

Abstract
AIM: To investigate the effect of vascular endothelial growth factor (VEGF) transfection on hepatic sinusoidal capillarization.
METHODS: Enhanced green fluorescent protein (EGFP)/VEGF transfection was confirmed by immunofluorescence microscopy and immunohistoche-mistry both in primary hepatocytes and in normal liver. Cirrhotic rats were generated by thioacetamide (TAA) administration and then divided into a treatment group, which received injections of 400 μg of plasmid DNA encoding an EGFP-VEGF fusion protein, and a blank group, which received an equal amount of normal saline through the portal vein. The portal vein pressure was measured in the normal and cirrhotic state, in treated and blank groups. The average number of fenestrae per hepatic sinusoid was determined using transmission electron microscopy (TEM), while the relative abundance of VEGF transcripts was examined by Gene array.
RESULTS: Green fluorescent protein was observed in the cytoplasms of liver cells under immunofluorescence microscopy 24 h after transfection with EGFP/VEGF plasmid in vitro. Staining with polyclonal antibodies against VEGF illustrated that hepatocytes expressed immunodetectable VEGF both in vitro and in vitro. There were significant differences in the number of fenestrae and portal vein pressures between normal and cirrhotic rats (7.40 ± 1.71 vs 2.30 ± 1.16 and 9.32 ± 0.85 cmH2O vs 17.92 ± 0.90 cmH2O, P < 0.01), between cirrhotic and treated rats (2.30 ± 1.16 cmH2O vs 4.60 ± 1.65 and 17.92 ± 0.90 cmH2O vs 15.52 ± 0.93 cmH2O, P < 0.05) and between the treatment group and the blank group (4.60 ± 1.65 cmH2O vs 2.10 ± 1.10 cmH2O and 15.52 ± 0.93 cmH2O vs 17.26 ± 1.80 cmH2O, P < 0.05). Gene-array analysis revealed that the relative abundance of transcripts of VEGF family members decreased in the cirrhotic state and increased after transfection.
CONCLUSION: Injection of a plasmid encoding VEGF through the portal vein is an effective method to induce the formation of fenestrae and decrease portal vein pressure in cirrhotic rats. Therefore, it may be a good choice for treating hepatic cirrhosis and portal hypertension.
Keywords: Liver cirrhosis, Hepatic sinusoid capillarization, Fenestrae, Vascular endothelial growth factor, Transmission electrical microscopy, Ultrastructure, Gene array
INTRODUCTION
Fenestrae are described as membrane-bound cytoplasmic holes with an average diameter of about 110 nm in transmission electrical microscopy (TEM) by Wisse in 1970[1,2]. Liver sinusoidal endothelial cells (LSECs) possess open fenestrae that perforate the hepatic endothelial lining, but lack a basal lamina. Fenestrae, vesicles and channels together control the bulk of trans-endothelial transport between blood and tissues[3]. Structural integrity of the fenestrated sinusoidal liver endothelium is believed to be essential for the maintenance of the normal exchange of fluids, solutes, particles and metabolites between the sinusoidal blood and hepatocytes[4,5]. Although there are various causes and morphologies of hepatic cirrhosis, all forms of cirrhosis are characterized by a defenestrated sinusoidal endothelium and the presence of a subendothelial basement membrane[6–9]. It has been demonstrated that the disappearance of the normal filtration barrier in cirrhotic livers results in an impaired bidirectional exchange between the sinusoidal blood and parenchymal cells[10]. As a result, capillarization of the sinusoidal endothelium may be a major contributor to hepatic failure in patients with cirrhosis.
Vascular endothelial growth factor (VEGF), which plays a role in regulating vasculogenesis, induces angiogenesis and endothelial cell proliferation. In recent years, it has been proven that VEGF is relevant to the increased number of fenestrae and endothelial permeability in endothelial cells[10–13] and renal glomerulus[14]. Injection of VEGF-D plasmid into both normal and ischemic rat liver resulted in an increased number of new capillaries around hepatic sinuses[15]. Many studies have shown that, in the state of hepatic fibrosis and cirrhosis, both in patients and experimental models, VEGF is increased with harmful effects[16,17]. By contrast, other studies have shown decreased expression of VEGF in cirrhotic patients, and suggested that VEGF makes a helpful contribution[18,19]. After hepatectomy in cirrhotic rats, VEGF was found to effectively promote liver regeneration[20,21]. Moreover, human urokinase-type plasminogen activator gene administration via an adenoviral (Ad)-vector induced cirrhosis regression and ameliorated hepatic dysfunction with up-regulation of VEGF in a model of experimental liver cirrhosis[22,23].
Because defenestration and basement membrane formation result in a disordered exchange between the sinusoidal blood and hepatocytes, it is necessary to restore the function of liver sinusoidal endothelial cells in order to reverse cirrhosis. VEGF provides the perfect means to achieve this, because of it promotes fenestration and permeability. On account of these ideas, we studied the effects of VEGF transfection in cirrhotic rat livers.
MATERIALS AND METHODS
EGFP/VEGF transfection of hepatocytes
EGFP/VEGF was obtained from the Medical School of Shandong University (Jinan, China). Human VEGF-D cDNA was inserted into pEGFP-N1 plasmid between the constitutive cytomegalovirus promoter (pCMV) and enhanced green fluorescent protein (EGFP) to produce a plasmid encoding an EGFP-VEGF fusion protein (EGFP/VEGF plasmid), which was used as a tool to express VEGF consistently. EGFP was used to detect expression of the plasmid, and the expression of VEGF was detected by immunohistochemistry.
Primary hepatocytes were isolated from the livers of male Wistar rats (150-200 g) by collagenase perfusion as previously described[24]. Cells were plated on collagen-coated 6-well plates at a density of 3 × 105 cells/well in Williams’ medium E supplemented with 10% fetal bovine serum, 2 mmol/L L-glutamine, 1% penicillin/streptomycin, and 100 nmol/L dexamethasone. The cells were incubated in 5% CO2 at 37°C to facilitate attachment, and the medium was exchanged after 4 h with serum-free Williams’ medium E supplemented with 2 mmol/L L-glutamine, 1% penicillin/streptomycin, and 100 nmol/L dexamethasone. After overnight incubation in 5% CO2 at 37°C, the medium was exchanged with serum-free Williams’ medium E supplemented with 2 mmol/L L-glutamine and 1% penicillin/streptomycin. The EGFP/VEGF plasmid (50 μg) was added to the culture medium and the cells were incubated for 24 h. We investigated 5 dishes for each treatment in the present study.
Immunofluorescence microscopy and immunohistochemistry
Immunofluorescence microscopy was used to visualize the distribution of EGFP in hepatocytes. At the same time, immunocytochemical staining of VEGF was performed. Cultured hepatocytes were washed with phosphate buffered saline (PBS) 3 times, and fixed with acetone at room temperature for 10 min. Then, they were blocked with goat serum and permeabilized for 30 min with 0.1% Triton X-100 in PBS containing 1% bovine serum albumin (BSA). The cells were incubated overnight with rabbit anti-human VEGF polyclonal antibody (Santa Cruz) at a 1:50 dilution at 4°C. Subsequently, they were incubated with tetramethylrhodamine isomer R1-conjugated swine anti-rabbit IgG (Santa Cruz) at room temperature for 1 h. The distribution of VEGF was visualized by light microscopy.
Transfection of normal liver
Twenty healthy male Wistar rats, weighing 200-220 g, bought from the experimental animal center of Shandong Agricultural Research Center (Jinan, China) were divided equally into two groups. In the study group, each rat received an injection of 400 μg of the EGFP/VEGF plasmid through the portal vein. In the control group, each rat received an equal amount of normal saline in the same manner. All rats were sacrificed eight days after the operation. Liver samples were collected and fixed in 10% neutral-buffered formalin, embedded in paraffin and cut into 4-6 μm sections.
Immunohistochemistry
Immunohistochemistry was performed according to Hong-Lei Weng[25]. The integral score method was used to evaluate the positive cell distribution and intensity[26]. First, a proportion score was assigned, which represented the estimated proportion of positive-staining cells (0, none; 1, 0 to 1/100; 2, 1/100 to 1/10; 3, 1/10 to 1/3; 4, 1/3 to 2/3; and 5, 2/3). Next, an intensity score was assigned, which represented the average intensity of positive cells (0, none; 1, weak, 2, intermediate; and 3, strong). The proportion and intensity scores were then added to obtain a total score, which ranged from 0 to 8. Then the score was compared between the study and control groups.
Transfection of cirrhotic liver
The portal vein pressures of 40 normal Wistar rats were measured and hepatic specimens were taken. Then, the rats received an oral administration of 0.03% thioacetamide (TAA) solution instead of feed water. After 5 wk, the concentration of TAA was increased to 0.04%. Ten wk later, 26 cirrhotic rats were identified and randomly divided into a treatment group and a blank group. The portal vein pressures of these rats were measured and cirrhotic liver specimens were obtained. Then, 400 μg of the VEGF/EGFP plasmid was infused through the portal veins of rats in the treatment group, while an equal amount of normal saline was given through the portal vein to rats in the blank group. After 2 wk, portal vein pressures were measured again; then, all rats were sacrificed and liver samples were collected. Comparisons of portal vein pressure were made between the normal and cirrhotic, treated and untreated rats. A randomized controlled trial was carried out to compare the treatment group and the blank group.
Transmission electron microscopy
Fresh specimens were first fixed in 3% glutaral, and then fixed in 1% glutaral after being washed with PBS 3 times for 60 min in total. The samples were dehydrated with an alcohol gradient, embedded in EPON812 epoxy resin, and then cut into 50-nm sections with an Ultrathin microtome. After being dyed with uranyl acetate and lead citrate for 30 min, the sections were observed under transmission electronic microscope (JEM-1200EX, Japan). Ten hepatic sinusoids with a diameter of 2-3 μm were randomly selected, and the average number of fenestrae per hepatic sinusoid in each state and group was determined. Valid fenestrae ran through LSECs.
Gene array
Angiogenesis microarrays were obtained from SuperArray Bio.Co. (Catalog No. ORN-024). Fresh specimens were put into 4°C of RNA later and incubated for one night. The experiment was performed according to the Oligo GEArray assay protocol. Briefly, total RNA was extracted from tissue using TRIzol reagent (Invitrogen Life Technologies, USA). The quantity and purity of RNA were estimated by measuring A260 and A280. A total of 3 μg of RNA was used to synthesize cDNA. The cRNA was then labeled by Biotin-16-dUTP (Roche Cat. No. 1-093-070) and amplified using a TrueLabeling-AMPTM linear RNA amplification kit. The membranes were hybridized with denatured cDNA probe and processed for chemiluminescent detection on X-ray film, and images were acquired using a flatbed desktop scanner. Subsequently, images of spots were converted into numerical data using the free ScanAlyze software and the raw data was saved as a Microsoft Excel file. All raw signal intensities were corrected for background signal by subtracting the minimum value to avoid the appearance of negative numbers, and were also normalized to the level of a housekeeping gene. These corrected, normalized signals were used to estimate the relative abundance of particular transcripts.
Statistical analysis
Results are expressed as mean ± SD, and statistical analysis was carried out using the Student’s t-test and Spearman’s rank correlation coefficient for paired data. A P value of less than 0.05 was considered to be significant.
RESULTS
VEGF expression in vitro and in vivo
Twenty-four hours after transfection of cultured hepatocytes with the EGFP/VEGF plasmid, green fluorescent protein was observed in the cytoplasms of liver cells under immunofluorescence microscopy, revealing expression of the plasmid (Figure 1). At the same time, staining of hepatocytes with polyclonal antibodies against VEGF illustrated that these cells expressed immunodetectable VEGF (Figure 2). Moreover, the transfection of liver cells following injection of the EGFP/VEGF plasmid into the portal veins of normal rats in vivo was also successful, because a considerable amount of VEGF was identified in the study group, but little was seen in the control group (Figure 3). VEGF was mainly observed in the cytoplasms of hepatocytes and some endothelial cells. The stain integral scores of the study group and control group were 6.0 ± 1.63 and 3.7 ± 2.31 respectively, allowing these groups to be differentiated from each other distinctively (Student’s t test, t = 2.74 > 2.62, P < 0.05) (Figure 3).
Figure 1.
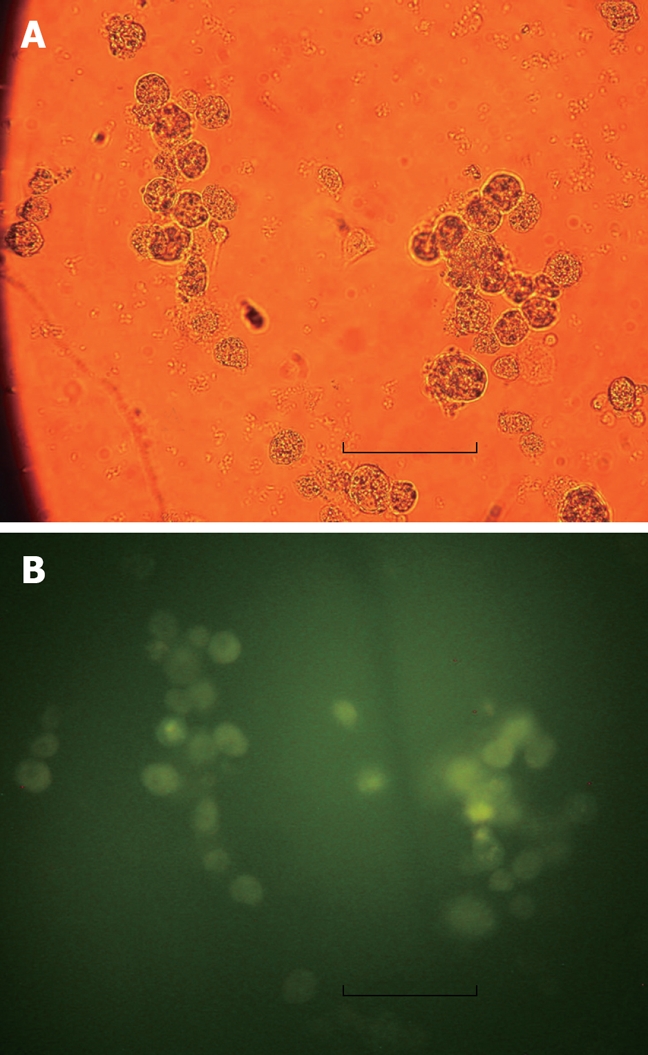
Hepatocytes incubated with EGFP/VEGF plasmid for 24 h. A: Light microscopy: no green fluorescent protein was found. Bar denotes 30 μm; B: Immunofluorescence microscopy: green fluorescent protein expression. Bar denotes 30 μm.
Figure 2.
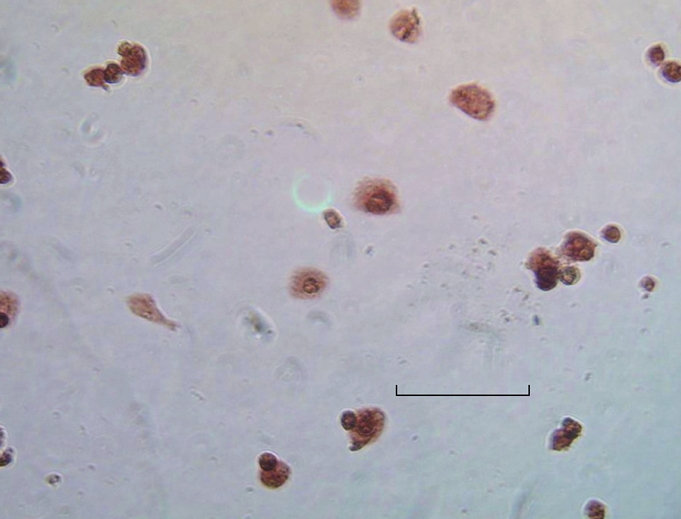
Immunohistochemistry: Positively stained hepatocytes for EGFP/VEGF. Bar denotes 30 μm.
Figure 3.

Immunohistochemistry × 200. A: Study group injected with EGFP/VEGF plasmid, VEGF-positive hepatocytes × 200; B: Control group injected with NS, VEGF-negative hepatocytes.
Fenestrae and portal vein pressures
A comparison was carried out between normal, cirrhotic and treated rats. The number of fenestrae per LSEC in cirrhotic and normal rats was 2.30 ± 1.16 and 7.40 ± 1.71, respectively (Table 1). The decrease in cirrhotic rats was significant (Student’s t-test, t = -7.965 < -6.548, P < 0.01). The portal vein pressures were 17.92 ± 0.90 in cirrhotic rats and 9.32 ± 0.85 in normal rats (Table 1), revealing an obvious difference (Student’s t-test, t = 27.32 > 9.30, P < 0.01).
Table 1.
The number of fenestrae and portal vein pressures in different groups (n = 11)
| Group | The number of fenestrae | Portal vein pressures (mean ± SD) (cmH2O) |
| Normal state | 7.40 ± 1.71d | 9.32 ± 0.85d |
| Cirrhotic state | 2.30 ± 1.16 | 17.92 ± 0.90 |
| Treated group | 4.60 ± 1.65a | 15.52 ± 0.93a |
| Blank group | 2.10 ± 1.10 | 17.26 ± 1.80 |
Paired samples t test. Significant differences are shown both in fenestrae and pressure.
P < 0.05 vs Cirrhotic state;
P < 0.01 vs Blank group.
After transfection with the EGFP/VEGF plasmid, the number of fenestrae in the treated group was 4.60 ± 1.65 (Table 1), representing a significant increase compared with cirrhotic rats (Student’s t-test, t = -4.14 < -3.56, P < 0.05). The portal vein pressure in the treated group was 15.52 ± 0.93 (Table 1), representing a significant decrease compared with cirrhotic rats (Student’s t-test, t = 6.04 > 3.29, P < 0.05).
A randomized controlled trial was performed to compare the treated group and the blank group. There was an obvious difference in the number of fenestrae (4.60 ± 1.65 vs 2.10 ± 1.10, Student’s t-test, t = 4.04 > 3.90, P < 0.05) and in portal vein pressure (15.52 ± 0.93 vs 17.26 ± 1.80, Student’s t-test, t = -3.97 < -3.46, P < 0.05), but there was no significant difference between the cirrhotic group and the blank group in these measures.
Ultrastructural changes in cirrhotic rats after EGFP/VEGF plasmid transfection
Transmission electron microscopy revealed hepatocellular apoptosis in rats with TAA-induced cirrhosis (Figure 4B); moreover, the fenestrae of endothelial cells disappeared and a basement membrane appeared (Figure 5C). Cell conjunction between hepatocytes was destroyed and particles of bilirubin overflowed into the cytoplasm of hepatocytes and LECs, even into the hepatic sinusoid (Figure 6B). The microvilli of hepatocytes in the space of Disse and the cholangiole were ablated in cirrhotic rats (Figure 5C and Figure 6B). However, this morphology changed after transfection with the EGFP/VEGF plasmid. The fenestrae, cell conjunctions, and microvilli of hepatocytes were restored, the basement membrane disappeared and cell apoptosis decreased. Newborn capillaries formed by a single liver endothelial cell emerged (Figure 6D).
Figure 4.
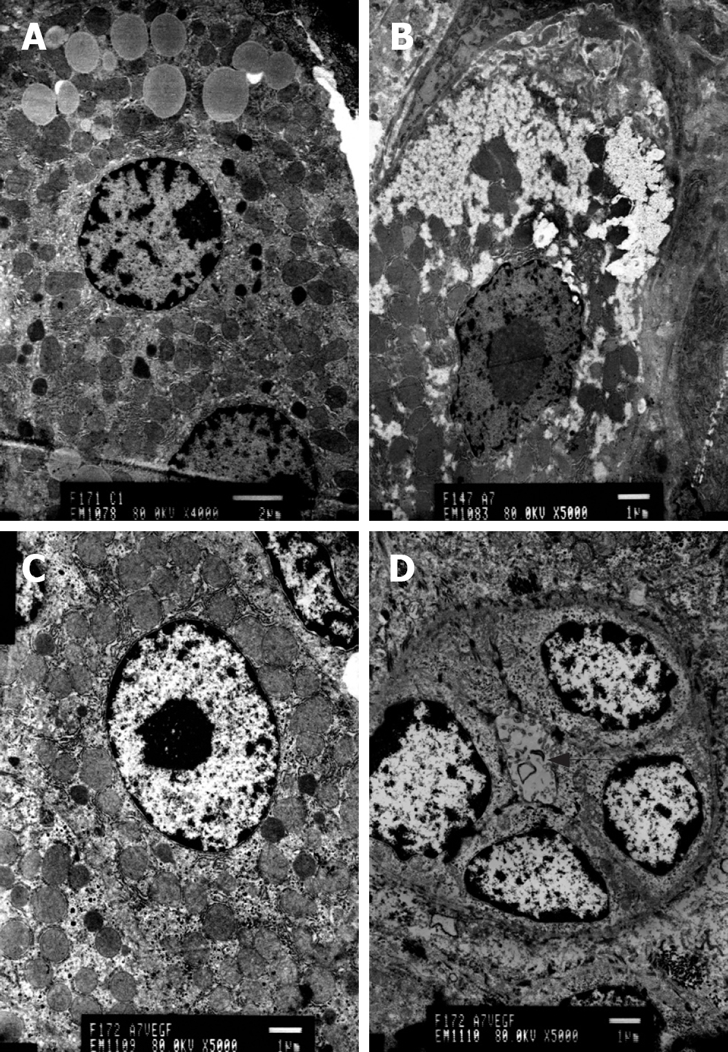
TEM. A: Normal hepatocytes, Bar denotes 2 μm; B: Cirrhotic state, hepatocyte apoptosis, Bar denotes 1 μm; C: Cirrhotic rats injected with the EGFP/VEGF plasmid, hepatocyte apoptosis decreased, Bar denotes 1 μm; D: Cirrhotic rats injected with EGFP/VEGF plasmid, Regenerated hepatocytes and cholangiole arrowhead, Bar denotes 1 μm.
Figure 5.

TEM. A: Normal state, LSECs and fenestrae (arrowhead). Bar denotes 200 nm; B: Normal state. Cell conjuction between LECs (small arrowhead) and microvilli of hepatocytes (large arrowhead). Bar denotes 100 nm; C: Cirrhotic state. Fenestrae, cell conjuction and microvilli of hepatocytes disappeared, and a basement membrane appeared (arrowhead). Bar denotes 500 nm; D: VEGF treated group. Fenestrae (small arrowhead), microvilli (large arrowhead) and cell conjunctions between LECs (triangle) appeared, Bar denotes 200 nm.
Figure 6.
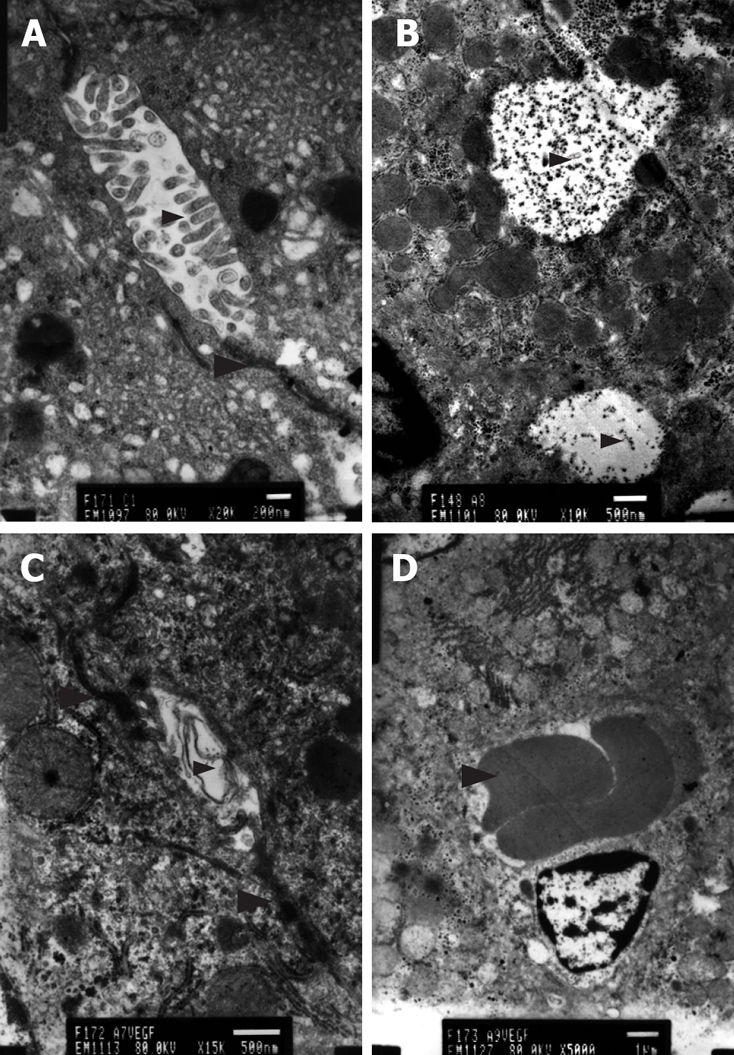
TEM. A: Normal state. Cholangiole, microvilli(small arrowhead) and cell conjuction between hepatocytes (large arrowhead). Bar denotes 200 nm; B: Cirrhotic state. Microvillus, cell conjuction disappeared and particles of bilirubin (arrowhead) overflowed. Bar denotes 500 nm; C: Treated group. Microvilli (small arrowhead) and cell conjunctions (large arrowhead) appeared and bilirubin overflow diminished. Bar denotes 500 nm; D: Treated group. Newborn capillary (arrowhead). Bar denotes 1 μm.
Gene array
A total of 96 genes involved in angiogenesis were identified on the microarray (Figure 7 and Table 2). The levels of 32 genes were decreased by more than 50% in cirrhotic rats, and only 8 genes were enhanced by two-fold or greater compared with normal rats. After EGFP/VEGF transfection, 56 genes were increased by two-fold or greater in treated rats; and only one gene was decreased by more than 50% in comparison with cirrhotic rats. In cirrhotic rats, the expression levels of members of the VEGF family were decreased, and the expression levels of members of the VEGF receptor family were increased compared with the levels in normal liver. However, after administration of VEGF, expression of VEGF family members increased and that of receptor family members decreased. Furthermore, the level of VEGF-D was increased by 22-fold, while levels of VEGF, VEGF-B and VEGF-C were increased by more than 2-fold after EGFP/VEGF transfection (Table 3).
Figure 7.

Representative expression profile of angiogenesis-related genes in the liver. The gene microarray included angiogenesis-related genes (1-96), negative control genes (97-99), blank controls (100-102), positive control genes (glyceraldehyde-3-phosphate dehydrogenase (103, 104), cyclophilin A (105-108), ribosomal protein L13a (109, 110) and b-actin (111,112). A: Normal state; B: Cirrhotic state; C: VEGF treated group.
Table 2.
Angiogenesis-related genes on the angiogenesis microarray
| Position | Description | Gene Name |
| 1 | A disintegrin-like and metalloprotease (reprolysin type) with thrombospondin type I motif, 1 | Adamts1 |
| 2 | Mus musculus a disintegrin-like and metalloprotease (reprolysin type) with thrombospondin type 1 mot | Adamts8 |
| 3 | Angiopoietin | Angiopoietin 1 |
| 4 | Mus musculus angiopoietin 2 (Agpt2) | Angiopoietin2 |
| 5 | Angiogenin | ANG |
| 6 | CD36 antigen | CD36 |
| 7 | Cadherin 5 | Cadherin 5 |
| 8 | Chromogranin A | Chromogranin A |
| 9 | Procollagen, type XVIII, alpha 1 | COL18A1 |
| 10 | Colony stimulating factor 3 (granulocyte) | G-CSF |
| 11 | Connective tissue growth factor | Fisp12 |
| 12 | Endothelial differentiation sphingolipid G-protein-coupled receptor 1 | Edg1 |
| 13 | Mus musculus ephrin A2 (Efna2) | Ephrin A2 |
| 14 | Ephrin A5 | Ephrin A receptor |
| 15 | Ephrin B2 | Ephrin B2 |
| 16 | Epidermal growth factor | EGF |
| 17 | Epidermal growth factor receptor | EGFR |
| 18 | Endoglin | Endoglin |
| 19 | Eph receptor B4 | Ephrin B4 |
| 20 | V-erb-b2 erythroblastic leukemia viral oncogene homolog 2, neuro/glioblastoma derived oncogene homolog (avian) | Neu/HER2 |
| 21 | E26 avian leukemia oncogene 1, 5’ domain | Ets-1 |
| 22 | Mus musculus coagulation factor II (F2) | Prothrombin kringle-1 |
| 23 | Fibroblast growth factor 1 | aFGF |
| 24 | Fibroblast growth factor 16 | FGF16 |
| 25 | Fibroblast growth factor 2 | bFGF |
| 26 | Fibroblast growth factor 4 | FGF4 |
| 27 | Fibroblast growth factor 6 | FGF6 |
| 28 | Fibroblast growth factor 7 | FGF7/KGF |
| 29 | Fibroblast growth factor receptor 1 | FLG |
| 30 | Fibroblast growth factor receptor 3 | FGFR3 |
| 31 | Fibroblast growth factor receptor 4 | FGFR4 |
| 32 | C-fos induced growth factor | VEGF-D/FIGF |
| 33 | Kinase insert domain protein receptor | VEGFR2/FLK 1 |
| 34 | FMS-like tyrosine kinase 1 | VEGFR |
| 35 | Mouse fibronectin (FN) mRNA | Fn1 |
| 36 | Chemokine (C-X-C motif) ligand 1 | Gro1 |
| 37 | Hepatocyte growth factor | HGF |
| 38 | Hypoxia inducible factor 1, alpha subunit | Hif1a |
| 39 | Inhibitor of DNA binding 1 | ID1 |
| 40 | Inhibitor of DNA binding 3 | ID3 |
| 41 | Interferon alpha family, gene 1 | IFNA1 |
| 42 | Interferon beta, fibroblast | IFN-b1 |
| 43 | Interferon gamma | IFN r |
| 44 | Insulin-like growth factor 1 | IGF-1 |
| 45 | Interleukin 10 | IL-10 |
| 46 | Interleukin 12A | IL-12A |
| 47 | Integrin alpha 5 (fibronectin receptor alpha) | Integrin a5 |
| 48 | Integrin alpha V | CD51 |
| 49 | Integrin beta 3 | CD61 |
| 50 | MAD homolog 1 (Drosophila) | Smad1 |
| 51 | Midkine | Midkine |
| 52 | Matrix metalloproteinase 2 | Gelatinase A |
| 53 | Matrix metalloproteinase 9 | Gelatinase B |
| 54 | Macrophage scavenger receptor 1 | SR-A |
| 55 | Nitric oxide synthase 3, endothelial cell | NOS3 |
| 56 | Neuropilin | Neuropilin |
| 57 | Platelet derived growth factor, alpha | PDGF a |
| 58 | Platelet derived growth factor, B polypeptide | PDGF b |
| 59 | Platelet derived growth factor receptor, alpha polypeptide | PDGFRa |
| 60 | Platelet derived growth factor receptor, beta polypeptide | PDGFRb |
| 61 | Platelet/endothelial cell adhesion molecule | PECAM1 |
| 62 | Chemokine (C-X-C motif) ligand 4 | PF4 |
| 63 | Placental growth factor | Placental growth factor |
| 64 | Plasminogen activator, urokinase | PLAU |
| 65 | Prostaglandin-endoperoxide synthase 1 | PTGS1 |
| 66 | Prostaglandin-endoperoxide synthase 2 | Cox-2 |
| 67 | Pleiotrophin | PTN |
| 68 | Restin (Reed-Steinberg cell-expressed intermediate filament-associated protein) | Restin |
| 69 | Chemokine (C-C motif) ligand 2 | Scya2 |
| 70 | Serine (or cysteine) proteinase inhibitor, clade B, member 5 | Maspin |
| 71 | Serine (or cysteine) proteinase inhibitor, clade E, member 1 | PAI-1 |
| 72 | Serine (or cysteine) proteinase inhibitor, clade B, member 2 | PAI-2 |
| 73 | Serine (or cysteine) proteinase inhibitor, clade F), member 1 | Pedf |
| 74 | Secreted acidic cysteine rich glycoprotein | BM-40 |
| 75 | Secreted phosphoprotein 1 | Osteopontin |
| 76 | Endothelial-specific receptor tyrosine kinase | Tie-2 |
| 77 | Transforming growth factor alpha | TGF-a |
| 78 | Transforming growth factor, beta 1 | TGFb1 |
| 79 | Transforming growth factor, beta 2 | TGF b2 |
| 80 | Transforming growth factor, beta 3 | TGF b3 |
| 81 | Transforming growth factor, beta receptor I | ALK-5 |
| 82 | Transforming growth factor, beta receptor II | TGFbR2 |
| 83 | Transforming growth factor, beta receptor III | Betaglycan |
| 84 | Thrombospondin 1 | THBS1 |
| 85 | Thrombospondin 2 | THBS2 |
| 86 | Mus musculus thrombospondin 3 (Thbs-3) | THBS3 |
| 87 | Mus musculus thrombospondin-4 mRNA | THBS4 |
| 88 | Tyrosine kinase receptor 1 | Tie1 |
| 89 | Tissue inhibitor of metalloproteinase 1 | Timp |
| 90 | Tissue inhibitor of metalloproteinase 2 | TIMP2 |
| 91 | Tenascin C | Tenascin C |
| 92 | Tumor necrosis factor | TNFa |
| 93 | Vascular cell adhesion molecule 1 | VCAM-1 |
| 94 | Vascular endothelial growth factor A | VEGF/VEGI |
| 95 | Vascular endothelial growth factor B | VEGF-B |
| 96 | Vascular endothelial growth factor C | VEGF-C |
Table 3.
Relative abundance of transcripts of VEGF and receptor family members
| Gene | Normal state | Cirrhotic state | Treated group | Cirrhotic/normal | Treated/cirrhotic |
| VEGF-D/FIGF | 5.790E-2 | 1.337E-2 | 2.936E-1 | 0.2309 | 21.950 |
| VEGF-C | 1.700E-1 | 1.099E-1 | 2.807E-1 | 0.6467 | 2.5530 |
| VEGF-B | 8.275E-2 | 5.057E-2 | 4.079E-1 | 0.6111 | 8.0650 |
| VEGF/VEGI | 8.344E-3 | 8.914E-4 | 2.536E-3 | 0.1068 | 2.8450 |
| VEGFR | 2.054E+0 | 2.368E+0 | 2.237E+0 | 1.1530 | 0.9448 |
| VEGFR2/FLK1 | 1.892E-2 | 1.586E-1 | 1.338E-1 | 8.3800 | 0.8438 |
DISCUSSION
VEGF-D was first cloned by Yamada in 1997 from a human lung cDNA library[27]. VEGF-D binds VEGF receptor 2 (VEGF R2/Flk-1/KDR) and VEGF R3 (Flt-4)[28]. VEGF R2 and VEGF R3 are localized to vascular and lymphatic endothelial cells and are involved in signaling mediating angiogenesis and lymphangiogenesis[29]. There are numerous techniques and approaches that have been investigated for gene transfer to the liver. For gene therapy of hepatic diseases in animal experiments, exogenous genes were usually delivered to the liver through the portal vein and bile duct[30–33]. In our studies, EGFP/VEGF plasmid injection through portal vein was found to be an effective method.
The EGFP/VEGF plasmid was specifically designed for mammalian expression, and was constructed using a human VEGF-D cDNA and pEGFP-N1 plasmid. In the first stage of our studies, we tried to ascertain its expression efficiency, both in vitro and in vivo. In vitro, enhanced green fluorescent protein was observed in the cytoplasm of liver cells under immunofluorescence microscopy. At the same time the staining of hepatocytes with polyclonal antibodies against VEGF illustrated that the cells expressed immunodetectable VEGF. In vivo, transfection resulted in a comparatively high expression of VEGF protein. These findings demonstrate the efficient transfection of liver cells with the EGFP/VEGF plasmid in vitro and in vivo.
VEGF is an important regulator of angiogenesis and vascular permeability, whose expression in the adult is correlated with low permeability of the blood-brain barrier endothelium and high permeability of the fenestrated glomerular endothelium[14,34]. Fenestrae are critical for the maintenance of the high hydraulic conductivity of the glomerular capillary wall, and their loss results in a reduction in the glomerular filtration rate[35]. Defenestration is an early event in the pathogenesis of cirrhosis, and precedes the initiation of fibrosis. Sinusoidal capillarization is believed to be important in the initiation of perisinusoidal fibrosis by altering retinol metabolism[7]. In rats with TAA-induced cirrhosis, the fenestrae of endothelial cell disappeared and a basement membrane appeared. As a result, the microvilli of hepatocytes in the space of Disse were ablated, indicating that material exchange between hepatocytes and hepatic sinusoidal blood failed. Hepatocytes could not obtain the necessary nutrition and eliminate metabolites, which resulted in cell apoptosis. Cell conjunction between hepatocytes was damaged and particles of bilirubin overflowed into the cytoplasm of hepatocytes and endothelial cells, and even into the hepatic sinusoid, resulting in jaundice.
VEGF can promote fenestration and permeability of liver sinusoidal endothelial cells; therefore, it can improve the exchange between hepatic sinusoidal blood and hepatocytes, which argues for the development of VEGF gene therapy for cirrhosis. Roberts and Palade[36,37] showed that topical administration of VEGF-165 induced fenestrations in continuous microvascular endothelia of muscle and skin, and that tumor neovasculature induced by VEGF is fenestrated. Epithelial cells stably transfected with VEGF cDNA secreted a high level of VEGF and induced a seven- or eight-fold increase in the number of fenestrae and fused clustered vesicles in co-cultured endothelial cells, thus providing direct evidence of a role for VEGF in fenestrae induction. Endothelial cells pretreated with VEGF developed fenestrae and showed increased endothelial permeability[10]. However, to date, there have been few reports on using VEGF to heal cirrhosis in vivo. In our study, after injection of EGFP/VEGF plasmid into cirrhotic rats through the portal vein, the number of fenestrae increased obviously, accompanied with a decrease in portal vein pressure. Hepatocytes and microvillus regeneration demonstrated that the material exchange between hepatocytes and hepatic sinusoidal blood recovered. Furthermore, cell conjunction of hepatocytes was restored and overflow of particles of bilirubin lessened. Meanwhile, the basement membrane disappeared and cell apoptosis decreased. We suspect that the fenestrae and proliferation of LECs played important roles in this change.
At present, there are three points of view on the use of VEGF in cirrhotic disease. The first is that there is decreased VEGF in cirrhosis, and that administration of exogenous VEGF would have a helpful effect[18,19]. The second is that there is increased VEGF in cirrhosis, and that this VEGF has a harmful effect[16,17]. The last point of view is that the increased VEGF in cirrhosis is compensative, having a helpful effect[38]. Our angiogenic gene array supports the first point of view. We can see clearly that in rats with TAA-induced cirrhosis, the levels of VEGF family members decreased and the levels of VEGF receptors increased, as a reflective compensation. After VEGF/EGFP transfection, the abundance of VEGF family members increased, and that of VEGF receptors decreased, explaining the newborn capillaries we observed by transmission electron microscopy. In our previous study, the density of capillaries was also increased after VEGF-D plasmid hepatic situ injection[15].
Thus, the levels of transcripts of VEGF family members decreased in TAA-induced cirrhotic livers. Administration of a plasmid encoding an EGFP-VEGF fusion protein attenuated sinusoidal capillarization through increasing the number of fenestrae and the permeability of LECs, which improved the exchange between hepatocytes and sinusoidal blood. Consequently, hepatocytes had more nutrition and oxygen and thus preserved liver function to some extent. Therefore, VEGF gene transfer by injection through the portal vein might be an ideal method for the treatment of cirrhosis.
COMMENTS
Background
Hepatic sinusoidal capillarization has been thought to be a major contributor to hepatic failure in cirrhosis. It includes a defenestrated sinusoidal endothelium and the presence of a subendothelial basement membrane. However vascular endothelial growth factor (VEGF) has been proven to increase the number of fenestrae and permeability of liver sinusoidal endothelial cells (LECs).
Research frontiers
This study was designed to investigate the effect of VEGF transfection on hepatic sinusoidal capillarization by observing the ultrastructural change of cirrhotic endothelium as well as the relationship between the number of fenestrae and portal vein pressure.
Innovations and breakthroughs
Our study shows effective endothelial growth factor (EGFP)/VEGF transfection via injection of plasmid DNA into the portal vein decreases portal pressure in experimental cirrhosis induced by thioacetamide. This effect is mediated by an increase in the formation of fenestrae of liver sinusoids. Previous studies on the role of VEGF in portal hypertension were performed in models of non-cirrhotic pre-hepatic portal hypertension obtained through part portal vein ligation, and showed remarkable angiogenesis effect. The ultra-microstructure in the liver of those rats has not been specifically investigated. This study is more related to what happens in cirrhotic patients.
Applications
Portal hypertension is an almost unavoidable complication of cirrhosis, and it is responsible for the more lethal complications of this syndrome. Previous research has shown the role of hepatic sinusoidal capillarization in cirrhosis. To understand the mechanisms underlying hepatic sinusoidal capillarization and reverse them will be of benefit to developing a new therapeutic method.
Peer review
This paper describes the effect of VEGF transfection on hepatic sinusoidal capillarization in cirrhotic animals as compared to normal rats. The authors have shown histological evidence of partial reversal of cirrhosis.
Supported by The National Natural Science Fundation of China, No. 30300341; The Natural Science Fundation of Shandong Province, No. Y2004Z12
Peer reviewer: Kunissery A Balasubramanian, Professor, Christian Medical College, Gastrointestinal Sciences, Ida Scudder Road, Vellore 632004, India
S- Editor Zhu WL L- Editor McGowan D E- Editor Yin DH
References
- 1.Wisse E. An electron microscopic study of the fenestrated endothelial lining of rat liver sinusoids. J Ultrastruct Res. 1970;31:125–150. doi: 10.1016/s0022-5320(70)90150-4. [DOI] [PubMed] [Google Scholar]
- 2.Braet F. How molecular microscopy revealed new insights into the dynamics of hepatic endothelial fenestrae in the past decade. Liver Int. 2004;24:532–539. doi: 10.1111/j.1478-3231.2004.0974.x. [DOI] [PubMed] [Google Scholar]
- 3.Irie S, Tavassoli M. Transendothelial transport of macromo-lecules: the concept of tissue-blood barriers. Cell Biol Rev. 1991;25:317–333, 340-341. [PubMed] [Google Scholar]
- 4.Braet F, Spector I, Shochet N, Crews P, Higa T, Menu E, de Zanger R, Wisse E. The new anti-actin agent dihydrohali-chondramide reveals fenestrae-forming centers in hepatic endothelial cells. BMC Cell Biol. 2002;3:7. doi: 10.1186/1471-2121-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fraser R, Dobbs BR, Rogers GW. Lipoproteins and the liver sieve: the role of the fenestrated sinusoidal endothelium in lipoprotein metabolism, atherosclerosis, and cirrhosis. Hepatology. 1995;21:863–874. [PubMed] [Google Scholar]
- 6.Braet F, Fraser R, McCuskey RS. Thirty-five years of liver sinusoidal cells: Eddie Wisse in retirement. Hepatology. 2003;38:1056–1058. doi: 10.1053/jhep.2003.50454. [DOI] [PubMed] [Google Scholar]
- 7.Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol. 2002;1:1. doi: 10.1186/1476-5926-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu GF, Wang XY, Ge GL, Li PT, Jia X, Tian DL, Jiang LD, Yang JX. Dynamic changes of capillarization and peri-sinusoid fibrosis in alcoholic liver diseases. World J Gastroenterol. 2004;10:238–243. doi: 10.3748/wjg.v10.i2.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franceschini B, Ceva-Grimaldi G, Russo C, Dioguardi N, Grizzi F. The complex functions of mast cells in chronic human liver diseases. Dig Dis Sci. 2006;51:2248–2256. doi: 10.1007/s10620-006-9082-8. [DOI] [PubMed] [Google Scholar]
- 10.Yokomori H, Oda M, Yoshimura K, Nagai T, Ogi M, Nomura M, Ishii H. Vascular endothelial growth factor increases fenestral permeability in hepatic sinusoidal endothelial cells. Liver Int. 2003;23:467–475. doi: 10.1111/j.1478-3231.2003.00880.x. [DOI] [PubMed] [Google Scholar]
- 11.Esser S, Wolburg K, Wolburg H, Breier G, Kurzchalia T, Risau W. Vascular endothelial growth factor induces endothelial fenestrations in vitro. J Cell Biol. 1998;140:947–959. doi: 10.1083/jcb.140.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Braet F, Brodsky S, Weinstein T, Romanov V, Noiri E, Goligorsky MS. VEGF-induced mobilization of caveolae and increase in permeability of endothelial cells. Am J Physiol Cell Physiol. 2002;282:C1053–C1063. doi: 10.1152/ajpcell.00292.2001. [DOI] [PubMed] [Google Scholar]
- 13.Nagy JA, Feng D, Vasile E, Wong WH, Shih SC, Dvorak AM, Dvorak HF. Permeability properties of tumor surrogate blood vessels induced by VEGF-A. Lab Invest. 2006;86:767–780. doi: 10.1038/labinvest.3700436. [DOI] [PubMed] [Google Scholar]
- 14.Ballermann BJ. Contribution of the endothelium to the glomerular permselectivity barrier in health and disease. Nephron Physiol. 2007;106:p19–p25. doi: 10.1159/000101796. [DOI] [PubMed] [Google Scholar]
- 15.Shi BM, Wang XY, Mu QL, Wu TH, Liu HJ, Yang Z. Angiogenesis effect on rat liver after administration of expression vector encoding vascular endothelial growth factor D. World J Gastroenterol. 2003;9:312–315. doi: 10.3748/wjg.v9.i2.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makhlouf MM, Awad A, Zakhari MM, Fouad M, Saleh WA. Vascular endothelial growth factor level in chronic liver diseases. J Egypt Soc Parasitol. 2002;32:907–921. [PubMed] [Google Scholar]
- 17.Giatromanolaki A, Kotsiou S, Koukourakis MI, Sivridis E. Angiogenic factor expression in hepatic cirrhosis. Mediators Inflamm. 2007;2007:67187. doi: 10.1155/2007/67187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akiyoshi F, Sata M, Suzuki H, Uchimura Y, Mitsuyama K, Matsuo K, Tanikawa K. Serum vascular endothelial growth factor levels in various liver diseases. Dig Dis Sci. 1998;43:41–45. doi: 10.1023/a:1018863718430. [DOI] [PubMed] [Google Scholar]
- 19.Genesca J, Gonzalez A, Mujal A, Cereto F, Segura R. Vascular endothelial growth factor levels in liver cirrhosis. Dig Dis Sci. 1999;44:1261–1262. doi: 10.1023/a:1026613332003. [DOI] [PubMed] [Google Scholar]
- 20.Oe H, Kaido T, Furuyama H, Mori A, Imamura M. Simultaneous transfer of vascular endothelial growth factor and hepatocyte growth factor genes effectively promotes liver regeneration after hepatectomy in cirrhotic rats. Hepatogastroenterology. 2004;51:1641–1647. [PubMed] [Google Scholar]
- 21.Oe H, Kaido T, Mori A, Onodera H, Imamura M. Hepatocyte growth factor as well as vascular endothelial growth factor gene induction effectively promotes liver regeneration after hepatectomy in Solt-Farber rats. Hepatogastroenterology. 2005;52:1393–1397. [PubMed] [Google Scholar]
- 22.Ueno T, Nakamura T, Torimura T, Sata M. Angiogenic cell therapy for hepatic fibrosis. Med Mol Morphol. 2006;39:16–21. doi: 10.1007/s00795-006-0311-1. [DOI] [PubMed] [Google Scholar]
- 23.Bueno M, Salgado S, Beas-Zarate C, Armendariz-Borunda J. Urokinase-type plasminogen activator gene therapy in liver cirrhosis is mediated by collagens gene expression down-regulation and up-regulation of MMPs, HGF and VEGF. J Gene Med. 2006;8:1291–1299. doi: 10.1002/jgm.961. [DOI] [PubMed] [Google Scholar]
- 24.Seglen PO. Preparation of isolated rat liver cells. Methods Cell Biol. 1976;13:29–83. doi: 10.1016/s0091-679x(08)61797-5. [DOI] [PubMed] [Google Scholar]
- 25.Weng HL, Ciuclan L, Liu Y, Hamzavi J, Godoy P, Gaitantzi H, Kanzler S, Heuchel R, Ueberham U, Gebhardt R, et al. Profibrogenic transforming growth factor-beta/activin receptor-like kinase 5 signaling via connective tissue growth factor expression in hepatocytes. Hepatology. 2007;46:1257–1270. doi: 10.1002/hep.21806. [DOI] [PubMed] [Google Scholar]
- 26.Shiota G, Okubo M, Noumi T, Noguchi N, Oyama K, Takano Y, Yashima K, Kishimoto Y, Kawasaki H. Cyclooxygenase-2 expression in hepatocellular carcinoma. Hepatogastroenterology. 1999;46:407–412. [PubMed] [Google Scholar]
- 27.Yamada Y, Nezu J, Shimane M, Hirata Y. Molecular cloning of a novel vascular endothelial growth factor, VEGF-D. Genomics. 1997;42:483–488. doi: 10.1006/geno.1997.4774. [DOI] [PubMed] [Google Scholar]
- 28.Yan J, Chen W, Ma Y, Sun X. Expression of vascular endothelial growth factor in liver tissues of hepatitis B. Zhonghua Ganzangbing Zazhi. 2000;8:150–152. [PubMed] [Google Scholar]
- 29.Vale PR, Isner JM, Rosenfield K. Therapeutic angiogenesis in critical limb and myocardial ischemia. J Interv Cardiol. 2001;14:511–528. doi: 10.1111/j.1540-8183.2001.tb00367.x. [DOI] [PubMed] [Google Scholar]
- 30.Byzova TV, Goldman CK, Jankau J, Chen J, Cabrera G, Achen MG, Stacker SA, Carnevale KA, Siemionow M, Deitcher SR, et al. Adenovirus encoding vascular endothelial growth factor-D induces tissue-specific vascular patterns in vivo. Blood. 2002;99:4434–4442. doi: 10.1182/blood.v99.12.4434. [DOI] [PubMed] [Google Scholar]
- 31.Lee LY, Patel SR, Hackett NR, Mack CA, Polce DR, El-Sawy T, Hachamovitch R, Zanzonico P, Sanborn TA, Parikh M, et al. Focal angiogen therapy using intramyocardial delivery of an adenovirus vector coding for vascular endothelial growth factor 121. Ann Thorac Surg. 2000;69:14–23. doi: 10.1016/s0003-4975(99)01102-9. [DOI] [PubMed] [Google Scholar]
- 32.Leotta E, Patejunas G, Murphy G, Szokol J, McGregor L, Carbray J, Hamawy A, Winchester D, Hackett N, Crystal R, et al. Gene therapy with adenovirus-mediated myocardial transfer of vascular endothelial growth factor 121 improves cardiac performance in a pacing model of congestive heart failure. J Thorac Cardiovasc Surg. 2002;123:1101–1113. doi: 10.1067/mtc.2002.121044. [DOI] [PubMed] [Google Scholar]
- 33.Deodato B, Arsic N, Zentilin L, Galeano M, Santoro D, Torre V, Altavilla D, Valdembri D, Bussolino F, Squadrito F, et al. Recombinant AAV vector encoding human VEGF165 enhances wound healing. Gene Ther. 2002;9:777–785. doi: 10.1038/sj.gt.3301697. [DOI] [PubMed] [Google Scholar]
- 34.Risau W. Development and differentiation of endothelium. Kidney Int Suppl. 1998;67:S3–S6. doi: 10.1046/j.1523-1755.1998.06701.x. [DOI] [PubMed] [Google Scholar]
- 35.Ballermann BJ. Contribution of the endothelium to the glomerular permselectivity barrier in health and disease. Nephron Physiol. 2007;106:p19–p25. doi: 10.1159/000101796. [DOI] [PubMed] [Google Scholar]
- 36.Roberts WG, Palade GE. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J Cell Sci. 1995;108(Pt 6):2369–2379. doi: 10.1242/jcs.108.6.2369. [DOI] [PubMed] [Google Scholar]
- 37.Roberts WG, Palade GE. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 1997;57:765–772. [PubMed] [Google Scholar]
- 38.Corpechot C, Barbu V, Wendum D, Kinnman N, Rey C, Poupon R, Housset C, Rosmorduc O. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology. 2002;35:1010–1021. doi: 10.1053/jhep.2002.32524. [DOI] [PubMed] [Google Scholar]