

Abstract
Colloidal nanoparticles of Fe3O4 (4 nm) were synthesized by high-temperature hydrolysis of chelated iron (II) and (III) diethylene glycol alkoxide complexes in a solution of the parent alcohol (H2DEG) without using capping ligands or surfactants: [Fe(DEG)Cl2]2- + 2[Fe(DEG)Cl3]2- + 2H2O + 2OH- → Fe3O4 + 3H2DEG + 8Cl- The obtained particles were reacted with different small-molecule polydentate ligands, and the resulting adducts were tested for aqueous colloid formation. Both the carboxyl and α-hydroxyl groups of the hydroxyacids are involved in coordination to the nanoparticles’ surface. This coordination provides the major contribution to the stability of the ligand-coated nanoparticles against hydrolysis.
Keywords: magnetic oxide nanoparticles, synthesis, colloid and surface chemistry, dynamic light scattering, magnetization, hydroxycarboxylic acid
Biomedical applications of magnetic metal oxide nanoparticles require them to form stable aqueous colloids and therefore largely rely on their organic coatings. The function of the coating is to stabilize the particles in colloidal form and prevent them from degradation, to decrease toxicity, and to act as a linker for the nanoparticles’ biofunctionalization. It is also imperative that the coating be resistant to hydrolysis. The most popular currently used coatings are biocompatible polymers that cloister on the nanoparticles, but inevitably bring a large diamagnetic contribution and thus decrease the resulting nanocomposites’ magnetic response. Using a small-molecule capping ligand addresses the problem of excessive size, however requires the careful tuning of several parameters. Polymers [1-3], dendrimers [4-6], cyclodextrins [7-9] and large capping ligands [10] stabilize nanoparticles in colloidal form mostly through the steric suppression of interparticle interaction, however the small-molecule ligands like carboxylic, phosphonic and hydroxamic [11] acids engage the electrostatic mechanism in aqueous colloids. Functionalized polymers are efficient in colloid stabilization due to the cooperative effects of the binding of their polar substituents or in-chain heteroatoms to the nanocrystal’s surface at multiple sites. Polydentate small-molecule capping ligands are efficient stabilizers for the same cause, however the nanoparticles coated with them, are more prone to entropy-driven dissociation. Therefore, in order to provide better colloidal stability for the polymer-free nanoparticle systems, the issues of molecular geometry and binding mode of the small-molecule capping ligands, must be addressed in greater detail.
The adsorption of small-molecule ligands on inorganic surfaces has been in focus with regard to their influence on crystal growth [12,13], colloid stabilization [14-16] and catalysis [17]. In this work we report the synthesis of magnetite and maghemite nanoparticles with chemically active surfaces, studies of their complexation reactions with small-molecule polyfunctional carboxylic acids, and studies of their relative aqueous colloidal stability. One of the aims was to determine the structural features and the binding mode of the most efficient stabilizing agents. The organic substances selected for this study have significantly different molecular geometries and therefore different potential binding capabilities. The common feature however, is their ability to bind to the same surface at several sites (polydentate bridging ligands). The studied compounds are citric, tartaric, malic, pyromellitic and 5-hydroxyisophthalic acid and β-cyclodextrin.
Experimental
Reagents and solvents from commercial sources were: anhydrous iron(II) chloride (99.5%) and iron(III) chloride (98%) from Alfa Aesar, diethylene glycol (99%), 1,2,4,5-benezenetetracarboxylic and 5-hydroxyisophthalic acid from Aldrich, sodium metal from Fisher Scientific, anhydrous citric, malic and L(+)-tartaric acids from ACROS Organics. All reagents were used without additional purification. Prior to use, the solvent diethylene glycol (DEG) was degassed on a vacuum under stirring for at least 1 hour. Synthesis and manipulations with air-sensitive materials were performed in the atmosphere of ultra pure nitrogen in a glovebox of the Vacuum Atmospheres Company (VAC) and by using Schlenk technique. Dynamic Light Scattering (DLS) and electrophoretic mobility experiments were performed on Malvern Zetasizer Nano ZS (ZEN-3600) instrument. Powder X-ray diffraction studies were performed on Rigaku Miniflex II diffractometer (CuKα radiation). Transmission Electron Microscopy studies were performed on JEOL 2011 TEM instrument. Dynamic magnetic measurements on colloids were carried out using the DynoMag AC susceptometer by IMEGO in a frequency range of 5-200 kHz. The ZFC FC data was measured with Quantum Design MPMS XL (SQUID) and the hysteresis loops were measured with Quantum Design PPMS-ACMS option-using DC extraction.
For synthesis, stock solutions of FeCl3 (0.500 mol/kg), FeCl2 (0.500 mol/kg) and H2O (1.00 mol/kg) were prepared in the degassed diethylene glycol. Prior to using, the iron salts solutions were purified by centrifuging or filtration. In the first step, the concentrated solution containing all precursors was prepared in a glove box. The following components in form of stock solutions were mixed: 3 mmols of FeCl3, 1.5 mmols of FeCl2 and 7 mmols of H2O (this is a solution A). Sodium metal in a flattened form or as small pieces (13 mmols) was dissolved in 10 mL of diethylene glycol under intensive stirring (this is a solution B). Both solutions (A and B) were mixed and transferred into addition funnel equipped with a gas inlet stopcock. Next, a 500 mL 3-necked reactor connected via gas inlet to the Schlenk line, was set over magnetic stirrer and heating mantle and equipped with spinbar and thermometer. DEG (160 mL) was added to the reactor and degassed. The addition funnel with the reaction solution was connected to the Schlenk line and attached to the reactor. The reactor temperature was raised to 235 °C and with intensive stirring, the precursor solution was injected rapidly (1-2 sec) into DEG using the higher pressure of the nitrogen line. The temperature of 220-230 °C was maintained for 5 to 60 minutes and the solution was allowed to cool while stirring was continued. The resulting solution contained no precipitate, looked black in bulk, but transparent in thin layer, and showed no light scattering when illuminated with strong incident beam. Colloids exposed to heat for only 5 minutes and colloids heated at 225 °C for 30-60 min. showed similar results in DLS spectra (peak at 7-8 nm).
In order to isolate the product in a powdery form for X-ray diffraction, magnetic measurements and for IR spectrometry, aliquots of the colloid were precipitated with 1.5 volumes of ethyl acetate (assisted with centrifugation or magnetic separation), the solid then decanted, washed with methanol 2-3 times and dried at room temperature under a flow of nitrogen.
Results and discussion
Synthesis and characterization of magnetite nanoparticles
As we have shown previously [18,19], magnetite and other transition metal ferrites can be conveniently prepared by high-temperature hydrolysis of the chelated diethylene glycol alkoxide complexes in a solution of the parent alcohol, according to the following reaction:
The advantage of this method is that it allows production of the surfactant-, capping ligand- and polymer-free non-aggregated particles with high yield. Surface modification with organic molecules requiring gentle conditions, can be done as a separate step from the rough conditions of the inorganic synthesis. Moreover, since organic ligands dramatically influence kinetics and mechanism of the crystal nucleation and growth, using and changing the desired organic compounds in the first step of the synthesis would afford no reproducible results in terms of composition and morphology of the resulting nanoparticles. The known practice of ligand exchange however, is associated with lower yields, contamination and also using larger volumes of solvents and sometimes expensive chemicals.
Our original method for the synthesis of ferrite nanoparticles [18, 19] uses metal chloride hydrates and sodium hydroxide as main reagents. Composition of these substances is not exact due to their hygroscopic nature and sensitivity of FeCl2·4H2O to oxygen. These factors lead to some stoichiometrical uncertainty, which can be critical for colloid and surface chemistry. Aiming the development of a new method, capable of providing reproducible results in colloidal and surface chemistry studies, we used the anhydrous salts and the exact amount of water according to the reaction stoichiometry. We also enhanced the protection from moisture and oxygen at all steps of the synthesis. All reagents, including water, were weighed, dissolved and quantitatively mixed into a final concentrated solution in dry glove box to assure the exact stoichiometry of the reaction, which was performed then using Schlenk technique. We also used injection technique, which we did not use in [18, 19], however we did not find evidence that this change was beneficial.
Crystal structure of the obtained products was examined by powder X-ray diffractometry. Samples of magnetite powders isolated from colloids after a short (10 min) and long (1 hour) heating at 225 °C showed almost identical diffractograms (Fig. 1) that were consistent with the literature cubic inversed spinel pattern. The average crystal size calculated using Scherrer’s formula, was 5.3 nm.
Fig. 1.
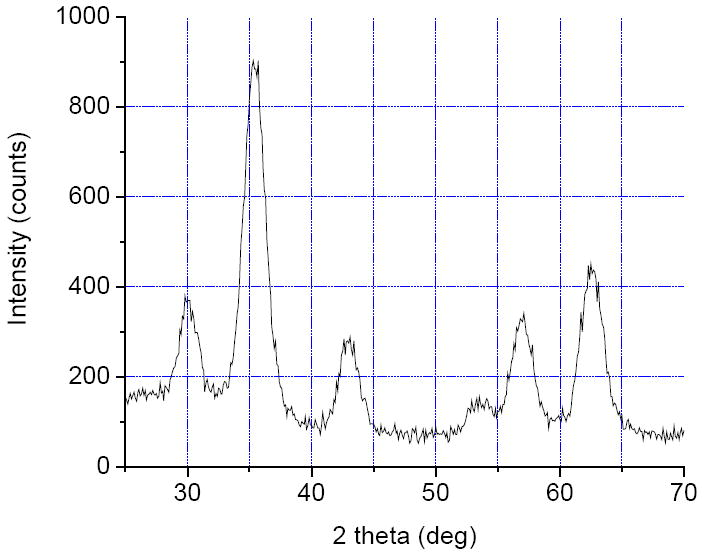
X-ray diffractogram for the Fe3O4 DEG colloid obtained after 15 minute of heating at 225 °C.
Upon exposure to open air the optical density of the original magnetite colloids decreases in several days, and the originally black solutions of magnetite become dark brown. This is attributed to oxidation of magnetite into maghemite. We examined this reaction in more details by passing oxygen through the colloid for several hours and comparing the DLS spectra of the original and resulting fluid. No substantial difference in the position and intensity of the peak at 7-8 nm was found. Calculations of the expected size change, assuming that iron content in each particle is constant, and using the bulk density of Fe3O4 equal to 5.18 g/cm3 and Fe2O3 equal to 4.87 g/cm3, show that 4 nm particles of maghemite originate from 3.87 nm particles of magnetite. This difference is below the resolution limit of the DLS method.
The low-frequency AC susceptibility was measured at room temperature for the original diethylene glycol Fe3O4 colloid (containing ~1.1×1016 particles/ml) and the same colloid after its exposure to oxygen. The measurements showed higher volume susceptibility of 5.0×10-4 SI units for magnetite colloid than for the oxidized colloid (3.6×10-4 SI units). The same trend is observed in bulk magnetite versus maghemite, but the magnitude of the susceptibility difference in their nanocolloids is more significant than in their bulk form. This could be explained by lower crystal ordering in the oxidized materials, because the oxidation took place at room temperature. In order to improve quality of the nanocrystals, the oxidized colloid was degassed and heated in nitrogen atmosphere at 220 °C for 15 minutes. As it could be expected, DLS spectra did not detect any difference in the particle size. The X-ray diffractogram of the powder obtained from the oxidized and re-heated DEG colloid, was indistinguishable from the diffractograms of the samples described above. This was expected, because both magnetite and maghemite have the same crystal structure with only slightly different spacing. Any difference was obscured by the peak broadening due to small crystal size.
TEM studies were performed on samples prepared from slowly oxidized magnetite colloids and also from colloids that were rapidly oxidized and then re-heated at 220 °C for 15 minutes. Large-area image (Fig. 2) indicates the uniformity of the colloid and shows that it is free from agglomerates. High-resolution image (Fig. 3) of the nanoparticles obtained after 15 minute-long heating, indicates that most of the nanoparticles have a size of 4 nm and the crystal lattice fringes can be seen for some of them. These results show that oxidation of magnetite into maghemite can be performed without interparticle mass transfer.
Fig. 2.

Large-area TEM image of the nanoparticles obtained from the oxidized and re-heated colloid. Scale bar = 100 nm.
Fig.3.
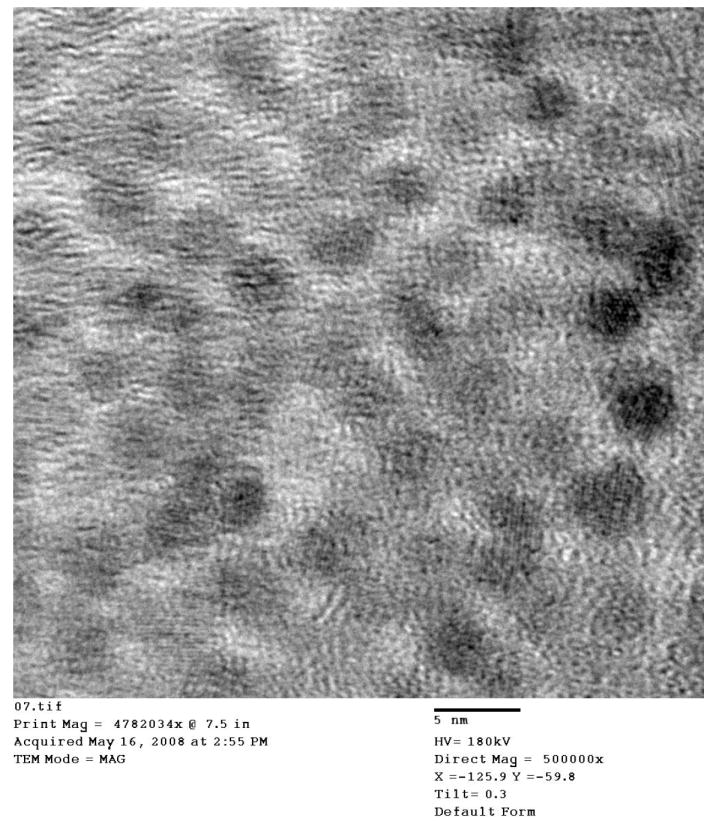
High-resolution TEM image obtained from the oxidized colloid. Scale bar = 5 nm.
Magnetization measurements were performed on powders isolated from the original Fe3O4 colloid and from another aliquot of this colloid subjected to oxygen treatment and re-heating. Product isolation was done at presence of pyridine presumably assisting with stabilization of FeII on the nanoparticle surface. The hysteresis curves obtained at 300 K (Fig. 4) show that saturation of magnetization is complete at 5 T with the calculated values of 84 emu/g for Fe3O4 and 80 emu/g for the substance treated with oxygen and annealed. The numbers are given per pure oxides whose content in the nanopowders was determined by the combustion analysis. The values of magnetization are higher than the values typically observed for small iron oxide nanocrystals [20, 21] indicating a high degree of their magnetic ordering. The difference in magnetization of both oxidized and non-oxidized samples is small, and it probably indicates that the oxidation of magnetite into maghemite is incomplete. It is also possible that maghemite was reduced by DEG at high temperature during annealing. The ZFC and FC plots look very similar for both samples (Fig. 5). Both curves obtained at 50 Oe merge at the blocking temperature of 52K corresponding to transition from superparamagnetic to ferromagnetic state. High value of the blocking temperature for the nanocrystals as small as 4 nm in diameter is probably the result of magnetic interaction between them.
Fig. 4.
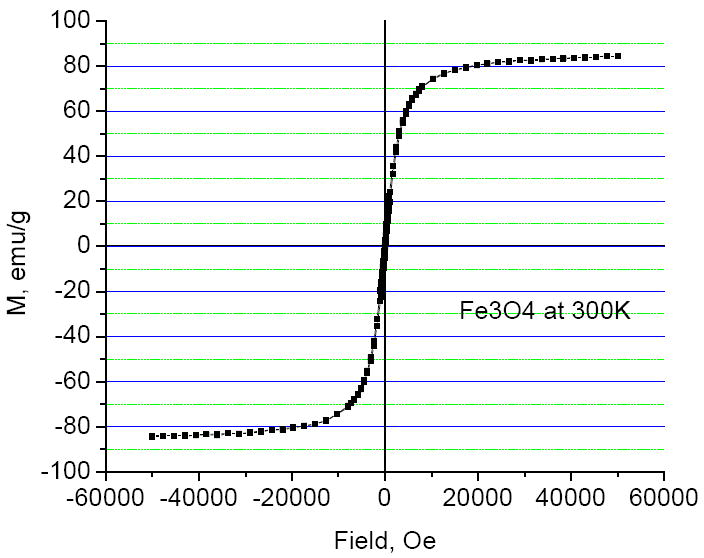
Hysteresis plot obtained at 300 K for the original magnetite nanoparticles.
Fig. 5.
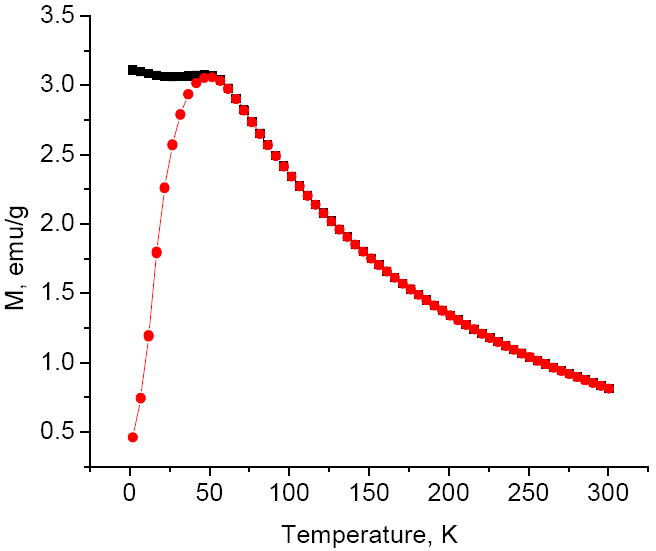
Zero field cooled and field cooled magnetization measurement curves obtained at 50 Oe for the oxidized nanoparticles.
Interaction of iron oxide nanoparticles with polydentate carboxylic acids
In order to identify small organic molecules which would strongly bind to the surface of iron oxide nanocrystals, efficiently stabilize their aqueous colloids and still contain a free functional group capable for chemical derivatization, we studied reactions of maghemite nanoparticles with citric, tartaric, malic, pyromellitic and 5-hydroxyisophthalic acid and β-cyclodextrin. Selection of these compounds was based on their ability to act as polydentate bridging ligands with different geometries and therefore different potential binding modes.
The Dynamic Light Scattering (DLS) peak corresponding to a hydrodynamic diameter of the nanoparticles in the “as-synthesized” DEG colloids is typically centered at 7-8 nm with the peak width of 0.5-0.7 nm. No purification was necessary because the abundance of these particles in the DLS spectra was greater than 99.3% by volume. In order to study the reactions of these nanoparticles with carboxylic acids, the 0.10 M solutions of these acids were added dropwise directly into the DEG colloids while stirring. Assuming the nanoparticles were spherical, the amounts of the acids added were estimated from their size determined by TEM (d = 4 nm; V = 33.5 nm3), and density of magnetite (5.18 g/cm3) or maghemite (4.87 g/cm3), as follows. The number of moles of Fe3O4 or Fe2O3 and the number of formula units per each particle were determined and used to compute the total number of atoms per particle, and then the fraction of the surface atoms was calculated using the known formula of F = 4/n1/3, where F = fraction of the surface atoms, n = total number of atoms. Since it was planned that each iron ion on the surface will be ligated with a carboxylic acid, further calculation was done to determine the molarity of the iron oxide in the reaction solution and the concentration of the nanoparticles. In order to make sure the reaction goes to completion, the actually added amount of acids was 50% greater than the calculated one.
Addition of citric, L(+)-tartaric and malic acid (as a 0.1 M methanol solution) and β-cyclodextrin (as a DEG solution) did not cause any change in the DLS spectra. On the other hand, the addition of pyromellitic and 5-hydroxyisophthalic acids caused a substantial increase in light scattering, and the DLS spectra showed that the original particles disappeared and larger particles formed. The agglomeration appears to be reversible, and the original 8 nm particles formed after a methanol solution of lithium methoxide was added. The solution thus became basic and the particles’ surface negatively charged, which helped to redisperse them.
In order to further characterize new adducts of the nanoparticles with carboxylic acids and study their aqueous colloid formation, the solutes were isolated from the DEG colloids. The DEG colloids were mixed with 5 volumes of deionized water and acidified with 0.01 M HCl to induce coagulation. The resulting precipitates were separated with a magnet or by centrifuging and used with no additional rinsing, to avoid any losses. The cyclodextrin-capped nanopowder was isolated from DEG solution by adding 1.5 volumes of ethyl acetate followed by washing the precipitate with methanol. For a reference purpose, the ligand-free nanopowder was also isolated from DEG colloid by the addition of ethyl acetate and washing the resultant precipitate with methanol.
IR spectra of the ligated particles clearly show presence of the coordinated carboxylates. Since all samples were carefully washed with methanol, the presence of the free acids was excluded. The observed shift of stretching C-O vibrations to lower wave numbers, compared to similar vibrations in free acids also supported this conclusion. The ligand-free nanopowder showed presence of the coordinated DEG in IR-spectrum.
Aqueous colloids of the obtained solids were studied by DLS and electrophoretic mobility methods. The experiments were performed in a rather non-traditional manner (for a colloidal chemist) as we never used excess of acids in solutions, since they cannot be added in the patient’s blood to stabilize the imaging agent. The initial (before the titration) concentration of Fe2O3 was 4.17×10-3 M or 5.6×1018 particles/L; the calculated concentration of the bound ligand was 1.70×10-3 M. The particle size and ζ-potentials were obtained as a function of pH for citrate-, tartrate- and malate-ligated nanoparticles.
The ligand-free nanopowder was insoluble in water and in alkaline solutions. It formed cloudy dispersions however, when the pH was lowered using hydrochloric acid. Cyclodextrin-capped nanopowder was soluble in water, however the initially clear solution (Tyndall test) rapidly turned into a light scattering colloid containing the aggregated particles. In contrast to this behavior, the nanoparticles whose surface was covered with coordinated acids showed strong pH dependence of their solubility in water, from cloudy dispersions in slightly acidic solutions to clear colloids in basic solutions. The pyromellitate and 5-hydroxyisophthalate basic colloids turned out to be unstable and aggregated spontaneously, which was a sign of their de-coordination. We performed titration of acidic aqueous suspensions of maghemite citrate, tartrate, and malate nanoparticles with NaOH, monitored by DLS and ζ–potential measurements.
Addition of 0.01 M NaOH to the acidic suspensions caused rapid formation of the clear colloids, and according to DLS studies, the particle size gradually decreased as pH was increased. For comparability, we recorded the lowest value of pH at which the DLS peak centered at 7-9 nm appeared with the intensity greater than 90% by volume (Table 1, line 1). This peak corresponds to hydrodynamic size of the non-aggregated colloidal particles. When pH of 9.2 was reached, the titration was reversed by using 0.01 M HCl. As one can see from the table (second line), the aggregation in citrate and tartrate colloids became significant at pH of 4.9 and 7.2 respectively, as opposed to pH 7.4 and 7.8, when acidic suspension was titrated with base (first line). Upon further lowering the pH, the citrate compound aggregated slowly, and only at pH of 4.5 the reference peak was finally absent. Aggregation of tartrate compound took place much more rapidly, and at pH of 6.9 the reference peak disappeared. The observed discrepancies in pH values are most likely resulting from not allowing enough time for the particle size to reach the equilibrium during titration. The reversed titration of the malate-maghemite colloid caused a very rapid irreversible aggregation indicating de-ligation of the nanoparticles. Slow de-ligation of the nanoparticles was observed for citrate and tartrate colloids that were exposed to high pH of 9.8, however at pH of 7.5 these colloids were stable for at least several weeks.
Table 1.
The pH values representing substantial aggregation and de-aggregation events during titration of aqueous colloids with 0.01 M HCl and 0.01 M NaOH (monitored by DLS).
| Citrate | Tartrate | Malate | |
|---|---|---|---|
| The reference peak* intensity turned > 90% (pH↑) | 7.4 | 7.8 | 8.8 |
| The reference peak* intensity is still > 90% (pH↓) | 4.9 | 7.2 | decomposes |
| The reference peak* intensity turned 0% (pH↓) | 4.5 | 6.9 | decomposes |
| Isoelectric point | 3.6 | 4.4 | 4.3 |
reference peak at 7-9 nm in the DLS spectra
pH↑ - titration with base
pH↓ - titration with acid
As pH was increased, ζ-potential turned more negative until it reached the values close to -45 mV for all samples, however this was not a smooth transition as there were many positive and negative spikes in the curves (not shown). Possible explanation for ζ-potential not changing smoothly with the increased pH is that breaking the aggregates apart was taking place until the original 7-8 nm particles became discrete, and this process was disordered. Isoelectric points for citrate, tartrate and malate colloids are 3.6, 4.4 and 4.3 respectively.
The observed differences in behavior of the three colloids allow speculating about the coordination mode of the three acids used as capping ligands. The most likely scenario is that the carboxyl and the adjacent α-OH groups of these acids are involved in coordination with the nanoparticle surface and they are the major contributors to the stability of the resulting adducts against hydrolysis. This hypothesis explains the significantly higher stability of the tartrate adduct compared to malate adduct, as malic acid differs from the tartaric acid only by the absence of one of the α-OH groups. Both carboxylic and both α-OH groups of tartaric acid are covalently attached to the nanoparticles. One of the carboxylic groups of the coordinated citric acid is either labile or uncoordinated. This explains the difference in solubility and isoelectric points of the citrate and tartrate adducts and also the difference in stability of the tartrate and malate adducts. Idealized geometries for the bound acids are shown in Fig. 6.
Fig. 6.
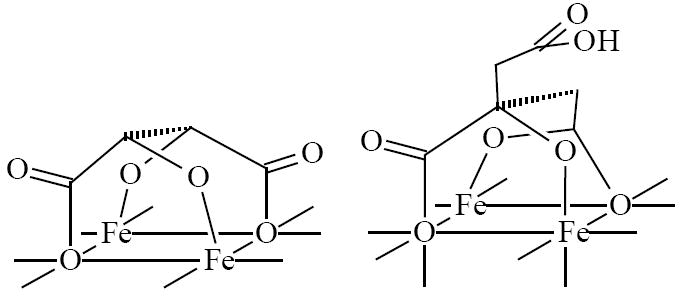
Idealized geometries of bound tartaric and citric acids.
The proposed binding mode of tartaric acid with all of its hydroxyl groups coordinated to the surface is different from previously reported model [14, 17] in which one or more functional groups of the acid remain uncoordinated. We believe the observed deviation can be attributed to different conditions of the complexation reactions. Ligation in [14, 17] takes place on positively charged particles in aqueous acidic solution, while in our system nanoparticles were reacted with tartaric acid in non-aqueous solution, and their surface was negatively charged (ζ = - 28 mV in H2O).
Conclusions
This study revealed that efficiency of the small-molecule ligands in nanoparticle colloid stabilization depends on their molecular structure. The most efficient ligands used in this work were di-and triprotic α-hydroxyacids, namely tartaric and citric acids. Compounds binding with only carboxylic or alcohol-type OH-groups were more prone to desorption from the nanoparticle surface, which was observed for 5-hydroxyisophthalic and pyromellitic acids and β-cyclodextrin.
Acknowledgments
We acknowledge financial support of this work by the National Institutes of Health, the Xavier NSF MIE program, and the UNCF Foundation via Henry McBay grant. We also would like to thank the following people who performed the measurements: Jibao He for TEM characterization, Leonard Spinu and Cosmin Radu for magnetization measurements, Christer Johansson, Andrea Prieto Astalan and Karolina Petersson for dynamic magnetic measurements on colloids.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hu F, Li Z, Tu C, Gao M. J Colloid Interface Sci. 2007;311:469. doi: 10.1016/j.jcis.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 2.Acar HY, Garaas RS, Syud F, et al. J Magn Magn Mater. 2005;293:1. [Google Scholar]
- 3.Carmen Bautista M, Bomati-Miguel O, Del Puerto Morales M, Serna CJ. J Magn Magn Mater. 2005;293:20. [Google Scholar]
- 4.Shi X, Thomas TP, Myc LA, et al. Phys Chem Chem Phys. 2007;9:5712. doi: 10.1039/b709147h. [DOI] [PubMed] [Google Scholar]
- 5.Gao F, Pan B-F, Zheng W-M, et al. J Magn Magn Mater. 2005;293:48. [Google Scholar]
- 6.Mornet S, Portier J, Duguet E. J Magn Magn Mater. 2005;293:127. [Google Scholar]
- 7.Hou Y, Kondoh H, Shimojo M, et al. J Phys Chem B. 2005;109:4845. doi: 10.1021/jp0476646. [DOI] [PubMed] [Google Scholar]
- 8.Bocanegra-Diaz A, Mohallem N, Novak M, Sinisterra R. J Magn Magn Mater. 2004;272-276:2395. [Google Scholar]
- 9.Bonacchi D, Caneschi A, Dorignac D, et al. Chem Mater. 2004;16:2016. [Google Scholar]
- 10.Boal AK, Das K, Gray M, Rotello V. Chem Mater. 2002;14:2628. [Google Scholar]
- 11.Baldi G, Bonacchi D, Comes Franchini M, et al. Langmuir. 2007;23:4026. doi: 10.1021/la063255k. [DOI] [PubMed] [Google Scholar]
- 12.Neveu S, Bee A, Robineau M, Talbot D. J Colloid Interface Sci. 2002;255:293. doi: 10.1006/jcis.2002.8679. [DOI] [PubMed] [Google Scholar]
- 13.Franger S, Berthet P, Berthon J. J Solid State Electrochem. 2004;8:218. [Google Scholar]
- 14.Fauconnier N, Bee A, Roger J, Pons JN. J Mol Liquids. 1999;83:232. [Google Scholar]
- 15.Racuciu M, Creanga D, Airinei A. Eur Phys J E: Soft Matter. 2006;21:117. doi: 10.1140/epje/i2006-10051-y. [DOI] [PubMed] [Google Scholar]
- 16.Johnson S, Brown G, Healy T, Scales P. Langmuir. 2005;21:6356. doi: 10.1021/la047030q. [DOI] [PubMed] [Google Scholar]
- 17.Barlow SM, Raval R. Surf Sci Reports. 2003;50:201. [Google Scholar]
- 18.Caruntu D, Remond Y, Chou NH, et al. Inorg Chem. 2002;41:6137. doi: 10.1021/ic025664j. [DOI] [PubMed] [Google Scholar]
- 19.Caruntu D, Caruntu G, Chen Y, et al. Chem Mater. 2004;16:5527. [Google Scholar]
- 20.Taboada E, Rodriguez E, Roig A, et al. Langmuir. 2007;23:4583. doi: 10.1021/la063415s. [DOI] [PubMed] [Google Scholar]
- 21.Ahniyaz A, Seisenbaeva GA, Haeggstroem L, et al. J Magn Magn Mater. 2008;320:781. [Google Scholar]