

Abstract
UDP-GlcNAc: lysosomal enzyme N-acetylglucosamine-1-phosphotransferase (GlcNAc-1-phosphotransferase) is an α2β2γ2 hexameric enzyme that catalyzes the first step in the synthesis of the mannose 6-phosphate targeting signal on lysosomal hydrolases. In humans, mutations in the gene encoding the α/β subunit precursor give rise to mucolipidosis II (MLII), whereas mutations in the gene encoding the γ subunit cause the less severe mucolipidosis IIIC (MLIIIC). In this study we describe the phenotypic, histologic, and serum lysosomal enzyme abnormalities in knockout mice lacking the γ subunit and compare these findings to those of mice lacking the α/β subunits and humans with MLII and MLIIIC. We found that both lines of mutant mice had elevated levels of serum lysosomal enzymes and cytoplasmic alterations in secretory cells of several exocrine glands; however, lesions in γ-subunit deficient (Gnptg−/−) mice were milder and more restricted in distribution than in α/β-subunit deficient (Gnptab−/−) mice. We found that onset, extent, and severity of lesions that developed in these two different knockouts correlated with measured lysosomal enzyme activity; with a more rapid, widespread, and severe storage disease phenotype developing in Gnptab−/− mice. In contrast to mice deficient in the α/β subunits, the mice lacking the γ subunits were of normal size, lacked cartilage defects, and did not develop retinal degeneration. The milder disease in the γ-subunit deficient mice correlated with residual synthesis of the mannose 6-phosphate recognition marker. Of significance, neither strain of mutant mice developed cytoplasmic vacuolar inclusions in fibrocytes or mesenchymal cells (I-cells), the characteristic lesion associated with the prominent skeletal and connective tissue abnormalities in humans with MLII and MLIII. Instead, the predominant lesions in both lines of mice were found in the secretory epithelial cells of several exocrine glands, including the pancreas, and the parotid, submandibular salivary, nasal, lacrimal, bulbourethral, and gastric glands. The absence of retinal and chondrocyte lesions in Gnptg−/− mice might be attributed to residual β-glucuronidase activity. We conclude that mice lacking either α/β or γ subunits displayed clinical and pathologic features that differed substantially from those reported in humans having mutations in orthologous genes.
Keywords: Knockout mice, lysosomal storage disease, mucolipidosis
Introduction
Most lysosomal storage diseases are caused by reduced activity of a single lysosomal enzyme, but in mucolipidosis II (MLII) and mucolipidosis type III (MLIII) a functional deficiency of multiple enzymes results from faulty intracellular trafficking of lysosomal hydrolases. In many eukaryotic cells, the proper targeting of newly synthesized acid hydrolases to lysosomes is mediated by the mannose 6-phosphate (Man-6-P) recognition system.24 The addition of Man-6-P recognition markers on newly synthesized lysosomal hydrolases is mediated by a multimeric enzyme, UDP-N-acetylglucosamine-1-phosphotransferase (GlcNAc-1-phosphotransferase) in the network Golgi.24 GlcNAc-1-phosphotransferase is a hexamic enzyme consisting of 3 subunits: α2, β2, and γ2.3 The α and β subunits, which contain the catalytic activity, are encoded by a single gene, GNPTAB.34,43 The γ subunit facilitates the recognition of protein determinants on acid hydrolase substrates and is encoded by the GNPTG gene.36,37
The added Man-6-P residues serve as recognition markers that bind lysosomal proenzymes to high-affinity Man-6-P-specific receptors (MPRs)21; which include a cation-dependent Man-6-P receptor (CD-MPR) and an insulin-like growth factor II/cation-independent Man-6-P receptor (IGF-II/CI-MPR).40 The resulting proenzyme-receptor complexes are transported from the trans-Golgi network to prelysosomal endosomes, where the proenzyme-receptor complexes dissociate in the low pH environment. In contrast, newly synthesized lysosomal proenzymes that lack Man-6-P recognition markers are released directly into the extracellular medium instead of being incorporated into lysosomes. As a result, lysosomes are enzyme deficient and cells accumulate undigested storage material, while elevated lysosomal enzyme levels are present in the serum.25 In humans, deficient GlcNAc-1-phosphotransferase activity gives rise to the autosomal recessive lysosomal storage diseases termed mucolipidosis II (MLII, I-cell disease) and mucolipidosis III (MLIII, pseudo-Hurler polydys-trophy).25 In human patients with MLII and MLIII, the striking elevations in serum levels of lysosomal acid hydrolases result from the lack of synthesis of the Man-6-P recognition markers that are required for proper targeting of these enzymes to lysosomes. As a result, the enzymes are secreted into the bloodstream where their activity can be measured.25
All forms of these disorders are characterized by high levels of multiple acid hydrolases in plasma and low levels of the same enzymes in mesenchymal fibroblasts. The fibroblasts develop numerous cytoplasmic inclusions, thus the name I (inclusion) cells.31 MLII is a severe, rapidly progressive disorder that is generally fatal during the first decade of life. All patients with MLII that have been characterized by genetic analysis have mutations in the GNPTAB gene.4,27,34,43 MLIII has a milder phenotype, likely because of some residual GlcNAc-1-phosphotransferase activity. Within MLIII, 2 variants have been recognized: MLIIIA and MLIIIC. MLIIIA results from mutations in GNPTAB, while MLIIIC is due to mutations in GNPTG 26,27,45 The GlcNAc-1-phosphotransferase in the cells of patients with MLIIIC exhibits normal levels of activity toward the simple sugar a-methylmannose, but decreased activity toward acid hydrolase substrates due to an impaired ability to recognize the protein determinants on the acid hydrolases, a key step in the high affinity interaction with these substrates.26,45
Animal models of lysosomal storage diseases have greatly facilitated studies to clarify the pathophysiology and treatment of these disorders. A few such animal models with clinical, biochemical, and pathologic similarities to human MLII have been described. GlcNAc-1-phosphotransferase-deficient cats display several clinical and radiographic features similar to those seen in human MLII disease, although affected kittens also developed diffuse retinal degeneration by 4 months of age, a finding not described in human MLII.8,32 Similarly, triple knockout mice, deficient for insulin growth factor 2 (Igf2) and the 2 Man-6-P receptors display coarse facial features, skeletal dysplasia, stiffened joints and skin, growth retardation, and psychomotor retardation, thereby resembling human MLII.9 However, this triple knockout mouse is not a true model of MLII. We recently reported on mice made deficient in GlcNAc-1-phosphotransferase by inactivation of the α/β gene.15 These mice exhibit some features of human MLII, such as growth retardation and elevated plasma lysosomal enzymes, but differ in developing retinal degeneration, having prominent secretory cell lesions, and lacking inclusions in fibrocyte/mesenchymal cells. To date, no animal models for MLIIIC have been reported.
Here we present and compare the phenotypic, histopathologic, and serum lysosomal enzyme abnormalities in γ-subunit deficient (Gnptg−/−) mice with those α/β-subunit deficient (Gnptab−/−) mice and humans with MLIIIC. We find that mice lacking either α/β or γ subunits displayed clinical and pathologic features that differed substantially from those reported in humans having mutations in orthologous genes.
Materials and Methods
Mouse production and husbandry
We previously reported the methods used to generate both the γ-subunit deficient mice30 and the α/β-subunit deficient mice15 used in this study. In brief, for both knockout lines, 129S5/SvEvBrd (Lex-2) embryonic stem cells were microinjected into C57BL/6-TyrBrd (albino) blastocysts to generate chimeric animals, which were bred to C57BL/6-TyrBrd (albino) females. The homozygous F2 knockout mice used in phenotyping studies were produced by intercrossing the F1 heterozygous knockout (−/+) offspring of chimeric founder parents. In both knockout lines, wild-type, heterozygous, and homozygous F2 littermates were produced in a normal 1 : 2 : 1 Mendelian ratio. All mice were housed in a barrier facility at 24°C on a fixed 12-hour light and 12-hour dark cycle and were fed rodent chow #5001 (Purina, St. Louis, MO) ad libitum. Procedures involving animals were conducted in conformity with the Institutional Animal Care and Use Committee guidelines that are in compliance with the state and federal laws and the standards outlined in the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996).20
Phenotypic screen
Wild-type and homozygous null mice were subjected to a comprehensive battery of phenotype screening exams as previously described.5,48 Screening assays included behavioral tests (such as circadian rhythm, open field, inverted screen, prepulse inhibition of the acoustic startle response, tail suspension, marble burying, and context trace conditioning), fundoscopy and retinal angiography exams, blood pressure and heart rate measurements, serum chemistries, insulin levels, glucose tolerance testing, hematology, peripheral blood fluorescence-activated cell sorting (FACS) analysis, urinalysis, Dual-energy x-ray absorptiometry (DEXA) scans, computerized axial tomography (CAT)-scans, Microcomputed tomography (Micro-CT) scans, fertility testing, and skin fibroblast proliferation assays, and pathology.
Histopathology
Tissues collected from knockout mice and age-matched wild-type control mice were immersion fixed in 10% neutral buffered formalin, except eyes, which were fixed for 24 hours in Davidson's fixative. All tissues were embedded in paraffin, sectioned at 4 μm, and mounted on positively charged glass slides (Superfrost Plus, Fisher Scientific, Pittsburgh, PA) and stained with hematoxylin and eosin (HE) for histopathologic examination. For immunohistochemistry, 4-μm sections were first deparaffinized in xylene and then rehydrated through graded alcohols to phosphate buffered saline (PBS); endogenous peroxidase activity was blocked by incubation in 3.0% hydrogen peroxide in PBS for 5 minutes. Rat anti-mouse F4/80 antibody (Abcam Inc, Cambridge, MA) was diluted 1 : 1,000 in PBS and applied for 1 hour. After rinsing, sections were incubated for 1 hour in the biotinylated rabbit anti-rat IgG secondary antibody (Vector Laboratories, Burlingame, CA) diluted 1 : 400 in PBS. Bound antibodies were detected by an avidin-biotin complex method with 3,3′-diaminobenzidine as chromogenic substrate following the manufacturer's instructions (Elite ABC Kit, Vector Laboratories).
Serum lysosomal enzyme assays
Acid hydrolase assays were performed as previously described with minor modifications.41,46 All reagents were obtained from Sigma (St. Louis, MO) unless otherwise noted. β-Hexosaminidase and β-galactosidase were assayed with 5 mM 4-methylumbelliferyl(MU)-N-acetyl-β-D-glucosaminide (Sigma M-2133) and 5 mM 4-MU-β-D-galactopyranoside (Calbiochem 474424, San Diego, CA), respectively, in 50 mM citrate buffer containing 0.5% Triton X-100 (pH 4.5). β-Glucuronidase was assayed with 5 mM 4-MU-β-D-glucuronide (Calbiochem 474427) in 0.1 M Na-acetate buffer containing 0.5% Triton X-100 (pH 4.6). β-Mannosidase was assayed with 5 mM 4-MU-β-D-mannopyranoside (Sigma M0905) in 50 mM Na-citrate buffer containing 0.5% Triton X-100 (pH 5.0). α-Mannosidase was assayed with 5 mM 4-MU-α-D-mannopyranoside (Sigma M3657) in 50 mM Na-citrate buffer containing 0.5% Triton X-100 (pH 4.0). The assay mixtures containing 90 μl of 5 mM substrate solution and 10 μl of sample were incubated for 1 to 5 hours at 37°C and then quenched with 900 μl of 0.4 M glycine-NaOH buffer, pH 10.8. The fluorescence was measured in a TURNER Model 450 Fluorometer (Barnstead Thermolyne Corp., Dubuque, IA) using excitation and emission wavelengths of 360 and 450 nm, respectively.
Man-6-P receptor affinity chromatography
The cation-independent mannose 6-phosphate receptor (CI-MPR)-affinity columns were provided by Walter Gregory (Washington University School of Medicine in St. Louis, MO). As described previously,11 50 μl of serum was diluted 10-fold with Buffer A (50 mM Imidazole-HCl, pH 7.25, 150 mM NaCl, 0.05% Tx-100) and loaded onto a 1-ml CI-MPR Sepharose column (0.86 mg/ml). The column was washed with 7 ml of Buffer A, then with 7 ml of 5 mM glucose 6-phosphate in Buffer A, and finally eluted with 5 ml of 10 mM Man-6-P in Buffer A.
Results
Phenotypic screening
No gross differences were noted between wild-type, heterozygous, and homozygous Gnptg−/− mice (8–14 weeks in age) subjected to a standardized array of tests in a comprehensive phenotypic analysis.5 In addition, Gnptg−/− mice that were monitored up to 2 years of age displayed normal growth, behavior, reproduction, and lifespan. There was no evidence of the skeletal or joint abnormalities that are so prominent in humans with MLII and MLIII.
In marked contrast, Gnptab−/− mice were easily differentiated from control littermates by their smaller size, having both decreased mean body weight and mean body length.15 DEXA analysis showed that homozygous mutant Gnptab−/− mice had decreased mean bone mineral content, volumetric bone density, and bone mineral density as well as decreased mean total tissue mass, lean body mass, and percentage total body fat. Micro-CT revealed notable decreases in vertebral trabecular bone volume, trabecular number, thickness, and connectivity density, and decreased mean femoral midshaft cortical thickness and cross-sectional area. In contrast, these studies were all within the normal limits in the Gnptg−/− mice. The Gnptab−/− mutant mice also had notably increased mean serum alkaline phosphatase levels compared with gender-matched Gnptab+/+ littermates and historic means. In addition, the Gnptg−/− mice had normal retinas by fundoscopy, whereas the Gnptab−/− mice exhibited multifocal retinal depigmentation with attenuated retinal vessels and decreased mean retinal artery-to-vein (A/V) ratios, which progressed to severe retinal degeneration by 3 months of age.15 The neurologic exams of both types of mice were unremarkable except that the Gnptab−/− mice showed increased pain tolerance.
Histopathology
Although Gnptg−/− mice were grossly normal, progressive histologic lesions developed in secretory cells in several glandular tissues, most notably the pancreas, parotid salivary glands, maxillary and lateral (Steno) nasal glands, and bulbourethral glands. A thorough survey of all sections showed fibroblasts to have a normal appearance at all time points in both knockout lines, clearly differentiating the disease in mice from the human mucolipidoses, where distended mesenchymal cells represent the key morphologic feature in both MLII and MLIII.31 In contrast, secretory cell lesions comprising the most common findings in both knockout mouse lines are not widely recognized or reported in human MLII or MLIII.13
The rapidity of onset, severity, and distribution of secretory cell lesions differed in the 2 knockout mouse lines, generally being slower developing, milder and less widely distributed in the Gnptg−/− mice (Table 1). Thus, at 14 weeks of age, lesions in the pancreas, parotid salivary gland, nasal glands, and bulbourethral glands of Gnptg−/− mice were relatively mild in comparison with those in Gnptab−/− mice of equivalent age. However, by 2 years of age, the lesions in these tissues had significantly progressed and in several tissues (pancreas, parotid salivary glands) were as severe as those found in the Gnptab−/− mice. Similarly, lesions were absent to mild in the submandibular salivary glands, lateral nasal glands, maxillary and lacrimal glands of 14-week-old Gnptg−/− mice, but by 2 years of age they were almost as severe as those of Gnptab−/− mice. In contrast, the lesions in the gastric glands, duodenal glands, chondrocytes, and retina of Gnptab−/− mice never developed even in aged Gnptg−/− mice.
Table 1.
Distribution and severity of lesions in Gnptg−/− and Gnptab−/− mice.*
|
Gnptg−/− |
Gnptab−/− |
||||
|---|---|---|---|---|---|
| 14 Weeks | 24 Months | 14 Weeks | 10 Months | WT | |
| Pancreas | ++ | ++++ | ++++ | ++++ | − |
| Parotid Salivary Glands | ++ | ++++ | ++++ | ++++ | − |
| Steno Glands | + | +++ | +++ | ++++ | − |
| Maxillary Glands | ++ | +++ | +++ | ++++ | |
| Submandibular Salivary Gland | + | ++ | +++ | +++ | − |
| Lacrimal Gland | − | ++ | +++ | +++ | − |
| Gastric Glands | − | − | ++++ | ++++ | − |
| Duodenal Glands | − | − | + | + | − |
| Bulbourethral Glands | ++ | ++ | ++++ | ++++ | − |
| Chondrocytes | − | − | ++ | ++ | − |
| Retina | − | − | +++ | +++ | − |
| Fibroblasts/Fibrocytes | − | − | − | − | − |
Gnptg−/− = γ-subunit deficient mice; Gnptab−/− = α/β-subunit deficient mice; WT = wild-type; + = minimal; ++ = mild; +++ = moderate; ++++ = marked.
Normal secretory acinar cells in the pancreas and parotid gland have a pyramidal shape, with the round nucleus centered in the broad cell base being surrounded by slightly basophilic cytoplasm that transitions sharply to the eosinophilic secretory granules that pack the apical zone of the cells (Fig. 1). In comparison, in Gnptg−/− mice at 14 weeks, many acinar cells in the pancreas and parotid salivary glands were moderately enlarged and had a pale zone of foamy cytoplasm between the basal nuclear region of the cell and the apical secretory granules (Fig. 2). Lesions in young Gnptab−/− mice were more severe; the normal cytoarchitecture of pancreatic acinar cells was disrupted by large cytoplasmic microvacuoles (Fig. 3) and contained indistinct granular material that stained positively with periodic acid Schiff (PAS) and Alcian Blue (AB) stains. With increasing age, the pancreatic lesions in Gnptg−/− mice progressed in severity to match those seen in both young and old Gnptab−/− mice. The more severe advanced lesions were characterized by marked disorganization of the acinar architecture and diffuse, marked cytoplasmic alteration and cytomegaly of acinar cells (Fig. 4). The ballooning vacuolar inclusions in affected cells typically effaced the zone of basophilia normally present in the basal perinuclear region of these cells and were associated with a marked decrease in the size and number of zymogen granules at the apex of the pancreatic acinar cells. Special stains showed that these ballooning vacuoles frequently contained small amounts of PAS and AB-positive material. Degenerating or necrotic acinar cells were very rarely detected among the tightly packed clusters of exocrine pancreatic cells but in aged Gnptg−/− and Gnptab−/− mice, the pancreatic interstitium was expanded multifocally by clusters of large round-to-polygonal histiocytic cells (top center of Fig. 4) that were filled with cytoplasmic granules that were both PAS (Fig. 5) and AB-positive (Fig. 6). Immunohistochemistry confirmed that these interstitial histiocytic cells were F4/80 positive macrophages (Fig. 7).
Fig. 1.

Pancreas; wild-type control mouse. Normal acinar cells have a pyramidal shape, with the round basal nucleus being surrounded by basophilic cytoplasm that transitions sharply to eosinophilic secretory granules that pack the apical zone. HE.
Fig. 2.

Pancreas; 14-week-old Gnptg−/− mouse. Acinar cells are moderately enlarged and contain a zone of pale foamy cytoplasm between the basal nuclear region of the cell and the apical secretory granules. HE.
Fig. 3.

Pancreas; 14-week-old Gnptab−/− mouse. Acinar cells are distorted by large cytoplasmic microvacuoles that compress basal nuclei and apical cytoplasm. HE.
Fig. 4.
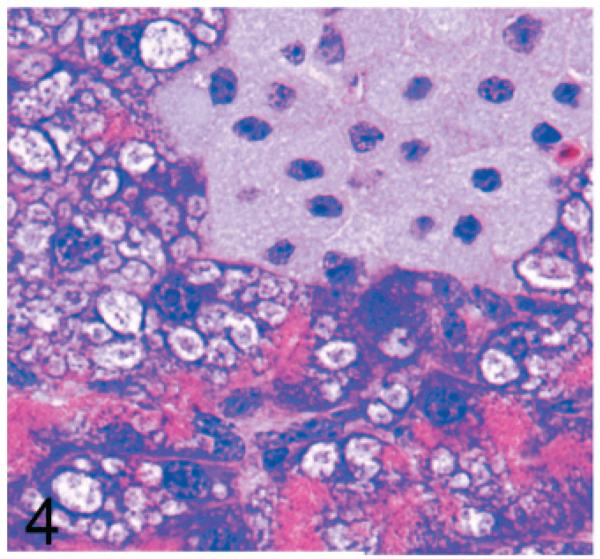
Pancreas; 2-year-old Gnptg−/− mouse. In older mice, there is disorganization of the acinar architecture and diffuse marked cytoplasmic alteration and cytomegaly of acinar cells. The ballooning vacuolar inclusions efface the basal perinuclear basophilia and are associated with decreased size and number of apical zymogen granules. The pancreatic interstitium was expanded multifocally by clusters of large round-to-polygonal pale histiocytic cells. HE.
Fig. 5.
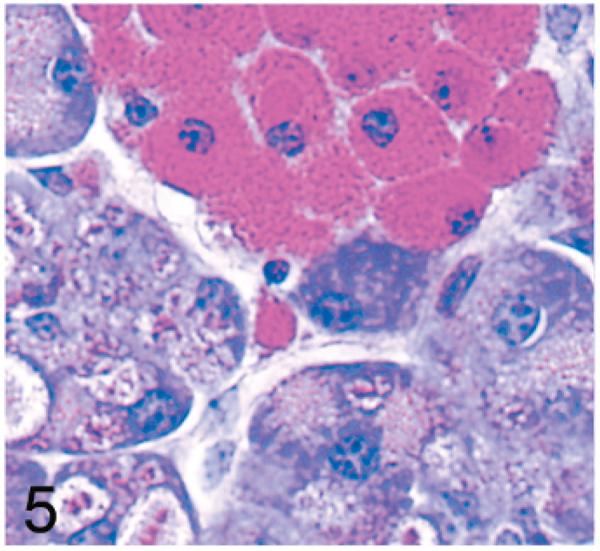
Pancreas; 2-year-old Gnptg−/− mouse. By periodic acid-Schiff (PAS)-stain reaction, cytoplasmic granules in acinar cells and filling interstitial histiocytes stain crimson red. PAS.
Fig. 6.
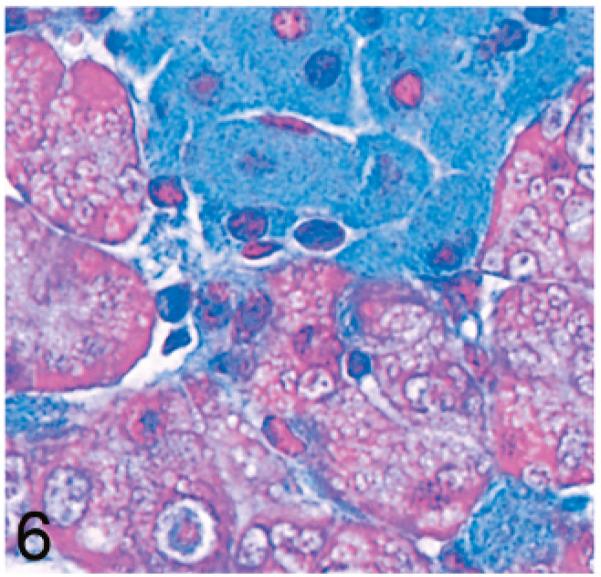
Pancreas; 2-year-old Gnptg−/− mouse. The blue cytoplasmic granules filling interstitial histiocytes are also AB positive. Alcian Blue stain (AB).
Fig. 7.
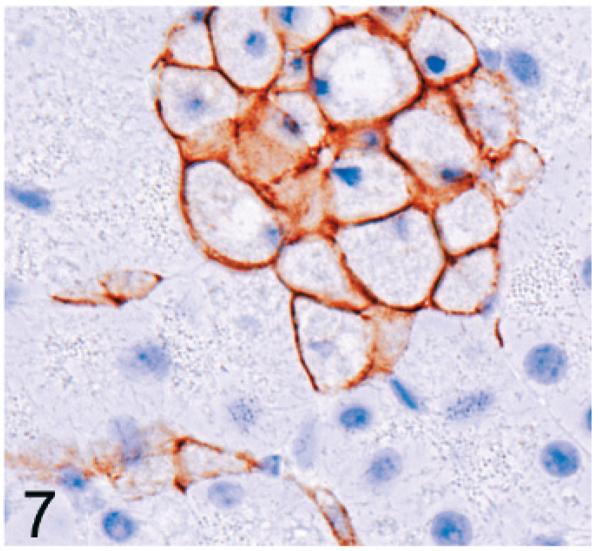
Pancreas; 2-year-old Gnptg−/− mouse. The clusters of pale interstitial cells are identified as macrophages by positive labeling for F4/80 antigen. Immunohistochemical staining: avidin-biotin complex methods, Mayer's hematoxylin counterstain.
Lesions in the pyramidal acinar secretory cells of the parotid gland were very similar to those seen in the pancreas. Again, cytoplasmic vacuolization in Gnptg−/− mice progressed from a midzonal microvacuolization at 14 weeks of age to a marked disruption of normal cellular and tissue structure by large ballooning vacuoles containing small amounts of fibrillogranular PAS positive material by 2 years of age (Fig. 8). In contrast, marked vacuolization of parotid gland secretory cells was present even at the earliest time points in Gnptab−/− mice (not shown).
Fig. 8.
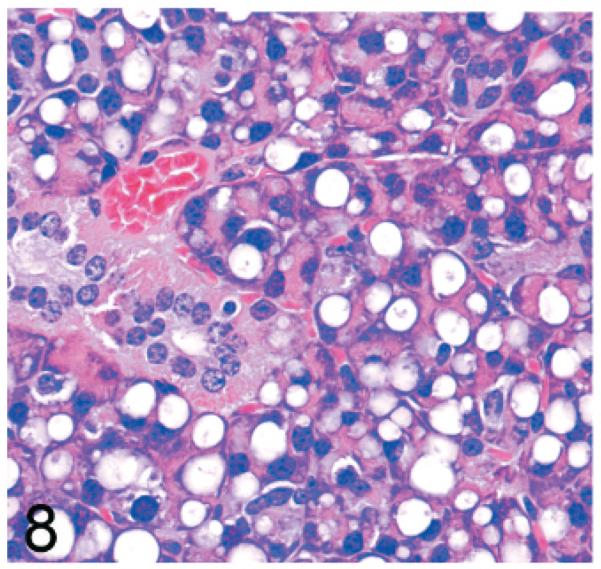
Parotid salivary gland; 2-year-old Gnptg−/− mouse. There is marked disruption of normal cellular and tissue structure by large ballooning vacuoles. HE.
The submandibular, maxillary, lateral nasal, lacrimal, and bulbourethral glands were affected in both knockout mouse lines; however, lesions were either absent or minimal in the 14-week-old Gnptg−/− mice. Even at 2 years of age, lesions in these tissues were less severe than in the Gnptab−/− mice. Typically, the mucus acinar cells of the submandibular salivary gland were enlarged by myriad cytoplasmic microvacuoles (Fig. 9). In aged Gnptg−/− mice, widespread cytoplasmic vacuolization was also present in the lacrimal glands, bulbourethral glands, and in some of the glands surrounding the maxillary sinus.
Fig. 9.
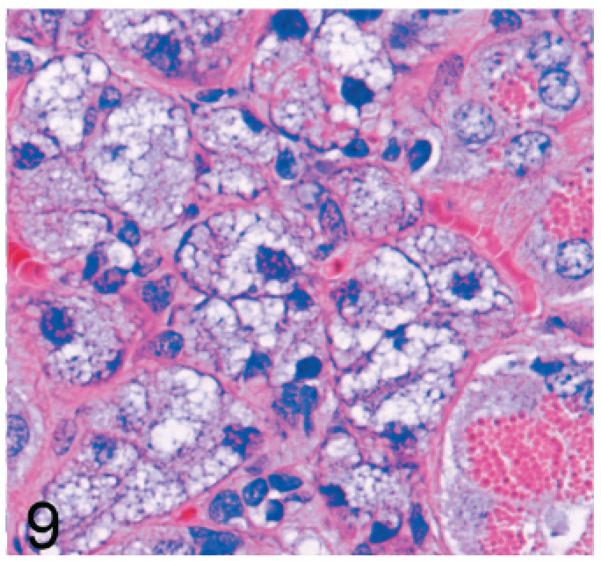
Submandibular salivary gland; 2-year-old Gnptg−/− mouse. The mucus acinar cells of the submandibular salivary gland were enlarged by myriad cytoplasmic microvacuoles. HE.
The mouse maxillary sinus is surrounded by the maxillary glands and the lateral nasal (Steno) glands, which can be distinguished by differences in apical granule size and staining characteristics even in HE-stained sections. With PAS staining, the intense crimson staining of the maxillary gland permits clear differentiation from the lateral nasal (Steno) gland (Fig. 10). In both knockout mouse lines, there was an earlier onset and more severe vacuolization affecting the secretory cells of the maxillary glands, which tend to be located dorsomedially, than of the lateral nasal (Steno) glands, which are generally ventrolateral to the maxillary sinus (Fig. 11).
Fig. 10.
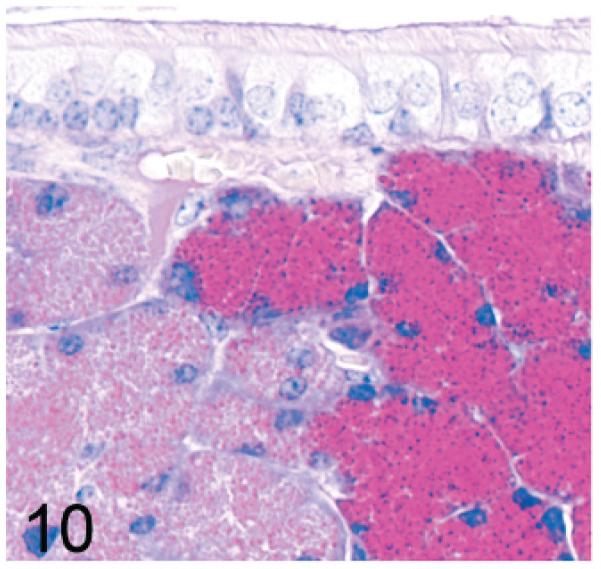
Maxillary sinus; wild-type control mouse. The intense crimson PAS staining of the maxillary gland on the right side is clearly differentiated from the pale lateral nasal (Steno) glands on the left side. PAS.
Fig. 11.
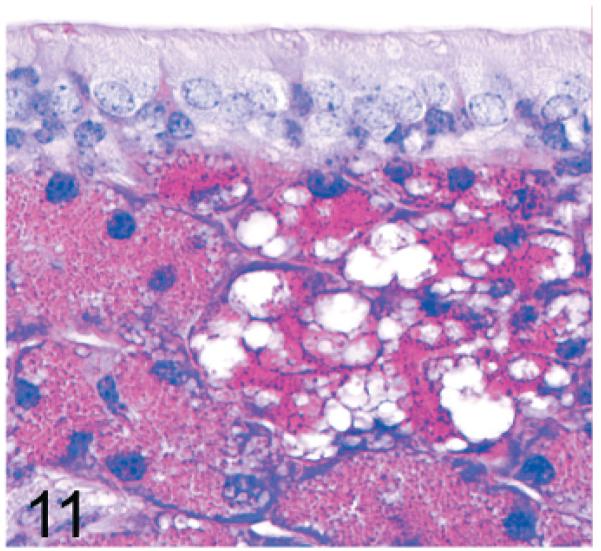
Maxillary sinus; 14-week-old Gnptg−/− mouse. Vacuolization is more severe in the secretory cells of the maxillary glands (right side) than in lateral nasal (Steno) glands (left side). PAS.
Of significance, some cell types and tissues that developed lesions in Gnptab−/− mice were apparently unaffected in Gnptg−/− mice. For example, chief cells in gastric glands of normal mice typically have basal nuclei with apical secretory granules (Fig 12). In contrast, gastric gland chief cells in Gnptab−/− mice were generally distended by cytoplasmic microvacuolization and ill-defined secretory granules (Fig. 13), while chief cells in Gnptg−/− mice remained essentially normal even in aged mice (Fig. 14). Normal chondrocytes typically contain a single, large, clear vacuole, and in routinely processed sections, their cytoplasm typically only partially fills the lacunar space (Fig. 15). In contrast, chondrocytes in Gnptab−/− mice were hypertrophic and swollen by myriad cytoplasmic vacuoles that completely filled enlarged cartilage lacunae (Fig. 16). These cytoplasmic changes affected both the round central chondrocytes and the spindle-shaped perichondral chondrocytes. As noted previously, the bones of Gnptab−/− mice were significantly shorter and narrower than those of wild-type mice; however, the perichondral osteoblasts, osteoclasts, and periostial fibroblasts appeared to be normal, and bone deformities or dysplasias were not detectable by gross or histologic examination. Chondrocytes appeared to be completely normal in the Gnptg−/− mice (Fig. 17).
Fig. 12.
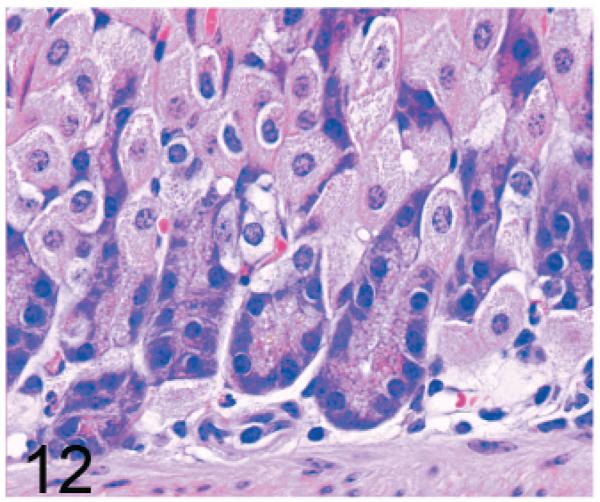
Stomach, gastric glands; wild-type control mouse. The chief cells in gastric glands of normal mice typically show basal nuclei with apical secretory granules. HE.
Fig. 13.
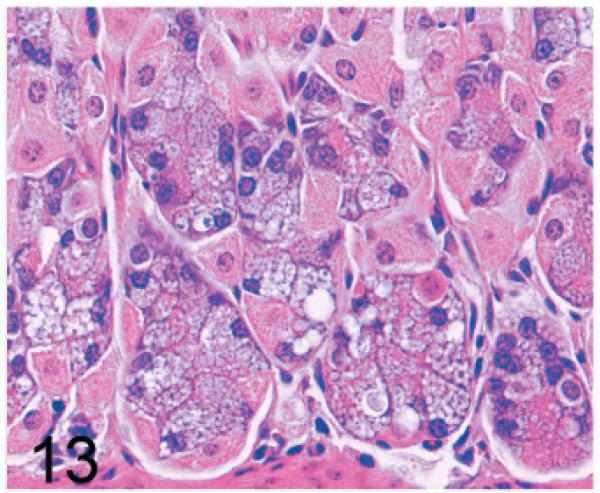
Stomach, gastric glands; 14-week-old Gnptab−/− mouse. Chief cells are distended by cytoplasmic microvacuolization and ill-defined secretory granules. HE.
Fig. 14.
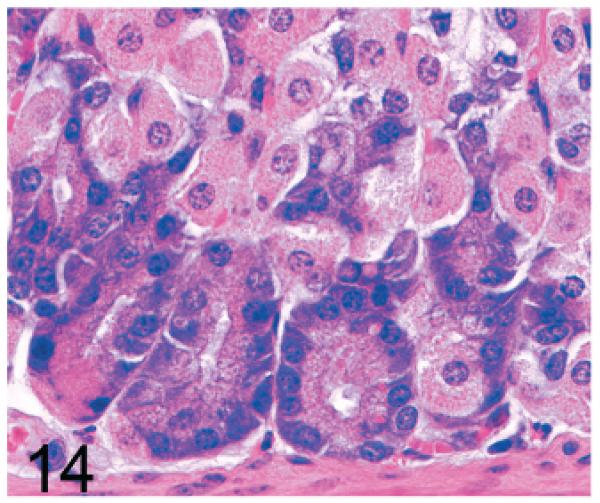
Stomach, gastric glands; 2-year-old Gnptg−/− mouse. Chief cells are essentially normal even in aged mice.
Fig. 15.
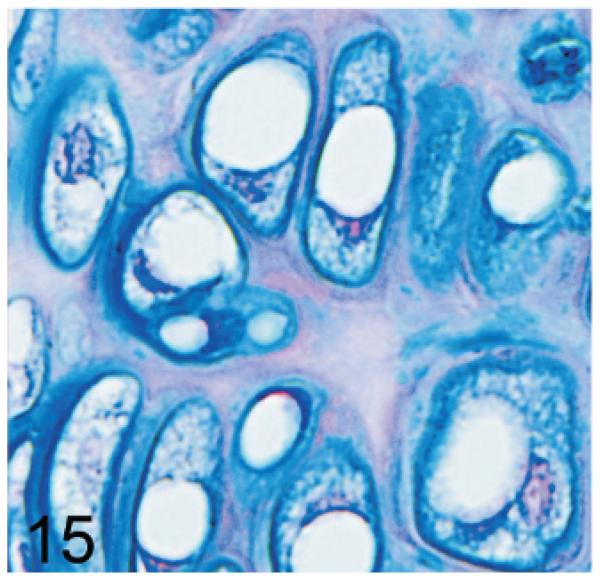
Sternum, cartilage; wild-type control mouse. Normal chondrocytes typically contain a single, large, clear vacuole, and their cytoplasm typically only partially fills the lacunar space. AB.
Fig. 16.
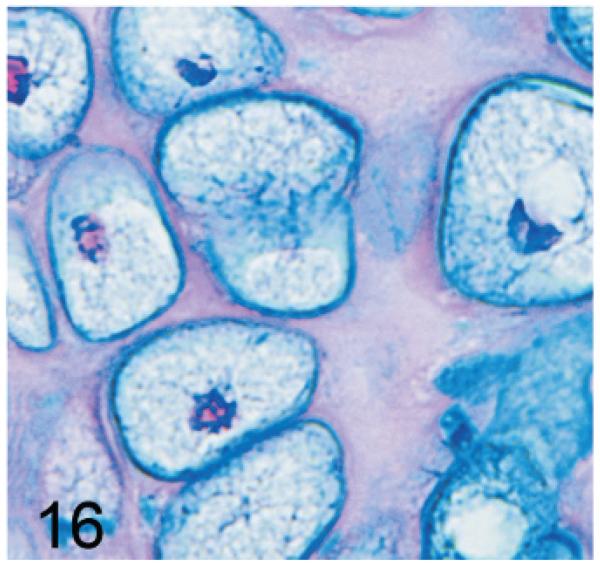
Sternum, cartilage; Gnptab−/− mouse. Chondrocytes are hypertrophic and distended by myriad cytoplasmic vacuoles to completely fill the enlarged cartilage lacunae. AB.
Fig. 17.
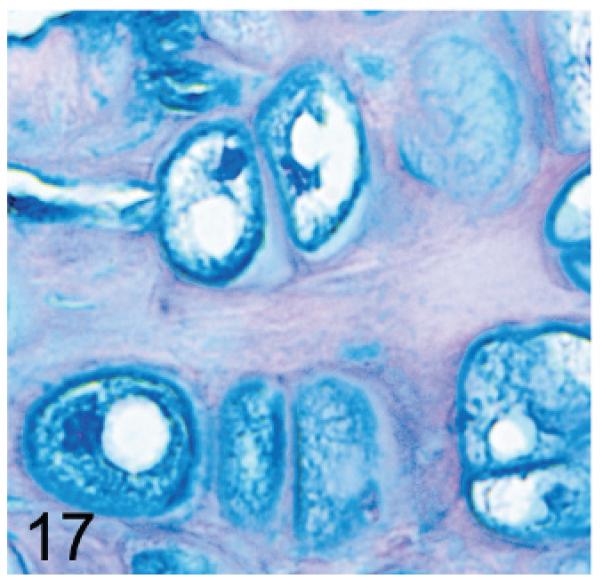
Sternum, cartilage; Gnptab−/− mouse. Chondrocytes appear to be completely normal in the Gnptg−/− mice. AB.
We previously reported that progressive retinal degeneration is an important lesion in Gnptab−/− mice.15 In Gnptab−/− mice, the retina appeared to be normal by light microscopy at 1 month of age, but by 3 months all mice were blind as a result of severe progressive retinal degeneration characterized by a loss of inner and outer segments of the photoreceptors as well as the outer nuclear layer (Fig. 18, top panel). In marked contrast, no evidence of retinal degeneration was present even in 2-year-old Gnptg−/− mice (Fig. 18, bottom panel).
Fig. 18.
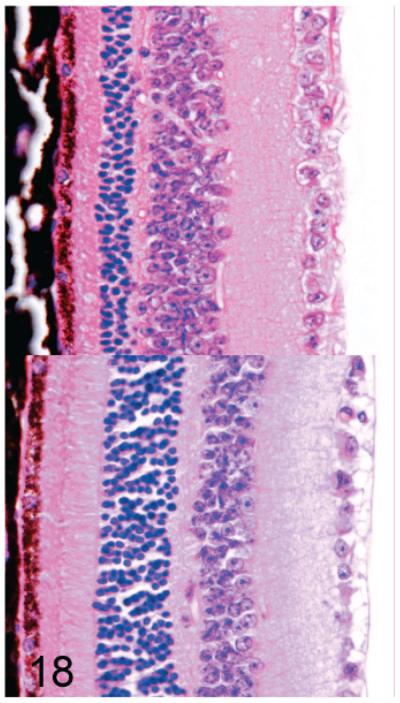
(upper panel). Retina; Gnptab−/− mouse. There is a severe reduction in the thickness of both the inner and outer segments of the photoreceptors as well as the outer nuclear layer. HE.
(lower panel). Retina; 2-year-old Gnptg−/− mouse. In marked contrast, there is no evidence of retinal degeneration in aged Gnptg−/− mice. HE.
Serum lysosomal enzyme activities
The serum levels of 5 different acid hydrolases were markedly elevated in both Gnptab−/− and Gnptg−/− mice (Table 2). Although elevations of 3 enzymes (β-hexosaminidase, β-mannosidase, and β-galactosidase) were similar in both knockout lines, serum levels of the other 2 measured enzymes (α-mannosidase and β-glucuronidase) were increased disproportionately in the Gnptab−/− mice. In Gnptg−/− mice, there was only a 16.5-fold increase in serum α-mannosidase versus a 40.5-fold increase in α/β subunit knockout mice, and an even larger difference was seen in serum β-glucuronidase levels (2.5-fold increase in Gnptg−/− versus 11.2-fold increase in Gnptab−/− mice).
Table 2.
Serum lysosomal enzyme activities in Gnptg−/− and Gnptab−/− mice.*
| Enzyme† |
|||||
|---|---|---|---|---|---|
| Genotype (No. of mice) | β- Hexosaminidase | α-Mannosidase | β-Mannosidase | β-Galactosidase | β-Glucuronidase |
| Wild-type (7) | 311 ± 114 | 990 ± 328 | 156 ± 33 | 20 ± 8 | 34 ± 14 |
| α,β-KO (5) | 2,087 ± 318 | 40,073 ± 5181 | 1,464 ± 211 | 277 ± 65 | 382 ± 92 |
| Fold Increase Over Normal | 6.7-fold | 40.5-fold | 9.4-fold | 13.9-fold | 11.2-fold |
| γ-KO (5) | 2,688 ± 878 | 16,313 ± 1351 | 1,826 ± 453 | 335 ± 122 | 86 ± 31 |
| Fold Increase Over Normal | 8.6-fold | 16.5-fold | 11.7-fold | 16.8-fold | 2.5-fold |
Gnptg−/− = γ-subunit deficient mice; Gnptab−/− = α/β-subunit deficient mice; KO = knock-out.
Data are expressed as nmol/ml/hr.
Binding of serum lysosomal hydrolases to CI-MPR-affinity column
Aliquots of diluted serum were passed over a CI-MPR-affinity column and then the run-through and Man-6-P-eluted fractions were assayed for 5 different acid hydrolase activities. The percentage of the total activity that bound to the column and was eluted with Man-6-P provides a measure of the content of phosphorylated high mannose oligosaccharides on these enzymes (Table 3). With the wild-type sera, between 27 and 52% of the activities bound specifically to the CI-MPR column. All 5 acid hydrolases of the Gnptg−/− mice bound to the affinity column to some extent, with high binding occurring with β-glucuronidase (89% of wild-type levels), intermediate binding with α-Mannosidase (17% of wild-type), and lower binding (2.5 to 10.6% of wild-type) for the 3 other enzymes. In contrast, less than 1% of the various acid hydrolases from the Gnptab−/− mice bound to the affinity column, which represents the lower limit of detection in this assay. These findings demonstrate that the serum acid hydrolases of the Gnptg−/− mice contain decreased but readily detectable levels of Man-6-P, whereas the hydrolases of the Gnptab−/− mice have no detectable mannose phosphorylation.
Table 3.
Man-6-P receptor affinity chromatography: binding of serum acid hydrolases to CI-MPR-affinity columns.*
| % Enzyme bound to CI-MPR column |
|||
|---|---|---|---|
| Enzymes | Controls (wild-type) | Gnptg−/− | Gnptab−/− |
| β-Hexosaminidase | 26.7 ± 5.5 | 1.3 ± 0.7 | 0.25 ± 0.18 |
| α-Mannosidase | 30.6 ± 9.5 | 5.2 ± 1.6 | 0.11 ± 0.16 |
| β-Mannosidase | 31.0 ± 7.7 | 3.3 ± 1.9 | 0.79 ± 0.77 |
| β-Galactosidase | 51.8 ± 9.2 | 1.3 ± 0.6 | 0.56 ± 0.41 |
| β-Glucuronidase | 46.5 ± 7.6 | 41.6 ± 6.2 | 0.66 ± 0.43 |
Man-6-P = mannose 6-phosphate; CI-MPR = cation-independent Man-6-P receptor; Gnptg−/− = γ-subunit deficient mice; Gnptab−/− = α/β-subunit deficient mice.
Discussion
Although both the α/β and the γ subunits of GlcNAc-1-phosphotransferase have the same biochemical properties in mice as in their human counterparts, inactivation of these genes resulted in a clinical course of disease and histopathologic and radiologic findings in mice that were markedly different from those reported in human patients. Despite these differences in disease severity and presentation, Gnptab−/− and Gnptg−/− mice illustrate important general features of lysosomal storage disease pathogenesis and provide valuable insights into several important biologic processes.
Previously we reported the phenotype of mice with a disruption of the Gnptab gene that encodes the catalytic subunits of GlcNAc-1-phosphotransferase and compared these findings with humans with MLII, the autosomal recessive disorder also due to mutations in the GNPTAB gene.15 While both disorders are characterized by greatly increased levels of multiple serum lysosomal hydro-lases, several critical differences emerged. First, the phenotype of the Gnptab−/− mice was not as severe as that seen in humans. The knockout mice exhibited growth retardation and a decrease in body weight but failed to develop the coarse facial features, severe skeletal abnormalities, or the psychomotor retardation that characterizes the human disease. Further, the Gnptab−/− mice had a normal life span, whereas most humans with MLII die in their first decade. Curiously, the affected mice developed severe retinal degeneration by 3 months of age, whereas this complication has not been reported in humans with MLII. However, cats with MLII exhibit a phenotype closer to that of humans and do develop diffuse retinal degeneration by 4 months of age.8
A second major difference between the Gnptab−/− mice and human MLII concerned the cell types that exhibited morphologic abnormalities. Fibroblasts obtained from the Gnptab−/− mice did not show the typical inclusion bodies that are characteristic of MLII in humans. However, the secretory cells of multiple exocrine glands of the knockout mice exhibited striking morphologic alterations. These were most notable in the pancreatic acinar cells and parotid salivary glands. Similar pathologic changes have been described in the secretory cells of exocrine glands of some infants with MLII.13
In this study we have determined the phenotype of mice with a disruption of the Gnptg gene that encodes the γ subunit of GlcNAc-1-phosphotransferase. This subunit functions in the recognition of acid hydrolases as specific substrates, and in its absence the phosphorylation of mannose residues on these substrates is significantly impaired, but not completely lost.30 As a result, mutations in this gene give rise to a clinical phenotype (termed MLIIIC) that is considerably less severe than occurs with MLII. This is what we observed in the mouse models of MLII and MLIIIC, where loss of the catalytic α/β subunits of the GlcNAc-1-phosphotransferase resulted in a more severe phenotype than occurs with loss of the γ subunit. While both types of mice had elevations in their serum lysosomal hydrolase levels, the Gnptg−/− mice were of normal size, did not develop retinal degeneration or chondrocyte involvement, and showed a milder, more slowly progressive and a more restricted secretory cell involvement. The most significant and unexpected finding in both knockout mouse lines was the almost complete absence of lesions in mesenchymal cells. Of significance, fibroblasts/fibrocytes appeared to be completely normal in both lines, clearly differentiating the disease in these knockout mice from the human mucolipidoses, where distended mesenchymal cells (mostly fibrocytes) comprise the single most characteristic morphologic feature in both ML II and ML III.25 The critical role of the M-6-P markers in the proper trafficking of lysosomal enzymes in human fibroblasts has been clearly demonstrated, and bone changes are a prominent feature of mucolipidosis II in human patients, where intracellular, membrane-bound vacuoles accumulate in the chondrocytes, osteoblasts, osteocytes, and stromal fibroblasts of affected children.35 The relatively mild lesions we observed in chondrocytes of Gnptab−/− mice probably contributed to the stunted growth of those animals, but grossly apparent skeletal dysplasia was absent. However, even in aged Gnptab−/− mice, storage disease inclusions were not detected in fibrocytes.
This absence of fibrocyte involvement is in marked contrast to the central role that fibroblasts/fibrocytes play in the pathogenesis of mucolipidoses in humans and other animal models, which essentially develop a spectrum of connective tissue disorders. Similarly, the Gnptg-deficient mice did not exhibit any inclusions in mesenchymal cells, demonstrating a clear difference between these knockout mice and human MLII/MLIII. The lack of fibroblast/fibrocyte involvement may account for the failure of the mice to develop the connective tissue abnormalities that are characteristic of the human diseases. The severe connective tissue lesions in human mucolipidoses suggest that human fibroblasts/fibrocytes are more susceptible to Man-6-P pathway deficiencies. Several reports indicate that some cell types use alternative pathways that bypass the Man-6-P recognition system for the delivery of acid hydrolases to lysosomes.14,33,44 It has been observed that several cell types (e.g., hepatocytes, Kupffer cells, leukocytes) and tissues (e.g., liver, kidney, brain) contain near normal levels of most lysosomal enzymes in patients with I-cell disease.10,17 It is therefore possible that mouse fibroblasts/fibrocytes might use similar alternative pathways for lysosomal enzyme trafficking. Further studies and comparison of fibroblasts obtained from the triple null mice and our mice may enhance understanding of alternative trafficking routes for lysosomal enzymes.
Of interest, triple knockout mice that carry null alleles for IGF-II and the CD- and CI- Man-6-P receptors provide an indirect form of MLII that is more severe than the mouse models we have analyzed.9,10 Many of these triple deficient mice die at birth, but those that survive go on to develop a phenotype closely resembling human MLII; characterized by stunted growth, facial dysmorphism, waddling gait, dysostosis multiplex, and elevated levels of lysosomal hydrolases in their serum. Further, in this model, the fibroblasts exhibit prominent storage vesicles with neurons and hepatocytes also being affected. However, the triple-null mice did not appear to develop retinal degeneration. This triple knockout mouse model is complex, as the loss of IGF-II alone results in growth retardation, and the CI-MPR has been reported to participate in a number of functions in addition to mediating lysosomal enzyme trafficking.16
The lesions we observed in secretory cells illustrate the importance of the Man-6-P sorting pathway in the process of secretory granule maturation. In secretory cells specialized for regulated exocytosis, enzymes normally precipitate into dense, insoluble protein complexes as secretory granules.2 In both parotid and pancreatic exocrine cells, it is known that some lysosomal proenzymes fail to bind to Man-6-P receptors but instead enter the regulated secretory pathway in the trans-Golgi network (TGN) and become packaged with digestive enzymes in the secretory zymogen granules.19,22 Normally, most of the missorted proenzymes are removed from immature secretory granules during granule maturation via binding to Man-6-P receptors that, in turn, are packaged into adaptor protein 1 (AP-1)-containing clathrin-coated vesicles that bud from the maturing secretory granules.22 However, lysosomal hydrolases lacking the Man-6-P residues needed for binding to the Man-6-P receptors are not removed from immature granules but are incorporated into mature secretory granules.28 In addition, since packaging of the acid hydrolases into transport vesicles for delivery to lysosomes would be impaired in the knockout mice, a greater fraction of the hydrolases may enter the secretory granules than occurs under normal circumstances. As a consequence, increased amounts of lysosomal hydrolases remain in the secretory granules where they could eventually alter the contents, perhaps accounting for the observed abnormal morphology and histochemical staining characteristics of secretory granules in our knockout mice. Unlike normal zymogen granules, the granules in vacuolated secretory cells and large interstitial macrophages of knockout mice were PAS- and alcian blue–positive. The long-term persistence of these granules in macrophages may be due to lysosomal enzyme deficiencies in these phagocytic cells or to the altered chemical composition of the granule contents.
A major difference between the Gnptab−/− and Gnptg−/− mice is that the former develop diffuse retinal degeneration, whereas the latter do not. It is known that lysosomes help maintain normal retinal structure and function.6 Perhaps the defective trafficking of lysosomal hydrolases in the Gnptab−/− mice prevents the normal processing or release of photoreceptor proteins, resulting in the accumulation of toxic metabolic byproducts. Lysosomal enzymes secreted by the retinal pigment epithelium into the interphotoreceptor matrix have been proposed to have an important role in digesting shed outer segments1 with cathepsin D believed to be the major lysosomal protease in this process.18,23,38,39,47,49 The absence of any detectable retinal lesions in aged Gnptg−/− mice, which retain residual GlcNAc-1-phosphotransferase activity, indicates that a relatively low critical threshold level of lysosomal hydrolases delivery to the correct destination in the retina may be all that is required to prevent the development of photoreceptor degeneration. In particular, the relatively high level of phosphorylation of β-glucuronidase in the Gnptg−/− mice raises the possibility that this enzyme might be critical in maintaining normal retinal structure and function in mice.29,42 This possibility is supported by findings in mice lacking β-glucuronidase activity; these mice develop a lysosomal storage disease that is very similar to human mucopolysaccharidosis type VII (MPS VII) except for the development of progressive retinal degeneration in the affected mice.7 It has been shown that the retinal pigmented epithelium utilizes β-glucuronidase to degrade glycosaminoglycans in the retinal interphotoreceptor matrix (IPM) and thus maintains photoreceptor viability.29,42 Recently it was shown that restoration of low levels of β-glucuronidase expression (1 to 5% of normal) can significantly reduce the biochemical and histopathologic manifestations of MPS VII in affected mice.12 Based on these findings, we speculate that the lack of retinal (and chondrocyte) lesions in the Gnptg−/− mice may be due to the maintenance of a critical level of β-glucuronidase activity in the tissues of these mice.
In conclusion, the identification and characterization of lysosomal storage diseases in animal models are valuable because these models can offer important insights into fundamental biologic processes and mechanisms of disease. In particular, additional studies of fibroblasts and secretory exocrine cells from these mice should prove useful in better characterizing alternative molecular mechanisms involved in trafficking of lysosomal enzymes to lysosomes and the functional role of Man-6-P pathways in secretory granules development. Future studies in these mouse models may also enhance understanding of the role of lysosomes and lysosomal enzymes in the maintenance and function of the normal retina and secretory cells.
Acknowledgements
We thank Walter Gregory of the Washington University School of Medicine for the affinity chromatography columns and June Wingert, Mary Thiel, Kathy Henze, and Lindsey Guthrie for histology support. We are also grateful to Joe Shaw for his helpful suggestions and thorough review of the manuscript. This work was supported in part by National Institutes of Health Grant No. CA08759 (to SK).
References
- 1.Adler AJ. Selective presence of acid hydrolases in the interphotoreceptor matrix. Exp Eye Res. 1989;49:1067–1077. doi: 10.1016/s0014-4835(89)80027-2. [DOI] [PubMed] [Google Scholar]
- 2.Arvan P, Castle D. Sorting and storage during secretory granule biogenesis: looking backward and looking forward. Biochem J. 1998;332(Pt 3):593–610. doi: 10.1042/bj3320593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bao M, Elmendorf BJ, Booth JL, Drake RR, Canfield WM. Bovine UDP-N-acetylglucosamine:-lysosomal-enzyme N-acetylglucosamine-1-phosphotransferase. II. Enzymatic characterization and identification of the catalytic subunit. J Biol Chem. 1996;271:31446–31451. doi: 10.1074/jbc.271.49.31446. [DOI] [PubMed] [Google Scholar]
- 4.Bargal R, Zeigler M, Abu-Libdeh B, Zuri V, Mandel H, Ben Neriah Z, Stewart F, Elcioglu N, Hindi T, Le Merrer M, Bach G, Raas-Rothschild A. When mucolipidosis III meets mucolipidosis II: GNPTA gene mutations in 24 patients. Mol Genet Metab. 2006;88:359–363. doi: 10.1016/j.ymgme.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Beltrandelrio H, Kern F, Lanthorn T, Oravecz T, Piggott J, Powell D, Ramirez-Solis R, Sands AT, Zambrowicz BP. Saturation screening of the drug-gable mammalian genome. In: Carroll PM, Fitzgerald K, editors. Model Organisms in Drug Discovery. John Wiley & Sons; Chichester, UK: 2003. pp. 251–279. [Google Scholar]
- 6.Birch DG. Retinal degeneration in retinitis pigmentosa and neuronal ceroid lipofuscinosis: an overview. Mol Genet Metab. 1999;66:356–366. doi: 10.1006/mgme.1999.2829. [DOI] [PubMed] [Google Scholar]
- 7.Birkenmeier EH, Davisson MT, Beamer WG, Ganschow RE, Vogler CA, Gwynn B, Lyford KA, Maltais LM, Wawrzyniak CJ. Murine mucopolysaccharidosis type VII: characterization of a mouse with beta-glucuronidase deficiency. J Clin Invest. 1989;83:1258–1266. doi: 10.1172/JCI114010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bosshard NU, Hubler M, Arnold S, Briner J, Spycher MA, Sommerlade HJ, von Figura K, Gitzelmann R. Spontaneous mucolipidosis in a cat: an animal model of human I-cell disease. Vet Pathol. 1996;33:1–13. doi: 10.1177/030098589603300101. [DOI] [PubMed] [Google Scholar]
- 9.Dittmer F, Hafner A, Ulbrich EJ, Moritz JD, Schmidt P, Schmahl W, Pohlmann R, Figura KV. I-cell disease-like phenotype in mice deficient in mannose 6-phosphate receptors. Transgenic Res. 1998;7:473–483. doi: 10.1023/a:1008823315416. [DOI] [PubMed] [Google Scholar]
- 10.Dittmer F, Ulbrich EJ, Hafner A, Schmahl W, Meister T, Pohlmann R, von Figura K. Alternative mechanisms for trafficking of lysosomal enzymes in mannose 6-phosphate receptor-deficient mice are cell type-specific. J Cell Sci. 1999;112(Pt 10):1591–1597. doi: 10.1242/jcs.112.10.1591. [DOI] [PubMed] [Google Scholar]
- 11.Do H, Lee WS, Ghosh P, Hollowell T, Canfield W, Kornfeld S. Human mannose 6-phosphate-uncovering enzyme is synthesized as a proenzyme that is activated by the endoprotease furin. J Biol Chem. 2002;277:29737–29744. doi: 10.1074/jbc.M202369200. [DOI] [PubMed] [Google Scholar]
- 12.Donsante A, Levy B, Vogler C, Sands MS. Clinical response to persistent, low-level beta-glucuronidase expression in the murine model of mucopolysaccharidosis type VII. J Inherit Metab Dis. 2007;30:227–238. doi: 10.1007/s10545-007-0483-4. [DOI] [PubMed] [Google Scholar]
- 13.Elleder M, Martin JJ. Mucolipidosis type II with evidence of a novel storage site. Virchows Arch. 1998;433:575–578. doi: 10.1007/s004280050292. [DOI] [PubMed] [Google Scholar]
- 14.Gabel CA, Goldberg DE, Kornfeld S. Identification and characterization of cells deficient in the mannose 6-phosphate receptor: evidence for an alternate pathway for lysosomal enzyme targeting. Proc Natl Acad Sci USA. 1983;80:775–779. doi: 10.1073/pnas.80.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gelfman CM, Vogel P, Issa TM, Turner CA, Lee WS, Kornfeld S, Rice DS. Mice lacking alpha/beta subunits of GlcNAc-1-phosphotransferase exhibit growth retardation, retinal degeneration, and secretory cell lesions. Invest Ophthalmol Vis Sci. 2007;48:5221–5228. doi: 10.1167/iovs.07-0452. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh P, Dahms NM, Kornfeld S. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol. 2003;4:202–212. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]
- 17.Glickman JN, Kornfeld S. Mannose 6-phosphate-independent targeting of lysosomal enzymes in I-cell disease B lymphoblasts. J Cell Biol. 1993;123:99–108. doi: 10.1083/jcb.123.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayasaka S, Hara S, Mizuno K. Degradation of rod outer segment proteins by cathepsin D. J Biochem (Tokyo) 1975;78:1365–1367. doi: 10.1093/oxfordjournals.jbchem.a131034. [DOI] [PubMed] [Google Scholar]
- 19.Hirano T, Manabe T, Kyogoku T, Ando K, Tobe T. Pancreatic lysosomal enzyme secretion via gut-hormone-regulated pathway in rats. Nippon Geka Hokan. 1991;60:415–423. [PubMed] [Google Scholar]
- 20.Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council . Guide for the care and use of laboratory animals. National Academy Press; Washington, DC: 1996. [Google Scholar]
- 21.Kaplan A, Achord DT, Sly WS. Phosphohexosyl components of a lysosomal enzyme are recognized by pinocytosis receptors on human fibroblasts. Proc Natl Acad Sci USA. 1977;74:2026–2030. doi: 10.1073/pnas.74.5.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klumperman J, Kuliawat R, Griffith JM, Geuze HJ, Arvan P. Mannose 6-phosphate receptors are sorted from immature secretory granules via adaptor protein AP-1, clathrin, and syntaxin 6-positive vesicles. J Cell Biol. 1998;141:359–371. doi: 10.1083/jcb.141.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koike M, Shibata M, Ohsawa Y, Nakanishi H, Koga T, Kametaka S, Waguri S, Momoi T, Kominami E, Peters C, Figura K, Saftig P, Uchiyama Y. Involvement of two different cell death pathways in retinal atrophy of cathepsin D-deficient mice. Mol Cell Neurosci. 2003;22:146–161. doi: 10.1016/s1044-7431(03)00035-6. [DOI] [PubMed] [Google Scholar]
- 24.Kornfeld S. Trafficking of lysosomal enzymes in normal and disease states. J Clin Invest. 1986;77:1–6. doi: 10.1172/JCI112262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kornfeld S, Sly WS. I-cell disease and pseudo-hurler polydystrophy: disorders of lysosomal enzyme phosphorylation and localization. In: Scriver C, Sly W, Childs B, Beaudet A, Valle D, Kinzler K, Vogelstein B, editors. The Metabolic and Molecular Basis of Inherited Disease. McGraw-Hill Professional; New York, NY: 2000. pp. 3469–3482. [Google Scholar]
- 26.Kudo M, Bao M, D'Souza A, Ying F, Pan H, Roe BA, Canfield WM. The alpha- and beta-subunits of the human UDP-N-acetylglucosamine:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase [corrected] are encoded by a single cDNA. J Biol Chem. 2005;280:36141–36149. doi: 10.1074/jbc.M509008200. [DOI] [PubMed] [Google Scholar]
- 27.Kudo M, Brem MS, Canfield WM. Mucolipidosis II (I-Cell Disease) and mucolipidosis IIIA (classical pseudo-Hurler polydystrophy) are caused by mutations in the GlcNAc-phosphotransferase α/β–sub-units precursor gene. Am J Hum Genet. 2006;78:451–463. doi: 10.1086/500849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuliawat R, Klumperman J, Ludwig T, Arvan P. Differential sorting of lysosomal enzymes out of the regulated secretory pathway in pancreatic beta-cells. J Cell Biol. 1997;137:595–608. doi: 10.1083/jcb.137.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lazarus HS, Sly WS, Kyle JW, Hageman GS. Photoreceptor degeneration and altered distribution of interphotoreceptor matrix proteoglycans in the mucopolysaccharidosis VII mouse. Exp Eye Res. 1993;56:531–541. doi: 10.1006/exer.1993.1067. [DOI] [PubMed] [Google Scholar]
- 30.Lee WS, Payne BJ, Gelfman CM, Vogel P, Kornfeld S. Murine UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase lacking the gamma-subunit retains substantial activity toward acid hydrolases. J Biol Chem. 2007;282:27198–27203. doi: 10.1074/jbc.M704067200. [DOI] [PubMed] [Google Scholar]
- 31.Leroy JG, Spranger JW. I-cell disease. N Engl J Med. 1970;283:598–599. [PubMed] [Google Scholar]
- 32.Mazrier H, Van Hoeven M, Wang P, Knox VW, Aguirre GD, Holt E, Wiemelt SP, Sleeper MM, Hubler M, Haskins ME, Giger U. Inheritance, biochemical abnormalities, and clinical features of feline mucolipidosis II: the first animal model of human I-cell disease. J Hered. 2003;94:363–373. doi: 10.1093/jhered/esg080. [DOI] [PubMed] [Google Scholar]
- 33.Owada M, Neufeld EF. Is there a mechanism for introducing acid hydrolases into liver lysosomes that is independent of mannose 6-phosphate recognition? Evidence from I-cell disease. Biochem Biophys Res Commun. 1982;105:814–820. doi: 10.1016/0006-291x(82)91042-7. [DOI] [PubMed] [Google Scholar]
- 34.Paik KH, Song SM, Ki CS, Yu HW, Kim JS, Min KH, Chang SH, Yoo EJ, Lee IJ, Kwan EK, Han SJ, Jin DK. Identification of mutations in the GNPTA (MGC4170) gene coding for GlcNAc-phosphotransferase alpha/beta subunits in Korean patients with mucolipidosis type II or type IIIA. Hum Mutat. 2005;26:308–314. doi: 10.1002/humu.20205. [DOI] [PubMed] [Google Scholar]
- 35.Pazzaglia UE, Beluffi G, Castello A, Coci A, Zatti G. Bone changes of mucolipidosis II at different ages: postmortem study of three cases. Clin Orthop Relat Res. 1992:283–290. [PubMed] [Google Scholar]
- 36.Raas-Rothschild A, Bargal R, Goldman O, Ben-Asher E, Groener JE, Toutain A, Stemmer E, BenNeriah Z, Flusser H, Beemer FA, Penttinen M, Olender T, Rein AJ, Bach G, Zeigler M. Genomic organization of the UDP-N-acetylglucosamine-1-phosphotransferase gamma subunit (GNPTAG) and its mutations in mucolipidosis III. J Med Genet. 2004;41:e52. doi: 10.1136/jmg.2003.015222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raas-Rothschild A, Cormier-Daire V, Bao M, Genin E, Salomon R, Brewer K, Zeigler M, Mandel H, Toth S, Roe B, Munnich A, Canfield WM. Molecular basis of variant pseudo-Hurler polydystrophy (mucolipidosis IIIC) J Clin Invest. 2000;105:673–681. doi: 10.1172/JCI5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rakoczy PE, Sarks SH, Daw N, Constable IJ. Distribution of cathepsin D in human eyes with or without age-related maculopathy. Exp Eye Res. 1999;69:367–374. doi: 10.1006/exer.1999.0700. [DOI] [PubMed] [Google Scholar]
- 39.Rakoczy PE, Zhang D, Robertson T, Barnett NL, Papadimitriou J, Constable IJ, Lai CM. Progressive age-related changes similar to age-related macular degeneration in a transgenic mouse model. Am J Pathol. 2002;161:1515–1524. doi: 10.1016/S0002-9440(10)64427-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roberts DL, Weix DJ, Dahms NM, Kim JJ. Molecular basis of lysosomal enzyme recognition: three-dimensional structure of the cation-dependent mannose 6-phosphate receptor. Cell. 1998;93:639–648. doi: 10.1016/s0092-8674(00)81192-7. [DOI] [PubMed] [Google Scholar]
- 41.Steet R, Chung S, Lee WS, Pine CW, Do H, Kornfeld S. Selective action of the iminosugar isofagomine, a pharmacological chaperone for mutant forms of acid-beta-glucosidase. Biochem Pharmacol. 2007;73:1376–1383. doi: 10.1016/j.bcp.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stramm LE, Wolfe JH, Schuchman EH, Haskins ME, Patterson DF, Aguirre GD. Beta-glucuronidase mediated pathway essential for retinal pigment epithelial degradation of glycosaminoglycans: disease expression and in vitro disease correction using retroviral mediated cDNA transfer. Exp Eye Res. 1990;50:521–532. doi: 10.1016/0014-4835(90)90041-r. [DOI] [PubMed] [Google Scholar]
- 43.Tiede S, Storch S, Lubke T, Henrissat B, Bargal R, Raas-Rothschild A, Braulke T. Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phosphotransferase. Nat Med. 2005;11:1109–1112. doi: 10.1038/nm1305. [DOI] [PubMed] [Google Scholar]
- 44.Tsuji A, Omura K, Suzuki Y. I-cell disease: evidence for a mannose 6-phosphate independent pathway for translocation of lysosomal enzymes in lymphoblastoid cells. Clin Chim Acta. 1988;176:115–121. doi: 10.1016/0009-8981(88)90181-7. [DOI] [PubMed] [Google Scholar]
- 45.Varki AP, Reitman ML, Kornfeld S. Identification of a variant of mucolipidosis III (pseudo-Hurler polydystrophy): a catalytically active N-acetylglucosaminylphosphotransferase that fails to phosphorylate lysosomal enzymes. Proc Natl Acad Sci USA. 1981;78:7773–7777. doi: 10.1073/pnas.78.12.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Varki A, Reitman ML, Vannier A, Kornfeld S, Grubb JH, Sly WS. Demonstration of the heterozygous state for I-cell disease and pseudo-Hurler polydystrophy by assay of N-acetylglucosaminylphosphotransferase in white blood cells and fibroblasts. Am J Hum Genet. 1982;34:717–729. [PMC free article] [PubMed] [Google Scholar]
- 47.Yamada T, Hara S, Tamai M. Immunohistochemical localization of cathepsin D in ocular tissues. Invest Ophthalmol Vis Sci. 1990;31:1217–1223. [PubMed] [Google Scholar]
- 48.Zambrowicz BP, Abuin A, Ramirez-Solis R, Richter LJ, Piggott J, Beltrandelrio H, Buxton EC, Edwards J, Finch RA, Friddle CJ, Gupta A, Hansen G, Hu Y, Huang W, Jaing C, Key BW, Jr, Kipp P, Kohlhauff B, Ma ZQ, Markesich D, Payne R, Potter DG, Qian N, Shaw J, Schrick J, Shi ZZ, Sparks MJ, Van Sligtenhorst I, Vogel P, Walke W, Xu N, Zhu Q, Person C, Sands AT. Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc Natl Acad Sci USA. 2003;100:14109–14114. doi: 10.1073/pnas.2336103100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang D, Brankov M, Makhija MT, Robertson T, Helmerhorst E, Papadimitriou JM, Rakoczy PE. Correlation between inactive cathepsin D expression and retinal changes in mcd2/mcd2 transgenic mice. Invest Ophthalmol Vis Sci. 2005;46:3031–3038. doi: 10.1167/iovs.04-1510. [DOI] [PubMed] [Google Scholar]