

Abstract
PPARβ/δ (peroxisome-proliferator-activated receptor β/δ) is one of three PPARs in the nuclear hormone receptor superfamily that are collectively involved in the control of lipid homoeostasis among other functions. PPARβ/δ not only acts as a ligand-activated transcription factor, but also affects signal transduction by interacting with other transcription factors such as NF-κB (nuclear factor κB). Constitutive expression of PPARβ/δ in the gastrointestinal tract is very high compared with other tissues and its potential physiological roles in this tissue include homoeostatic regulation of intestinal cell proliferation/differentiation and modulation of inflammation associated with inflammatory bowel disease and colon cancer. Analysis of mouse epithelial cells in the intestine and colon has clearly demonstrated that ligand activation of PPARβ/δ induces terminal differentiation. The PPARβ/δ target genes mediating this effect are currently unknown. Emerging evidence suggests that PPARβ/δ can suppress inflammatory bowel disease through PPARβ/δ-dependent and ligand-independent down-regulation of inflammatory signalling. However, the role of PPARβ/δ in colon carcinogenesis remains controversial, as conflicting evidence suggests that ligand activation of PPARβ/δ can either potentiate or attenuate this disease. In the present review, we summarize the role of PPARβ/δ in gastrointestinal physiology and disease with an emphasis on findings in experimental models using both high-affinity ligands and null-mouse models.
Keywords: colon cancer, differentiation, gastrointestinal tract, inflammatory bowel disease, nuclear receptor, peroxisome-proliferator-activated receptor β/δ (PPARβ/δ)
Introduction
The seminal work identifying PPARα (peroxisome-proliferator-activated receptor α) in 1990 [1] led to the subsequent identification of a family of three related receptors in the nuclear receptor superfamily that can modulate biological function by acting as transcription factors or through protein-protein interactions. PPARα, PPARβ/δ and PPARγ are each encoded by separate genes and exhibit different tissue distribution patterns. The functions of PPARα and PPARγ are well characterized as central regulators of lipid and glucose homoeostasis. Although considerably less is known about the biological roles of PPARβ/δ, ligand activation of PPARβ/δ can effectively increase fatty acid catabolism in skeletal muscle and modulate insulin sensitivity, which makes this receptor a promising drug target for treatment and prevention of metabolic syndrome (reviewed in [2]). Additionally, there is evidence that PPARβ/δ can also modulate gastrointestinal function and disease. There are a number of different compounds that have been shown to activate PPARβ/δ, ranging from essential dietary fatty acids, endogenous prostaglandins and synthetic pharmaceuticals currently in use or under development (Table 1). The specificity of these compounds is dependent not only on the relative affinity for the receptor, but also receptor expression and subsequent transcriptional/molecular activity. The mechanisms underlying the biological effects induced by activation of PPARβ/δ are mediated through a number of potential pathways.
Table 1. Natural and synthetic activators/ligands of PPARβ/δ.
| Compound | Comment | Relative affinity for PPARβ/δ* | Reference |
|---|---|---|---|
| Linoleic acid | Essential dietary fatty acid; also activates PPARα | Micromolar | [165–167] |
| Oleic acid | Dietary fatty acid; also activates PPARα and PPARγ | Micromolar | [110,167,168] |
| Arachidonic acid | Essential dietary fatty acid; also activates PPARγ | Micromolar | [165] |
| Eicosapentaenoic acid | Essential dietary fatty acid; also activates PPARα | Micromolar | [165] |
| Docosahexaenoic acid | Essential dietary fatty acid | Micromolar | [165] |
| Prostaglandin A1 | Endogenous prostaglandin | Micromolar | [165,169] |
| Carbaprostacyclin | Synthetic stable PGI2 analogue; also activates PPARα | Micromolar | [165,169] |
| Iliprost | Antihypertensive drug; prostacyclin analogue | Micromolar | [165] |
| L165,041 | Synthetic high-affinity ligand; can activate PPARα and PPARγ at high concentration | Nanomolar | [170] |
| GW501516 | Synthetic high-affinity ligand; can activate PPARα and PPARγ at high concentration | Nanomolar | [171] |
| GW0742 | Synthetic high-affinity ligand; can activate PPARα and PPARγ at high concentration | Nanomolar | [171] |
Relative concentration range required to activate the receptor based on the relative ability to transactivate reporter constructs. PGI2, prostacyclin.
There are at least three primary mechanisms by which PPARβ/δ can regulate biological functions (Figure 1). PPARβ/δ is thought to exist in the cell in a complex containing the receptor, auxiliary proteins and co-repressors. The histone deacetylase activity of the co-repressors maintains the tightly bound chromatin structure preventing transcription. In response to ligand binding, PPARβ/δ undergoes a conformational change, leading to release of auxiliary proteins and co-repressors and recruitment of co-activators that contain histone acetylase activity. Acetylation of histones by co-activators bound to the ligand-PPARβ/δ complex leads to nucleosome remodelling, allowing for recruitment of RNA polymerase II causing target gene transcription. In addition to this direct ligand-dependent transcriptional up-regulation of target genes that modulates biological processes such as terminal differentiation (Figure 1A), PPARβ/δ can interfere with NF-κB (nuclear factor κB) signalling (Figure 1B) [3–5], leading to anti-inflammatory activities associated with PPARβ/δ and its ligands [3–23]. The anti-inflammatory activities of PPARβ/δ may be mediated in part by interactions with STAT3 (signal transducer and activator of transcription 3) and ERK5 (extracellular-signal-regulated kinase 5) [24,25]. It is worth noting that the inhibition of STAT3 signalling due to interacting with PPARβ/δ [24] may also be important in carcinogenesis, as STAT3 is often overexpressed in many cancers, including colon cancer, and is associated with increasing anti-apoptotic signalling and increased c-myc expression [26]. As there is evidence that PPARα can interfere with AP1 (activator protein 1) signalling [27,28], it remains a possibility that PPARβ/δ could also function in concert with AP1 signalling. Thus PPARβ/δ can modulate inflammatory signalling and possibly cell growth by directly interfering with a number of transcription factors (Figure 1B). It was also shown that PPARβ/δ can repress the expression of some genes by associating with co-repressors [29] (Figure 1C). Cell lines with stable overexpression of PPARβ/δ have diminished induction of PPARα and PPARγ target genes in response to ligand activation of these receptors [29]; however, this effect may be cell-specific. For example, PPARα target gene expression is not increased in PPARβ/δ-null mouse liver, white adipose tissue or small intestine, and ligand activation of PPARα in the absence of PPARβ/δ expression does not lead to enhanced expression of PPARα target genes in these tissues [30]. In contrast, ligand activation of PPARγ in the colon in the absence of PPARβ/δ expression does not cause enhanced expression of PPARγ target genes, yet ligand activation of PPARα in the colon in the absence of PPARβ/δ expression leads to enhanced expression of PPARα target genes [31]. Thus there is in vivo evidence that PPARβ/δ can repress the transcription of PPARα target genes in the colon, but this does not appear to occur in the liver, adipose tissue or small intestine. Additionally, the idea that PPARβ/δ represses PPARγ target gene expression in the colon is not supported by these findings and is also inconsistent with relative expression patterns. PPARβ/δ is expressed at significantly higher levels in the colon as compared with PPARγ (described below), and it is well known that ligand activation of PPARγ in the colon can attenuate diseases of this tissue. Taken together, there are a number of important pathways that can be modulated by PPARβ/δ, and all of these are likely to have some function in gastrointestinal physiology and disease, as PPARβ/δ is expressed at relatively high levels in both the small and large intestine.
Figure 1. PPARβ/δ-mediated regulation of transcription.
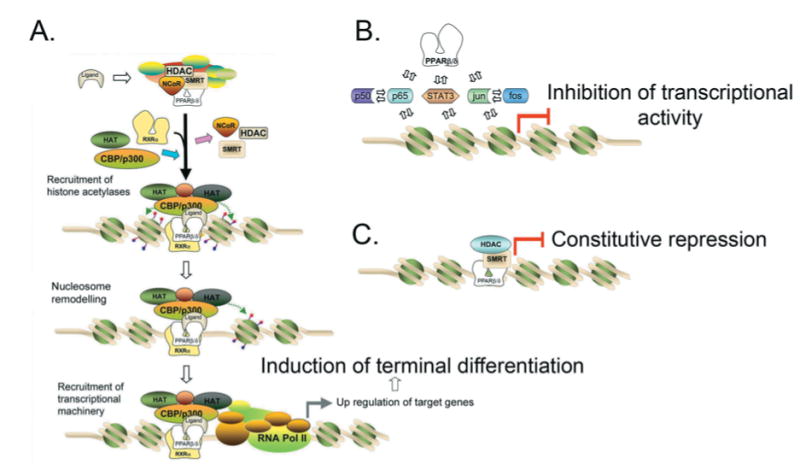
PPARβ/δ can directly up-regulate target genes in response to ligand activation (A), interact with other transcription factors such as NF-κB and possibly others such as AP1 (B), or repress gene expression (C). CBP, CREB (cAMP-response-element-binding protein)-binding protein; HAT, histone acetylase; HDAC, histone deacetylase; NCoR, nuclear receptor co-repressor; RNA Pol II, RNA polymerase II; RXRα, retinoid X receptor α; SMRT, silencing mediator for retinoic acid receptor and thyroid-hormone receptor.
PPARβ/δ Expression in the Gastrointestinal Tract
There are a number of reports describing the expression of PPARβ/δ in the gastrointestinal tract (Table 2), with the majority of them examining the mRNA encoding the receptor. When compared with the other PPARs, expression of PPARβ/δ mRNA is considerably higher in both the small and large intestine in rats [32]. When compared with most major tissues, such as the liver, adipose tissue, kidney and muscle, the expression of PPARβ/δ mRNA is markedly elevated in the gastrointestinal tract [32]. The cis elements in the promoter responsible for this high level of expression have not been delineated, but a strong enhancer sequence in the PPARβ/δ promoter has been described [33]. In humans, there is a developmental increase in the expression of PPARβ/δ in the small and large intestine, with the adult colon expressing more protein compared with neonates [34]. Furthermore, expression of PPARβ/δ in the neonatal human intestine has a cytoplasmic localization early on and a more nuclear localization in the later fetal stages and, in particular, in the adult [34]. Note that the reliability of this work depends in large part on the quality of the antibody used, as it is very difficult to detect the expression of low-abundance nuclear receptors in the absence of a highly specific antibody. Hepatic expression of PPARα is increased upon fasting [35,36], probably due to the requirement of this receptor in activating the expression of enzymes involved in fatty acid catabolism. In contrast, no significant variation in PPARβ/δ expression in the small or large intestine is observed after feeding or fasting, suggesting that the high level of PPARβ/δ expression is independent of the energy status of the cell [32]. Although the gastrointestinal tract is composed of both similar and unique cells, including enterocytes, goblet cells, Paneth cells and stem cells, an extensive analysis of PPARβ/δ expression in the different cell types has not been performed to date. However, it is known that many cryptic cells of the colon express PPARβ/δ protein [37]. The expression of PPARβ/δ may also be markedly changed during cancer progression, an issue that will be addressed in greater detail in a later section. Given the relatively high expression of PPARβ/δ in the small and large intestine, it is clear that this nuclear receptor must have an important role in the gastrointestinal tract in both maintaining normal homoeostasis and in disease.
Table 2. Relative expression of PPARβ/δ in the small and large intestine.
| Species | Specimen | Relative expression | Method | Reference |
|---|---|---|---|---|
| Rat | 300 g Sprague–Dawley | Relatively high expression of mRNA encoding PPARβ/δ in the small intestine and colon | In situ hybridization | [172] |
| Mouse | 7-week-old NMRI | Modest expression of mRNA encoding PPARβ/δ in the colon, less expression of mRNA encoding PPARβ/δ in the small intestine | Ribonuclease protection assay | [173] |
| Human | 7–22-week fetuses; adult controls | Expression of PPARβ/δ protein is higher in the adult colon compared with fetal colon; appears to move from cytoplasm to nucleus with age | Western blotting and immunohistochemistry | [34] |
| Rat | 8–9-weeks-old (approx. 200 g) Sprague–Dawley | Markedly high expression of mRNA encoding PPARβ/δ compared with other tissues and other PPARs | Ribonuclease protection assay | [32] |
| Rat | 5 and 25 days postnatally; Sprague–Dawley | High expression of mRNA encoding PPARβ/δ compared with other PPARs in the small intestine; increased expression of mRNA encoding PPARβ/δ by postnatal day 25. | Northern blotting | [174] |
| Mouse | 5–6-weeks-old CD-1 | Relatively high expression of PPARβ/δ protein in intestinal epithelial cells; expressed in cryptic cells | Tissue microarray-based immunohistochemistry | [37] |
Roles of PPARβ/δ in The Gastrointestinal Tract
A significant role for PPARβ/δ in regulating intestinal cell differentiation has recently been described by two independent laboratories [31,38]. In the absence of PPARβ/δ expression in the small intestine, the number of Paneth cells is significantly decreased [38]. Ligand activation of PPARβ/δ causes an increase in the number of duodenal Paneth cells coincident with an increase in markers of Paneth cell differentiation [38]. Induction of Paneth cell differentiation in the small intestine is mediated by PPARβ/δ-dependent down-regulation of hedgehog signalling [38]. As these effects are not found in similarly treated PPARβ/δ-null mice, this demonstrates that PPARβ/δ is required to mediate the differentiation of intestinal Paneth cells. In another study, ligand activation of PPARβ/δ also induced terminal differentiation of colonic epithelial cells [31]. Administration of a potent PPARβ/δ ligand increased the expression of genes associated with terminal differentiation of colon epithelium and an increase in the relative state of differentiation of colonic epithelial cells [31]. It is of interest to note that one of the mRNA markers, cathepsin E, is also associated with the induction of M cell differentiation, which functions to process antigens in the colon epithelium. Collectively, these two reports demonstrate that PPARβ/δ has an important role in terminal differentiation of both the small and large intestine. This is consistent with a larger body of evidence from multiple laboratories showing an association between PPARβ/δ and PPARβ/δ ligands and terminal differentiation in epithelial cells [13,21,31,38–43]. This has led to a working hypothesis that constitutive expression and activation of PPARβ/δ is maintained at a high level in the intestinal epithelium to mediate differentiation of cell populations that are turning over at a relatively high rate (Figure 2).
Figure 2. Functional role of PPARβ/δ in the intestine and a working hypothesis of how PPAR β/δ functions when expression is increased.

Constitutive expression of PPARβ/δ is high in the epithelial cells of the small and large intestine. Activation of PPARβ/δ by endogenous ligands probably serves to maintain a population of terminally differentiated cells allowing for normal absorption function and/or modulation of inflammation. Expression of PPARβ/δ can be increased by inflammation (e.g. in response to TNFα and AP1 signalling). It is a working hypothesis that this increase in PPARβ/δ-mediated signalling will lead to increased differentiation, inhibition of cell proliferation and anti-inflammatory activity that will culminate by resolution in the affected cell(s).
Role of PPARβ/δ in Inflammatory Bowel Disease
Inflammatory bowel disease refers collectively to both Crohn's disease and ulcerative colitis and is characterized by the overexpression of inflammatory signalling molecules, including TNFα (tumour necrosis factor κ), NF-κB and IL (interleukin) 1β. PPARβ/δ is known to have strong anti-inflammatory activities (Figure 1B), which may be due, in part, to interference with NF-κB signalling. Coupled with the fact that PPARβ/δ is expressed at high levels in the intestinal epithelium, the rationale for examining the potential role of PPARβ/δ in these diseases is compelling. Consistent with the idea that PPARβ/δ may inhibit inflammatory bowel disease, dietary administration of CLA (conjugated linoleic acid) increases the intestinal expression of PPARβ/δ target genes and was shown to protect against experimentally induced colitis [7], which is in agreement with a study showing that CLA isomers can activate PPARβ/δ [44]. However, although that study [7] suggested a possible role for PPARβ/δ in ameliorating inflammatory bowel disease because of the association between the up-regulation of PPARβ/δ target genes and CLA-induced inhibition of experimentally induced colitis, this effect of CLA was probably more dependent on PPARγ as suggested. That activation of PPARβ/δ does not influence inflammatory bowel disease is also supported by a more recent finding reporting that, in the absence of PPARβ/δ expression, experimentally induced colitis is significantly exacerbated, as revealed by enhanced clinical symptoms, increased expression of inflammatory signalling molecules (e.g. IL6 and TNFα) and histopathological analysis [11]. Although this observation suggests that ligand activation may prevent experimentally induced colitis, surprisingly administration of a highly specific PPARβ/δ ligand (GW0742; 5 mg/kg of body weight) did not ameliorate any of these effects [11]. However, it remains a possibility that higher concentrations of the PPARβ/δ ligand could prove to be inhibitory in this model system, since the concentration used in that study [11] was shown to specifically activate PPARβ/δ in vivo and falls in the mid-concentration range of effective doses, thus suggesting that PPARβ/δ can inhibit inflammation in the bowel through a ligand-independent mechanism. The idea that the anti-inflammatory activities of PPARβ/δ and its ligands may be of use for the treatment of inflammatory bowel disease is also supported by the recent findings that chemically induced colitis is exacerbated in stearoyl-CoA desaturase-1-null mice, which have decreased concentrations of oleic acid and linoleic acid [45]: two fatty acids that can activate PPARβ/δ (Table 1). It is likely that PPARβ/δ-dependent modulation of experimentally induced colitis is mediated through protein–protein interactions, such as interfering with NF-κB signalling (Figure 1B). It is also possible that PPARβ/δ ligands could cause SUMOylation of the receptor, similar to that observed with PPARγ, leading to repression of inflammatory responses by interfering with clearance of co-repressor complexes (reviewed in [46]). Alternatively, inhibition of Paneth cell differentiation in the intestine that occurs in the absence of PPARβ/δ expression [47] could contribute to exacerbated experimentally induced colitis in PPARβ/δ-null mice [11], due to decreased expression of antimicrobial peptides such as α-defensins produced by Paneth cells, which serve to protect against intestinal inflammation [47].
In contrast with these findings, administration of GW0742 (5 mg/kg of body weight) caused enhanced colitis in a genetic model of colitis (IL10-null mice) [48]. The reason why mice expressing IL10 in a chemically induced colitis model do not exhibit exacerbated colitis in response to a PPARβ/δ ligand, whereas those lacking expression of IL10 do, remains to be determined. It is tempting to speculate that this difference could be related to PPARβ/δ inhibition of STAT3 signalling. As IL10 activation of STAT3 leads to an anti-inflammatory response, but IL6 activation of STAT3 does not [49], it is possible that ligand activation of PPARβ/δ interferes with the anti-inflammatory response associated with IL10 by interfering with STAT3. This view deserves further consideration.
Role of PPARβ/δ in Colon Cancer
By far the most controversial issue regarding PPARβ/δ in the intestine is the potential roles of this receptor in colon cancer. Indeed, there are two opposing hypotheses, with one suggesting that ligand activation of PPARβ/δ potentiates colon tumorigenesis through a variety of mechanisms, whereas the other suggests that ligand activation of PPARβ/δ inhibits colon tumorigenesis (Figure 3).
Figure 3. Hypothetical roles of PPARβ/δ in colon carcinogenesis.
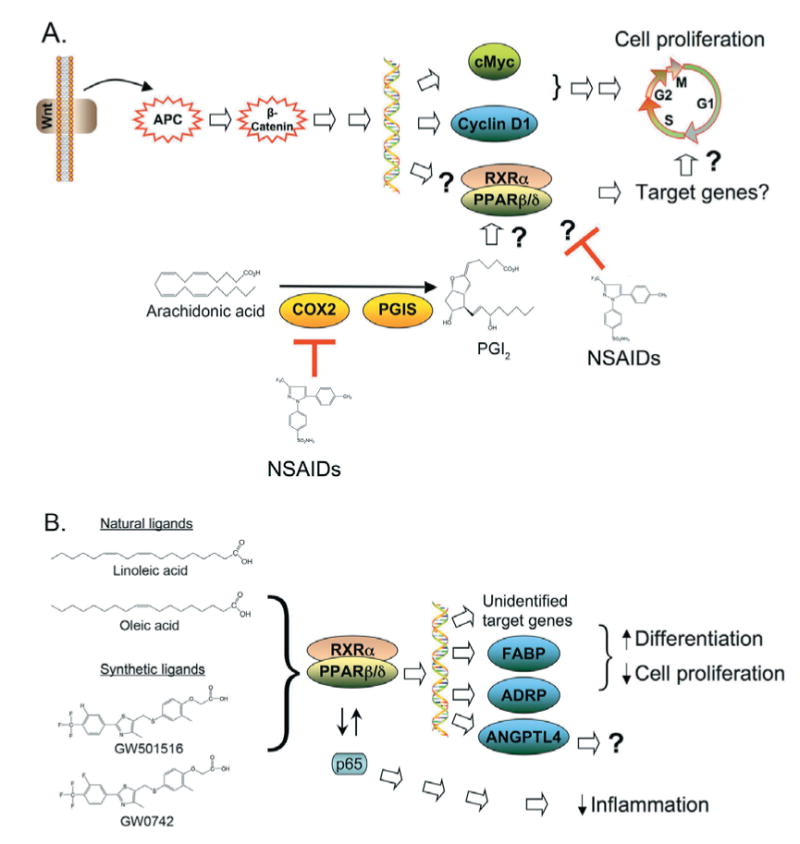
(A) Ligand activation of PPARβ/δ potentiates colon cancer. In this model, PPARβ/δ is a downstream target of the APC/β-catenin/Tcf4 pathway and, through unknown target genes, leads to enhanced cell proliferation. COX-2-derived prostacyclin (PGI2) may be an endogenous ligand for PPARβ/δ in this model, and inhibition of COX2 by NSAIDs may prevent activation of PPARβ/δ and unknown target genes. Alternatively, there is also evidence that NSAIDs may inhibit PPARβ/δ expression leading to reduced anti-apoptotic activity mediated through ill-defined mechanisms. ?, pathways of uncertainty. PGIS, prostaglandin I2 synthase. (B) Ligand activation of PPARβ/δ attenuates colon cancer. In this model, ligand activation of PPARβ/δ leads to the induction of well-characterized target genes, including FABP and ADRP, that are associated with terminal differentiation and probably other unidentified target genes that participate. PPARβ/δ can also interfere with NF-κB and AP1 signalling, causing anti-inflammatory activities. The induction of terminal differentiation and anti-inflammatory activities are associated with inhibition of cell proliferation. RXRα, retinoid X receptor α.
Less than ten years ago, a hypothetical mechanism (Figure 3A) was described linking the expression and function of PPARβ/δ to colon carcinogenesis [50]. In this model, PPARβ/δ is a downstream target of the APC (adenomatous polyposis coli)/β-catenin/Tcf4 (T-cell transcription factor 4) pathway, similar to the oncogene c-myc [51]. Reduced function of APC due to familial or somatic mutations leads to increased activity of β-catenin and Tcf4 transcriptional activity, which causes an up-regulation of target genes such as c-myc and reportedly PPARβ/δ [50]. The increased presence of PPARβ/δ could then potentially be activated by endogenous ligands such as COX (cyclo-oxygenase)-derived prostacyclin [52]. It was proposed further that ligand-activated PPARβ/δ would then drive the expression of target genes, which still remain to be identified, and potentiate cell growth. In support of this model, PPARβ/δ-null HCT116 cells have reduced tumorigenicity in a xenograft model [53]. Taken together, an attractive model that presumably explained how COX2 inhibition prevents colon cancer was developed (Figure 3A). In addition to this model, a couple of related models were proposed suggesting that higher expression of PPARβ/δ in colon cancer prevents activation of PPARγ [54,55], and that NSAIDs (non-steroidal anti-inflammatory drugs) may prevent colon cancer by inhibiting the increased expression of PPARβ/δ reported in colon cancer [56] (Figure 3A). Subsequent findings from other laboratories using both in vitro and in vivo models have provided results in support of these models, whereas others have not. Indeed, there is an alternative hypothesis that ligand activation of PPARβ/δ can attenuate colon cancer (Figure 3B). This mechanism is supported by the observations that colon carcinogenesis is exacerbated in both a genetic APCmin mouse model and a chemically induced azoxymethane treatment model in the absence of PPARβ/δ expression [57,58], that silencing PPARβ/δ expression in human colon cancer cells promotes cell proliferation [59], that PPARβ/δ has potent anti-inflammatory activities [3–23], that PPARβ/δ mediates terminal differentiation in a number of models (reviewed later) and that inhibition of azoxymethane-induced colon cancer by ligand activation of PPARβ/δ requires a functional PPARβ/δ [31]. As noted above, some findings in the literature support this model whereas others do not. The following sections will address the most central regulatory mechanisms of these pathways with an emphasis on the expression of PPARβ/δ in colon cancer and the influence of ligand administration in colon cancer models.
Is PPARβ/δ an APC/β-catenin/Tcf4 Target Gene that is Up-regulated during Colon Cancer?
A fundamental question that has remained unanswered in the PPAR literature is whether PPARβ/δ is a downstream target of the APC/β-catenin/Tcf4 pathway. It is important to stress that constitutive expression of PPARβ/δ is relatively high in the gastrointestinal tract and that this high expression probably facilitates terminal differentiation. Recent evidence also suggests that PPARβ/δ may be constitutively active in the presence of endogenous fatty acids, as fatty acids are tightly bound to the ligand-binding domain [60]. Despite the relatively high constitutive expression, there are a number of studies suggesting that PPARβ/δ is up-regulated in colon tumours in both human and rodent models (Table 3a). These findings are consistent with the idea that PPARβ/δ is a downstream target of the APC/β-catenin/Tcf4 pathway. This hypothesis is based on the original study showing that PPARβ/δ expression is diminished with overexpression of APC in a human colon cancer cell line, coupled with additional studies, including the demonstration that PPARβ/δ mRNA is higher in colon tumours from human cancer patients [50]. Six independent studies from other laboratories support these findings, some with greater experimental and statistical strength than others. For example, two studies show that expression of PPARβ/δ mRNA is higher in colon tumours compared with normal mucosa in human patients [52,61], whereas findings from two other laboratories report relatively higher expression of PPARβ/δ mRNA in only a small cohort of the examined colon tumour samples [62,63]. One study also showed relatively high expression of PPARβ/δ in one patient with FAP (familial adenomatous polyposis) using immunohistochemistry [64], but follow-up Western blot analyses indicate that the immunohistochemical analysis was influenced by non-specific immunoreactivity, a point alluded to above. There is also evidence from animal studies showing relatively higher expression of PPARβ/δ in colon tumours from rats treated with the colon-specific carcinogen azoxymethane, intestinal adenomas from APCmin mice treated with azoxymethane and polyps from APCmin mice [52,64,65]. However, as noted above, Western blot analysis with the same antibody used in the immunohistochemical analysis revealed no change in PPARβ/δ, even though expression of β-catenin and cyclin D1 were elevated [64,65]. This indicates that the results from immunohistochemical analysis suggesting increased expression of PPARβ/δ could be an incorrect interpretation resulting from non-specific immunoreactivity, as the same antibody was used for both studies [64,65]. In contrast with these studies, there are publications from 18 independent laboratories that do not support the view that PPARβ/δ is up-regulated in colon carcinogenesis by the APC/β-catenin/Tcf4 pathway (Table 3b). Expression of mRNA encoding PPARβ/δ is lower in colon adenomas and adenocarcinomas compared with normal tissue in human cancer patients [66]. In patients with a family history of sporadic colon cancer, expression of PPARβ/δ is decreased in normal mucosa compared with mucosa from controls with no family history of colon cancer [67]. In two other independent studies, expression of mRNA was lower in more than half of the colon tumours examined from patients [63] or was unchanged in more than half of the colon tumours examined from another cohort of cancer patients [62]. Similarly, expression of PPARβ/δ mRNA is lower in colon mucosa from cancer patients compared with mucosa from non-cancer patients [68]. In yet another study examining human colon tumours, mRNA encoding PPARβ/δ was high in some patients, but, in general, the expression was not different between tumours and normal mucosa [69]. Studies in human cancer cell lines show that the relative expression of PPARβ/δ is either similar or lower in SW480 cells that have a mutant APC gene and a wild-type β-catenin gene compared with HCA7 cells that have a wild-type APC gene and a β-catenin gene [52,70]. Knocking down the expression of PPARβ/δ in HT29 human colon cancer cells has no effect on cell growth or apoptosis [71], but silencing PPARβ/δ expression in HCT116 human colon cancer cells promotes cell growth [59]. Studies in animal models provide another body of evidence indicating that PPARβ/δ is not increased during colon carcinogenesis and that PPARβ/δ is not up-regulated by the APC/β-catenin/Tcf4 pathway. Expression of mRNA encoding PPARβ/δ is lower in polyps from APC mutant mice and in polyps from azoxymethane-treated mice compared with normal mucosa [31,58,68]. However, the relative expression of PPARβ/δ protein is reportedly unchanged in polyps compared with mucosa in APCmin mice [72], and expression of the PPARβ/δ protein is not different in the wild-type colon compared with the colon from APCmin mice [57]. This is consistent with another study showing increased expression of β-catenin and cyclin D1 in intestinal adenomas from azoxymethane-treated APCmin mice, but no change in PPARβ/δ expression [64]. This suggests that some of the reported differences in mRNA expression of PPARβ/δ may not cause functional changes in protein levels. Some of the most convincing evidence to date indicating that PPARβ/δ is not up-regulated by the APC/β-catenin/Tcf4 pathway is provided from transgenic mouse models. Complete deletion of the APC gene in mouse intestine causes increased expression of the well-characterized oncogene c-myc, but down-regulation of mRNA encoding PPARβ/δ [58,73]. Similarly, targeted deletion of β-catenin in mouse intestine causes the down-regulation of c-myc mRNA but no change in the expression of PPARβ/δ mRNA [74]. Gene expression analysis of a human colon cancer cell line expressing a dominant-negative form of Tcf4 also showed a significant decrease in the expression of c-myc mRNA but no change in the expression of mRNA encoding PPARβ/δ [75]. Taken together, there are some studies demonstrating that PPARβ/δ is up-regulated in colon cancer, but many other studies that are inconsistent with this result. Regardless of whether the expression of PPARβ/δ is increased, decreased or unchanged during colon carcinogenesis, it is important to note that the biological role of this receptor in colon carcinogenesis is still unknown. To better understand the function of PPARβ/δ in this disease, the effect of ligand activation and/or targeted disruption of PPARβ/δ has provided some clues towards this goal.
Table 3. Expression of PPARβ/δ in intestinal cancer models.
| (a) Evidence suggesting that PPARβ/δ is up-regulated in colon cancer by the APC/β-catenin/Tcf4 pathway | ||||
|---|---|---|---|---|
| Species | Specimen | Relative expression | Method | Reference |
| Human | Normal colon epithelium and primary colorectal cancer cells from four patients | Relative expression of mRNA encoding PPARβ/δ is higher in primary colorectal cancer cells compared with normal epithelium. | Northern blotting | [50] |
| Human | Normal mucosa and colorectal carcinomas from six patients | Relative expression of mRNA encoding PPARβ/δ is higher in colorectal carcinomas compared with normal mucosa. | Northern blotting | [52] |
| Rat | Normal colon tissue and colon tumours from six rats | Relative expression of mRNA encoding PPARβ/δ is higher in colon tumours compared with normal colon tissue. | Northern blotting | [52] |
| Human | Colon cancer cell lines | Variable expression, but decreased expression of PPARβ/δ in cells with mutant APC and functional β-catenin (SW480) compared with cells with wild-type APC and functional β-catenin (HCA7). | Northern blotting and Western blotting | [52] |
| Human | Colorectal cancer tissue from 17 patients | Relative expression of mRNA encoding PPARβ/δ is higher in four out of 17 tumour samples compared with normal mucosa. | Quantitative real-time PCR | [62] |
| Mouse | Adenoma and normal tissue from azoxymethane-treated APCmin mice | Relative expression of PPARβ/δ is higher in flat aberrant crypt foci and adenoma compared with normal tissue. | Immunohistochemistry | [64] |
| Human | One adenoma from one patient with FAP | Relative expression of PPARβ/δ is higher in adenoma, but no control tissue presented. | Immunohistochemistry | [64] |
| Mouse | Normal mucosa and polyps from APCmin mice | Relative expression of PPARβ/δ is higher in normal mucosa and polyps from APCmin mice compared with wild-type mucosa. | Immunohistochemistry | [65] |
| Human | Colorectal tumours from 32 patients | Relatively higher expression of PPARβ/δ correlates with malignant morphology. Relative expression of PPARβ/δ is high in 15 patients but low in 17 patients. | Immunohistochemistry and quantitative real-time PCR | [63] |
| Human | Tumour and non-tumour samples from 20 patients | Relative expression of mRNA encoding PPARβ/δ is higher in tumour samples compared with non-tumour samples | Quantitative real-time PCR | [61] |
| (b) Evidence suggesting that PPARβ/δ is not up-regulated in colon cancer by the APC/β-catenin/Tcf4 pathway | ||||
| Species | Specimen | Relative expression | Method | Reference |
| Human | Colon cancer cell lines | Variable expression, but decreased expression of PPARβ/δ in cells with mutant APC and functional β-catenin (SW480) compared with cells with wild-type APC and functional β-catenin (HCA7). | Northern blotting and Western blotting | [52] |
| Human | Normal colonic mucosa from 12 patients with a family history of colon cancer | Relative expression of mRNA encoding PPARβ/δ is decreased compared with patients with no family history of colon cancer. | Quantitative real-time PCR | [67] |
| Human | Colon adenocarcinomas and normal tissue from 18 patients | Relative expression of mRNA encoding PPARβ/δ is lower in adenocarcinomas samples as compared with normal tissue. | Affymetrix microarray | [66] |
| Human | Colon adenomas and normal tissue from four patients | Relative expression of mRNA encoding PPARβ/δ is lower in adenocarcinomas samples compared with normal tissue. | Affymetrix microarray | [66] |
| Human | HCA7 and SW480 colon cancer cell lines | Relative expression of PPARβ/δ is lower in SW480 cells that have a mutant APC compared with HCA7 cells that have a wild-type APC gene. | Northern blotting and Western blotting | [52] |
| Human | HCA7 and SW480 colon cancer cell lines | Relative expression of mRNA encoding PPARβ/δ is not different in SW480 cells that have a mutant APC gene compared with HCA7 cells that have a wild-type APC gene. | Quantitative real-time PCR | [101] |
| Human | HCA7 and SW480 colon cancer cell lines | Relative expression of PPARβ/δ is not different in SW480 cells that have a mutant APC gene compared with HCA7 cells that have a wild-type APC gene. | Western blotting | [70] |
| Human | HT29 colon cancer cell line | Knockdown of PPARβ/δ expression does not affect cell growth or apoptosis | Western blotting | [71] |
| Mouse | Normal mucosa and polyps from APCmin mice | Relative expression of PPARβ/δ is not different between normal mucosa and polyps from APCmin mice | Western blotting | [72] |
| Mouse | Adenoma and normal tissue from azoxymethane-treated APCmin mice | No difference in expression of PPARβ/δ in flat aberrant crypt foci and adenoma compared with normal tissue | Western blotting | [64] |
| Mouse | Normal mucosa from wild-type and APCmin mice | Relative expression of PPARβ/δ is not different between wild-type and APCmin normal mucosa | Western blotting | [57] |
| Mouse | Normal mucosa and polyps from APCmin mice | Relative expression of mRNA encoding PPARβ/δ is lower in polyps from APCmin mice compared with wild-type normal mucosa | Quantitative real-time PCR | [68] |
| Human | Normal mucosa from non-cancer patients and mucosa from cancer patients | Relative expression of mRNA encoding PPARβ/δ is lower in mucosa from cancer patients compared with non-cancer patients | Quantitative real-time PCR | [68] |
| Mouse | Conditional deletion of β-catenin in mouse intestine | Relative expression of mRNA encoding PPARβ/δ is not changed in the absence of β-catenin expression, despite a decreased expression of c-myc | Microarray | [74] |
| Human | Colorectal cancer cell line expressing a dominant-negative Tcf4 | Relative expression of PPARβ/δ is unchanged when Tcf4 activity is suppressed, despite a decrease in c-myc etc. | Microarray | [75] |
| Mouse | Conditional deletion of APC in mouse intestine | Relative expression of PPARβ/δ is lower in the absence of APC expression, despite the induction of c-myc | Reverse transcriptase–PCR and Western blotting | [58,73] |
| Mouse | Normal mucosa and colon polyps from azoxymethane-treated mice | Relative expression of mRNA encoding PPARβ/δ is lower in colon polyps compared with normal mucosa | Quantitative real-time PCR | [31] |
| Mouse | Normal mucosa and colon polyps from APCmin mice | Relative expression of mRNA encoding PPARβ/δ is lower in colon polyps compared with normal mucosa | Quantitative real-time PCR | [31] |
| Human | Colorectal tumours from 32 patients | Relative expression of PPARβ/δ is high in 15 patients but low in 17 patients; relative expression of PPARβ/δ does not correlate with Ki67 staining | Immunohistochemistry and quantitative real-time PCR | [63] |
| Human | Colorectal cancer tissue from 86 patients | Relative expression of mRNA encoding PPARβ/δ is high in some patients but, in general, the relative expression of PPARβ/δ is not different between tumours and normal mucosa | Quantitative real-time PCR | [69] |
| Human | Colorectal cancer tissue from 17 patients | Relative expression of mRNA encoding PPARβ/δ is unchanged in 12 out of 17 tumour samples compared with normal mucosa; relative expression of mRNA encoding PPARβ/δ does not correlate with Dukes stage | Quantitative real-time PCR | [62] |
FAP, familial adenomatous polyposis.
Effect of Ligand Activation of PPARβ/δ in Colon Cancer Models
Some of the earliest studies examining the effect of a PPARβ/δ ligand in cancer were performed before the link between the ligand and the receptor was described. Prior to the suggestion that prostacyclin may be an endogenous ligand for PPARβ/δ [52], a number of studies had been performed examining the effect of this COX-derived metabolite in cancer models, but did not focus on intestinal carcinogenesis. For example, in 1981 it was shown that prostacyclin inhibits cell growth of the B16 melanoma cell line [76]. This is some of the first evidence suggesting that a potential PPARβ/δ ligand can inhibit cell growth, although this was not specifically examined in this study as there was no reason at the time to link prostacyclin with activation of PPARβ/δ. The same investigators also demonstrated that prostacyclin is a potent antimetastatic agent [77], an observation that has been examined extensively since that time (reviewed in [78]). Indeed, prostacyclin can significantly inhibit cell adhesion molecule adherence of COLO 205 colon carcinoma cells to endothelial cells [79]. Whether the inhibitory effect or the antimetastatic effect of prostacyclin is mediated by PPARβ/δ is uncertain, but it is worth noting that, in a lung cancer cell line, inhibition of cell proliferation by carbaprostacyclin is mediated by PPARβ/δ as shown by siRNA (short interfering RNA) experiments [80]. There is some indication that inhibition of cell growth by prostacyclin could be influenced by increased apoptosis, as this has been shown in vascular smooth muscle cells [81]; however, as with many of the studies investigating the role of PPARβ/δ in cancer, this idea is inconsistent with other findings. Although there is good evidence that prostacyclin can inhibit cell growth and have antimetastatic activity in a variety of tumour models, there is also a study suggesting that prostacyclin produced in colonic fibroblasts can promote cell survival of colonocytes via the activation of PPARβ/δ [82]; however, this hypothesis was not examined extensively. Prostacyclin has also been shown to be chemoprotective for lung cancer [83,84] and, interestingly, the mechanism of this chemoprevention may involve the up-regulation of PPARγ [84]. It is also worth noting that there are high-affinity prostacyclin receptors (e.g. affinity in the nanomolar range [85,86]) on the cell surface. This suggests that any potential effect of prostacyclin that could be mediated by PPARβ/δ, which is localized in the cytosol and/or nucleus, would result in having bypassed binding to the cell-surface receptor. Collectively, the role of PPARβ/δ, if any, in modulating any effects of prostacyclin in colon cancer models remains to be determined.
The studies using prostacyclin are more suggestive of possible roles for PPARβ/δ, as it has not been conclusively demonstrated with any degree of certainty that prostacyclin is a true endogenous ligand of PPARβ/δ [87] and, in many cases, this possibility was not examined specifically. There are a number of studies that have more directly examined the effect of PPARβ/δ in intestinal carcinogenesis using low- and high-affinity ligands of PPARβ/δ and, in some cases, also controlling for PPARβ/δ expression. Unfortunately, these studies have also provided conflicting results that require discussion. Linoleic acid can activate PPARβ/δ (Table 1), and there is some evidence associating increased dietary intake of linoleic acid with increased colon cancer in experimental animal models [88–91]. Increased dietary intake of linoleic acid is also associated with increased risk of colon cancer in patients with a mutation in v-Ki-ras2 Kirsten rat sarcoma viral oncogene homologue [92]. However, linoleic acid is a relatively low-affinity activator of PPARβ/δ and other variables other than linoleic acid may have influenced these findings, including dietary fibre. The first study describing the effect of a potent PPARβ/δ ligand on intestinal carcinogenesis showed that GW501516 increased the average number (approx. 30 polyps in control compared with approx. 56 polyps in the GW501516-treated) and average size of small intestinal polyps in APCmin mice, which was due in part to modulation of apoptosis [93]. The number of colon polyps in response to administration of GW501516 to APCmin mice was not different in this study [93]. The same group went on to show that administration of GW501516 to APCmin mice increased the average number of polyps in the small intestine (approx. 60 polyps in control compared with approx. 120 polyps in GW501516-treated) and colon (approx. one polyp in control compared with approx. three polyps in GW501516-treated) through a PPARβ/δ-dependent mechanism, as this was not found in APCmin mice that lacked expression of PPARβ/δ [94]. It was hypothesized that the observed increase in intestinal polyps resulting from administration of the highly potent PPARβ/δ ligand was mediated, in part, by PPARβ/δ-dependent modulation of VEGF (vascular endothelial growth factor) expression, which presumably caused increased phosphorylation of Akt and inhibition of apoptosis [94].
In contrast with these studies suggesting that ligand activation of PPARβ/δ will potentiate colon carcinogenesis, a number of findings from independent laboratories using low- and high-affinity activators of PPARβ/δ do not support this view. Although some studies suggest that increased dietary linoleic acid, which can activate PPARβ/δ (Table 1), may be associated with increased risk of colon cancer [88–92], a careful review of multiple human studies indicates that high dietary intake of linoleic acid does not increase the risk of colon cancer [95]. Notably, there is a lack of an increased risk of colon cancer by high dietary intake of linoleic acid in patients with colon cancer with an APC mutation [92]. Other studies suggest that fatty acids, including linoleic acid, oleic acid and others, can inhibit colon cancer. For example, linoleic acid and oleic acid can inhibit cell proliferation and induce apoptosis in HT29 cells [96]. Consumption of olive oil, which is high in oleic acid that can activate PPARβ/δ (Table 1), has also been associated with a decreased risk of colon cancer [97,98]. However, other components of olive oil, such as phenolic compounds, may contribute to this chemopreventive association [98]. Chemopreventive associations between a high intake of eicosapentaenoic acid or docosahexaenoic acid and colon cancer are well documented [99], and both of these fatty acids can also activate PPARβ/δ (Table 1). Culturing human colonocytes with the high-affinity PPARβ/δ ligand GW501516 does not change cell viability but inhibits cell proliferation [100]. However, this finding was viewed with some uncertainty as the expression of PPARβ/δ mRNA was below the level of detection in these cells. It is difficult to reconcile this finding as others have clearly demonstrated relatively high expression of PPARβ/δ in human colon (Table 3a), so it remains a possibility that PPARβ/δ protein was present in their model or that technical problems prevented the detection of the mRNA. Relative cell proliferation of three human colon cancer cell lines (HT29, SW480 and HCA7) is unchanged in response to cell culture in the presence of a high-affinity PPARβ/δ ligand, although only one concentration was examined in that study [101]. As SW480 cells have a mutant APC and functional β-catenin, if PPARβ/δ was a target of the APC/β-catenin/Tcf4 pathway it might be expected that these cells would have higher PPARβ/δ expression and perhaps be more responsive than HCA7 cells, but this was not observed [101]. Administration of GW0742 to APCmin mice caused no change in the number of polyps in the small intestine (approx. 20 polyps in control compared with approx. 25 polyps in GW0742-treated) or colon (approx. three polyps in control compared with approx. two polyps in GW0742-treated), nor in the size of the polyps [31]. However, administration of GW0742 to mice treated with the colon-specific carcinogen azoxymethane did inhibit colon tumour multiplicity, and this effect required PPARβ/δ as this was not seen in similarly treated PPARβ/δ-null mice [31]. Results from that study also suggested that the observed inhibition of colon tumorigenesis is due to PPARβ/δ-dependent modulation of differentiation as terminal differentiation markers [e.g. ADRP (adipose differentiation-related protein) and FABP (fatty-acid-binding protein)] were increased by GW0742 treatment in colonocytes [31]. The contradiction with the observation that ligand activation of PPARβ/δ inhibits colon cancer resulting from the colon-specific carcinogen azoxymethane [31] by the studies that ligand activation potentiates colon cancer in the APCmin mouse model [93,94] may have been due to differences in the ligand used, as GW0742 was used for one study [31], whereas other studies used GW501516 [93,94]. This hypothesis was examined in a recent paper [102]. Critical analysis of both human colon cancer cell lines (HT29, HCT116 and LS-174T) and mice revealed many inconsistencies with previous studies. For example, inhibition of cell proliferation of HT29, HCT116 and LS-174T cell was observed in response to either GW0742 or GW501516 (1–10 μmol/l), and this effect was unaffected by the presence or absence of serum. The presence of serum was evaluated as a possible variable that could contribute to some of the reported disparities, because inhibition of apoptosis reported by others was found after serum removal [93,94]. In contrast with a previous study suggesting that VEGF is regulated by a PPARβ/δ-dependent mechanism leading to inhibition of apoptosis due to increased phosphorylation of Akt in LS-174T cells [94], no change in VEGF expression, phosphorylation of Akt or PARP (polyADP-ribose polymerase) cleavage was observed in either HT29, HCT116 and LS-174T cells, in the presence or absence of serum, in response to either GW0742 or GW501516 over a broad concentration range known to specifically activate PPARβ/δ (0.1–10 μmo/l) [102]. Furthermore, expression of VEGF and phosphorylation of Akt was not different in colon or colon polyps after administration of either GW0742 or GW501516, or GW0742 respectively [102]. The idea that PPARβ/δ could modulate Akt signalling stems from previous findings made in keratinocytes suggesting that PPARβ/δ directly up-regulates PDK1 (phosphoinositide-dependent kinase 1) and ILK (integrin-linked kinase) and inhibits PTEN (phosphatase and tensin homologue deleted on chromosome 10) expression, which combined cause increased phosphorylation of Akt and inhibition of apoptosis [103]. However, this pathway appears to be context-specific as it does not function in normal mouse or human keratinocytes in the absence of inflammatory signalling [40]. Additionally, this pathway was examined in mouse colon and human cancer cell lines and was found to be non-functional in response to ligand activation of PPARβ/δ [31,102]. Thus there is no convincing evidence that PPARβ/δ modulates apoptosis by either VEGF-dependent regulation of Akt signalling or the PDK/ILK/Akt pathway during colon carcinogenesis.
Collectively, these studies have contributed significantly to the current controversy regarding the effect of ligand activation in colon carcinogenesis. There are a number of possible explanations for these differences that should be discussed. One difference is the model used: the APCmin mouse compared with the chemical induction of colon tumours using azoxymethane. The APCmin mouse, although useful for studying intestinal tumorigenesis, has the drawback that the majority of tumours are found in the small intestine rather than the colon. APC genetically mutant mice can also have a large variation in polyp distribution due to simple differences in housing conditions [104], which might be influenced by differences in gut flora. The studies examining the effect of GW501516 in APCmin mice also used mice on a mixed genetic background [94], such that the results could be influenced by the presence of modifiers of min alleles, which can significantly modulate the incidence of intestinal tumorigenesis in the APCmin mouse model [105–107]. Additionally, the variation observed in the average number of intestinal polyps in control and ligand-treated APCmin mice from one laboratory has a large variation, such that control tumour multiplicity in one study is similar to tumour multiplicity in the ligand-treated group in a separate study [93,94,108]. Thus the studies using azoxymethane are clearly more specific for determining the effect of ligand activation of PPARβ/δ in colon cancer. The hypothesis that ligand activation of PPARβ/δ up-regulates VEGF expression leading to phosphorylation of Akt and inhibition of apoptosis during colon carcinogenesis [94] is inconsistent with findings using the same human colon cancer cell line, as well as comparative in vivo analysis using two different PPARβ/δ ligands demonstrating no changes in phosphorylation of Akt or VEGF expression [31,102]. The hypothesis that ligand activation of PPARβ/δ leads to anti-apoptotic signalling, as suggested by some studies [93,94,108], is inconsistent with a larger body of evidence showing that PPARβ/δ induces terminal differentiation and/or is associated with inhibition of cell growth in a variety of models [13,15,17,18,21,31,38–43,59,80,100,102,109–121]. Despite the controversies, these combined observations shed light on the complexities of PPARβ/δ signalling and emphasize the need to critically examine current working hypotheses for the role of this receptor in colon carcinogenesis.
Modulation of PPARβ/δ Activity by NSAIDS During Colon Carcinogenesis
In addition to the putative differences in PPARβ/δ expression and ligand activation of PPARβ/δ during colon cancer, a few related alternative pathways involving PPARβ/δ have also been postulated as possible targets for colon cancer chemoprevention. One recently hypothesized mechanism suggested that NSAID-mediated inhibition of colon tumorigenesis is facilitated by down-regulating PPARβ/δ expression and function [54]. The reported PPARβ/δ-dependent inhibitory activity induced by NSAIDs was due to the increased expression of LOX1 (lipoxygenase 1)-derived 13-S-HODE [13(S)-hydroxyoctadecadienoic acid] that surprisingly binds to PPARβ/δ but decreased, rather than increased, transcriptional activity of PPARβ/δ as well as decreased the expression of PPARβ/δ [54]. This down-regulation of PPARβ/δ was linked to increased apoptosis, suggesting that PPARβ/δ mediates anti-apoptotic activity. This is similar to another hypothesis linking NSAID-dependent down-regulation of both 14-3-3 and PPARβ/δ with increased apoptosis in human cancer cell lines [56]. It was also suggested that NSAID/LOX1-dependent down-regulation of PPARβ/δ allowed increased activity of PPARγ [55]. This indicates that NSAID-dependent inhibition of colon cancer by NSAIDs could be due, in part, to increased activity of PPARγ, which is known to be a chemopreventive target (reviewed in [122]), in addition to the inhibition of COX2-dependent pathways. Although the findings from these studies indicate a potential novel regulatory pathway for NSAIDs, they are also highly inconsistent with other findings. The first level of inconsistency is the idea that PPARβ/δ is increased during colon cancer, as described above. Although NSAIDs are reported to decrease PPARβ/δ expression in human cancer cell lines, there are other findings indicating that NSAIDs increase the expression of PPARβ/δ (Table 4). If expression of PPARβ/δ is not changed or is decreased as suggested by some studies, then the former hypotheses are infeasible. It is also unclear, as outlined above, whether PPARβ/δ modulates apoptosis and, if so, the mechanisms of this modulation are unknown. The idea that a PPARβ/δ ligand (e.g. 13-S-HODE) can bind to and repress PPARβ/δ activity is unprecedented and inconsistent with other work showing increased PPARβ/δ-dependent transcriptional activity by 13-S-HODE [123]. The relevance of the findings made in cancer cell lines are also not consistent with what is seen in vivo (e.g. PPARβ/δ does not interfere with PPARγ signalling [31,124] and NSAIDs do not change expression of PPARβ/δ; Table 4b). It is also worth noting that administration of the NSAID nimesulide does not change the expression of PPARβ/δ in the mouse colon and effectively inhibits colon cancer in both wild-type and PPARβ/δ-null mice [112] and that NSAID-induced apoptosis in HCT116 cells does not require PPARβ/δ [53]. Taken together, the hypothesis that NSAIDs inhibit colon tumorigenesis through down-regulation of PPARβ/δ is not supported by many observations.
Table 4. Expression of PPARβ/δ in response to NSAIDs.
| (a) Evidence that PPARβ/δ is down-regulated by NSAIDs | ||||
|---|---|---|---|---|
| NSAID | Specimen | Relative expression | Method | Reference |
| Celecoxib | RKO colon cancer cell line | Decreased expression of mRNA encoding PPARβ/δ | Northern blotting | [54] |
| Sulindac sulfide | HT29 colon cancer cell line | Decreased expression of PPARβ/δ | Western blotting | [56] |
| Indomethacin | HT29 colon cancer cell line | Decreased expression of PPARβ/δ | Western blotting | [56] |
| NS-398 | MKN45 gastric cancer cell line | Decreased expression of mRNA encoding PPARβ/δ | Quantitative real-time PCR | [175] |
| Sulindac sulfide/sulindac sulfone | SW480 colon cancer cell line | Decreased expression of PPARβ/δ | Western blotting | [70] |
| (b) Evidence that PPARβ/δ is unchanged or up-regulated by NSAIDs | ||||
| NSAID | Specimen | Relative expression | Method | Reference |
| Nimesulide | HCT116 colon cancer cell line | Increased expression of mRNA encoding PPARβ/δ | Quantitative real-time PCR | [112] |
| Indomethacin | HCT116 colon cancer cell line | Increased expression of mRNA encoding PPARβ/δ | Quantitative real-time PCR | [112] |
| Indomethacin | SW480 colon cancer cell line | Increased expression of mRNA encoding PPARβ/δ | Northern blotting | [176] |
| NS-398 | Human fibroblasts | Increased expression of mRNA encoding PPARβ/δ | Quantitative real-time PCR | [177] |
| Indomethacin | Renal carcinoma | Increased expression of PPARβ/δ | Western blotting | [178] |
| Aspirin | Jurkat (human T-cell lymphoblast-like cell line) | Increased expression of mRNA encoding PPARβ/δ | Northern blotting | [179] |
| Sulindac sulfide/sulindac sulfone | LS174 and Caco2 colon cancer cell lines | No change in the expression of PPARβ/δ | Western blotting | [70] |
| NS-398 | MKN28 gastric cancer cell line | No change in the expression of mRNA encoding PPARβ/δ | Quantitative real-time PCR | [175] |
| Indomethacin | HCT116 colon cancer cell line | No change in the expression of mRNA encoding PPARβ/δ | Northern blotting | [176] |
| Sulindac | APCmin mouse colon and polyps | No change in the expression of PPARβ/δ | Western blotting | [72] |
| Nimesulide | Mouse colon | No change in the expression of mRNA encoding PPARβ/δ | Quantitative real-time PCR | [112] |
| Sulindac sulfide | SW480 and HCT116 colon cancer cell lines | No change in the expression of mRNA encoding PPARβ/δ | Northern blotting | [50] |
Concluding Comments and Data Gaps
In the present review, we have summarized the controversial issues surrounding the role of PPARβ/δ in intestinal physiology and disease. In particular, there remains considerable controversy regarding the role of PPARβ/δ during colon cancer. A number of major points of emphasis should be explored further to more definitively determine how PPARβ/δ modulates colon cancer, including issues related to expression levels, identification of direct PPARβ/δ target genes and the potential roles of polymorphisms in the PPARβ/δ gene in human cancer.
It is clear that there are many inconsistencies in the literature in support of the view that PPARβ/δ is up-regulated by the APC/β-catenin/Tcf4 pathway [50]. Indeed, there are many examples indicating that PPARβ/δ expression is not significantly changed in the absence of APC-dependent signalling, including complete disruption of APC and β-catenin and/or inhibiting Tcf4 signalling. However, there are also examples in some patients indicating that, in some cases, expression does appear to be higher than normal. Does this difference in expression reflect differences or interactions with other transcriptional regulators? Perhaps the APC/β-catenin/Tcf4 pathway modulates PPARβ/δ in some individuals/models, but not others due to unidentified mechanisms such as expression of modifier genes? Given the large differences in expression pattern and lack of change in PPARβ/δ expression in some models, it is not possible to conclude that PPARβ/δ is up-regulated by the APC/β-catenin/Tcf4 pathway. Regardless of whether PPARβ/δ is increased or decreased during colon carcinogenesis, one must also come to terms with the fact that PPARβ/δ expression is already very high in the intestine. There is good evidence that the high expression of PPARβ/δ is due to the requirement for maintenance of terminally differentiated cell types, including Paneth cells. Additionally, recent evidence suggests that PPARβ/δ may be constitutively active in the presence of endogenous fatty acids, which can be found tightly bound to the ligand-binding domain [60]. Thus it is curious that if PPARβ/δ is increased during colon cancer and is causally related, then given its relatively high expression in colonic epithelium, why does colon cancer not affect the majority of humans? One idea that has not been considered to date is that if PPARβ/δ is increased during colon carcinogenesis the increased expression of PPARβ/δ may serve as a protective mechanism. For example, the higher expression of PPARβ/δ could inhibit inflammation and/or mediate the induction of terminal differentiation to prevent proliferation of cells that would normally be predisposed for tumorigenesis. This view is supported by a large body of evidence from a number of independent laboratories showing that PPARβ/δ and/or ligand activation of PPARβ/δ have anti-inflammatory activities [3–23] and is associated or causally linked to the induction of terminal differentiation and/or inhibition of cell proliferation [13,15,17,18,21,31,38–43,59,80,100,102,109–121]. The idea that increased expression of PPARβ/δ may be a protective mechanism is also supported by comparable findings showing increased expression of PPARγ in colon tumours [125] and that PPARγ ligands inhibit or prevent colon carcinogenesis by inducing terminal differentiation, inhibiting cell proliferation and/or increasing apoptosis through both receptor-dependent and receptor-independent mechanisms [122,126]. Although the induction of terminal differentiation is typically considered a good approach to prevent or treat cancer, there is evidence that increased Paneth cell differentiation is associated with increased APC/β-catenin/Tcf4 activity [127]. This could be viewed as suggesting that increased PPARβ/δ-dependent Paneth cell differentiation may be causally linked to colon cancer. Alternatively, as noted above, it remains a possibility that the increased PPARβ/δ-dependent Paneth cell differentiation resulting from enhanced APC/β-catenin/Tcf4 activity is a protective mechanism. Lastly, although the expression of PPARβ/δ was decreased during colon cancer progression in some models, the significance of this change remains uncertain. Further studies are necessary to differentiate between all of these possibilities.
The notion that the presence of PPARβ/δ could mechanistically explain how COX2 inhibitors prevent colon cancer was indeed an attractive hypothesis [50,52]. If COX2-derived metabolites served as ligands for PPARβ/δ that lead to increased signalling for enhanced cell proliferation and if PPARβ/δ expression were elevated similar to c-myc and cyclin D1 via the APC/β-catenin/Tcf4 pathway, then inhibiting PPARβ/δ activity might explain how NSAIDs prevent this disease. Unfortunately, since the original hypothesis was proposed, this idea has been met with significant inconsistencies. For example, suppression of azoxymethane-induced colon cancer by the COX2 inhibitor nimesulide occurs in the absence of PPARβ/δ expression [112]. It was suggested that COX2-derived PGE2 (prostaglandin E2) and Wnt signalling interact with PPARβ/δ leading to a shift from cell death to cell survival [108], but these findings are not supported by the observed PPARβ/δ-independent decrease in colon-specific carcinogenesis in response to COX2 inhibition [112]. Similarly, there is one study suggesting a relationship between a polymorphism in the human PPARβ/δ gene and the protective effect of NSAIDs on colorectal cancer risk [128], whereas another study found no correlation [129]. Given these inconsistencies, it is not surprising that, since originally hypothesized, other related but different hypotheses proposing that NSAIDs up-regulate PPARβ/δ expression leading to chemopreventive effects have also been postulated [54–56]. Similarly, these ideas are also inconsistent on many levels as outlined above. For example, other models have increased expression of PPARβ/δ, rather than decreased expression, and, more importantly, the relevance of increased PPARβ/δ expression in the intestine is very unclear given its relatively high constitutive expression. Further work is necessary to determine whether NSAIDs do in fact up-regulate PPARβ/δ expression during colon cancer progression in both cell culture and in vivo models, and to definitively determine what the specific biological role of PPARβ/δ is in this tissue. In particular, the identification and characterization of critical PPARβ/δ target genes and their specific functional roles is of the highest interest.
There are a number of PPARβ/δ-responsive gene targets described in the literature. Some have been verified and characterized by independent laboratories, whereas others have not. For example, ADRP was initially shown to be a functional PPARβ/δ target gene in macrophages [130]. Since this time, this has been confirmed in other cell types, such as keratinocytes and colonocytes, including the demonstration that this requires PPARβ/δ, as shown by the lack of induction in PPARβ/δ-null mice [21,31,40,131,132]. Similarly, expression of ANGPTL4 (angiopoietin-like protein 4) is increased by treatment with PPARβ/δ ligands [133], due to direct PPARβ/δ-dependent modulation of a cluster of PPREs (PPAR-responsive elements) in an intron [134,135]. Thus it is not surprising that increased expression of ANGPTL4 also occurs via a PPARβ/δ-dependent mechanism in epithelial cells, including keratinocytes, and the colon [21,112]. Expression of L-FABP (liver FABP) is also increased by the activation of PPARβ/δ in the small intestine [136], liver [16] and colon [31] and does not occur in response to ligand activation in PPARβ/δ-null mouse colon [31]. The precise biological role(s) of ADRP, FABP and ANGPTL4 in normal intestinal homoeostasis is unclear, although there is evidence that expression of ADRP and FABP is associated with terminal differentiation of intestinal epithelium [31,137,138]. As the induction of terminal differentiation is one approach suitable for chemoprevention and chemotherapy, this suggests that increased expression of ADRP and L-FABP by ligand activation of PPARβ/δ could be viewed as beneficial. This is consistent with a number of findings including: (i) L-FABP expression is decreased in human colon tumours compared with normal tissue [137,139,140], (ii) disruption of β-catenin/Tcf4 signalling in a human colon cancer cell line causes an up-regulation of terminal differentiation markers including L-FABP [75], (iii) loss of L-FABP expression occurs in colon adenomas, suggesting that this occurs during early tumour progression [139], (iv) increased expression of the related I-FABP (intestinal FABP) inhibits cell growth of a human colon cancer cell line [141], (v) expression of ANGPTL4 is reduced in gastric cancer [142], and (vi) studies in other models suggesting that ANGPTL4 inhibits cell proliferation, angiogenesis and metastasis [143–145]. Although these observations suggest that PPARβ/δ-dependent activation of these proteins would be of benefit for colon cancer chemoprevention, it is important to note that there is also evidence that increased expression of L-FABP is found in some gastric adenocarcinomas [146] and can increase cell proliferation in hepatocytes [147]. Additionally, enhanced expression of ANGPTL4 is found in a prostate cancer cell line [148] and may promote angiogenesis in renal carcinogenesis [149]. Thus, although there is good reason to suggest that increased expression of L-FABP, ADRP and ANGPTL4 by ligand activation of PPARβ/δ could be chemopreventive, there are other observations indicating that this may not be true. Other putative PPARβ/δ target genes with potential links to cancer have also been described, but verification from other laboratories has raised some uncertainty. For example, PPARβ/δ can up-regulate PTEN expression and up-regulate expression of PDK1 and ILK1 leading to increased phosphorylation of Akt and inhibition of apoptosis during skin wound healing [103]. However, this pathway does not function in normal keratinocytes, human colon cancer cell lines or in colonocytes during colon cancer progression [31,40,102]. Expression of VEGF has also been postulated to occur in response to ligand activation of PPARβ/δ in colon cancer cell lines [94], but this effect is not consistently observed [102]. Importantly, there is currently no convincing evidence to date linking a direct PPARβ/δ target gene with oncogenesis. This is somewhat surprising given the strong link between PPARβ/δ and colon cancer. This is clearly a major area of research that is currently needed to strengthen the body of evidence linking PPARβ/δ with colon cancer. Given the fact that FABP, ADRP and ANGPTL4 can be activated by ligand activation of PPARα, PPARβ/δ and PPARγ, due to the presence of essentially redundant response elements in these target genes, it is also of great interest to determine why, or if, there are differential effects of the different PPAR ligands on colon cancer. This is especially true as activation of PPARα and PPARγ can both inhibit intestinal tumorigenesis [150–154]. Alternatively, the consequence of ligand-independent effects of PPARβ/δ (e.g. interactions with NF-κB etc.) and off-target receptor-independent effect of PPARβ/δ ligands should also be examined in more detail.
The recent finding that PPARβ/δ may be a target of the VDR (vitamin D receptor) also raises some interesting questions. It is fairly well accepted that activating the VDR can be chemopreventive for intestinal carcinogenesis and/or that vitamin D deficiency exacerbates intestinal carcinogenesis [155]. As a VDR ligand can increase the expression of PPARβ/δ in human breast and prostate cancer cell lines [156], this might suggest that the increase in PPARβ/δ may participate in the chemopreventive effects of VDR ligands. This idea deserves further evaluation in colon cancer models. Potential participation of PPARβ/δ in modulating colon cancer is also suggested by a recently described antithrombotic role for PPARβ/δ [157]. COX2 can facilitate production of endocannabinoid metabolites that activate PPARβ/δ, which, in turn, negatively regulates the expression of tissue factor, an essential element required for blood coagulation [157]. As ligand activation of PPARβ/δ by endocannabinoids is antithrombotic, this is of interest because antithrombotic agents are being examined for their potential use in preventing tumour growth and metastasis [158–160]. Determining whether endogenous or exogenous endocannabinoids via activation of PPARβ/δ could be used for targeting during colon cancer is another area that deserves investigation.
One of the primary purposes of the present review is to highlight both sides of the controversy regarding the role of PPARβ/δ in colon cancer given the large disparities in the published literature. It is clear that there are currently large data gaps in our understanding of what the role of PPARβ/δ is during intestinal tumorigenesis and how ligand activation of this receptor influences these diseases. It should be noted that there remains some controversy regarding the role of a related receptor, PPARγ, in colon cancer. Previous studies have shown that administration of PPARγ ligands may enhance colon cancer in experimental mouse models [161–163], which was hypothesized to be due to increased expression of PPARγ and activation of this receptor by COX-derived metabolites [125]. The hypothesis that COX-derived metabolites activate PPARγ and potentiate colon cancer [125] turned out to be incorrect; however, subsequent studies still support this contention that PPARγ ligands promote colon cancer [163,164]. Nevertheless, there is currently a larger body of evidence indicating that ligand activation of PPARγ may actually be chemopreventive (reviewed in [122]), which supports past and present clinical trials examining the efficacy of this approach. Until a larger body of evidence is obtained delineating the role of PPARβ/δ in normal intestinal homoeostasis and diseases such as colon cancer, the controversy for this PPAR isoform will remain.
Acknowledgments
We gratefully acknowledge Dr Pallavi Devchand for critical review and suggestions for this manuscript prior to submission. This work is supported, in part, by the National Institutes of Health R01 CA97999 and R01 CA124533 (J.M.P.)
Abbreviations
- ADRP
adipose differentiation-related protein
- ANGPTL4
angiopoietin-like protein 4
- AP1
activator protein 1
- APC
adenomatous polyposis coli
- CLA
conjugated linoleic acid
- COX
cyclo-oxygenase
- FABP
fatty-acid-binding protein
- 13-S-HODE
13(S)-hydroxyoctadecadienoic acid
- IL
interleukin
- ILK
integrin-linked kinase
- L-FABP
liver FABP
- LOX1
lipoxygenase 1
- NF-κB
nuclear factor κB
- NSAID
non-steroidal anti-inflammatory drug
- PDK1
phosphoinositide-dependent kinase 1
- PPAR
peroxisome-proliferator-activated receptor
- PTEN
phosphatase and tensin homologue deleted on chromosome 10
- STAT3
signal transducer and activator of transcription 3
- Tcf4
T-cell transcription factor 4
- TNFα
tumour necrosis factor α
- VDR
vitamin D receptor
- VEGF
vascular endothelial growth factor
References
- 1.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 2.Grimaldi PA. Regulatory functions of PPARβ in metabolism: implications for the treatment of metabolic syndrome. Biochim Biophys Acta. 2007;1771:983–990. doi: 10.1016/j.bbalip.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Ding G, Cheng L, Qin Q, Frontin S, Yang Q. PPARδ modulates lipopolysaccharide-induced TNFα inflammation signaling in cultured cardiomyocytes. J Mol Cell Cardiol. 2006;40:821–828. doi: 10.1016/j.yjmcc.2006.03.422. [DOI] [PubMed] [Google Scholar]
- 4.Rival Y, Beneteau N, Taillandier T, et al. PPARα and PPARδ activators inhibit cytokine-induced nuclear translocation of NF-κB and expression of VCAM-1 in EAhy926 endothelial cells. Eur J Pharmacol. 2002;435:143–151. doi: 10.1016/s0014-2999(01)01589-8. [DOI] [PubMed] [Google Scholar]
- 5.Shan W, Nicol CJ, Ito S, et al. Peroxisome proliferator-activated receptor-β/δ protects against chemically induced liver toxicity in mice. Hepatology. 2008;47:225–235. doi: 10.1002/hep.21925. [DOI] [PubMed] [Google Scholar]
- 6.Arsenijevic D, de Bilbao F, Plamondon J, et al. Increased infarct size and lack of hyperphagic response after focal cerebral ischemia in peroxisome proliferator-activated receptor β-deficient mice. J Cereb Blood Flow Metab. 2006;26:433–445. doi: 10.1038/sj.jcbfm.9600200. [DOI] [PubMed] [Google Scholar]
- 7.Bassaganya-Riera J, Reynolds K, Martino-Catt S, et al. Activation of PPARγ and δ by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology. 2004;127:777–791. doi: 10.1053/j.gastro.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 8.Dyroy E, Rost TH, Pettersen RJ, et al. Tetradecylselenoacetic acid, a PPAR ligand with antioxidant, antiinflammatory, and hypolipidemic properties. Arterioscler Thromb Vasc Biol. 2007;27:628–634. doi: 10.1161/01.ATV.0000255950.70774.d5. [DOI] [PubMed] [Google Scholar]
- 9.Fan Y, Wang Y, Tang Z, et al. Suppression of pro-inflammatory adhesion molecules by PPAR-δ in human vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:315–321. doi: 10.1161/ATVBAHA.107.149815. [DOI] [PubMed] [Google Scholar]
- 10.Graham TL, Mookherjee C, Suckling KE, Palmer CN, Patel L. The PPARδ agonist GW0742X reduces atherosclerosis in LDLR−/− mice. Atherosclerosis. 2005;181:29–37. doi: 10.1016/j.atherosclerosis.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 11.Hollingshead HE, Morimura K, Adachi M, et al. PPARβ/δ protects against experimental colitis through a ligand-independent mechanism. Dig Dis Sci. 2007;52:2912–2919. doi: 10.1007/s10620-006-9644-9. [DOI] [PubMed] [Google Scholar]
- 12.Jakobsen MA, Petersen RK, Kristiansen K, Lange M, Lillevang ST. Peroxisome proliferator-activated receptor α, δ, γ1 and γ2 expressions are present in human monocyte-derived dendritic cells and modulate dendritic cell maturation by addition of subtype-specific ligands. Scand J Immunol. 2006;63:330–337. doi: 10.1111/j.1365-3083.2006.01745.x. [DOI] [PubMed] [Google Scholar]
- 13.Kim DJ, Bility MT, Billin AN, Willson TM, Gonzalez FJ, Peters JM. PPARβ/δ selectively induces differentiation and inhibits cell proliferation. Cell Death Differ. 2006;13:53–60. doi: 10.1038/sj.cdd.4401713. [DOI] [PubMed] [Google Scholar]
- 14.Kim HJ, Ham SA, Kim SU, et al. Transforming growth factor-β1 is a molecular target for the peroxisome proliferator-activated receptor δ. Circ Res. 2008;102:193–200. doi: 10.1161/CIRCRESAHA.107.158477. [DOI] [PubMed] [Google Scholar]
- 15.Man MQ, Barish GD, Schmuth M, et al. Deficiency of PPARβ/δ in the epidermis results in defective cutaneous permeability barrier homeostasis and increased inflammation. J Invest Dermatol. 2007;128:370–377. doi: 10.1038/sj.jid.5701026. [DOI] [PubMed] [Google Scholar]
- 16.Nagasawa T, Inada Y, Nakano S, et al. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARδ agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol. 2006;536:182–191. doi: 10.1016/j.ejphar.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 17.Peters JM, Lee SST, Li W, et al. Growth, adipose, brain and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor β(δ) Mol Cell Biol. 2000;20:5119–5128. doi: 10.1128/mcb.20.14.5119-5128.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Planavila A, Rodriguez-Calvo R, Jove M, et al. Peroxisome proliferator-activated receptor β/δ activation inhibits hypertrophy in neonatal rat cardiomyocytes. Cardiovasc Res. 2005;65:832–841. doi: 10.1016/j.cardiores.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 19.Ravaux L, Denoyelle C, Monne C, Limon I, Raymondjean M, El Hadri K. Inhibition of interleukin-1β-induced group IIA secretory phospholipase A2 expression by peroxisome proliferator-activated receptors (PPARs) in rat vascular smooth muscle cells: cooperation between PPARβ and the proto-oncogene BCL-6. Mol Cell Biol. 2007;27:8374–8387. doi: 10.1128/MCB.00623-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riserus U, Sprecher D, Johnson T, et al. Activation of peroxisome proliferator-activated receptor (PPAR)δ promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes. 2008;57:332–339. doi: 10.2337/db07-1318. [DOI] [PubMed] [Google Scholar]
- 21.Schmuth M, Haqq CM, Cairns WJ, et al. Peroxisome proliferator-activated receptor (PPAR)-β/δ stimulates differentiation and lipid accumulation in keratinocytes. J Invest Dermatol. 2004;122:971–983. doi: 10.1111/j.0022-202X.2004.22412.x. [DOI] [PubMed] [Google Scholar]
- 22.Sheng L, Ye P, Liu YX, Han CG, Zhang ZY. Peroxisome proliferator-activated receptor β/δ activation improves angiotensin II-induced cardiac hypertrophy in vitro. Clin Exp Hypertens. 2008;30:109–119. doi: 10.1080/10641960801945840. [DOI] [PubMed] [Google Scholar]
- 23.Welch JS, Ricote M, Akiyama TE, Gonzalez FJ, Glass CK. PPARγ and PPARδ negatively regulate specific subsets of lipopolysaccharide and IFN-γ target genes in macrophages. Proc Natl Acad Sci USA. 2003;100:6712–6717. doi: 10.1073/pnas.1031789100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kino T, Rice KC, Chrousos GP. The PPARδ agonist GW501516 suppresses interleukin-6-mediated hepatocyte acute phase reaction via STAT3 inhibition. Eur J Clin Invest. 2007;37:425–433. doi: 10.1111/j.1365-2362.2007.01796.x. [DOI] [PubMed] [Google Scholar]
- 25.Woo CH, Massett MP, Shishido T, et al. ERK5 activation inhibits inflammatory responses via peroxisome proliferator-activated receptor δ (PPARδ) stimulation. J Biol Chem. 2006;281:32164–32174. doi: 10.1074/jbc.M602369200. [DOI] [PubMed] [Google Scholar]
- 26.Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502–2512. doi: 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 27.Delerive P, De Bosscher K, Besnard S, et al. Peroxisome proliferator-activated receptor α negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-κB and AP-1. J Biol Chem. 1999;274:32048–32054. doi: 10.1074/jbc.274.45.32048. [DOI] [PubMed] [Google Scholar]
- 28.Delerive P, Martin-Nizard F, Chinetti G, et al. Peroxisome proliferator-activated receptor activators inhibit thrombin- induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ Res. 1999;85:394–402. doi: 10.1161/01.res.85.5.394. [DOI] [PubMed] [Google Scholar]
- 29.Shi Y, Hon M, Evans RM. The peroxisome proliferator-activated receptor δ, an integrator of transcriptional repression and nuclear receptor signaling. Proc Natl Acad Sci USA. 2002;99:2613–2618. doi: 10.1073/pnas.052707099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peters JM, Aoyama T, Burns AM, Gonzalez FJ. Bezafibrate is a dual ligand for PPARα and PPARβ: studies using null mice. Biochim Biophys Acta. 2003;1632:80–89. doi: 10.1016/s1388-1981(03)00065-9. [DOI] [PubMed] [Google Scholar]
- 31.Marin HE, Peraza MA, Billin AN, et al. Ligand activation of peroxisome proliferator-activated receptor β/δ (PPARβ/δ) inhibits colon carcinogenesis. Cancer Res. 2006;66:4394–4401. doi: 10.1158/0008-5472.CAN-05-4277. [DOI] [PubMed] [Google Scholar]
- 32.Escher P, Braissant O, Basu-Modak S, Michalik L, Wahli W, Desvergne B. Rat PPARs: quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology. 2001;142:4195–4202. doi: 10.1210/endo.142.10.8458. [DOI] [PubMed] [Google Scholar]
- 33.Larsen LK, Amri EZ, Mandrup S, Pacot C, Kristiansen K. Genomic organization of the mouse peroxisome proliferator-activated receptor β/δ gene: alternative promoter usage and splicing yield transcripts exhibiting differential translational efficiency. Biochem J. 2002;366:767–775. doi: 10.1042/BJ20011821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huin C, Corriveau L, Bianchi A, et al. Differential expression of peroxisome proliferator-activated receptors (PPARs) in the developing human fetal digestive tract. J Histochem Cytochem. 2000;48:603–611. doi: 10.1177/002215540004800504. [DOI] [PubMed] [Google Scholar]
- 35.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor α mediates the adaptive response to fasting. J Clin Invest. 1999;103:1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor α (PPARα) in the cellular fasting response: the PPARα-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci USA. 1999;96:7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Higashiyama H, Billin AN, Okamoto Y, Kinoshita M, Asano S. Expression profiling of peroxisome proliferator-activated receptor-δ (PPAR-δ) in mouse tissues using tissue microarray. Histochem Cell Biol. 2007;127:485–494. doi: 10.1007/s00418-007-0279-5. [DOI] [PubMed] [Google Scholar]
- 38.Varnat F, Heggeler BB, Grisel P, et al. PPARβ/δ regulates paneth cell differentiation via controlling the hedgehog signaling pathway. Gastroenterology. 2006;131:538–553. doi: 10.1053/j.gastro.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 39.Aung CS, Faddy HM, Lister EJ, Monteith GR, Roberts-Thomson SJ. Isoform specific changes in PPARα and β in colon and breast cancer with differentiation. Biochem Biophys Res Commun. 2006;340:656–660. doi: 10.1016/j.bbrc.2005.12.061. [DOI] [PubMed] [Google Scholar]
- 40.Burdick AD, Bility MT, Girroir EE, et al. Ligand activation of peroxisome proliferator-activated receptor-β/δ(PPARβ/δ) inhibits cell growth of human N/TERT-1 keratinocytes. Cell Signaling. 2007;19:1163–1171. doi: 10.1016/j.cellsig.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nadra K, Anghel SI, Joye E, et al. Differentiation of trophoblast giant cells and their metabolic functions are dependent on peroxisome proliferator-activated receptor β/δ. Mol Cell Biol. 2006;26:3266–3281. doi: 10.1128/MCB.26.8.3266-3281.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan NS, Michalik L, Noy N, et al. Critical roles of PPARβ/δ in keratinocyte response to inflammation. Genes Dev. 2001;15:3263–3277. doi: 10.1101/gad.207501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Westergaard M, Henningsen J, Svendsen ML, et al. Modulation of keratinocyte gene expression and differentiation by PPAR-selective ligands and tetradecylthioacetic acid. J Invest Dermatol. 2001;116:702–712. doi: 10.1046/j.1523-1747.2001.01329.x. [DOI] [PubMed] [Google Scholar]
- 44.Moya-Camarena SY, Van den Heuvel JP, Belury MA. Conjugated linoleic acid activates peroxisome proliferator-activated receptor α and β subtypes but does not induce hepatic peroxisome proliferation in Sprague-Dawley rats. Biochim Biophys Acta. 1999;1436:331–342. doi: 10.1016/s0005-2760(98)00121-0. [DOI] [PubMed] [Google Scholar]
- 45.Chen C, Shah YM, Morimura K, et al. Metabolomics reveals that hepatic stearoyl-CoA desaturase 1 downregulation exacerbates inflammation and acute colitis. Cell Metab. 2008;7:135–147. doi: 10.1016/j.cmet.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 47.Wehkamp J, Salzman NH, Porter E, et al. Reduced Paneth cell α-defensins in ileal Crohn's disease. Proc Natl Acad Sci USA. 2005;102:18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JW, Bajwa PJ, Carson MJ, et al. Fenofibrate represses interleukin-17 and interferon-γ expression and improves colitis in interleukin-10-deficient mice. Gastroenterology. 2007;133:108–123. doi: 10.1053/j.gastro.2007.03.113. [DOI] [PubMed] [Google Scholar]
- 49.Murray PJ. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr Opin Pharmacol. 2006;6:379–386. doi: 10.1016/j.coph.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 50.He TC, Chan TA, Vogelstein B, Kinzler KW. PPARδ is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335–345. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 52.Gupta RA, Tan J, Krause WF, et al. Prostacyclin-mediated activation of peroxisome proliferator-activated receptor δ in colorectal cancer. Proc Natl Acad Sci USA. 2000;97:13275–13280. doi: 10.1073/pnas.97.24.13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park BH, Vogelstein B, Kinzler KW. Genetic disruption of PPARδ decreases the tumorigenicity of human colon cancer cells. Proc Natl Acad Sci USA. 2001;98:2598–2603. doi: 10.1073/pnas.051630998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shureiqi I, Jiang W, Zuo X, et al. The 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid down-regulates PPAR-δ to induce apoptosis in colorectal cancer cells. Proc Natl Acad Sci USA. 2003;100:9968–9973. doi: 10.1073/pnas.1631086100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zuo X, Wu Y, Morris JS, et al. Oxidative metabolism of linoleic acid modulates PPAR-β/δ suppression of PPAR-γ activity. Oncogene. 2006;25:1225–1241. doi: 10.1038/sj.onc.1209160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liou JY, Ghelani D, Yeh S, Wu KK. Nonsteroidal anti-inflammatory drugs induce colorectal cancer cell apoptosis by suppressing 14-13-3ϵ. Cancer Res. 2007;67:3185–3191. doi: 10.1158/0008-5472.CAN-06-3431. [DOI] [PubMed] [Google Scholar]
- 57.Harman FS, Nicol CJ, Marin HE, Ward JM, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-δ attenuates colon carcinogenesis. Nat Med. 2004;10:481–483. doi: 10.1038/nm1026. [DOI] [PubMed] [Google Scholar]
- 58.Reed KR, Sansom OJ, Hayes AJ, et al. PPARδ status and Apc-mediated tumourigenesis in the mouse intestine. Oncogene. 2004;23:8992–8996. doi: 10.1038/sj.onc.1208143. [DOI] [PubMed] [Google Scholar]
- 59.Yang L, Zhou ZG, Zheng XL, et al. RNA interference against peroxisome proliferator-activated receptor δ gene promotes proliferation of human colorectal cancer cells. Dis Colon Rectum. 2008;51:318–328. doi: 10.1007/s10350-007-9145-8. [DOI] [PubMed] [Google Scholar]
- 60.Fyffe SA, Alphey MS, Buetow L, et al. Reevaluation of the PPAR-β/δ ligand binding domain model reveals why it exhibits the activated form. Mol Cell. 2006;21:1–2. doi: 10.1016/j.molcel.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 61.Delage B, Rullier A, Capdepont M, Rullier E, Cassand P. The effect of body weight on altered expression of nuclear receptors and cyclooxygenase-2 in human colorectal cancers. Nutr J. 2007;6:20. doi: 10.1186/1475-2891-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feilchenfeldt J, Brundler MA, Soravia C, Totsch M, Meier CA. Peroxisome proliferator-activated receptors (PPARs) and associated transcription factors in colon cancer: reduced expression of PPARγ-coactivator 1 (PGC-1) Cancer Lett. 2004;203:25–33. doi: 10.1016/j.canlet.2003.08.024. [DOI] [PubMed] [Google Scholar]
- 63.Takayama O, Yamamoto H, Damdinsuren B, et al. Expression of PPARδ in multistage carcinogenesis of the colorectum: implications of malignant cancer morphology. Br J Cancer. 2006;95:889–895. doi: 10.1038/sj.bjc.6603343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Knutsen HK, Olstorn HB, Paulsen JE, et al. Increased levels of PPARβ/δ and cyclin D1 in flat dysplastic ACF and adenomas in ApcMin/+ mice. Anticancer Res. 2005;25:3781–3789. [PubMed] [Google Scholar]
- 65.Ouyang N, Williams JL, Rigas B. NO-donating aspirin isomers downregulate peroxisome proliferator-activated receptor (PPAR)δ expression in APCmin/+ mice proportionally to their tumor inhibitory effect: implications for the role of PPARδ in carcinogenesis. Carcinogenesis. 2006;27:232–239. doi: 10.1093/carcin/bgi221. [DOI] [PubMed] [Google Scholar]
- 66.Notterman DA, Alon U, Sierk AJ, Levine AJ. Transcriptional gene expression profiles of colorectal adenoma, adenocarcinoma, and normal tissue examined by oligonucleotide arrays. Cancer Res. 2001;61:3124–3130. [PubMed] [Google Scholar]
- 67.Hao CY, Moore DH, Wong P, Bennington JL, Lee NM, Chen LC. Alteration of gene expression in macroscopically normal colonic mucosa from individuals with a family history of sporadic colon cancer. Clin Cancer Res. 2005;11:1400–1407. doi: 10.1158/1078-0432.CCR-04-1942. [DOI] [PubMed] [Google Scholar]
- 68.Chen LC, Hao CY, Chiu YS, et al. Alteration of gene expression in normal-appearing colon mucosa of APCmin mice and human cancer patients. Cancer Res. 2004;64:3694–3700. doi: 10.1158/0008-5472.CAN-03-3264. [DOI] [PubMed] [Google Scholar]
- 69.Yang L, Zhou ZG, Luo HZ, Zhou B, Xia QJ, Tian C. Quantitative analysis of PPARδ mRNA by real-time RT-PCR in 86 rectal cancer tissues. Eur J Surg Oncol. 2006;32:181–185. doi: 10.1016/j.ejso.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 70.Chang WC, Everley LC, Pfeiffer GR, II, Cooper HS, Barusevicius A, Clapper ML. Sulindac sulfone is most effective in modulating β-catenin-mediated transcription in cells with mutant APC. Ann NY Acad Sci. 2005;1059:41–55. doi: 10.1196/annals.1339.020. [DOI] [PubMed] [Google Scholar]
- 71.Schaefer KL, Takahashi H, Morales VM, et al. PPARγ inhibitors reduce tubulin protein levels by a PPARγ, PPARδ and proteasome-independent mechanism, resulting in cell cycle arrest, apoptosis and reduced metastasis of colorectal carcinoma cells. Int J Cancer. 2007;120:702–713. doi: 10.1002/ijc.22361. [DOI] [PubMed] [Google Scholar]
- 72.Orner GA, Dashwood WM, Blum CA, Diaz GD, Li Q, Dashwood RH. Suppression of tumorigenesis in the Apcmin mouse: down-regulation of β-catenin signaling by a combination of tea plus sulindac. Carcinogenesis. 2003;24:263–267. doi: 10.1093/carcin/24.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sansom OJ, Reed KR, Hayes AJ, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18:1385–1390. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fevr T, Robine S, Louvard D, Huelsken J. Wnt/β-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol Cell Biol. 2007;27:7551–7559. doi: 10.1128/MCB.01034-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van de Wetering M, Sancho E, Verweij C, et al. The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–250. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 76.Honn KV, Meyer J. Thromboxanes and prostacyclin: positive and negative modulators of tumor growth. Biochem Biophys Res Commun. 1981;102:1122–1129. doi: 10.1016/s0006-291x(81)80128-3. [DOI] [PubMed] [Google Scholar]
- 77.Honn KV, Cicone B, Skoff A. Prostacyclin: a potent antimetastatic agent. Science. 1981;212:1270–1272. doi: 10.1126/science.7015512. [DOI] [PubMed] [Google Scholar]
- 78.Schneider MR, Tang DG, Schirner M, Honn KV. Prostacyclin and its analogues: antimetastatic effects and mechanisms of action. Cancer Metastasis Rev. 1994;13:349–364. doi: 10.1007/BF00666104. [DOI] [PubMed] [Google Scholar]
- 79.Daneker GW, Lund SA, Caughman SW, Staley CA, Wood WC. Anti-metastatic prostacyclins inhibit the adhesion of colon carcinoma to endothelial cells by blocking E-selectin expression. Clin Exp Metastasis. 1996;14:230–238. doi: 10.1007/BF00053896. [DOI] [PubMed] [Google Scholar]
- 80.Fukumoto K, Yano Y, Virgona N, et al. Peroxisome proliferator-activated receptor δ as a molecular target to regulate lung cancer cell growth. FEBS Lett. 2005;579:3829–3836. doi: 10.1016/j.febslet.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 81.Li RC, Cindrova-Davies T, Skepper JN, Sellers LA. Prostacyclin induces apoptosis of vascular smooth muscle cells by a cAMP-mediated inhibition of extracellular signal-regulated kinase activity and can counteract the mitogenic activity of endothelin-1 or basic fibroblast growth factor. Circ Res. 2004;94:759–767. doi: 10.1161/01.RES.0000121568.40692.97. [DOI] [PubMed] [Google Scholar]
- 82.Cutler NS, Graves-Deal R, LaFleur BJ, et al. Stromal production of prostacyclin confers an antiapoptotic effect to colonic epithelial cells. Cancer Res. 2003;63:1748–1751. [PubMed] [Google Scholar]
- 83.Keith RL, Miller YE, Hoshikawa Y, et al. Manipulation of pulmonary prostacyclin synthase expression prevents murine lung cancer. Cancer Res. 2002;62:734–740. [PubMed] [Google Scholar]
- 84.Keith RL, Miller YE, Hudish TM, et al. Pulmonary prostacyclin synthase overexpression chemoprevents tobacco smoke lung carcinogenesis in mice. Cancer Res. 2004;64:5897–5904. doi: 10.1158/0008-5472.CAN-04-1070. [DOI] [PubMed] [Google Scholar]
- 85.Siegl AM, Smith JB, Silver MJ, Nicolaou KC, Ahern D. Selective binding site for [3H]prostacyclin on platelets. J Clin Invest. 1979;63:215–220. doi: 10.1172/JCI109292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Narumiya S. Molecular diversity of prostanoid receptors; subtypes and isoforms of prostaglandin E receptor. Adv Exp Med Biol. 1997;400A:207–213. doi: 10.1007/978-1-4615-5325-0_30. [DOI] [PubMed] [Google Scholar]
- 87.Fauti T, Müller-Brüsselbach S, Kreutzer M, et al. Induction of PPARβ and prostacyclin (PGI2) synthesis by Raf signaling: failure of PGI2 to activate PPARβ. FEBS J. 2005;273:170–179. doi: 10.1111/j.1742-4658.2005.05055.x. [DOI] [PubMed] [Google Scholar]
- 88.Hendrickse CW, Keighley MR, Neoptolemos JP. Dietary omega-3 fats reduce proliferation and tumor yields at colorectal anastomosis in rats. Gastroenterology. 1995;109:431–439. doi: 10.1016/0016-5085(95)90330-5. [DOI] [PubMed] [Google Scholar]
- 89.Minoura T, Takata T, Sakaguchi M, et al. Effect of dietary eicosapentaenoic acid on azoxymethane-induced colon carcinogenesis in rats. Cancer Res. 1988;48:4790–4794. [PubMed] [Google Scholar]
- 90.Reddy BS, Burill C, Rigotty J. Effect of diets high in omega-3 and omega-6 fatty acids on initiation and postinitiation stages of colon carcinogenesis. Cancer Res. 1991;51:487–491. [PubMed] [Google Scholar]
- 91.Reddy BS, Sugie S. Effect of different levels of omega-3 and omega-6 fatty acids on azoxymethane-induced colon carcinogenesis in F344 rats. Cancer Res. 1988;48:6642–6647. [PubMed] [Google Scholar]
- 92.Weijenberg MP, Luchtenborg M, de Goeij AF, et al. Dietary fat and risk of colon and rectal cancer with aberrant MLH1 expression, APC or KRAS genes. Cancer Causes Control. 2007;18:865–879. doi: 10.1007/s10552-007-9032-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gupta RA, Wang D, Katkuri S, Wang H, Dey SK, DuBois RN. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-δ accelerates intestinal adenoma growth. Nat Med. 2004;10:245–247. doi: 10.1038/nm993. [DOI] [PubMed] [Google Scholar]
- 94.Wang D, Wang H, Guo Y, et al. Crosstalk between peroxisome proliferator-activated receptor δ and VEGF stimulates cancer progression. Proc Natl Acad Sci USA. 2006;103:19069–19074. doi: 10.1073/pnas.0607948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zock PL, Katan MB. Linoleic acid intake and cancer risk: a review and meta-analysis. Am J Clin Nutr. 1998;68:142–153. doi: 10.1093/ajcn/68.1.142. [DOI] [PubMed] [Google Scholar]
- 96.Llor X, Pons E, Roca A, et al. The effects of fish oil, olive oil, oleic acid and linoleic acid on colorectal neoplastic processes. Clin Nutr. 2003;22:71–79. doi: 10.1054/clnu.2002.0627. [DOI] [PubMed] [Google Scholar]
- 97.Escrich E, Moral R, Grau L, Costa I, Solanas M. Molecular mechanisms of the effects of olive oil and other dietary lipids on cancer. Mol Nutr Food Res. 2007;51:1279–1292. doi: 10.1002/mnfr.200700213. [DOI] [PubMed] [Google Scholar]
- 98.Hashim YZ, Eng M, Gill CI, McGlynn H, Rowland IR. Components of olive oil and chemoprevention of colorectal cancer. Nutr Rev. 2005;63:374–386. doi: 10.1111/j.1753-4887.2005.tb00374.x. [DOI] [PubMed] [Google Scholar]
- 99.Theodoratou E, McNeill G, Cetnarskyj R, et al. Dietary fatty acids and colorectal cancer: a case-control study. Am J Epidemiol. 2007;166:181–195. doi: 10.1093/aje/kwm063. [DOI] [PubMed] [Google Scholar]
- 100.Matthiessen MW, Pedersen G, Albrektsen T, Adamsen S, Fleckner J, Brynskov J. Peroxisome proliferator-activated receptor expression and activation in normal human colonic epithelial cells and tubular adenomas. Scand J Gastroenterol. 2005;40:198–205. doi: 10.1080/00365520410009573. [DOI] [PubMed] [Google Scholar]
- 101.Stephen RL, Gustafsson MC, Jarvis M, et al. Activation of peroxisome proliferator-activated receptor δ stimulates the proliferation of human breast and prostate cancer cell lines. Cancer Res. 2004;64:3162–3170. doi: 10.1158/0008-5472.can-03-2760. [DOI] [PubMed] [Google Scholar]
- 102.Hollingshead HE, Killins RL, Borland MG, et al. Peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) ligands do not potentiate growth of human cancer cell lines. Carcinogenesis. 2007;28:2641–2649. doi: 10.1093/carcin/bgm183. [DOI] [PubMed] [Google Scholar]
- 103.Di-Poi N, Tan NS, Michalik L, Wahli W, Desvergne B. Antiapoptotic role of PPARβ in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol Cell. 2002;10:721–733. doi: 10.1016/s1097-2765(02)00646-9. [DOI] [PubMed] [Google Scholar]
- 104.Colnot S, Niwa-Kawakita M, Hamard G, et al. Colorectal cancers in a new mouse model of familial adenomatous polyposis: influence of genetic and environmental modifiers. Lab Invest. 2004;84:1619–1630. doi: 10.1038/labinvest.3700180. [DOI] [PubMed] [Google Scholar]
- 105.Dietrich WF, Lander ES, Smith JS, et al. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell. 1993;75:631–639. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- 106.Moser AR, Dove WF, Roth KA, Gordon JI. The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. J Cell Biol. 1992;116:1517–1526. doi: 10.1083/jcb.116.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Silverman KA, Koratkar R, Siracusa LD, Buchberg AM. Identification of the modifier of Min 2 (Mom2) locus, a new mutation that influences Apc-induced intestinal neoplasia. Genome Res. 2002;12:88–97. doi: 10.1101/gr.206002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang D, Wang H, Shi Q, et al. Prostaglandin E2 promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor δ. Cancer Cell. 2004;6:285–295. doi: 10.1016/j.ccr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 109.Ali FY, Egan K, Fitzgerald GA, et al. Role of prostacyclin receptor versus PPARβ with treprostinil sodium on lung fibroblast proliferation. Am J Respir Cell Mol Biol. 2005;34:242–246. doi: 10.1165/rcmb.2005-0289OC. [DOI] [PubMed] [Google Scholar]
- 110.Di Loreto S, D'Angelo B, D'Amico MA, et al. PPARβ agonists trigger neuronal differentiation in the human neuroblastoma cell line SH-SY5Y. J Cell Physiol. 2007;211:837–847. doi: 10.1002/jcp.20996. [DOI] [PubMed] [Google Scholar]
- 111.Girroir EE, Hollingshead HE, Billin AN, et al. Peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) ligands inhibit growth of UACC903 and MCF7 human cancer cell lines. Toxicology. 2008;243:236–243. doi: 10.1016/j.tox.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hollingshead HE, Borland MG, Billin AN, Willson TM, Gonzalez FJ, Peters JM. Ligand activation of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) and inhibition of cyclooxygenase 2 (COX2) attenuate colon carcinogenesis through independent signaling mechanisms. Carcinogenesis. 2008;29:169–176. doi: 10.1093/carcin/bgm209. [DOI] [PubMed] [Google Scholar]
- 113.Kim DJ, Akiyama TE, Harman FS, et al. Peroxisome proliferator-activated receptor β (δ)-dependent regulation of ubiquitin C expression contributes to attenuation of skin carcinogenesis. J Biol Chem. 2004;279:23719–23727. doi: 10.1074/jbc.M312063200. [DOI] [PubMed] [Google Scholar]
- 114.Kim DJ, Murray IA, Burns AM, Gonzalez FJ, Perdew GH, Peters JM. Peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) inhibits epidermal cell proliferation by down-regulation of kinase activity. J Biol Chem. 2005;280:9519–9527. doi: 10.1074/jbc.M413808200. [DOI] [PubMed] [Google Scholar]
- 115.Martinasso G, Maggiora M, Trombetta A, Canuto RA, Muzio G. Effects of di(2-ethylhexyl) phthalate, a widely used peroxisome proliferator and plasticizer, on cell growth in the human keratinocyte cell line NCTC 2544. J Toxicol Environ Health A. 2006;69:353–365. doi: 10.1080/15287390500227522. [DOI] [PubMed] [Google Scholar]
- 116.Matsuura H, Adachi H, Smart RC, Xu X, Arata J, Jetten AM. Correlation between expression of peroxisome proliferator-activated receptor β and squamous differentiation in epidermal and tracheobronchial epithelial cells. Mol Cell Endocrinol. 1999;147:85–92. doi: 10.1016/s0303-7207(98)00214-7. [DOI] [PubMed] [Google Scholar]
- 117.Otsuyama KI, Ma Z, Abroun S, et al. PPARβ-mediated growth suppression of baicalein and dexamethasone in human myeloma cells. Leukemia. 2007;21:187–190. doi: 10.1038/sj.leu.2404462. [DOI] [PubMed] [Google Scholar]
- 118.Saluja I, Granneman JG, Skoff RP. PPAR δ agonists stimulate oligodendrocyte differentiation in tissue culture. Glia. 2001;33:191–204. [PubMed] [Google Scholar]
- 119.Teunissen BE, Smeets PJ, Willemsen PH, De Windt LJ, Van der Vusse GJ, Van Bilsen M. Activation of PPARδ inhibits cardiac fibroblast proliferation and the transdifferentiation into myofibroblasts. Cardiovasc Res. 2007;75:519–529. doi: 10.1016/j.cardiores.2007.04.026. [DOI] [PubMed] [Google Scholar]
- 120.Vosper H, Khoudoli GA, Palmer CN. The peroxisome proliferator activated receptor δ is required for the differentiation of THP-1 monocytic cells by phorbol ester. Nucl Recept. 2003;1:9. doi: 10.1186/1478-1336-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Werling U, Siehler S, Litfin M, Nau H, Gottlicher M. Induction of differentiation in F9 cells and activation of peroxisome proliferator-activated receptor δ by valproic acid and its teratogenic derivatives. Mol Pharmacol. 2001;59:1269–1276. doi: 10.1124/mol.59.5.1269. [DOI] [PubMed] [Google Scholar]
- 122.Peraza MA, Burdick AD, Marin HE, Gonzalez FJ, Peters JM. The toxicology of ligands for peroxisome proliferator-activated receptors (PPAR) Toxicol Sci. 2006;90:269–295. doi: 10.1093/toxsci/kfj062. [DOI] [PubMed] [Google Scholar]
- 123.Coleman JD, Prabhu KS, Thompson JT, et al. The oxidative stress mediator 4-hydroxynonenal is an intracellular agonist of the nuclear receptor peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) Free Radical Biol Med. 2007;42:1155–1164. doi: 10.1016/j.freeradbiomed.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Matsusue K, Peters JM, Gonzalez FJ. PPARβ/δ potentiates PPARγ-stimulated adipocyte differentiation. FASEB J. 2004;18:1477–1479. doi: 10.1096/fj.04-1944fje. [DOI] [PubMed] [Google Scholar]
- 125.DuBois RN, Gupta R, Brockman J, Reddy BS, Krakow SL, Lazar MA. The nuclear eicosanoid receptor, PPARγ, is aberrantly expressed in colonic cancers. Carcinogenesis. 1998;19:49–53. doi: 10.1093/carcin/19.1.49. [DOI] [PubMed] [Google Scholar]
- 126.Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. 2004;4:61–70. doi: 10.1038/nrc1254. [DOI] [PubMed] [Google Scholar]
- 127.Andreu P, Colnot S, Godard C, et al. Crypt-restricted proliferation and commitment to the Paneth cell lineage following Apc loss in the mouse intestine. Development. 2005;132:1443–1451. doi: 10.1242/dev.01700. [DOI] [PubMed] [Google Scholar]
- 128.Siezen CL, Tijhuis MJ, Kram NR, et al. Protective effect of nonsteroidal anti-inflammatory drugs on colorectal adenomas is modified by a polymorphism in peroxisome proliferator-activated receptor δ. Pharmacogenet Genomics. 2006;16:43–50. doi: 10.1097/01.fpc.0000182778.03180.f3. [DOI] [PubMed] [Google Scholar]
- 129.McGreavey LE, Turner F, Smith G, et al. No evidence that polymorphisms in CYP2C8, CYP2C9, UGT1A6, PPARδ and PPARγ act as modifiers of the protective effect of regular NSAID use on the risk of colorectal carcinoma. Pharmacogenet Genomics. 2005;15:713–721. doi: 10.1097/01.fpc.0000174786.85238.63. [DOI] [PubMed] [Google Scholar]
- 130.Chawla A, Lee CH, Barak Y, et al. PPARδ is a very low-density lipoprotein sensor in macrophages. Proc Natl Acad Sci USA. 2003;100:1268–1273. doi: 10.1073/pnas.0337331100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Piqueras L, Reynolds AR, Hodivala-Dilke KM, et al. Activation of PPARβ/δ induces endothelial cell proliferation and angiogenesis. Arterioscler Thromb Vasc Biol. 2007;27:63–69. doi: 10.1161/01.ATV.0000250972.83623.61. [DOI] [PubMed] [Google Scholar]
- 132.Tachibana K, Kobayashi Y, Tanaka T, et al. Gene expression profiling of potential peroxisome proliferator-activated receptor (PPAR) target genes in human hepatoblastoma cell lines inducibly expressing different PPAR isoforms. Nucl Recept. 2005;3:3. doi: 10.1186/1478-1336-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ge H, Cha JY, Gopal H, et al. Differential regulation and properties of angiopoietin-like proteins 3 and 4. J Lipid Res. 2005;46:1484–1490. doi: 10.1194/jlr.M500005-JLR200. [DOI] [PubMed] [Google Scholar]
- 134.Heinaniemi M, Uski JO, Degenhardt T, Carlberg C. Meta-analysis of primary target genes of peroxisome proliferator-activated receptors. Genome Biol. 2007;8:R147. doi: 10.1186/gb-2007-8-7-r147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mandard S, Zandbergen F, Tan NS, et al. The direct peroxisome proliferator-activated receptor target fasting-induced adipose factor (FIAF/PGAR/ANGPTL4) is present in blood plasma as a truncated protein that is increased by fenofibrate treatment. J Biol Chem. 2004;279:34411–34420. doi: 10.1074/jbc.M403058200. [DOI] [PubMed] [Google Scholar]
- 136.Poirier H, Niot I, Monnot MC, et al. Differential involvement of peroxisome-proliferator-activated receptors α and δ in fibrate and fatty-acid-mediated inductions of the gene encoding liver fatty-acid-binding protein in the liver and the small intestine. Biochem J. 2001;355:481–488. doi: 10.1042/0264-6021:3550481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Davidson NO, Ifkovits CA, Skarosi SF, et al. Tissue and cell-specific patterns of expression of rat liver and intestinal fatty acid binding protein during development and in experimental colonic and small intestinal adenocarcinomas. Lab Invest. 1993;68:663–675. [PubMed] [Google Scholar]
- 138.Rubin DC, Ong DE, Gordon JI. Cellular differentiation in the emerging fetal rat small intestinal epithelium: mosaic patterns of gene expression. Proc Natl Acad Sci USA. 1989;86:1278–1282. doi: 10.1073/pnas.86.4.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lawrie LC, Dundas SR, Curran S, Murray GI. Liver fatty acid binding protein expression in colorectal neoplasia. Br J Cancer. 2004;90:1955–1960. doi: 10.1038/sj.bjc.6601828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Melis R, White R. Characterization of colonic polyps by two-dimensional gel electrophoresis. Electrophoresis. 1999;20:1055–1064. doi: 10.1002/(SICI)1522-2683(19990101)20:4/5<1055::AID-ELPS1055>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 141.Darimont C, Gradoux N, Persohn E, Cumin F, De Pover A. Effects of intestinal fatty acid-binding protein overexpression on fatty acid metabolism in Caco-2 cells. J Lipid Res. 2000;41:84–92. [PubMed] [Google Scholar]
- 142.Kaneda A, Kaminishi M, Yanagihara K, Sugimura T, Ushijima T. Identification of silencing of nine genes in human gastric cancers. Cancer Res. 2002;62:6645–6650. [PubMed] [Google Scholar]
- 143.Galaup A, Cazes A, Le Jan S, et al. Angiopoietin-like 4 prevents metastasis through inhibition of vascular permeability and tumor cell motility and invasiveness. Proc Natl Acad Sci USA. 2006;103:18721–18726. doi: 10.1073/pnas.0609025103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ito Y, Oike Y, Yasunaga K, et al. Inhibition of angiogenesis and vascular leakiness by angiopoietin-related protein 4. Cancer Res. 2003;63:6651–6657. [PubMed] [Google Scholar]
- 145.Li KQ, Li WL, Peng SY, Shi XY, Tang HL, Liu YB. Anti-tumor effect of recombinant retroviral vector-mediated human ANGPTL4 gene transfection. Chin Med J. 2004;117:1364–1369. [PubMed] [Google Scholar]
- 146.Hashimoto T, Kusakabe T, Watanabe K, et al. Liver-type fatty acid-binding protein is highly expressed in intestinal metaplasia and in a subset of carcinomas of the stomach without association with the fatty acid synthase status in the carcinoma. Pathobiology. 2004;71:115–122. doi: 10.1159/000076465. [DOI] [PubMed] [Google Scholar]
- 147.Sorof S. Modulation of mitogenesis by liver fatty acid binding protein. Cancer Metastasis Rev. 1994;13:317–336. doi: 10.1007/BF00666102. [DOI] [PubMed] [Google Scholar]
- 148.Ifon ET, Pang AL, Johnson W, et al. U94 alters FN1 and ANGPTL4 gene expression and inhibits tumorigenesis of prostate cancer cell line PC3. Cancer Cell Int. 2005;5:19. doi: 10.1186/1475-2867-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Le Jan S, Amy C, Cazes A, et al. Angiopoietin-like 4 is a proangiogenic factor produced during ischemia and in conventional renal cell carcinoma. Am J Pathol. 2003;162:1521–1528. doi: 10.1016/S0002-9440(10)64285-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jackson L, Wahli W, Michalik L, et al. Potential role for peroxisome proliferator activated receptor (PPAR) in preventing colon cancer. Gut. 2003;52:1317–1322. doi: 10.1136/gut.52.9.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mutoh M, Niho N, Wakabayashi K. Concomitant suppression of hyperlipidemia and intestinal polyp formation by increasing lipoprotein lipase activity in Apc-deficient mice. Biol Chem. 2006;387:381–385. doi: 10.1515/BC.2006.051. [DOI] [PubMed] [Google Scholar]
- 152.Niho N, Takahashi M, Kitamura T, et al. Concomitant suppression of hyperlipidemia and intestinal polyp formation in Apc-deficient mice by peroxisome proliferator-activated receptor ligands. Cancer Res. 2003;63:6090–6095. [PubMed] [Google Scholar]
- 153.Niho N, Takahashi M, Shoji Y, et al. Dose-dependent suppression of hyperlipidemia and intestinal polyp formation in Min mice by pioglitazone, a PPARγ ligand. Cancer Sci. 2003;94:960–964. doi: 10.1111/j.1349-7006.2003.tb01385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Osawa E, Nakajima A, Wada K, et al. Peroxisome proliferator-activated receptor γ ligands suppress colon carcinogenesis induced by azoxymethane in mice. Gastroenterology. 2003;124:361–367. doi: 10.1053/gast.2003.50067. [DOI] [PubMed] [Google Scholar]
- 155.Harris DM, Go VL. Vitamin D and colon carcinogenesis. J Nutr. 2004;134:3463S–3471S. doi: 10.1093/jn/134.12.3463S. [DOI] [PubMed] [Google Scholar]
- 156.Dunlop TW, Vaisanen S, Frank C, Molnar F, Sinkkonen L, Carlberg C. The human peroxisome proliferator-activated receptor δ gene is a primary target of 1α,25-dihydroxyvitamin D3 and its nuclear receptor. J Mol Biol. 2005;349:248–260. doi: 10.1016/j.jmb.2005.03.060. [DOI] [PubMed] [Google Scholar]
- 157.Ghosh M, Wang H, Ai Y, et al. COX-2 suppresses tissue factor expression via endocannabinoid-directed PPARδ activation. J Exp Med. 2007;204:2053–2061. doi: 10.1084/jem.20070828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fernandez PM, Patierno SR, Rickles FR. Tissue factor and fibrin in tumor angiogenesis. Semin Thromb Hemostasis. 2004;30:31–44. doi: 10.1055/s-2004-822969. [DOI] [PubMed] [Google Scholar]
- 159.Mousa SA. Anti-thrombotics in thrombosis and cancer. Future Oncol. 2005;1:395–403. doi: 10.1517/14796694.1.3.395. [DOI] [PubMed] [Google Scholar]
- 160.Rickles FR. Mechanisms of cancer-induced thrombosis in cancer. Pathophysiol Haemostasis Thromb. 2006;35:103–110. doi: 10.1159/000093551. [DOI] [PubMed] [Google Scholar]
- 161.Lefebvre AM, Chen I, Desreumaux P, et al. Activation of the peroxisome proliferator-activated receptor γ promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med. 1998;4:1053–1057. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- 162.Saez E, Tontonoz P, Nelson MC, et al. Activators of the nuclear receptor PPARγ enhance colon polyp formation. Nat Med. 1998;4:1058–1061. doi: 10.1038/2042. [DOI] [PubMed] [Google Scholar]
- 163.Yang K, Fan KH, Lamprecht SA, et al. Peroxisome proliferator-activated receptor γ agonist troglitazone induces colon tumors in normal C57BL/6J mice and enhances colonic carcinogenesis in Apc1638N/+Mlh1+/− double mutant mice. Int J Cancer. 2005;116:495–499. doi: 10.1002/ijc.21018. [DOI] [PubMed] [Google Scholar]
- 164.Pino MV, Kelley MF, Jayyosi Z. Promotion of colon tumors in C57BL/6J-APCmin/+ mice by thiazolidinedione PPARγ agonists and a structurally unrelated PPARγ agonist. Toxicol Pathol. 2004;32:58–63. doi: 10.1080/01926230490261320. [DOI] [PubMed] [Google Scholar]
- 165.Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc Natl Acad Sci USA. 1997;94:4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kliewer SA, Forman BM, Blumberg B, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA. 1994;91:7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Solanes G, Pedraza N, Iglesias R, Giralt M, Villarroya F. Functional relationship between MyoD and peroxisome proliferator-activated receptor-dependent regulatory pathways in the control of the human uncoupling protein-3 gene transcription. Mol Endocrinol. 2003;17:1944–1958. doi: 10.1210/me.2002-0395. [DOI] [PubMed] [Google Scholar]
- 168.Poirier H, Braissant O, Niot I, Wahli W, Besnard P. 9-cis-retinoic acid enhances fatty acid-induced expression of the liver fatty acid-binding protein gene. FEBS Lett. 1997;412:480–484. doi: 10.1016/s0014-5793(97)00830-2. [DOI] [PubMed] [Google Scholar]
- 169.Yu K, Bayona W, Kallen CB, et al. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J Biol Chem. 1995;270:23975–23983. doi: 10.1074/jbc.270.41.23975. [DOI] [PubMed] [Google Scholar]
- 170.Berger J, Leibowitz MD, Doebber TW, et al. Novel peroxisome proliferator-activated receptor (PPAR) γ and PPARδ ligands produce distinct biological effects. J Biol Chem. 1999;274:6718–6725. doi: 10.1074/jbc.274.10.6718. [DOI] [PubMed] [Google Scholar]
- 171.Sznaidman ML, Haffner CD, Maloney PR, et al. Novel selective small molecule agonists for peroxisome proliferator-activated receptor δ (PPARδ)-synthesis and biological activity Bioorg. Med Chem Lett. 2003;13:1517–1521. doi: 10.1016/s0960-894x(03)00207-5. [DOI] [PubMed] [Google Scholar]
- 172.Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-α, -β, and -γ in the adult rat. Endocrinology. 1996;137:354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- 173.Mansen A, Guardiola-Diaz H, Rafter J, Branting C, Gustafsson JA. Expression of the peroxisome proliferator-activated receptor (PPAR) in the mouse colonic mucosa. Biochem Biophys Res Commun. 1996;222:844–851. doi: 10.1006/bbrc.1996.0832. [DOI] [PubMed] [Google Scholar]
- 174.Mochizuki K, Suruga K, Yagi E, Takase S, Goda T. The expression of PPAR-associated genes is modulated through postnatal development of PPAR subtypes in the small intestine. Biochim Biophys Acta. 2001;1531:68–76. doi: 10.1016/s0167-4889(01)00071-4. [DOI] [PubMed] [Google Scholar]
- 175.Yu J, Leung WK, Chen J, Ebert MP, Malfertheiner P, Sung JJ. Expression of peroxisome proliferator-activated receptor δ in human gastric cancer and its response to specific COX-2 inhibitor. Cancer Lett. 2005;223:11–17. doi: 10.1016/j.canlet.2004.09.052. [DOI] [PubMed] [Google Scholar]
- 176.Hawcroft G, D'Amico M, Albanese C, Markham AF, Pestell RG, Hull MA. Indomethacin induces differential expression of β-catenin, γ-catenin and T-cell factor target genes in human colorectal cancer cells. Carcinogenesis. 2002;23:107–114. doi: 10.1093/carcin/23.1.107. [DOI] [PubMed] [Google Scholar]
- 177.Diamond MP, Saed G. Modulation of the expression of peroxisome proliferators-activated receptors in human fibroblasts. Fertil Steril. 2007;87:706–709. doi: 10.1016/j.fertnstert.2006.07.1513. [DOI] [PubMed] [Google Scholar]
- 178.Ou YC, Yang CR, Cheng CL, Raung SL, Hung YY, Chen CJ. Indomethacin induces apoptosis in 786-O renal cell carcinoma cells by activating mitogen-activated protein kinases and AKT. Eur J Pharmacol. 2007;563:49–60. doi: 10.1016/j.ejphar.2007.01.071. [DOI] [PubMed] [Google Scholar]
- 179.Lopez JM, Fernandez MA, Pique M, Gil J. Aspirin-induced apoptosis in Jurkat cells is not mediated by peroxisome proliferator-activated receptor δ. Mol Cell Biochem. 2004;266:57–63. doi: 10.1023/b:mcbi.0000049138.67699.7b. [DOI] [PubMed] [Google Scholar]