

Abstract
Inherited deficiency of galactose-1-phosphate uridyltransferase (GALT) can result in a potentially lethal disorder called classic galactosemia. Although the neonatal lethality associated with this disease can be prevented through early diagnosis and a galactose-restricted diet, the lack of effective therapy continues to have consequences: developmental delay, neurological disorders, and premature ovarian failure are common sequelae in childhood and adulthood. Several lines of evidence indicate that an elevated level of galactose-1-phosphate (gal-1-p), the product of galactokinase (GALK), is a major, if not sole, pathogenic mechanism in patients with classic galactosemia. The authors hypothesize that elimination of gal-1-p production by inhibiting GALK will relieve GALT-deficient cells from galactose toxicity. To test this hypothesis, they obtained human GALK using a bacterial expression system. They developed a robust, miniaturized, high-throughput GALK assay (Z′ factor =0.91) and used this assay to screen against libraries composed of 50,000 chemical compounds with diverse structural scaffolds. They selected 150 compounds that, at an average concentration of 33.3 μM, inhibited GALK activity in vitro more than 86.5% and with a reproducibility score of at least 0.7 for a confirmatory screen under identical experimental conditions. Of these 150 compounds, 34 were chosen for further characterization. Preliminary results indicated that these 34 compounds have potential to serve as leads to the development of more effective therapy of classic galactosemia.
Keywords: classic galactosemia, galactose-1-phosphate uridyltransferase (GALT), galactokinase (GALK), galactose-1-phosphate, GHMP small-molecule kinase, high-throughput screening, small-molecule inhibitors
INTRODUCTION
Classic galactosemia (OMIM 230400) is an inherited metabolic condition caused by deficiency of an galactose-1-phosphate uridyltransferase (GALT, EC 2.7.7.12) activity (Fig. 1).1 GALT is the second enzyme in the evolutionarily conserved galactose (Leloir) metabolic pathway and facilitates the simultaneous conversion of uridine diphosphoglucose (UDP-glucose) and galactose-1-phosphate (gal-1-p) to uridine diphosphogalactose (UDP-galactose) and glucose-1-phosphate.2 Consequently, GALT deficiency leads to the unique accumulation of gal-1-p and deficiency of UDP-galactose in patient cells.3-5 If untreated, classic galactosemia (CG) can result in severe disease in the newborn period, including liver dysfunction, quickly progressing to liver failure, coagulopathy, coma, and death.6 Ever since most states in the United States have included CG in the newborn screening panel, neonatal morbidity and mortality have decreased considerably.7 The current mainstay of treatment is the withdrawal of (ga-)lactose from the diet.6
FIG. 1.
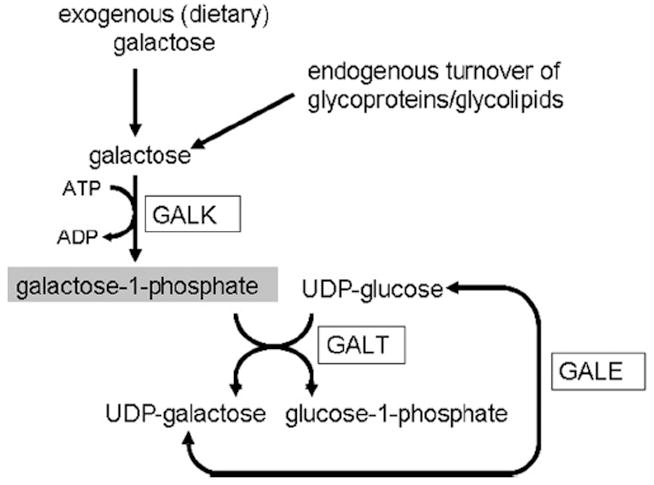
The human galactose metabolic pathway. Exogenous and endogenously produced α-D-galactose is phosphorylated by galactokinase (GALK) to form galactose-1-phosphate (gal-1-p) (highlighted). In the presence of galactose-1-phosphate uridyltransferase (GALT), galactose-1-phosphate will react with uridine diphosphoglucose (UDP-glucose) to form glucose-1-phosphate and uridine diphosphogalactose (UDP-galactose). UDP-galactose can also be formed from UDP-glucose via the UDP galactose-4′-epimerase (GALE) reaction. ATP, adenosine triphosphate; ADP, adenosine diphosphate.
However, it has become clear that despite early identification and intervention, including optimal dietary management, chronic complications such as IQ deficits, ataxia, speech dyspraxia, and premature ovarian failure develop in patients with CG.8-10 The pathogenic mechanisms for these chronic complications remain uncertain, but several lines of evidence suggest that chronically elevated gal-1-p is the major contributor to the long-term sequelae.4,8-11 First, except for cataracts, patients with inherited deficiency of galactokinase (GALK, E.C. 2.7.1.6) (Fig. 1), a member of the family of GHMP small-molecule kinases,12,13 do not accumulate gal-1-p and do not experience the complications observed in GALT-deficient patients.6,14-16 Second, although a ga17-deleted (i.e., GALT-deficient) yeast stops growing upon addition of galactose to the growth medium, a ga17 ga11 double knockout strain deficient in both GALT and GALK enzyme activities is no longer sensitive to galactose and grows well.17,18 Third, our laboratory recently demonstrated that galactose challenge to isogenic GALT-deficient (but not GALK-deficient) yeast led to overt manifestation of environmental stress response (ESR).19 All these studies indicate that gal-1-p is the major, if not sole, culprit for the galactose toxicity observed in GALT-deficient cells.
This raises the question about the origin of gal-1-p, the enzymatic product of GALK upon α-D-galactose, in a galactosemic patient who refrains from dairy products. It has been demonstrated that galactose moieties converted to gal-1-p can also be derived from nondairy sources (e.g., galactose-containing fruits and vegetables amounting to as much as 30 mg per day).20-22 Moreover, galactose moieties can also be produced endogenously from UDP-glucose via the UDP-4-galactose epimerase (GALE) reaction, as well as from the natural turnover of glycolipids and glycoproteins (Fig. 1), which may amount to 1.2 g in a 50-kg adult man.23,24 Because endogenous galactose production is not amenable to dietary manipulation, there is a need for innovative, nondietary therapy.
As patients with galactokinase deficiency do not experience the chronic sequelae of CG,16 a medically induced galactokinase deficiency could provide protection from the conversion of galactose to gal-1-p, regardless of the source of galactose. We, therefore, hypothesize that in conjunction with dietary therapy, an inhibitor of GALK can prevent the sequelae of chronic gal-1-p exposure in patients with CG. To enable us to test this hypothesis, we searched for small-molecule inhibitors of galactokinase and report our findings in this study.
MATERIALS AND METHODS
Overexpression and purification of human galactokinase
cDNA coding for the human GALK1 gene was obtained from the I.M.A.G.E. consortium (Clone ID: 3501788). This sequence was amplified by PCR using specific primers designed to introduce an NcoI restriction site and an His6 epitope tag at the 5′ end, as well as a 3′ BamHI restriction site. The resulting fragment was then subcloned into plasmid pET15b (Novagen, Madison, WI) using the NcoI and BamHI cloning sites. The nucleotide sequence of the GALK1 cDNA insert was confirmed by DNA sequencing using the T7 forward and reverse primers (Novagen). Subsequently, the plasmid containing the designed insert was transformed into Escherichia coli HMS174 (DE3) (Novagen) cells. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added at a final concentration of 1 mM to cell culture in LBAmp upon reaching OD600 = 0.6 at 37 °C to induce overexpression of GALK for 3 h, and the pellet was subsequently stored at −80 °C.
Protein purification was conducted at 4 °C throughout. Briefly, cell pellets were resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, and 100 mM galactose, pH 8). Cells were then lysed using a microfluidizer and clarified by centrifugation, and the lysate was loaded onto a chromatography column containing Nickel affinity resin. The resin was washed, and bound GALK was eluted using an imidazole concentration gradient. Final purified GALK was concentrated to 1 mg/mL, dialyzed into phosphate-buffered saline (PBS), aliquoted, and stored frozen at −80 °C. Galactose-dependent adenosine triphosphatase (ATPase) activity was verified using the standard pyruvate kinase/ lactate dehydrogenase-coupled assay.25 To expedite the purification process, we subcontracted part of the work described above to Paragon BioServices, Inc. (Baltimore, MD), on a fee-for-service basis.
Development of high-throughput screening assay for GALK
Through a written agreement between the principal investigator (Dr. Kent Lai) and the high-throughput screening (HTS) facility of the Broad Institute at Harvard University/Massachusetts Institute of Technology (MIT), whose setup was partly funded by a grant from the National Institutes of Health, we were granted access to use the facility to develop an HTS assay for GALK and the subsequent screening for small-molecule inhibitors.
Using the Kinase-Glo™ reagent (Promega, Madison, WI), we developed a miniaturized, 2-step HTS assay for GALK. This assay measures GALK activity indirectly by determining the amount of adenosine triphosphate (ATP) remaining after completion of the GALK-mediated reaction (step 1): galactose + ATP → gal-1-p + adenosine diphosphate (ADP). If there is ample GALK activity, most ATP will be used up in step 1, and little will be left for the luciferase reaction (step 2): ATP + luciferin and luciferase (Kinase-Glo™) → oxyluciferin + light.
Final assay conditions in a total volume of 30 μL were as follows: 0.15 μg GALK, 5 mM MgCl2, 60 mM NaCl, 20 mM HEPES, 1 mM dithiothreitol (DTT), 0.5% DMSO, 0.01% bovine serum albumin (BSA), 1 mM α-D-galactose, and 5 μM ATP. After 60 min at room temperature (22 °C), 30 μL of Kinase-Glo™ was added, and luminescence was measured within 30 min using an Envision Multilabel Plate Reader (PerkinElmer, Waltham, MA). The Wellmate® reagent dispenser (Matrix Technologies, Hudson, NH) was used to dispense biochemicals and Kinase-Glo™.
Z′ factor for the assays with GALK (samples) and assays without GALK (controls) was determined using the following formula: Z′ factor = 1 − [3 × (σp + σn)/|μp − μn|].26 The term |μp− μn| denotes the absolute difference between the mean of the samples and the mean of the controls, whereas σp and σn correspond to standard deviations for the samples and controls, respectively.
As a proof of principle, we used the ATP analog adenosine 5′-O-(3-thio-)triphosphate (ATP-γ-S) to demonstrate increasing inhibition of GALK when increasing ATP-γ-S from 0 to 13.3 μM, keeping ATP constant at 5 μM.
HTS of small-molecule inhibitors of GALK
As we prepared for the primary screen of small-molecule inhibitors of GALK, we realized that it might take months with this low-yield bacterial expression system to obtain enough GALK to screen against 50,000 small molecules. Consequently, we subcontracted the overproduction and the subsequent purification of GALK to Paragon BioServices, Inc. on a fee-for-service basis. Using the same expression system, we harvested sufficient GALK enzyme from 200 liters of bacterial culture but also noted that the GALK enzyme purified had higher specific activity. Therefore, only 0.015 μg of GALK protein per well was used in each screening assay. We screened 50,000 compounds from various libraries of small molecules with diverse structural scaffolds for inhibitory properties of GALK activity in vitro. Briefly, 100 nL of a solution of compounds dissolved in DMSO at a concentration of 10 mM was pinned, using CyBi™ Well 384/1536 (CyBio, Inc., Woburn, MA), into 384-well plates containing 20 μL of Master Mix #1 (0.015 μg GALK, 5 mM MgCl2, 60 mM NaCl, 20 mM HEPES, 1 mM DTT, and 0.01% BSA). After allowing the single compound to equilibrate with GALK for 60 min, the GALK reaction was started by adding 10 μL of Master Mix #2 (5 mM MgCl2, 60 mM NaCl, 20 mM HEPES, 1 mM DTT, 3 mM α-D-galactose, 0.01% BSA, and 15 μM ATP). The reaction was then allowed to proceed at room temperature (22 °C) for 60 min. Then, 30 μL of Kinase-Glo™ was added to stop the reaction, and luminescence was measured 15 to 30 min later using the Envision Multilabel Plate Reader (PerkinElmer). We screened, in duplicate, 50,000 small molecules for inhibitory properties against purified GALK. The Z′ factor assay was included each day as part of the quality control process.
Data analysis
Raw data were submitted to the data analysis team of the HTS facility for further analysis. In short, duplicate luminescence measurements were corrected for background measurements using the method described by Seiler et al.27 For each compound, the cosine correlation of the duplicate pair (A and B) of dimensionless z-score values yielded a composite z-score value representing the final primary screening result as previously described.27 Reproducibility of the 2 z-scores was defined as the cosine of the replicate vector [z-scoreA, z-scoreB] and the vector [1, 1] representing perfect reproducibility. Details regarding the reproducibility score calculation can be found at the Chembank Web site.28
IC50 determination
As the percentage of substrate conversion can have significant impact on the accuracy of IC50 determination, the preferred approach is to determine reaction rates at initial velocity (i.e., zero substrate conversion).29 Initially, when we determined IC50 with the luminescence-based assay, we found that the percentages of substrate conversion were relatively high (~50%). We therefore analyzed the dose response of the selected compounds using the standard pyruvate kinase/lactate dehydrogenase-coupled assay.25 In this assay, the amount of galactose present in the reaction is kept in excess, whereas ATP consumed by the galactokinase reaction is recycled by the pyruvate kinase reaction. As a result, the reaction rates calculated will be close to initial velocity.29 IC50 values were determined on normalized data from this enzyme-linked assay using the values obtained for the corresponding controls (no inhibitor) as 100%. These data were fitted with a standard dose-response inhibition model using GraphPad Prism 5.01 software (San Diego, CA), and a log(inhibitor) versus normalized response model, y = 100/[1 + 10(x−logIC50)], was established for each inhibitor. The models demonstrated adequate fit, with R2 values of all curves greater than 0.90.
RESULTS AND DISCUSSION
The 2 main objectives for this work are the development of a robust HTS assay for human GALK activity and the use of this established assay to identify small molecules that inhibited human GALK activity in vitro. Being the first enzyme of the Leloir pathway of galactose metabolism, the level of GALK activity dictates the amount of galactose entering into the galactose metabolic pathway, making GALK a significant biological target for overall galactose metabolism. Hence, discovery of inhibitors for this important biological target will not only provide an opportunity for novel therapy for CG but also advance our understanding of the role played by galactose catabolism in cellular metabolism.
Overexpression and purification of human GALK
Initial estimation showed that a minimum of 5 mg of purified GALK would be required for the development of the HTS assay, as well as the subsequent screening of 50,000 compounds in duplicate. Because overexpression and purification of active recombinant galactokinases had been successfully performed via affinity-chromatography using both E. coli and yeast as hosts,30,31 we decided to adopt one of the published protocols to prepare sufficient human GALK enzyme for our screening needs. As shown in Figure 2, we achieved a typical yield of 0.1 mg of purified GALK protein from 1 liter of bacterial culture. Galactose-dependent ATPase activity of the purified enzyme was verified using the standard pyruvate kinase/lactate dehydrogenase-coupled assay.25 We confirmed the presence of purified recombinant His6-tagged GALK protein using anti-His6-tag antibody (data not shown).
FIG. 2.
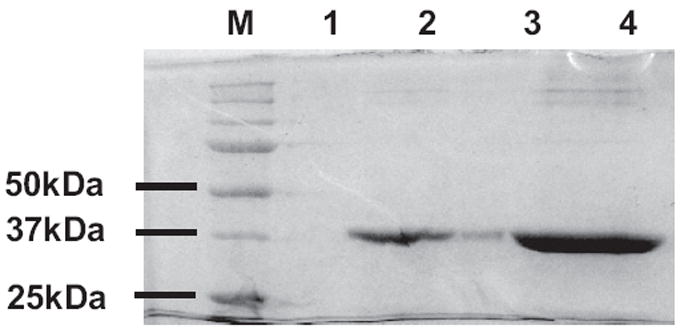
Purification of human galactokinase (GALK). Different amounts of Ni2+-NTA affinity-purified GALK protein were loaded on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) as follows: M, molecular weight markers; 1, unloaded; 2, 1.25 μg GALK; 3, unloaded but some spillover of GALK from lane 4 was seen; 4, 6.25 μg GALK.
Using the standard pyruvate kinase/lactate dehydrogenase-coupled assay, we established that the KM for ATP was 42 μM and KM for galactose was 210 μM in the purified human GALK. Timson and Reece,31 using the same assay, established that the KM for ATP was 34 μM, and the KM for galactose was 970 μM.31 The differences in the KM values for galactose remain as yet unexplained but could be due to differences in ionic strength, temperature, and the percentage of the α-anomeric form (vs. β-anomeric form) of the D-galactose used.
Development of HTS assay for purified GALK
Despite the relatively low yield of GALK from our bacterial expression system, we harvested sufficient GALK enzyme to establish a high-throughput biochemical assay for GALK activity in a 384-well microplate format at the HTS facility at the Broad Institute. As described in Materials and Methods, the 2-step luminescence-based high-throughput assay we developed measured GALK activity indirectly by quantifying the amount of ATP that remained in the GALK reaction. Therefore, the luminescent signal is inversely proportional to the amount of kinase activity.
During the assay development phase, we aimed to identify reaction conditions that would use the minimal amount of GALK enzyme, give a wide sensitivity range (i.e., high signal-to-noise ratio), and ensure that the reaction kinetics in the presence of a strong GALK inhibitor is within linear range. Using ATP ranging from 5 to 20 μM, we were unable to produce a low end-of-reaction value at 10 μM or higher of ATP, thus compromising dynamic separation. In fact, we established that the largest signal-to-noise ratio achieved among all tested conditions was 96-fold, which took place with 0.15 μg GALK protein in the presence of 5 μM ATP over a time course of 60 min at room temperature (Fig.3a). We therefore believe that this signal-to-noise ratio indicates good dynamic range, and we conclude that this assay provided the optimal conditions to identify strong inhibitors of GALK.
FIG. 3.

Galactokinase (GALK) high-throughput screening (HTS) optimization. (a) Effect of varying amounts of adenosine triphosphate (ATP) and enzyme on GALK activity. GALK reaction was carried out at different ATP concentrations (5, 10, 20 μM) and different GALK amount (0, 0.05, 0.1, 0.15, 0.3 μg) in a final volume of 30 μL. Each condition was carried out in 48 replicates (n = 48). At the end of the reaction time (60 min), 30 μL of Kinase-Glo™ reagent was added. Luminescence was recorded as relative luminescence units (RLU) 10 min thereafter. Absolute luminescence values for different amounts of ATP standards (without GALK and galactose) were also determined using the Kinase-Glo™ reagent, and this information was used to convert the raw RLU to the amount of ATP remaining at the end of the reaction. Each data point represents the mean of 48 replicates. Error bars represent ± 1 SD. The experiment was repeated twice to confirm reproducibility. (b) Z′ factor determination. A 384-well plate was divided into 176 wells each for samples and controls, leaving the 2 center columns (32 wells) empty to prevent crossover of luminescence signals. To the sample wells, we added 0.15 μg GALK in the presence of 5 μM ATP and 3 mM galactose in a final volume of 30 μL. In the control wells, GALK was omitted, but other reaction conditions remained identical. Reaction was carried out at room temperature for 60 min. At the end of the reaction, 30 μL of Kinase-Glo™ reagent was added. Luminescence was recorded as RLU after 10 min. Each data point on the graph represents the luminescence recorded for each well. The mean and the standard deviation were calculated for the samples and controls. Sample/control ± 3 SD were plotted around each data point. The experiment was repeated 3 times to confirm reproducibility. (c) Inhibition of GALK activity by ATP-γ-S. GALK activity was assayed in HTS format in the presence of varying amount of ATP-γ-S (pink squares). To control for potential inhibition of luciferase reaction by ATP-γ-S, a separate GALK assay with GALK omitted was performed (blue diamonds). Error bars represent ± 1 SD (n =32).
To test the robustness of the HTS assay, we performed Z′ factor analysis of the results. As illustrated in Figure 3b, a Z′ factor of 0.91 was determined from our assay. Therefore, we are confident that we have established a very robust, miniaturized, 2-step in vitro HTS assay for recombinant human GALK. As a proof of concept, we showed that an ATP mimetic, ATP-γ-S, acts as a GALK inhibitor by demonstrating decreased GALK activity in the presence of an increasing amount of ATP-γ-S (Fig. 3c).
It is noteworthy that the 2-step luminescence-based assay executed in this case is unique because it measures the rate of disappearance of a substrate, rather than the formation of the product(s). Thus, the more luminescence detected at the endpoint, the less GALK activity was inferred. In addition, inhibitors that compete with ATP for luciferase, and therefore result in false negatives, will not be identified by this assay. Although these false negatives may be an undesirable outcome, one must realize that the compounds we are going to “miss out” in these cases are likely to be the less selective inhibitors that compete for ATP binding sites in other kinases. As a result, this assay may reduce, in the long run, the number of nonselective hits for GALK.
HTS of small-molecule inhibitors of GALK
Because of the mission of the HTS facility at the Broad Institute, it should be emphasized that the libraries of compounds we screened were not necessarily “drug-like.” Moreover, most compounds in the libraries were stored at a concentration of 10 mM. Consequently, the final concentration of the compounds in the assay was in the micromolar range. A Z′ factor assay was included each day as part of the quality control process, with a Z′ factor of 0.9 or above observed throughout all screening days. From the standard curves constructed with varying amount of GALK, we found that a composite z-score of 7.68 was equivalent to 86.3% inhibition of GALK activity (data not shown). Hits were defined arbitrarily as a composite z-score above 7.68 in combination with a reproducibility score of at least 0.7 (Fig. 4). Based on these criteria, 200 compounds, representing 0.4% of the total 50,000 compounds in the libraries, were found to inhibit GALK activity by 86.3% or more. This percentage is in good agreement with published results of primary HTS.32-34
FIG. 4.
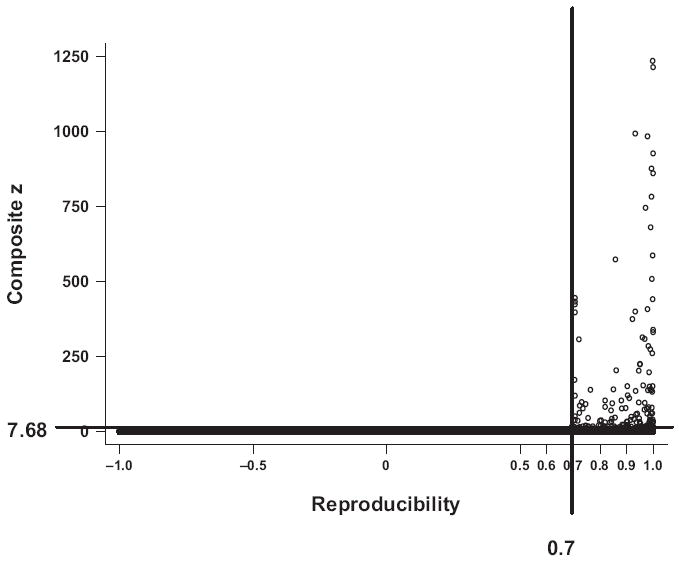
Selection of 150 compounds from primary screen. A composite z-score and a reproducibility score were assigned to each of the 50,000 compounds tested based on their ability to inhibit galactokinase (GALK) activity in vitro and the reproducibility of the 2 replicate results. The 150 compounds with a reproducibility score above 0.70 and a composite z-score greater than 7.68 were selected.
In total, 150 of the 200 compounds were subsequently selected for repeated testing (i.e., confirmatory screen) under identical experimental conditions. Most hits not selected for confirmatory screen were compounds with a molecular weight in excess of 500. As a validation of our screen, we also identified a few known kinase inhibitors among our hits, whereas most of the others are novel compounds that have not been characterized.
Confirmatory screen
Figure 5 shows the correlation results between the luminescence values recorded for each of the selected 150 compounds in the primary screen and the confirmatory screen. We were able to demonstrate a correlation coefficient of +0.48. Following completion of the confirmatory screen, 34 compounds were selected for further characterization (Fig. 6). The other 116 compounds were excluded either because inhibitory properties on GALK activity were not confirmed in the confirmatory screen or because of known toxicity.
FIG. 5.
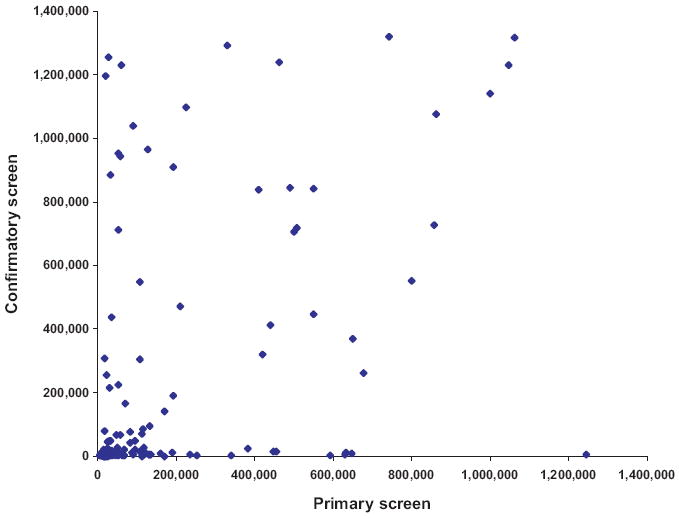
Primary versus confirmatory screen. Average (n = 2) luminescence values recorded for the top 150 compounds selected from the primary screen were plotted against average (n = 2) luminescence values of the corresponding compounds recorded at the confirmatory screen.
FIG. 6.
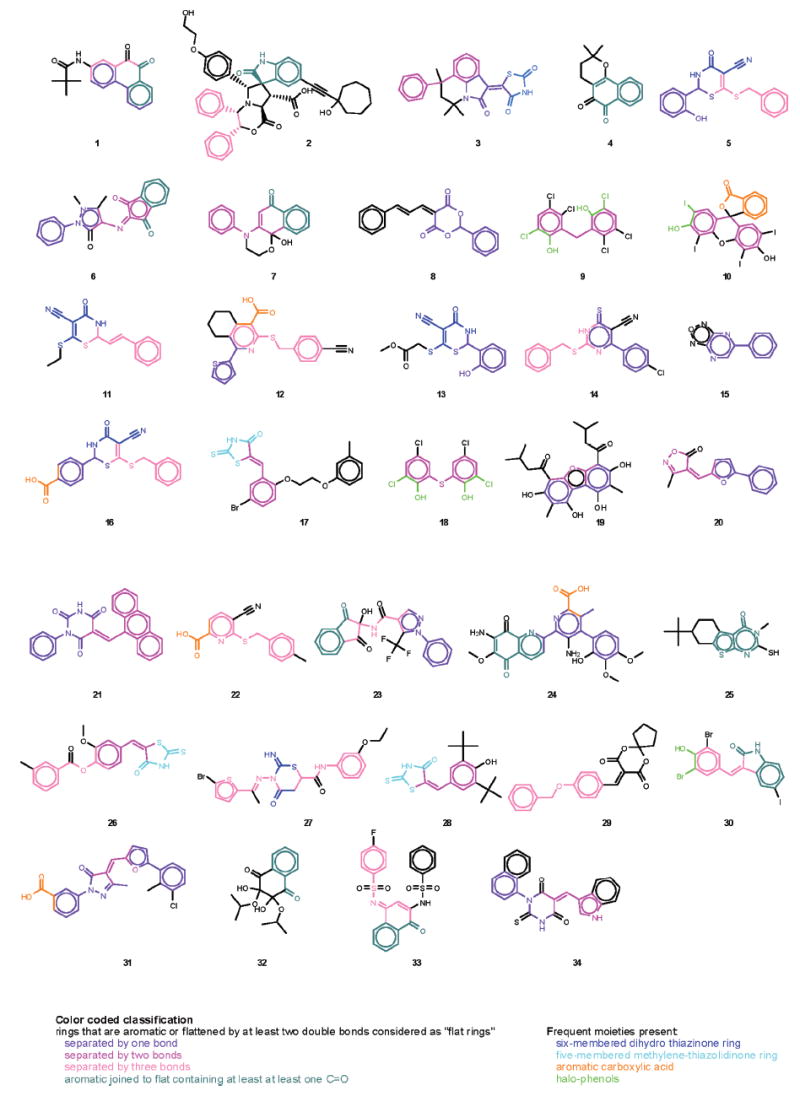
Structures of 34 selected compounds. Compounds are numbered by composite z-score in descending order.
Preliminary characterization of the selected compounds
The molecular weights of the 34 identified structures were between approximately 200 and 800. They cover a relatively wide range of lipophilicity based on calculated log octanol-water partition coefficients in the −1 to 8 range (CLOGP from ChemDraw 10, CambridgeSoft). Most compounds had no Lipinski violations, and 10 compounds had 1 violation, with a maximum of 2 violations in 3 compounds.
The IC50 of the compounds studied thus far using the established luminescence-based assay ranged from 200 nM to 33 μM (data not shown). The IC50 values of 4 selected representatives (compounds 1, 3, 9, and 24) have also been confirmed in the standard pyruvate kinase/lactate dehydrogenase-coupled assay (Fig. 7). In this assay, ATP consumed by the galactokinase reaction is recycled by the pyruvate kinase reaction, and the amount of galactose present in the reaction is in large excess; hence, calculated reaction rates are close to initial velocity. Values obtained were somewhat lower than in the luminescence assay, which was in agreement with Wu and coworkers’ observation that IC50s determined at conditions other than zero substrate conversion will be higher than the true rate.29
FIG. 7.
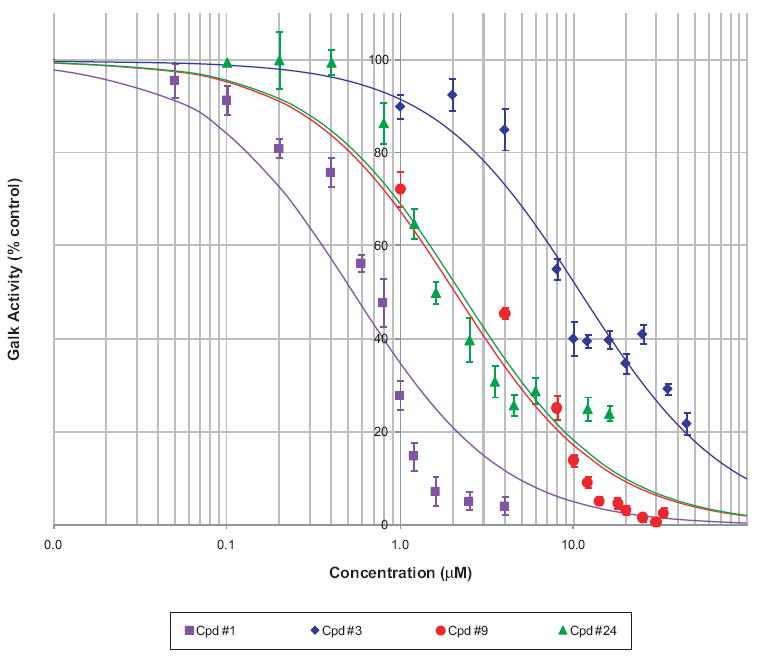
Determination of IC50 for selected inhibitors. Dose response of the inhibition of galactokinase (GALK) activity in the standard pyruvate kinase/lactate dehydrogenase-coupled assay for compounds 1, 3, 9, and 24. Data are shown as a percentage of the corresponding controls (symbols representing mean ± SD) and fitted with a standard log(inhibitor) versus response model (lines) to determine the median inhibitory IC50 values.
All 34 compounds contain at least 2 rings, at least 1 of which is aromatic, and all have at least 2 hydrogen-bond acceptor sites. A pair of aromatic rings separated by 1, 2, or 3 bonds is the most common recurring motif. If we also consider rings flattened by at least 2 double bonds (and some resonance effects) such as those in structures 3, 5, 7, 8, 11, 13, 14, 16, 17, 21, 26, 27, 28, 31, 33, and 34, as a part of these pairs, then such a pair of close, flat rings is present in the majority of structures (31 of 34; separation of 1 bond: 1, 3, 5, 6, 8, 12, 13, 14, 15, 16, 19, 20, 21, 23, 24, 31, 34; 2 bonds: 3, 5, 6, 7, 9, 10, 17, 18, 19, 20, 21, 26, 28, 31, 34; 3 bonds: 1, 2, 5, 11, 12, 14, 16, 22, 23, 26, 27, 29, 30, 33). The remaining structures (4, 25, and 32) as well as some of the other ones (e.g., 1, 2, 6, 7, 23, 24, 30, 33) all contain an aromatic ring joined to a flat ring containing at least 1 >C =O moiety. One of the most frequent motifs is a 6-membered dihydro-thiazinone ring, usually thio and cyano substituted (e.g., 5, 11, 13, 16, or 27; 14 having a similar ring), as well as a 5-membered methylene-thiazolidinone ring (e.g., 3, 17, 26, 28). A cyano substituent is present in 7 of the 34 structures. Most structures are neutral, nonionizable compounds: only 1 contains an ionizable aliphatic amine (2), but 6 contain an aromatic carboxylic acid (10—isomeric, 12, 16, 22, 24, and 31). There are also quite a few halo-substituted phenols such as 9, 10, 18, and 30. Because there is considerable structural similarity among these 34 most promising compounds identified here, they can help in identifying an active binding site and serve as good leads for the development of an effective drug therapy for CG.
Our report represents the starting point of a series of ongoing studies that aimed to identify bioactive, potent, and selective GALK inhibitors. Our goal is that such inhibitors eventually will result in novel, medical treatment complementary to dietary therapy for patients with CG.
Acknowledgments
Grants support to Kent Lai includes NIH grant 1R01 HD054744, American Heart Association South-East Affiliate Scientist Development grant 0435267B, and The Woman’s Cancer Association of the University of Miami. The University of Miami and the authors have filed a Provisional Patent application to USPTO for the screening methodology and the selected compounds described in this study. We also acknowledge the support of Dr. Frank An and the members of the HTS Facility at the Broad Institute of Harvard University/MIT.
References
- 1.Isselbacher KJ, Anderson EP, Kurahashi K, Kalckar HM. Congenital galactosemia, a single enzymatic block in galactose metabolism. Science. 1956;123:635–636. doi: 10.1126/science.123.3198.635. [DOI] [PubMed] [Google Scholar]
- 2.Leloir LF. The enzymatic transformation of uridine diphosphate glucose into a galactose derivative. Arch Biochem. 1951;33:186–190. doi: 10.1016/0003-9861(51)90096-3. [DOI] [PubMed] [Google Scholar]
- 3.Gitzelmann R. Estimation of galactose-I-phosphate in erythrocytes: a rapid and simple enzymatic method. Clin Chim Acta. 1969;26:313–316. doi: 10.1016/0009-8981(69)90385-4. [DOI] [PubMed] [Google Scholar]
- 4.Gitzelmann R, Curtius HC, Schneller I. Galactitol and galactose-1-phosphate in the lens of a galactosemic infant. Exp Eye Res. 1967;6:1–3. doi: 10.1016/s0014-4835(67)80047-2. [DOI] [PubMed] [Google Scholar]
- 5.Lai K, Langley SD, Khwaja FW, Schmitt EW, Elsas LJ. GALT deficiency causes UDP-hexose deficit in human galactosemic cells. Glycobiology. 2003;13:285–294. doi: 10.1093/glycob/cwg033. [DOI] [PubMed] [Google Scholar]
- 6.Segal S, Berry GT. Disorders of galactose metabolism. In: Scriver D, Sly W, Valle D, editors. The Metabolic Basis of Inherited Diseases. New York: McGraw-Hill; 1995. pp. 967–1000. [Google Scholar]
- 7.Kaye CI, Committee on Genetics. Accurso F, La Franchi S, Lane PA, Hope N, et al. Newborn screening fact sheets. Pediatrics. 2006;118:e934–e963. doi: 10.1542/peds.2006-1783. [DOI] [PubMed] [Google Scholar]
- 8.Gitzelmann R, Steinmann B. Galactosemia: how does long-term treatment change the outcome? Enzyme. 1984;32:37–46. doi: 10.1159/000469448. [DOI] [PubMed] [Google Scholar]
- 9.Waggoner D, Buist NRM, Donnell GN. Long-term prognosis in galactosemia: results of a survey of 350 cases. J Inherited Metab Disord. 1990;13:802–818. doi: 10.1007/BF01800204. [DOI] [PubMed] [Google Scholar]
- 10.Waggoner D, Buist NRM. Long-term complications in treated galactosemia: 175 U.S. cases. Int Pediatr. 1993;8:97–100. [Google Scholar]
- 11.Gitzelmann R. Galactose-1-phosphate in the pathophysiology of galactosemia. Eur J Pediatr. 1995;154(suppl 2):S45–S49. doi: 10.1007/BF02143803. [DOI] [PubMed] [Google Scholar]
- 12.Holden HM, Thoden JB, Timson DJ, Reece RJ. Galactokinase: structure, function and role in type II galactosemia. Cell Mol Life Sci. 2004;61:2471–2484. doi: 10.1007/s00018-004-4160-6. [DOI] [PubMed] [Google Scholar]
- 13.Zhou T, Daugherty M, Grishin NV, Osterman AL, Zhang H. Structure and mechanism of homoserine kinase: prototype for the GHMP kinase superfamily. Structure. 2000;8:1247–1257. doi: 10.1016/s0969-2126(00)00533-5. [DOI] [PubMed] [Google Scholar]
- 14.Gitzelmann R. Letter: additional findings in galactokinase deficiency. J Pediatr. 1975;87(pt 1):1007–1008. doi: 10.1016/s0022-3476(75)80937-1. [DOI] [PubMed] [Google Scholar]
- 15.Gitzelmann R, Wells HJ, Segal S. Galactose metabolism in a patient with hereditary galactokinase deficiency. Eur J Clin Invest. 1974;4:79–84. doi: 10.1111/j.1365-2362.1974.tb00376.x. [DOI] [PubMed] [Google Scholar]
- 16.Bosch AM, Bakker HD, van Gennip AH, van Kempen JV, Wanders RJ, Wijburg FA. Clinical features of galactokinase deficiency: a review of the literature. J Inherit Metab Disord. 2002;25:629–634. doi: 10.1023/a:1022875629436. [DOI] [PubMed] [Google Scholar]
- 17.Douglas HC, Hawthorne DC. Enzymatic expression and genetic linkage of genes controlling galactose utilization in saccharomyces. Genetics. 1964;49:837–844. doi: 10.1093/genetics/49.5.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Douglas HC, Hawthorne DC. Regulation of genes controlling synthesis of the galactose pathway enzymes in yeast. Genetics. 1966;54:911–916. doi: 10.1093/genetics/54.3.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slepak T, Tang M, Addo F, Lai K. Intracellular galactose-1-phosphate accumulation leads to environmental stress response in yeast model. Mol Genet Metab. 2005;86:360–371. doi: 10.1016/j.ymgme.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 20.Acosta PB, Gross KC. Hidden sources of galactose in the environment. Eur J Pediatr. 1995;154(suppl 2):S87–S92. doi: 10.1007/BF02143811. [DOI] [PubMed] [Google Scholar]
- 21.Berry GT, Palmieri M, Gross KC, Acosta PB, Henstenburg JA, Mazur, et al. The effect of dietary fruits and vegetables on urinary galactitol excretion in galactose-1-phosphate uridyltransferase deficiency. J Inherit Metab Disord. 1993;16:91–100. doi: 10.1007/BF00711320. [DOI] [PubMed] [Google Scholar]
- 22.Bosch AM, Bakker HD, Wenniger-Prick LJ, Wanders RJ, Wijburg FA. High tolerance for oral galactose in classical galactosaemia: dietary implications. Arch Dis Child. 2004;89:1034–1036. doi: 10.1136/adc.2003.037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berry GT, Moate PJ, Reynolds RA, Yager CT, Ning C, Boston RC, et al. The rate of de novo galactose synthesis in patients with galactose-1-phosphate uridyltransferase deficiency. Mol Genet Metab. 2004;81:22–30. doi: 10.1016/j.ymgme.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 24.Berry GT, Nissim I, Lin Z, Mazur AT, Gibson JB, Segal S. Endogenous synthesis of galactose in normal men and patients with hereditary galactosaemia. Lancet. 1995;346:1073–1074. doi: 10.1016/s0140-6736(95)91745-4. [DOI] [PubMed] [Google Scholar]
- 25.Heinrich MR, Howard SM. Galactokinase. Methods Enzymol. 1966;9:407–412. [Google Scholar]
- 26.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 27.Seiler KP, George GA, Happ MP, Bodycombe NE, Carrinski HA, Norton S, et al. ChemBank: a small-molecule screening and cheminformatics resource database. Nucleic Acids Res. 2007 doi: 10.1093/nar/gkm843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.CHEMBANK Web site [Online] Retrieved from http://chembank.broad.harvard.edu/details.htm?tag=Help.
- 29.Wu G, Yuan Y, Hodge CN. Determining appropriate substrate conversion for enzymatic assays in high-throughput screening. J Biomol Screen. 2003;8:694–700. doi: 10.1177/1087057103260050. [DOI] [PubMed] [Google Scholar]
- 30.Thoden JB, Timson DJ, Reece RJ, Holden HM. Molecular structure of human galactokinase: implications for type II galactosemia. J Biol Chem. 2005;280:9662–9670. doi: 10.1074/jbc.M412916200. [DOI] [PubMed] [Google Scholar]
- 31.Timson DJ, Reece RJ. Functional analysis of disease-causing mutations in human galactokinase. Eur J Biochem. 2003;270:1767–1774. doi: 10.1046/j.1432-1033.2003.03538.x. [DOI] [PubMed] [Google Scholar]
- 32.Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barberis A, Gunde T, Berset C, Audetat S, Luthi U. Yeast as a screening tool. Drug Discov Today. 2005;2:187–192. doi: 10.1016/j.ddtec.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 34.Perkins E, Sun D, Nguyen A, Tulac S, Francesco M, Tavana H, et al. Novel inhibitors of poly(ADP-ribose) polymerase/PARP1 and PARP2 identified using a cell-based screen in yeast. Cancer Res. 2001;61:4175–4183. [PubMed] [Google Scholar]