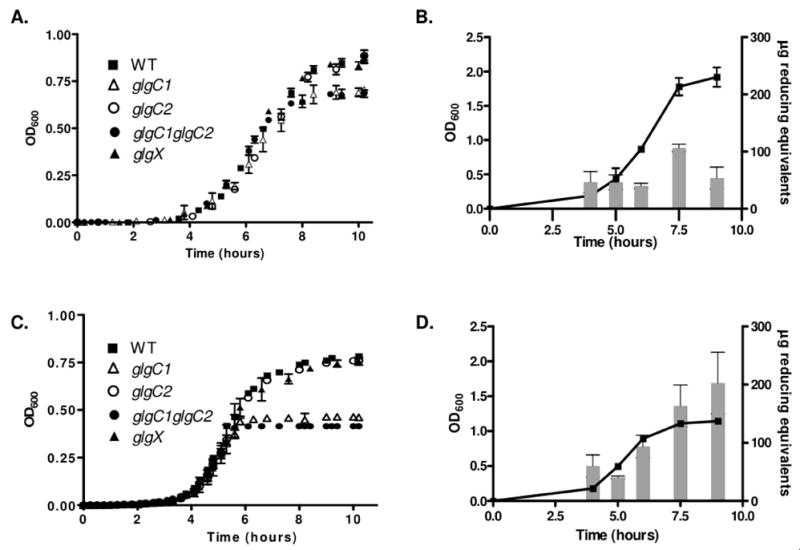
(A,C) Growth of WT, glgC1, glgC2, glgC1 glgC2 and glgX in M9-glucose in the presence of high nitrogen (A) or low nitrogen (C). Samples were grown at 37°C in M9-glucose with high or limiting ntirogen and the OD600 of each sample was read every 20 minutes for 10 hours. All samples were analyzed in triplicate. The mean OD600 and standard deviation at each time point from a representative experiment are shown. (B, D) Glycogen was assayed in WT V. cholerae during growth in M9-glucose in the presence of high (B) or low nitrogen (D). At each of the indicated time-points, samples were removed and the glycogen content was determined using the enzymatic hydrolysis assay. All samples were analyzed in triplicate. The mean and standard deviation of a representative experiment is shown.
V. cholerae utilizes glycogen stores to prolong survival in nutrient poor environments
The role of glycogen in prolonging survival under carbon starvation varies between bacterial species. Survival is diminished in nutrient poor environments for E. coli and S. Typhimurium mutants unable to accumulate glycogen (Strange, 1968, McMeechan et al., 2005). In contrast, glycogen stores attenuate survival of Sarcina lutea in a nutrient poor condition and glycogen accumulation is dispensable for stationary phase survival in Corynebacterium glutamicum (Burleigh & Dawes, 1967, Seibold et al., 2007). To investigate the role of glycogen on the survival of V. cholerae, experiments were performed to determine if glycogen provides a fitness advantage in nutrient poor environments. WT and each mutant strain were grown under nitrogen limiting conditions, mixed in a 1:1 ratio and inoculated into either M9 with no carbon source or in filter-sterilized pond water and survival over time was monitored. WT and glgC2, which store equivalent amounts of glycogen, were equally fit for survival in both media (Fig 5A–B). The viable counts of both strains dropped one to two orders of magnitude over a 48 hr period. In contrast, in M9 without carbon, glgC1 was 12-fold attenuated and the glgC1 glgC2 mutant was ~20-fold attenuated for survival by 48 hr (Fig 5A). The viable counts for the glgC1 and glgC1 glgC2 mutants dropped three orders of magnitude over a 48 hr period. In competitions between WT and glgX, glgX was nearly 6-fold attenuated after 2 hr incubation in M9 with no carbon source (compared to 2-fold for glgC1 glgC2) and was over 75-fold attenuated for survival compared to WT by 48 hr. The viable counts of glgX mutant dropped over three orders of magnitude in a 48 hr period. Similar trends were observed when competitions were performed in pond water, where glgC1 is 3.5-fold attenuated and glgC1 glgC2 and glgX were 45-fold and 100-fold attenuated compared to WT by 48 hr, respectively (Fig 5B). All strains were equally fit for survival when competed in rich media (data not shown).
Figure 5. Glycogen metabolism prolongs survival in nutrient poor environments.
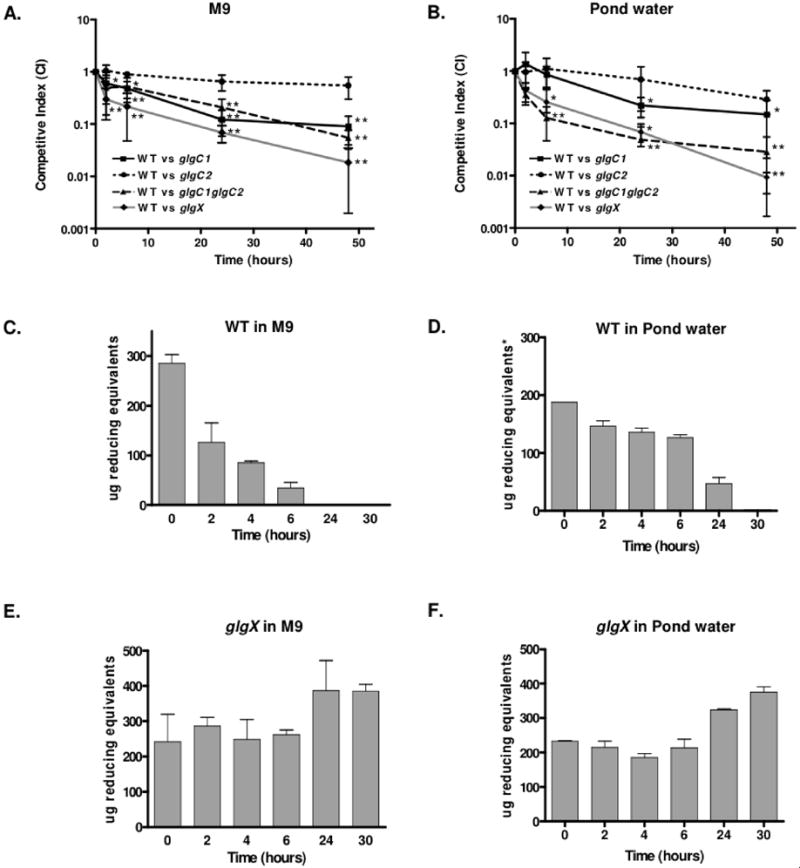
(A–B) M9 with no carbon source (A) or pond water (B) were inoculated with 106 cfu of a 1:1 mix of mutant (LacZ+) and wt (LacZ−) grown under nitrogen limiting conditions. At the indicated times, the ratio of viable bacteria was quantitated by blue:white colony screening. The competitive index (CI) is the ratio of mutant:wt corrected for the input ratio. Each data point is the geometric mean of the CI obtained from three independent samples, each done in triplicate. The asterisks indicate a significant decrease in the median CI compared to that from competitions between WT and glgC2 (*p<0.05 or ** p<0.001 using a One-way Anova). (C–F) Quantitation of glycogen in WT and glgX strains during incubation in M9 with no carbon source or in pond water as indicated. The inocula were grown under nitrogen limiting conditions. All samples were analyzed in triplicate. The mean and standard deviation at each time-point of a representative experiment is shown.
The survival advantage of the WT strain in nutrient poor environments is due to mobilization of internal glycogen stores. Glycogen stores were depleted over time in WT when incubated in M9 with no carbon source (Fig 5C) or in pond water (Fig 5D), although at a slower rate than in the latter as pond water contains a limited amount of carbon from environmental sources. In contrast, a glgX mutant was unable to degrade its internal glycogen stores in carbon poor environments and instead appeared to accumulate small amounts of glycogen during prolonged incubation in such environments (Fig 5E–F). Because the glgX mutant cannot degrade glycogen for utilization as a carbon source, glgX is dramatically attenuated for survival compared to WT in carbon poor environments (Fig 5A–B). These results demonstrate that, for V. cholerae, mobilization of internal glycogen stores dramatically prolongs survival in carbon poor environments including pond water, a known ecological niche of the bacteria.
Glycogen stores are protective and prolong survival in rice-water stool
When released from the small intestinal epithelium during the acute stage of cholera, V. cholerae must survive in rice-water stool, which is nutrient poor, prior to entering aquatic environments (Freter et al., 1961). To determine if glycogen provides a fitness advantage in rice-water stool, competition experiments were performed between WT and glgC1 glgC2 or glgX by growing each strain under low nitrogen conditions and incubating the bacteria in rice-water stool supernatants from three different cholera patients. We found that the glgC1 glgC2 double mutant was attenuated for survival ranging from 2–12 fold after a 2 hr incubation (Fig 6). In contrast, glgX remained fit for survival in two of the stool samples tested. In the third stool sample, which was the most hostile to bacterial survival (glgC1 glgC2 was 12-fold attenuated for survival), glgX was 6-fold attenuated. These data suggest that the presence of glycogen in a glgX mutant, regardless of its ability to be utilized as a carbon source, is protective against the hostile environment V. cholerae encounters in rice-water stool. The glgC1 glgC2 mutant, on the other hand, does not contain glycogen and therefore we hypothesize is more susceptible to the harsh effects of rice-water stool.
Figure 6. The glgC1 glgC2 mutant is attenuated for survival in cholera stool.
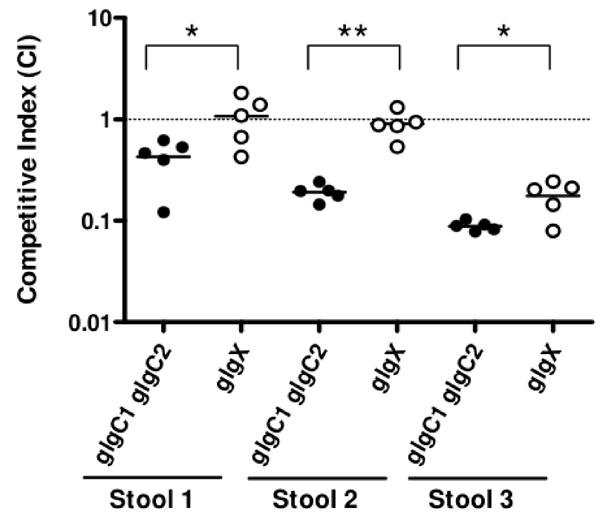
Three stool supernatants were inoculated with a 1:1 mix of WT and glgC1 glgC2 (filled circles), or WT and glgX (open circles) and incubated statically at 37°C. After 2 hr, the ratio of mutant:wt bacteria was determined by blue:white colony screening. Each data point indicates the CI from a single competition experiment. The horizontal line indicates the geometric mean of each data set. The glgC1 glgC2 mutant is significantly attenuated for survival compared to the respective competition between WT and glgX in each stool sample using a two-tailed student’s t-test (*p<0.05 and ** p<0.001).
RpoS negatively regulates glycogen accumulation during growth under nitrogen limitation
RpoS plays an important role in mediating the global response to starvation as well as other stresses in E. coli. RpoS encodes an alternative sigma factor, σS, that is induced during entry into stationary phase or under periods of nutrient deprivation or stress (Hengge-Aronis, 2002). RpoS has been shown to be required for normal glycogen accumulation in E. coli and in the plant pathogen Erwinia carotovora (Hengge-Aronis & Fischer, 1992, Andersson et al., 1999, Eydallin et al., 2007b). Genetic studies in E. coli revealed that RpoS does not affect glgC expression but controls expression of glgS. The exact function of glgS is not clear but a glgS null strain of E. coli accumulates less glycogen than WT (Hengge-Aronis & Fischer, 1992). V. cholerae does not contain a recognizable GlgS ortholog. However, V. cholerae rpoS mutants were impaired for survival in carbon poor environments and are more sensitive to environmental stress (Yildiz & Schoolnik, 1998), similar to phenotypes we observe with mutants that are unable to store or degrade glycogen.
To determine if RpoS is essential for glycogen metabolism in V. cholerae, glycogen was measured in an rpoS mutant. In contrast to E. coli, RpoS negatively regulates glycogen synthesis in V. cholerae as an rpoS mutant accumulated more glycogen than WT in low nitrogen conditions (Fig 7A). This appears to be due to elevated expression of glgC1, as shown by qRT-PCR analysis (Fig 7B). Interestingly, glgC2 was repressed in an rpoS mutant, suggesting that in V. cholerae, RpoS inversely regulates expression of the two glgC homologs. The pattern of glycogen accumulation is similar to WT in high nitrogen conditions. Glycogen stores were depleted similarly to WT when the rpoS mutant is incubated in carbon poor environments (data not shown). These results indicate that in V. cholerae, RpoS is a mild repressor of glycogen storage but does not regulate glycogen utilization.
Figure 7. RpoS negatively regulates glycogen synthesis in V. cholerae.
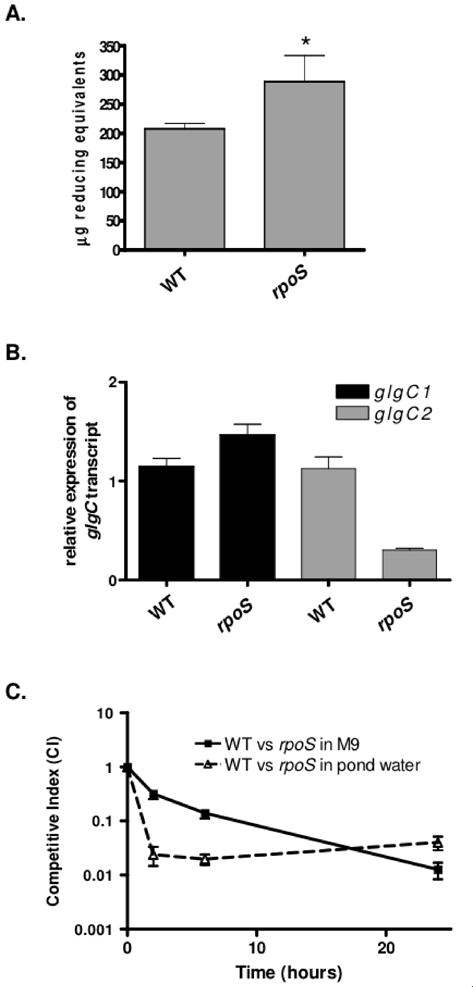
(A) Glycogen accumulation in WT and an rpoS mutant was quantified by enzymatically hydrolyzing glycogen stores into glucose monomers using amyloglucosidase during growth on low nitrogen M9 agar. The median and interquartile range of two independent experiments performed in quadruplicate are shown. An rpoS mutant was found to store significantly more glycogen than WT using a two-tailed student’s t-test (p=0.0014). (B) Quantitative RT-PCR (qRT-PCR) analysis of glgC1 and glgC2 expression during growth of WT and an rpoS mutant on M9-glucose under nitrogen limitation. Samples were collected during late- logarithmic growth (OD600 = 0.8). All samples were prepared in triplicate and expression levels were normalized to rpoB expression in each sample and expressed relative to glgC1 or glgC2 expression in WT. The mean and standard deviation of a representative experiment is shown. (C) M9 minimal media with no carbon source or pond water were inoculated with a 1:1 ratio of rpoS:wt that were grown under nitrogen limiting conditions. At the indicated time-points, bacteria were removed and differentiated by blue:white screening. Each data point is the geometric mean of the CI obtained from samples tested in quadruplicate. A representative experiment is shown.
In light of these findings, we wanted to determine if rpoS mutants that contain glycogen are able to survive normally in carbon poor environments. To test this, WT and the rpoS mutant were grown under nitrogen limiting conditions to induce glycogen storage. During incubation in M9 with no carbon source or pond water, we observed that the rpoS mutant was attenuated for survival compared to WT despite our findings that an rpoS mutant stores slightly more glycogen than WT and is able to degrade its glycogen stores (Fig 7C). Therefore, the near normal glycogen metabolism of the rpoS mutant is not sufficient to override the pleiotropic effects of the rpoS mutation in nutrient poor environments.
Glycogen-rich V. cholerae are more virulent in a transmission model of cholera infection
The role of glycogen metabolism in the virulence of V. cholerae was tested using competition experiments in an infant mouse model of cholera. When the inocula were grown under low nitrogen conditions to induce glycogen storage or under conditions that induce little to no glycogen storage (rich media), the glgC1 glgC2 double mutant showed only a modest attenuation of virulence (CI = 0.79) in competition with WT (Fig 8A). However, this experimental design is limiting since it is known that after exiting the human small intestine, V. cholerae are shed from the host into nutrient poor environments including rice-water stool and pond water. In light of our findings that glycogen protects V. cholerae from nutrient deprivation and environmental stress, we hypothesized that glycogen may enhance transmission of V. cholerae to new hosts. To test this, a transmission model of cholera was developed whereby WT and the glgC1 glgC2 double mutant were grown under low nitrogen conditions to induce glycogen storage and subsequently passaged through a pond water intermediate prior to inoculation of infant mice. Using this model of transmission, the glgC1 glgC2 mutant was nearly 2.5 fold attenuated for virulence compared to WT after 3 hr incubation in pond water and 4-fold attenuated after 24 hr incubation in pond water. Importantly, the glgC1 glgC2 mutant was not attenuated for survival when the pond water passaged inoculum was competed in parallel in M9-glucose, indicating that the attenuation of glgC1 glgC2 compared to WT is specific for in vivo conditions and not due to a general growth defect as a result of the period of pond water incubation.
Figure 8. Glycogen plays a role in the transmission of V. cholerae.
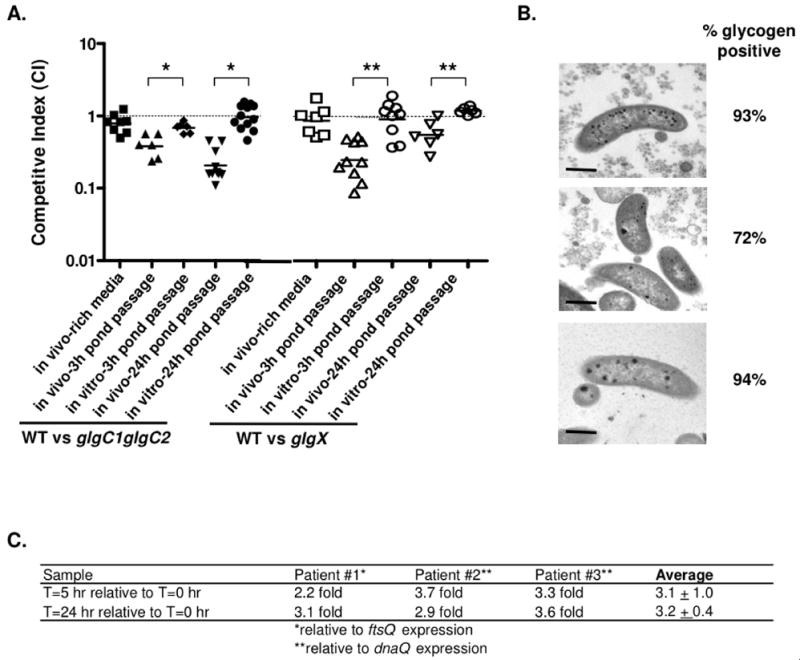
(A) Competition assays were done using the infant mouse model of cholera. WT was competed against glgC1 glgC2 (filled shapes) or glgX (open shapes) following growth on rich media (LB, squares) or low nitrogen M9 followed by pond water passage for 3 h (triangles) or 24 h (upside down triangles). In vitro control competition assays, in which the pond water passaged bacteria were grown for 20 hr in M9-glucose at 37°C with aeration, are shown. The CI was determined by calculating the mutant:wt corrected for the input ratio. Each data point represents the CI from an individual mouse or in vitro competition. The horizontal line represents the geometric mean of each data set. glgC1 glgC2 and glgX are significantly attenuated for virulence compared to WT after pond water passage compared to the respective in vitro competition (*p<0.001 and **p<0.005, by a student’s two-tailed t-test). (B) Representative TEM images of stool V. cholerae obtained from three cholera patients. The percentage of glycogen positive V. cholerae in each stool sample is noted next to a representative TEM image. Scale bar = 0.5 μm. (C) qRT-PCR analysis of glgX expression in stool V. cholerae from three cholera patients after incubation in pond water for 0, 5 and 24 h. Expression of the glgX transcript relative to t=0 h is shown. Transcript amounts were normalized to either ftsQ or dnaQ.
The fitness advantage conferred by glycogen in the above transmission model could be due to utilization of glycogen to provide carbon and energy or, alternatively, could simply be due to the physical presence of glycogen within the cells. To differentiate between these possibilities, we assayed the role of glycogen breakdown in virulence in the transmission model by competing the glgX mutant with WT. Similar to results obtained with competitions between WT and the glgC1 glgC2 mutant, the glgX mutant showed only a slight attenuation of virulence in competition with WT (CI = 0.87) when inoculated directly into mice without pond water passage (Fig 8A). In contrast, when WT and glgX are grown under glycogen storage inducing conditions and passaged through pond water in our transmission model of cholera, the glgX mutant was 4-fold attenuated for virulence compared to WT after 3 hr incubation in pond water and was 2-fold attenuated after 24 hr incubation in pond water. The glgX mutant was not attenuated for survival when the pond water passaged inoculum was competed in M9-glucose, suggesting that the virulence defect of the glgX mutant is specific for in vivo conditions. Thus, glycogen plays a role in the ability of V. cholerae to infect in a transmission model of cholera as mutants that do not contain glycogen or cannot degrade glycogen are attenuated for virulence in this assay.
A role for glycogen in the transmission of V. cholerae is further supported by the presence of glycogen granules in human shed rice-water stool V. cholerae collected from cholera patients (Fig 8B). The majority of V. cholerae cells in cholera stool (>72%) contain numerous glycogen granules, compared to the glgC1 glgC2 double mutant where <5% of the cells contained glycogen granules by TEM analysis. This indicates that V. cholerae synthesizes glycogen in the human small intestine prior to being shed. The sum of these findings suggest these internal stores of glycogen could, in turn, provide resistance against the harsh effects encountered in rice-water stool and be mobilized for survival in aquatic environments.
Analysis of the transcriptome of rice-water stool V. cholerae has revealed that glgC1 is induced whereas glgX is repressed (Merrell et al., 2002, Nelson E.J. & Camilli, 2008), suggesting stool V. cholerae conserve their glycogen stores as they are shed from the host. After exiting the host, glycogen stores that are synthesized in vivo are likely mobilized for survival and therefore, glycogen degradation genes should be induced in nutrient poor environments. To test this hypothesis, glgX expression was measured by qRT-PCR analysis in three freshly shed rice-water cholera stool samples that were incubated in pond water for up to 24 hr. This analysis revealed that glgX expression was induced 3-fold after 5 h and 24 h of incubation in pond water (Fig 8C). Increased expression of glgX after incubation of rice-water stool in pond water is consistent with our hypothesis that human shed V. cholerae degrade their internal stores of glycogen to prolong survival in aquatic environments.
Discussion
A major challenge that many facultative pathogens, including V. cholerae, must overcome is how to survive in the disparate environments it encounters between the host and the environment. After ingestion by the human host, enteric bacteria encounter numerous stresses including acidic pH, nitric oxide, osmotic stress, as well as nutrient and carbon starvation (El Hassani et al., 2005, Janoff et al., 1997, Nielsen et al., 2006, Slauch et al., 1997). In the aquatic environment, bacteria must adapt to lower temperatures, changes in pH, and nutrient and carbon limitation (Slauch et al., 1997). To adapt and survive such changes, it is likely that enteric pathogens, such as V. cholerae, have evolved mechanisms to enhance fitness during these transitions.
Here, we show that V. cholerae stores glycogen in vivo and that glycogen plays an important role in the transition of V. cholerae between the host and environment. Additionally, using model experimental systems, we report that glycogen prolongs environmental survival and is important for the transmission of V. cholerae to new hosts. Prior to this study, the ability of V. cholerae to accumulate glycogen and the role glycogen metabolism plays in the lifecycle of V. cholerae had not been addressed. Our findings implicate glycogen metabolism as an additional survival strategy employed by enteric bacteria to survive the dramatic changes encountered in the host and the environment.
We determined using quantitative assays that glycogen accumulation is induced in WT V. cholerae during growth under nitrogen limitation. This result raises the interesting possibility that V. cholerae may also store glycogen in natural aquatic environments as it is known that fixed nitrogen is limiting in pond water (Nelson et al., 2008). To study glycogen metabolism in V. cholerae, we constructed deletions in each of the glgC homologs. The glgC1 mutant showed a drastic decrease in glycogen accumulation compared to WT whereas glgC2 stored glycogen to similar levels as WT. We observed that the glgC1 glgC2 double mutant strain does not accumulate glycogen in any condition tested. This finding is in contrast to recent studies in E. coli, Salmonella enterica (serovar Typhimurium), S. coelicolor and Mycobacterium tuberculosis that showed that an additional, unidentified source of ADP-glucose exists in these bacteria (Moran-Zorzano et al., 2007, Eydallin et al., 2007a, Sambou et al., 2008, Martin et al., 1997). V. cholerae may lack this additional source of ADP-glucose or this source may not be induced under the conditions tested in this study.
Consistent with findings in glgX mutants of E. coli and C. glutamicum (Seibold & Eikmanns, 2007, Dauvillee et al., 2005), a V. cholerae glgX mutant accumulated glycogen to greater levels than WT under low nitrogen conditions. The glgX mutants are unable to degrade internal glycogen stores, illustrating that GlgX is essential for glycogen utilization in V. cholerae. We noted that glgX mutants accumulate more glycogen during prolonged incubation in nutrient poor environments. The mechanism for this increase in accumulation is unclear. One possibility is that glycogen continues to be synthesized, possibly by utilizing breakdown products of other bacteria as a carbon source.
The presence of two glgC homologs, each of which bears ADP-glucose pyrophosphorylase activity in V. cholerae, would suggest either differential expression or post-transcriptional regulation. In support of the former, we observed that glgC2 but not glgC1 is repressed in an rpoS mutant. This finding is contrary to studies in E. coli that showed RpoS is not involved in the transcription of glgC (Hengge-Aronis & Fischer, 1992). It has been suggested that RpoS accumulates during later stages of cholera infection (Nielsen et al., 2006). Although we have not tested glgC2 expression in vivo, it is plausible that rpoS may induce glgC2 expression during late stages of infection. These findings raise the possibility that the two glgC homologs play different roles throughout the life-cycle of V. cholerae. This hypothesis is supported by the finding that glgC mutants of S. coelicolor have defects in glycogen accumulation specifically in phase I of development, suggesting a second source of ADP-glucose exists for glycogen synthesis in phase II of development (Martin et al., 1997).
Glycogen stores were shown to enable V. cholerae to persist in nutrient poor environments for significantly longer time periods than mutants that are unable to store or degrade glycogen by catabolizing their internal glycogen stores for use as a carbon source. The enhanced fitness of glycogen-rich V. cholerae in aquatic environments likely contributes to the transmission of V. cholerae as bacteria that contain glycogen persist in aquatic environments for longer periods of time and are therefore more likely to be ingested by a host to initiate a new round of infection.
The role glycogen metabolism plays in bacterial pathogenesis is not clear. A number of reports suggest a role by demonstrating that glycogen aids colonization (Jones et al., 2008) acid production (Harris et al., 1992, Spatafora et al., 1999) and persistence at mucosal surfaces (Carlson et al., 2008). A role for glycogen in Salmonellae virulence is controversial as glycogen was suggested to have a role in S. enteritidis virulence (Bonafonte et al., 2000) but was found to play a minor role in S. enterica (McMeechan et al., 2005).
Here, we provide evidence that glycogen metabolism contributes to pathogenesis in a transmission model of cholera by demonstrating that V. cholerae mutants that are either unable to store or degrade glycogen are attenuated for transmission. The attenuation of glgC1 glgC2 and glgX compared to WT in vivo after pond water passage could be due to a number of factors. As noted above, the presence of glycogen prolongs survival in nutrient poor environments, and therefore, glycogen-rich WT V. cholerae are presumably more fit to initiate infection after pond water passage. Additionally, the presence of glycogen stores, regardless of their ability to be metabolized, appears to be protective against at least some environmental stresses as evident by the observation that the glgX mutant is more resistant to incubation in rice-water stool. At present, we do not understand the basis of this protection.
To further support a role for glycogen in V. cholerae transmission, glycogen granules were found in rice-water stool V. cholerae. While it is not known if nitrogen is limiting in the human small intestine, the presence of glycogen granules in rice-water stool V. cholerae combined with data that glgB and glgX are induced during infection in human volunteers indicates that glycogen metabolism occurs in vivo (Lombardo et al., 2007). Based on our findings that glycogen-rich V. cholerae survive longer in rice-water stool, we hypothesize glycogen stored in vivo may be protective against stresses V. cholerae encounters in vivo, including low pH or osmotic stress. This hypothesis is supported a study in E. coli showing that glycogen metabolism is important for colonization of the murine intestine by both commensal and pathogenic E. coli (Jones et al., 2008).
While numerous questions regarding the regulation and the role of glycogen metabolism in V. cholerae remain, the sum of these findings suggests that the role of glycogen in the lifecycle of V. cholerae is multifaceted. The presence of glycogen provides a carbon source that prolongs survival in various ecological niches and glycogen is protective against challenges faced in the environment and in vivo. We anticipate that glycogen metabolism in V. cholerae may serve as a model for the role of glycogen in the transmission of other facultative pathogens.
Experimental Procedures
Bacterial Growth Conditions
Bacteria were grown in Luria-Bertani (LB) or in M9 minimal media with or without 0.4% glucose (Dauvillee et al., 2005). For low nitrogen M9 media, the nitrogen concentration was reduced from 0.1% to 0.025% NH4Cl. Expression from the pTrc99A promoter was induced by the addition of isopropyl-B-D-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. Pond water was obtained from the Chandler Pond in Brighton, MA and filter-sterilized through a 0.22 μm filter (Corning). Antibiotics were added when appropriate at the following concentrations: 100 μg/ml streptomycin (Sm), 50 μg/ml ampicillin (Ap), and 50 μg/ml kanamycin (Km).
Strain and Plasmid Construction
All strains used in this study are listed in Table 1. Plasmids were propagated in E. coli DH5α except plasmids that contain oriR6k, which were propagated in either SM10λpir or DH5αλpir. To generate gene deletions, splicing by overlap extension PCR was used as previously described (Senanayake & Brian, 1995). In-frame gene deletions were made by allelic exchange with plasmid pCVD442 as previously described (Donnenberg & Kaper, 1991). For complementation, glgC1 and glgC2 were amplified by PCR from V. cholerae N16961 genomic DNA and the products were cloned into PCR-Script (Stratagene). Plasmids were transformed into the indicated V. cholerae strain background by electroporation.
Table 1.
Strains used in this study
| Description | Reference or source | |
|---|---|---|
| V. cholerae strains | ||
| AC304 | El Tor N16961, SmR | (Angelichio et al., 1999) |
| AC390 | El Tor N16961, SmR lacZ::res-tet-res | Laboratory strain |
| KFV21 | N16961 rpoS::KnR SmR | Gift of Karla Satchell |
| AC1526 | N16961 ΔglgC1 | This work |
| LB5 | N16961 ΔglgC1 (pTrc99A::glgC1) | This work |
| LB6 | N16961 ΔglgC1 (pTrc99A::glgC2) | This work |
| AC1527 | N16961 ΔglgC2 | This work |
| LB7 | N16961 ΔglgC2 (pTrc99A::glgC2) | This work |
| AC1528 | N16961 ΔglgC1 ΔglgC2 | This work |
| LB8 | N16961 ΔglgC1 ΔglgC2 (pTrc99A::glgC1) | This work |
| LB9 | N16961 ΔglgC1 ΔglgC2 (pTrc99A::glgC2) | This work |
| LB12 | N16961 ΔglgX | This work |
|
| ||
| E. coli strains | ||
| DH5α | F-Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 suupE44thi-1 gyrA96 relaA1 | Laboratory strain |
| DH5αλpir | F-Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 suupE44thi-1 gyrA96 relaA1 λ::pir | Laboratory strain |
| SM10λpir | thi recA thr leu tonA lacY supE RP4-2-Tc::Mu λ::pir | Laboratory strain |
| AC1516 | E. coli DH5α (pCR-Script AmpK+::ΔglgC1). ApR | This work |
| AC1517 | E. coli DH5α (pCR-Script AmpK+::ΔglgC2). ApR | This work |
| AC1518 | E. coli DH5αλpir (pCVD442lac::ΔglgC1). ApR | This work |
| AC1519 | E. coli DH5αλpir (pCVD442lac::ΔglgC2). ApR | This work |
| AC1522 | E. coli SM10λpir (pCVD442lac::ΔglgC2). ApR | This work |
| AC1523 | E. coli Sm10λpir (pCVD442lac::ΔglgC1). ApR | This work |
| AC2393 | E. coli DH5α (pTrc99a). ApR | Laboratory strain |
| LB1 | E. coli DH5α (pTrc99a::glgC1). ApR | This work |
| LB2 | E. coli DH5α (pTrc99a::glgC2). ApR | This work |
| LB3 | E. coli SM10λpir (pTrc99a::glgC1). ApR | This work |
| LB4 | E. coli SM10λpir (pTrc99a::glgC2). ApR | This work |
| LB10 | E. coli DH5α (pCR-Script AmpK+::ΔglgX). ApR | This work |
| LB11 | E. coli DH5αλpir (pCVD442lac::ΔglgX). ApR | This work |
Glycogen Quantitation
Strains were patched in triplicate on M9-glucose agar plates containing an appropriate antibiotic and either high or low NH4Cl and incubated overnight at 37°C. Glycogen content in each strain was determined by digesting with amyloglucosidase (Parrou & Francois, 1997) followed by quantitation of released glucose using tetrazolium blue (Jue & Lipke, 1985), with modifications to each procedure. Briefly, bacteria were scraped from each M9 agar plate into 1 mL phosphate buffered saline (PBS). Samples were adjusted to a final OD600 of 1.0 (~109 colony-forming units/mL), pelleted, resuspended in 125 μl 0.25M Na2CO3 and incubated at 95°C for 4 hours. Immediately after incubation, the pH of the samples was adjusted by addition of 75 μl 1 acetic acid and 300 μl of 0.2 M sodium acetate, pH 5.2. 1.5 μl of a 50 mg/mL (1.2U/mL) preparation of amyloglucosidase from Aspergillus niger (Fluka) was added. Samples were incubated overnight at 57°C with constant agitation and clarified by centrifugation for 3 min at 6,700 × g. 20 μl of supernatant was added to 2 mL of a freshly filtered (0.22 uM, Corning) solution containing 0.1% tetrazolium blue, 0.05 M NaOH, and 0.5 M sodium potassium tartrate. Samples were incubated at 100°C for 5 minutes, cooled by incubation in an ice-water bath, and the absorbance was determined at 660 nm. The absorbance of each sample was compared to a standard curve generated with known concentrations of glycogen from bovine liver, type IX (Sigma) that was treated in an analogous fashion to determine the concentration of reducing sugars (reducing equivalents) released following amyloglucosidase digestion in each sample. All samples were analyzed in triplicate and the mean microgram reducing equivalents in the total volume of each sample is plotted with the standard deviation.
To determine the glycogen content in WT and the glgX mutant during incubation in carbon-poor environments (M9 with no carbon source or pond water), each strain was patched on low nitrogen M9-glucose agar, scraped into 1 mL PBS, and the OD600 was determined. Each strain was inoculated into M9 with no carbon source or pond water with a starting OD600 = 1.0. At each of the indicated time-points, the OD600 of the culture was determined and an equivalent amount of bacteria equal to an OD600 = 1.0 were removed and the glycogen content was analyzed using the enzymatic hydrolysis assay as described above.
Transmission Electron Microscopy (TEM)
Strains were grown on M9-glucose agar with high or low nitrogen at 37°C, scraped from each plate into 1 mL PBS, pelleted and subsequently fixed with 2.5% glutaraldehyde buffered by 0.1 M sodium cacodylate, pelleted and washed with 0.1 M sodium cacodylate buffer. Samples were then post-fixed with 1% osmium tetroxide, washed with 0.1 M sodium cacodylate and double distilled H20, dehydrated and embedded in fresh Epon. Thin sections were placed on gold grids and stained by inversion on 1.5% H202 for 10 min and lead citrate for 5 min. Glycogen was observed under a Philips CM-10 electron microscope. Cholera patient stool samples used for TEM analysis were described previously (Merrell et al., 2002, Schild et al., 2007, Nelson et al., 2007). The stool samples and in vitro grown control strains were prepared for TEM analysis and stained as described above. Glycogen positive V. cholerae were defined as comma shaped bacteria of ~1 μM in length that contained greater than 3 electron dense granules per thin section analyzed.
RNA Purification and Quantitative RT-PCR (qRT-PCR)
RNA was isolated from mid- and late-exponential-phase cultures (OD600 = 0.4 and OD600 = O.8) incubated with aeration at 37°C in low nitrogen M9-glucose minimal media. RNAprotect (Qiagen) was added to each sample. RNA was purified and cDNA synthesized from 1 μg RNA as previously described (Schild et al., 2007). qRT-PCR experiments were performed using a 1:20 dilution of cDNA synthesized from 1 μg RNA, 300 nM primers, ROX reference dye, and iQ SYBR Green Supermix (Biorad). Primer sequences used in this study are listed in Table 2 and all primer pairs amplified with efficiencies of 90.8% or greater (data not shown). The mean cycle threshold (Ct) of each test transcript was normalized to the Ct of the reference transcript rpoB. Two independent in vitro samples for each strain were tested in triplicate.
Table 2.
Primers used in this study
| Primer name | Primer sequence |
|---|---|
| GlgC1F | 5′-CGCAGTTCAAATCGCAATCGCT-3′ |
| GlgC1R | 5′-TAAATCGCATCGGCAGTGCCTT-3′ |
| GlgC2F | 5′-TGCCTGCATTCTGGATTACG-3′ |
| GlgCR | 5′-GCGGTTCCTTCGTACCATTT-3′ |
| GlgXF | 5′-TCATGCTGGGCAGAAATACG-3′ |
| GlgXR | 5′-CTGTCAAATGGCGGAGCATA-3′ |
| RpoBF | 5′-CTGTCTCAAGCCGGTTACAA-3′ |
| RpoBR | 5′-TTTCTACCAGTGCAGAGATGC-3′ |
| FtsQF | 5′-GCTGATAGGTGGTTTGCTGT-3′ |
| FtsQR | 5′-TGCACACTCTCTTGCAGAAC-3′ |
| DnaQF | 5′-AGACGATTGAAGTTCACG-3′ |
| DnaQR | 5′-ATAAAGCCGACGTCAAACGG-3′ |
For qRT-PCR analysis of glgX expression in stool V. cholerae after incubation in pond water, previously described samples were utilized (Nelson et al., 2008). RNA and cDNA were prepared as described above. Transcript levels of glgX were determined by performing qRT-PCR with gene specific primers and normalizing transcript levels to either ftsQ or dnaQ. The expression of ftsQ and dnaQ were each shown to be expressed to similar levels in stool V. cholerae after 5 hr and 24 hr incubation in pond water (Nelson et al., 2008).
Growth Curves
Growth of each strain was determined by measuring growth on a Bio-Tek microplate reader. For each strain, colonies were scraped off of LB agar plates in 1 mL PBS and the OD600 was adjusted to 0.01 (~107 cfu/mL) in 200 μl of M9-glucose with either high or low nitrogen. Bacteria were grown with aeration at 37°C for 10 hours in the microplate reader. All growth experiments were performed in triplicate.
In vitro Competition Assays
Competition experiments between WT and each mutant strain were performed by growing each strain on low nitrogen M9-glucose agar plates at 37°C. Equivalent amounts of each strain were mixed to yield a final starting OD600 of 0.01 in 3 mL low nitrogen M9 with no carbon source or in 3 mL filter-sterilized pond water. Cultures were incubated at 37°C with aeration. At the indicated times, bacteria were removed and the ratio of mutant:wt bacteria was enumerated by plating bacteria on LB agar plates containing 5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside (X-gal) and Sm. The competitive index (CI) was determined by calculating the mutant:wt corrected for the input ratio at each time-point.
Survival in Rice-water Stool
Survival in stool supernatants was assayed by growing WT, glgC1 glgC2 and glgX on low nitrogen M9-glucose agar plates at 37°C. Bacteria were prepared as above, mixed in a 1:1 ratio to yield a final OD600 = 0.25 in 200 μl filter-sterilized stool supernatants that were pre-warmed to 37°C. The stool supernatants utilized were collected as previously described (Nelson et al., 2007). Cultures were incubated statically in 96-well plates at 37°C. After 2 hr incubation, bacteria were removed and the ratio of mutant:wt bacteria and the CI was determined as described above.
In vivo Competition Assays
Competition assays were performed using the infant mouse model of infection as previously described (Tischler & Camilli, 2005). For experiments without starvation of bacteria, each strain was scraped from LB or low nitrogen M9-glucose plates into 1 mL PBS and the OD600 was adjusted to 0.01 (~107 cfu/mL). Inocula were prepared by mixing each strain with WT in a 1:1 ratio. Five-day old CD-1 mice were inoculated intragastrically with 50 μl of this mixture. The infection was allowed to proceed for 20 hr and the CI was calculated as described above. For experiments with pond passaged bacteria, each strain was grown on low nitrogen M9-glucose agar plates at 37°C to induce glycogen accumulation, resuspended in 1 mL PBS and the OD600 was adjusted to 0.1 (~108 cfu/mL) in filter-sterilized pond water. Bacteria were incubated in pond water for 3 hr or overnight at 37°C with aeration. The OD600 was determined and equivalent amounts of each pond-passaged strain were mixed at a 1:1 ratio to yield inocula as described above. In vitro competitions were performed in parallel by inoculating the pond water passaged bacteria into M9-glucose and incubating for 20 hr at 37°C with aeration. The CI of the in vitro competitions was calculated as above.
Acknowledgments
We are grateful to Karla Satchell for the gift of the unpublished strain KFV21. We thank Ernesto Munoz and Jane M. Liu for critical reading of the manuscript. A. Camilli is a Howard Hughes Medical Institute investigator. The research was supported by the National Institutes of Health R01 AI 055058.
References
- Alonso-Casajus N, Dauvillee D, Viale AM, Munoz FJ, Baroja-Fernandez E, Moran-Zorzano MT, Eydallin G, Ball S, Pozueta-Romero J. Glycogen phosphorylase, the product of the glgP Gene, catalyzes glycogen breakdown by removing glucose units from the nonreducing ends in Escherichia coli. J Bacteriol. 2006;188:5266–5272. doi: 10.1128/JB.01566-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson RA, Koiv V, Norman-Setterblad C, Pirhonen M. Role of RpoS in virulence and stress tolerance of the plant pathogen Erwinia carotovora subsp. carotovora. Microbiology. 1999;145(Pt 12):3547–3556. doi: 10.1099/00221287-145-12-3547. [DOI] [PubMed] [Google Scholar]
- Angelichio MJ, Spector J, Waldor MK, Camilli A. Vibrio cholerae intestinal population dynamics in the suckling mouse model of infection. Infect Immun. 1999;67:3733–3739. doi: 10.1128/iai.67.8.3733-3739.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger AE, Hatfull GF. Exponential-phase glycogen recycling is essential for growth of Mycobacterium smegmatis. J Bacteriol. 1999;181:6670–6678. doi: 10.1128/jb.181.21.6670-6678.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonafonte MA, Solano C, Sesma B, Alvarez M, Montuenga L, Garcia-Ros D, Gamazo C. The relationship between glycogen synthesis, biofilm formation and virulence in salmonella enteritidis. FEMS Microbiol Lett. 2000;191:31–36. doi: 10.1111/j.1574-6968.2000.tb09315.x. [DOI] [PubMed] [Google Scholar]
- Burleigh IG, Dawes EA. Studies on the endogenous metabolism and senescence of starved Sarcina lutea. Biochem J. 1967;102:236–250. doi: 10.1042/bj1020236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson JH, Whitmire WM, Crane DD, Wicke L, Virtaneva K, Sturdevant DE, Kupko JJ, 3rd, Porcella SF, Martinez-Orengo N, Heinzen RA, Kari L, Caldwell HD. The Chlamydia trachomatis plasmid is a transcriptional regulator of chromosomal genes and a virulence factor. Infect Immun. 2008;76:2273–2283. doi: 10.1128/IAI.00102-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell RR. Global climate and infectious disease: the cholera paradigm. Science. 1996;274:2025–2031. doi: 10.1126/science.274.5295.2025. [DOI] [PubMed] [Google Scholar]
- Damotte M, Cattaneo J, Sigal N, Puig J. Mutants of Escherichia coli K 12 altered in their ability to store glycogen. Biochem Biophys Res Commun. 1968;32:916–920. doi: 10.1016/0006-291x(68)90114-9. [DOI] [PubMed] [Google Scholar]
- Dauvillee D, I, Kinderf S, Li Z, Kosar-Hashemi B, Samuel MS, Rampling L, Ball S, Morell MK. Role of the Escherichia coli glgX gene in glycogen metabolism. J Bacteriol. 2005;187:1465–1473. doi: 10.1128/JB.187.4.1465-1473.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg MS, Kaper JB. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect Immun. 1991;59:4310–4317. doi: 10.1128/iai.59.12.4310-4317.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hassani RA, Benfares N, Caillou B, Talbot M, Sabourin JC, Belotte V, Morand S, Gnidehou S, Agnandji D, Ohayon R, Kaniewski J, Noel-Hudson MS, Bidart JM, Schlumberger M, Virion A, Dupuy C. Dual oxidase2 is expressed all along the digestive tract. Am J Physiol Gastrointest Liver Physiol. 2005;288:G933–942. doi: 10.1152/ajpgi.00198.2004. [DOI] [PubMed] [Google Scholar]
- Eydallin G, Moran-Zorzano MT, Munoz FJ, Baroja-Fernandez E, Montero M, Alonso-Casajus N, Viale AM, Pozueta-Romero J. An Escherichia coli mutant producing a truncated inactive form of GlgC synthesizes glycogen: further evidences for the occurrence of various important sources of ADPglucose in enterobacteria. FEBS Lett. 2007a;581:4417–4422. doi: 10.1016/j.febslet.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Eydallin G, Viale AM, Moran-Zorzano MT, Munoz FJ, Montero M, Baroja-Fernandez E, Pozueta-Romero J. Genome-wide screening of genes affecting glycogen metabolism in Escherichia coli K-12. FEBS Lett. 2007b;581:2947–2953. doi: 10.1016/j.febslet.2007.05.044. [DOI] [PubMed] [Google Scholar]
- Freter R, Smith HL, Jr, Sweeney FJ., Jr An evaluation of intestinal fluids in the pathogenesis of cholera. J Infect Dis. 1961;109:35–42. doi: 10.1093/infdis/109.1.35. [DOI] [PubMed] [Google Scholar]
- Halpern M, Broza YB, Mittler S, Arakawa E, Broza M. Chironomid egg masses as a natural reservoir of Vibrio cholerae non-O1 and non-O139 in freshwater habitats. Microb Ecol. 2004;47:341–349. doi: 10.1007/s00248-003-2007-6. [DOI] [PubMed] [Google Scholar]
- Harris GS, Michalek SM, Curtiss R., 3rd Cloning of a locus involved in Streptococcus mutans intracellular polysaccharide accumulation and virulence testing of an intracellular polysaccharide-deficient mutant. Infect Immun. 1992;60:3175–3185. doi: 10.1128/iai.60.8.3175-3185.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge-Aronis R. Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol Mol Biol Rev. 2002;66:373–395. doi: 10.1128/MMBR.66.3.373-395.2002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge-Aronis R, Fischer D. Identification and molecular analysis of glgS, a novel growth-phase-regulated and rpoS-dependent gene involved in glycogen synthesis in Escherichia coli. Mol Microbiol. 1992;6:1877–1886. doi: 10.1111/j.1365-2958.1992.tb01360.x. [DOI] [PubMed] [Google Scholar]
- Jahid IK, Silva AJ, Benitez JA. Polyphosphate stores enhance the ability of Vibrio cholerae to overcome environmental stresses in a low-phosphate environment. Appl Environ Microbiol. 2006;72:7043–7049. doi: 10.1128/AEM.00924-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janoff EN, Hayakawa H, Taylor DN, Fasching CE, Kenner JR, Jaimes E, Raij L. Nitric oxide production during Vibrio cholerae infection. Am J Physiol. 1997;273:G1160–1167. doi: 10.1152/ajpgi.1997.273.5.G1160. [DOI] [PubMed] [Google Scholar]
- Jones SA, Jorgensen M, Chowdhury FZ, Rodgers R, Hartline J, Leatham MP, Struve C, Krogfelt KA, Cohen PS, Conway T. Glycogen and maltose utilization by Escherichia coli O157:H7 in the mouse intestine. Infect Immun. 2008;76:2531–2540. doi: 10.1128/IAI.00096-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jue CK, Lipke PN. Determination of reducing sugars in the nanomole range with tetrazolium blue. J Biochem Biophys Methods. 1985;11:109–115. doi: 10.1016/0165-022x(85)90046-6. [DOI] [PubMed] [Google Scholar]
- Lee SH, Hava DL, Waldor MK, Camilli A. Regulation and temporal expression patterns of Vibrio cholerae virulence genes during infection. Cell. 1999;99:625–634. doi: 10.1016/s0092-8674(00)81551-2. [DOI] [PubMed] [Google Scholar]
- Lipp EK, Huq A, Colwell RR. Effects of global climate on infectious disease: the cholera model. Clin Microbiol Rev. 2002;15:757–770. doi: 10.1128/CMR.15.4.757-770.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo MJ, Michalski J, Martinez-Wilson H, Morin C, Hilton T, Osorio CG, Nataro JP, Tacket CO, Camilli A, Kaper JB. An in vivo expression technology screen for Vibrio cholerae genes expressed in human volunteers. Proc Natl Acad Sci U S A. 2007;104:18229–18234. doi: 10.1073/pnas.0705636104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MC, Schneider D, Bruton CJ, Chater KF, Hardisson C. A glgC gene essential only for the first of two spatially distinct phases of glycogen synthesis in Streptomyces coelicolor A3(2) J Bacteriol. 1997;179:7784–7789. doi: 10.1128/jb.179.24.7784-7789.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMeechan A, Lovell MA, Cogan TA, Marston KL, Humphrey TJ, Barrow PA. Glycogen production by different Salmonella enterica serotypes: contribution of functional glgC to virulence, intestinal colonization and environmental survival. Microbiology. 2005;151:3969–3977. doi: 10.1099/mic.0.28292-0. [DOI] [PubMed] [Google Scholar]
- Merrell DS, Butler SM, Qadri F, Dolganov NA, Alam A, Cohen MB, Calderwood SB, Schoolnik GK, Camilli A. Host-induced epidemic spread of the cholera bacterium. Nature. 2002;417:642–645. doi: 10.1038/nature00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Zorzano MT, Alonso-Casajus N, Munoz FJ, Viale AM, Baroja-Fernandez E, Eydallin G, Pozueta-Romero J. Occurrence of more than one important source of ADPglucose linked to glycogen biosynthesis in Escherichia coli and Salmonella. FEBS Lett. 2007;581:4423–4429. doi: 10.1016/j.febslet.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Nelson EJ, Chowdhury A, Flynn J, Schild S, Bourassa L, Shao Y, LaRocque RC, Calderwood SB, Qadri F, Camilli A. Transmission of Vibrio cholerae is antagonized by lytic phage and entry into the aquatic environment. PLoS Pathog. 2008;4:e1000187. doi: 10.1371/journal.ppat.1000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson EJ, Chowdhury A, Harris JB, Begum YA, Chowdhury F, Khan AI, Larocque RC, Bishop AL, Ryan ET, Camilli A, Qadri F, Calderwood SB. Complexity of rice-water stool from patients with Vibrio cholerae plays a role in the transmission of infectious diarrhea. Proc Natl Acad Sci U S A. 2007;104:19091–19096. doi: 10.1073/pnas.0706352104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen AT, Dolganov NA, Otto G, Miller MC, Wu CY, Schoolnik GK. RpoS controls the Vibrio cholerae mucosal escape response. PLoS Pathog. 2006;2:e109. doi: 10.1371/journal.ppat.0020109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa N, Tzeng CM, Fraley CD, Kornberg A. Inorganic polyphosphate in Vibrio cholerae: genetic, biochemical, and physiologic features. J Bacteriol. 2000;182:6687–6693. doi: 10.1128/jb.182.23.6687-6693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrou JL, Francois J. A simplified procedure for a rapid and reliable assay of both glycogen and trehalose in whole yeast cells. Anal Biochem. 1997;248:186–188. doi: 10.1006/abio.1997.2138. [DOI] [PubMed] [Google Scholar]
- Preiss J, Romeo T. Physiology, biochemistry and genetics of bacterial glycogen synthesis. Adv Microb Physiol. 1989;30:183–238. doi: 10.1016/s0065-2911(08)60113-7. [DOI] [PubMed] [Google Scholar]
- Preiss J, Romeo T. Molecular biology and regulatory aspects of glycogen biosynthesis in bacteria. Prog Nucleic Acid Res Mol Biol. 1994;47:299–329. doi: 10.1016/s0079-6603(08)60255-x. [DOI] [PubMed] [Google Scholar]
- Reidl J, Klose KE. Vibrio cholerae and cholera: out of the water and into the host. FEMS Microbiol Rev. 2002;26:125–139. doi: 10.1111/j.1574-6976.2002.tb00605.x. [DOI] [PubMed] [Google Scholar]
- Sack DA, Sack RB, Chaignat CL. Getting serious about cholera. N Engl J Med. 2006;355:649–651. doi: 10.1056/NEJMp068144. [DOI] [PubMed] [Google Scholar]
- Sambou T, Dinadayala P, Stadthagen G, Barilone N, Bordat Y, Constant P, Levillain F, Neyrolles O, Gicquel B, Lemassu A, Daffe M, Jackson M. Capsular glucan and intracellular glycogen of Mycobacterium tuberculosis: biosynthesis and impact on the persistence in mice. Mol Microbiol. 2008;70:762–774. doi: 10.1111/j.1365-2958.2008.06445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild S, Tamayo R, Nelson EJ, Qadri F, Calderwood SB, Camilli A. Genes induced late in infection increase fitness of Vibrio cholerae after release into the environment. Cell Host Microbe. 2007;2:264–277. doi: 10.1016/j.chom.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibold G, Dempf S, Schreiner J, Eikmanns BJ. Glycogen formation in Corynebacterium glutamicum and role of ADP-glucose pyrophosphorylase. Microbiology. 2007;153:1275–1285. doi: 10.1099/mic.0.2006/003368-0. [DOI] [PubMed] [Google Scholar]
- Seibold GM, Eikmanns BJ. The glgX gene product of Corynebacterium glutamicum is required for glycogen degradation and for fast adaptation to hyperosmotic stress. Microbiology. 2007;153:2212–2220. doi: 10.1099/mic.0.2006/005181-0. [DOI] [PubMed] [Google Scholar]
- Senanayake SD, Brian DA. Precise large deletions by the PCR-based overlap extension method. Mol Biotechnol. 1995;4:13–15. doi: 10.1007/BF02907467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slauch J, Taylor R, Maloy S. Survival in a cruel world: how Vibrio cholerae and Salmonella respond to an unwilling host. Genes Dev. 1997;11:1761–1774. doi: 10.1101/gad.11.14.1761. [DOI] [PubMed] [Google Scholar]
- Spatafora GA, Sheets M, June R, Luyimbazi D, Howard K, Hulbert R, Barnard D, el Janne M, Hudson MC. Regulated expression of the Streptococcus mutans dlt genes correlates with intracellular polysaccharide accumulation. J Bacteriol. 1999;181:2363–2372. doi: 10.1128/jb.181.8.2363-2372.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange RE. Bacterial “glycogen” and survival. Nature. 1968;220:606–607. doi: 10.1038/220606a0. [DOI] [PubMed] [Google Scholar]
- Tischler AD, Camilli A. Cyclic diguanylate regulates Vibrio cholerae virulence gene expression. Infect Immun. 2005;73:5873–5882. doi: 10.1128/IAI.73.9.5873-5882.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watnick P, Kolter R. Biofilm, city of microbes. J Bacteriol. 2000;182:2675–2679. doi: 10.1128/jb.182.10.2675-2679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz FH, Schoolni GK. Role of rpoS in stress survival and virulence of Vibrio cholerae. J Bacteriol. 1998;180:773–784. doi: 10.1128/jb.180.4.773-784.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]