

Summary
Uroporphyrinogen Decarboxylase (URO-D; EC 4.1.1.37), the fifth enzyme of the heme biosynthetic pathway, is required for the production of heme, vitamin B12, siroheme, and chlorophyll precursors. URO-D catalyzes the sequential decarboxylation of the four acetate side chains on the pyrrole groups of uroporphyrinogen to produce coproporphyrinogen. URO-D is a stable homodimer with the active site clefts of the two subunits adjacent to each other. It has been hypothesized that the two catalytic centers interact functionally, perhaps by shuttling of reaction intermediates between subunits. We tested this hypothesis by construction of a single chain protein (scURO-D) in which the two subunits were connected by a flexible linker. The crystal structure of this protein was shown to be superimposible with wild-type activity and have comparable catalytic activity. Mutations that impaired one or the other of the two active sites of scURO-D resulted in approximately half of wild-type activity. The distribution of reaction intermediates was the same for mutant and wild-type sequences, and was unaltered in a competition experiment using the I and III isomer substrates. These observations indicate that communication between active sites is not required for enzyme function, and suggest that the dimeric structure of URO-D is required to achieve conformational stability and create a large active site cleft.
Keywords: Uroporphyrinogen decarboxylase, porphyria, heme biosynthesis
Introduction
Biosynthesis of heme requires a complex multi-enzyme process that is distributed between mitochondria and the cytosol. The fifth enzyme of this pathway, cytosolic uroporphyrinogen decarboxylase (URO-D; EC 4.1.1.37), catalyzes the decarboxylation of the four acetate side chains of uroporphyrinogen (uro'gen) to generate coproporphyrinogen (copro'gen). The biologically relevant URO-D substrate is the uro'gen III isomer although uro'gen I, which is produced in the absence of adequate activity of the preceding uroporphyrinogen III synthase enzyme, is also a substrate. The copro'gen I product, however, cannot be converted to protoporphyrinogen and ultimately heme. Mechanistic studies have shown that there is an ordered decarboxylation of uro'gen-III beginning at the asymmetric D ring and proceeding sequentially to rings A, B and C (Figure 1) (1). URO-D is encoded by a single gene (2) and is essential for the synthesis of heme and chlorophyll (3). Subnormal activity of URO-D in hepatocytes is the cause of the most common form of porphyria in humans, porphyria cutanea tarda (PCT) (4).
Figure 1. Reaction catalyzed by URO-D.

The four acetate groups of uroporphyrinogen are decarboxylated to form methyl groups and four molecules of CO2. Decarboxylation begins with the D ring and proceeds clockwise to the A, B, and C rings.
URO-D forms a stable homodimer (5) in which each subunit adopts a (β/α)8-barrel structure (6). Each subunit contains a deep active site cleft, and the two active sites in the dimer are juxtaposed such that the two clefts are largely contiguous. Crystal structures of URO-D in complex with the copro'gen reaction product revealed that the product tetrapyrrole macrocycle adopts a domed conformation that lies against a collar of conserved hydrophobic residues and allows formation of hydrogen bonding interactions between a carboxylate oxygen atom of the invariant Asp86 side chain and the four pyrrole NH groups (7). These structural data and associated biochemical analyses of mutant URO-D proteins indicate a critical role for Asp86 in substrate binding and in promoting catalysis by stabilizing a positive charge on a reaction intermediate (7). This model may explain why, unlike many decarboxylases, URO-D does not utilize a cofactor. Furthermore, the central coordination geometry of Asp86 allows the initial substrates and the various partially decarboxylated intermediates to bind with similar activating interactions. Although the precise site of decarboxylation remains uncertain, this binding geometry strongly suggests that all four of the substrate acetate groups are decarboxylated by the same mechanism.
Although the active site in each URO-D subunit appears capable of performing all of the decarboxylation reactions that are required to form the ultimate product, the apposition of active sites within the dimer suggested the possibility that partially decarboxylated reaction intermediates might shuttle between subunits. This idea was especially attractive for the second decarboxylation reaction of the III-isomer substrate because the asymmetric structure of 7-carboxy porphyrinogen III would not permit this structure to bind with the reactive pyrrole acetate, propionate, and NH groups at the active site by a simple rotation within a single active site. Decarboxylation of 7-carboxy porphyrinogen III could result from dissociation and rebinding with its opposite face against the enzyme, or it could dissociate and be repositioned in the adjacent active site without the need to flip and reorient the opposite face of the substrate in the active site. It has also been suggested that once substrate is bound all four of the decarboxylation events are performed using the same catalytic residues without the need for the intermediate to leave the substrate binding pocket. Once all four decarboxylation events had been performed the product would be able to exit the binding pocket (8).
To determine if enzyme activity requires that partially decarboxylated reaction intermediates shuttle between subunit active sites without completely dissociating from URO-D we expressed and purified a single chain form of a URO-D dimer. In this protein the two monomeric domains are joined by a linker from the C-terminus of one URO-D to the N-terminus of the second URO-D. This single chain dimer behaved as authentic wild-type dimer by gel filtration chromatography, analytical ultracentrifugation, crystal structure, and enzyme activity. Variants of the linked dimer in which either of the active sites were inactivated by site-directed mutagenesis maintained approximately half of wild-type catalytic activity. Moreover, wild-type and mutant proteins displayed very similar distributions of reaction intermediates. These data indicate that all four decarboxylations can be catalyzed at a single active site and that shuttling of intermediates between active sites of the URO-D dimer is not required.
Results and Discussion
A single chain URO-D dimer, scURO-D
To establish the catalytic requirement for a dimeric structure of URO-D we constructed a monomeric single subunit variant by mutating residues that stabilize the dimer interface. A total of six different mutant URO-D proteins were constructed but in all cases they were insoluble (data not shown), consistent with the model that dimer formation is important for URO-D folding and/or conformational stability. We next took the approach of expressing URO-D proteins with two different tags, a 10× histidine tag (HIS) or a cellulose binding domain (CBD), within the same host strain of E. coli. Both tagged versions of URO-D were expressed in approximately equal amounts and the recovery of the properly folded proteins were equal to that observed for the single vector system. However, the percentage of dimers containing both tags (HIS and CBD) was very small (data not shown). These data suggest that formation of the dimer is cotranslational (from the same polysome) as has been described for p53 (9) and that little subunit exchange takes place. These properties and interactions lead to formation of a stable homodimer with a half-life of more than 80 h (10). We then expressed and purified each of the differentially tagged proteins individually. Equal molar ratios of the two proteins were mixed and allowed to equilibrate at various temperatures and concentrations. Results from this were similar to the dual expression system in that very little of the re-purified protein contained both purification tags.
We therefore took an alternative approach in which two separate active sites are housed within a single dimeric URO-D molecule. Using this single polypeptide approach eliminated any confounding variables due to subunit exchange between wild type and mutant monomers. This was done by constructing single chain variants (scURO-D) in which two human URO-D monomers were connected by a flexible linker sequence. Linkers were designed based on inspection of the wild type URO-D crystal structure (6), which showed that the N- and C-termini of the two subunits in a dimer are separated by approximately 75 Å when measured by the shortest path along the surface of the molecule. Linkers containing the 10× His affinity tag and rich in Gly and Ser residues were designed that comprised a total of 29, 33, 35, and 37 residues. These constructs were expressed, and proteins purified and tested for enzymatic activity. Maximal activity was observed for the 35-residue linker protein. This optimally-active construct, hereafter referred to simply as scURO-D, and its point mutant variants were used for all of the subsequent biochemical and structural studies. scURO-D displayed 75% and 85% of the wild type protein activity when assayed against uro'gen-I and uro'gen-III isomer substrates, respectively (Table 1). This statistically significant (p=0.01) 25% loss of activity presumably results from a subtle conformational restriction but does not result from a fundamental change in mechanism, as indicated by the conserved profile of reaction intermediates and the effect of active site mutants (below).
Table 1. Activity of the scURO-D proteins.
| Uro'gen-I Activity | |||||
|---|---|---|---|---|---|
| Construct | # | activity | std err | % URO-D | % scURO-D |
| URO-D | 6 | 4364 | 188 | 100.0 | 133.9 |
| Y164G URO-D | 3 | 353 | 49 | 8.1 | 10.8 |
| F217Y URO-D | 4 | 114 | 18 | 2.6 | 3.5 |
| scURO-D | 6 | 3260 | 143 | 74.7 | 100.0 |
| scURO-D(FY) | 6 | 1802 | 40 | 41.3 | 55.3 |
| scURO-D(YF) | 6 | 2014 | 128 | 46.2 | 61.8 |
| scURO-D(YY) | 4 | 107 | 5 | 2.5 | 3.3 |
| scURO-D(GY) | 2 | 578 | 13.2 | 17.7 | |
| Uro'gen-III Activity | |||||
| Construct | # | activity | std err | % URO-D | % scURO-D |
| URO-D | 6 | 8028 | 913 | 100.0 | 117.7 |
| scURO-D | 6 | 6821 | 662 | 85.0 | 100.0 |
| scURO-D(FY) | 6 | 4087 | 320 | 50.9 | 59.9 |
| scURO-D(YF) | 6 | 4229 | 330 | 52.7 | 62.0 |
| scURO-D(YY) | 4 | 1163 | 62 | 14.5 | 17.1 |
Uro'gen-I, uroporphyrinogen-I; Uro'gen-III, uroporphyrinogen-III at 30 nM # number of independent assays performed in duplicate activity- average activity as nmol product/hr/mg protein std err- standard error
Purified scURO-D protein with ∼80% of wild-type activity migrates at almost the same position as wt URO-D on a sizing column. The scURO-D preparations are contaminated (10-20%) by protein that has been clipped in the linker region (Figure 2), as indicated by N-terminal sequencing of the two species present (data not shown). In order to rigorously verify the oligomeric state of scURO-D, the purified protein was analyzed by equilibrium analytical ultracentrifugation. scURO-D protein sedimented as a single homogeneous species of 86 kDa (Figure 3), consistent with equivalent data obtained for the wild type protein, and indicating that scURO-D adopts the designed structure.
Figure 2. Purified proteins.
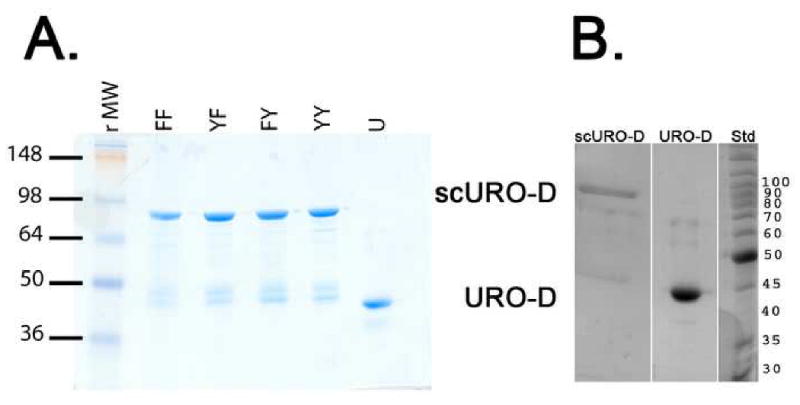
(A)1 μg sample of each of the purified proteins visualized on Coomassie-stained SDS-PAGE: scURO-D (YY), scURO-D(YF) (YF), scURO-D(FY) (FY), scURO-D(FF) (FF), and URO-D (U). The scURO-D proteins have an apparent molecular weight of 84 kDa while native URO-D runs at 42 kDa. Densitometry of the bands present in the scURO-D sample indicated that approximately 10 to 20% of the URO-D protein is present at the molecular weight corresponding to monomeric URO-D. (B) Crystals of scURO-D were removed from the crystallization well, washed and dissolved in 50 mM Tris pH 7.5. Proteins were separated by SDS page. Densitometry of the bands present in the scURO-D sample indicated that approximately 22% of the protein is present at a molecular weight corresponding to monomeric URO-D.
Figure 3. Analytical ultracentrifugation of scUROD.
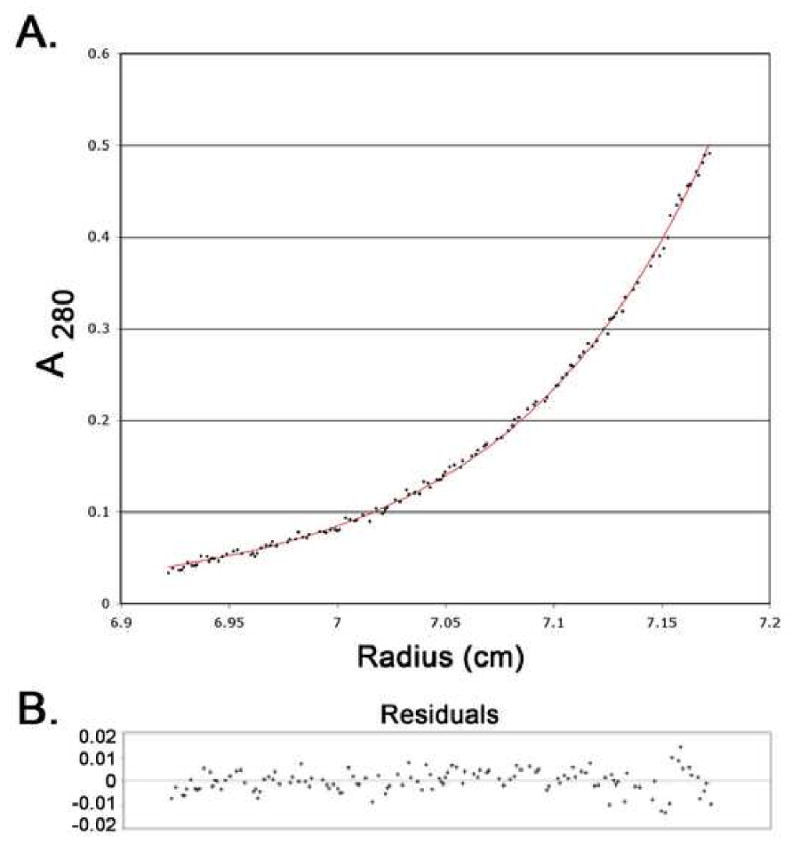
Samples of scUROD at 0.25 and 0.08 mg/mL were placed in two chambers of the centrifuge cell of a Beckman XL-A and spun at 12,000 rpm at 20° C to equilibrium. All data were fit globally to a monomer, resulting in a predicted molecular mass of 85,972 Da, consistent with the expected mass of the scUROD of 85,497 Da. (A) Points represent 268 averaged data values from the 5 scans (A280). The solid line is the best globally fit model. (B) Residuals for the scan shown in panel A.
Further evidence that the scURO-D structure is not substantially distorted from the wild-type by the linker was provided by crystal structure determination to a resolution of 2.1 Å and Rfree value of 0.245 (Table 2). Crystals were isomorphous with the wild-type URO-D crystals (Supplementary Figure 1). This was not expected as the space group, P3121, contains just one URO-D subunit in the asymmetric unit with the dimer built by application of a crystallographic two-fold axis, whereas the two domains of scURO-D differ by the presence of linker residues at their N or C termini. The structure was therefore built as if the two scURO-D domains were stochastically distributed on either side of the crystallographic two-fold, i.e., with full occupancy for groups that are present in both domains and half occupancy for groups that are unique to the linker region (almost all of which are disordered and lack density) (Supplementary Figure 2). This solution was supported by refinement and inspection of Fourier maps using the highly redundant data processed in the lower symmetry space group P3. The predominant presence of uncleaved scURO-D in the crystal was confirmed by visualization of washed crystals on SDS-PAGE (Figure 2). The P3121 space group is accommodated because, although the N- and C-terminal URO-D modules are distinguished by unique flanking sequences, they are otherwise identical and the crystal lattice contacts are mediated by segments common to both domains. The refined scURO-D and native URO-D models superimpose with an RMSD of 0.197 Å over 332/357 of the subunit Cα atoms.
Table 2. Data collection and refinement statistics for scURO-D proteins.
| Data set | scURO-D | scURO-D(GY) | scURO-D(FY) | scURO-D(YF) |
|---|---|---|---|---|
| Wavelength (Å) | 1.5418 | 1.5418 | 1.5418 | 1.5418 |
| Resolution (Å)a | 30.0 – 2.10 (2.16 – 2.10) | 30.0 – 2.20 (2.26 – 2.20) | 40.0 – 2.80 (2.90 – 2.80) | 40.0 – 2.80 (2.90 – 2.80) |
| # Reflections Measured | 129,445 | 135,250 | 131,959 | 83,299 |
| # Unique Reflections | 25,394 | 22,288 | 10,977 | 11,174 |
| Completeness (%) | 98.0 ( 86.3) | 99.3 ( 98.6) | 98.1 (85.9) | 100.0 (100.0) |
| <I/σI> | 10 (2.3) | 8.5 (2.0) | 9.5 (1.9) | 10.2 (1.9) |
| Mosaicity (°) | 0.91 | 1.10 | 0.39 | 0.85 |
| Rsymb (%) | 0.070 (0.553) | 0.110 (0.395) | 0.183 (0.455) | 0.086 (0.704) |
|
| ||||
| # Protein residues modeled | 11-366 | 11-366 | 11-366 | 11-366 |
| # Water molecules | 145 | 125 | 23 | 24 |
| <B>protein (Å2) | 40.0 | 39.8 | 48.4 | 68.6 |
| <B>main-chain (Å2) | 39.1 | 39.0 | 47.3 | 67.7 |
| <B>water (Å2) | 44.6 | 44.4 | 66.6 | 87.5 |
| Rcrystc (%) | 0.193 (0.244) | 0.193 (0.245) | 0.169 (0.264) | 0.193 (0.282) |
| Rfree (%)d | 0.231 (0.321) | 0.234 (0.297) | 0.223 (0.396) | 0.239 (0.338) |
| RMSD bond lengths | 0.014 / 1.265 | 0.012 / 1.293 | 0.020 / 1.922 | 0.023 / 2.204 |
| (Å) / angles (°) | ||||
Values in parentheses refer to the high-resolution shell.
Rsym = Σ|I-<I>|/ΣI where I is the intensity of an individual measurement and <I> is the corresponding mean value.
Crystallographic R value (Rcryst) = Σ‖Fo|-|Fc‖/Σ|Fo|, where |Fo| is the observed and |Fc| the calculated structure factor amplitude.
Rfree is the same as Rcryst calculated with a randomly selected test set of reflections that were never used in refinement calculations. For each refinement the following number of reflections were chosen for the test set; scURO-D, 1298; scURO-D(GY), 1145; scURO-D(FY), 1067; scURO-D(YF), 1086.
Design and characterization of inactive mutants
It was expected that if substrate shuttling between active sites was required, mutations that prevented binding of substrate or any of the intermediates would be less informative than constructing mutants that could bind substrate but that were catalytically inactive. We previously identified phenylalanine 217 and tyrosine 164 as active site residues (7). Enzymatic activity of mutant proteins showed that F217Y URO-D possesses negligible (2.6%) activity when assayed with uro'gen-I and that Y164G URO-D retains only 8.1% activity when assayed with uro'gen-I (Table 1). The single chain dimer construct was reengineered to encode the F217Y mutation in the active site of the first module [scURO-D(YF)], in the active site of the second module [scURO-D(FY)], and in the active sites of both modules [scURO-D(YY)]. Another single chain dimer construct was reengineered to encode the Y164G mutation in the active site of the first module [scURO-D(YG)]. As discussed below, scURO-D(YG) was especially valuable in confirming crystallographic assignment of the mutant proteins. In common with other mutant URO-D proteins (data not shown), it was noticed that F217Y URO-D proteins fluoresced upon illumination by UV light, suggesting that porphyrins were bound. A sample of each F217Y protein was denatured by heating and the eluted porphyrins were analyzed by HPLC. Coproporphyrin was found to be the major porphyrin detected along with minor amounts of partially decarboxylated porphyrins. This ability of the F217Y mutant proteins to bind reaction intermediates and product is consistent with the retention of small but measurable levels of enzyme activity.
In order to rigorously verify that mutation of F217 or Y164 does not inactivate scURO-D by gross disruption of the structure, we determined the crystal structures of scURO-D(GY), scURO-D(YF) and scURO-D(FY). The structures were refined at 2.2, 2.8 and 2.8 Å resolution respectively and Rfree values were better than 0.24 (Table 2). The conditions and analyses were the same as for the unmodified scURO-D protein described above. The scURO-D(GY) structure superimposes on 332/357 pairs of Cα atoms of scURO-D with a RMSD of 0.280 Å. The scURO-D(FY) and scURO-D(YF) structures superimpose on 332/357 pairs of Cα atoms of scURO-D with RMSDs of 0.237 Å and 0.283 Å, respectively. Inspection of electron density maps indicated, as expected, that all three structures are identical with the exception of the mutated residue, which is best modeled as a half occupied Gly and a half occupied Tyr (scURO-D(GY) and as a half occupied Tyr and a half occupied Phe (scURO-D(YF) and scURO-D(FY). F217Y is the more debilitating mutation and these variants were used for the primary enzymatic analyses (below), although the Y164G mutation robustly validated the crystallographic assignment of two superimposible scURO-D orientations due to the large number of electrons eliminated in changing from tyrosine to glycine (Supplementary Figure 2). These structures support the earlier crystallographic assignments and indicate that the mutations do not have unanticipated structural consequences that might complicate interpretation of the biochemical data.
Activities of scURO-D proteins and implications for mechanism
Compared to scURO-D, scURO-D(FY) and scURO-D(YF) display 55 and 62% of full activity, respectively (Table 1). ANOVA analysis indicated that the difference between these values was statistically insignificant. Activity of the scURO-D(YY) mutant was 3% and 15% of scURO-D when assayed against uro'gen-I and uro'gen-III respectively. The higher activity observed when uro'gen-III was used as a substrate is likely due to tighter binding of the III isomer substrate (11, 12). The reaction mechanism is unchanged in the mutants as indicated by a conserved profile of reaction intermediates (Figure 4, Table 3). If substrate shuttling was required and one active site was inactive, it is possible that an altered profile of the partially decarboxylated intermediates would accumulate. The peak heights of the species quantified in this HPLC assay change depending on catalytic activity of the sample tested, but the ratio of product to intermediate substrates remains constant in all cases for both uro'gen-I and uro'gen-III substrates, when there is a single active site present. These data indicate that mutation of F217 does not affect substrate binding and release for any of the intermediate products in the URO-D assay when juxtaposed to a wild type active site, and that the two catalytic centers of the URO-D dimer are functionally independent. This implies that the dimeric structure has been conserved evolutionarily for reasons other than the catalytic chemistry, with one possibility being that formation of the dimer interface stabilizes the active site cleft geometry because residues at the interface also line the active site cleft.
Figure 4. Product and intermediate profiles of the URO-D reaction.
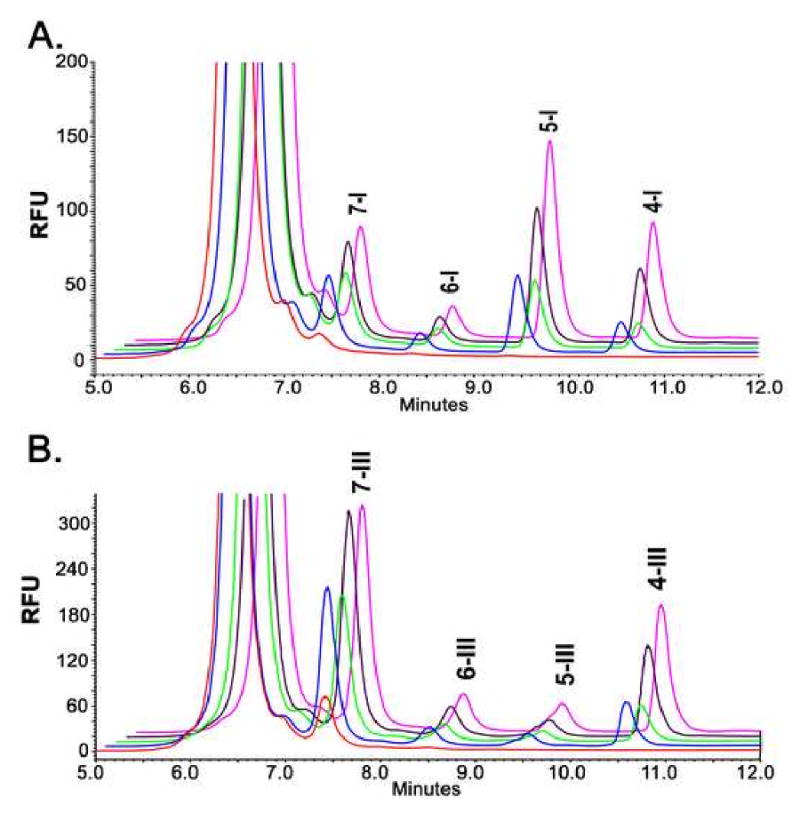
Reaction products of either uroporphyrinogen-I (A) or -III (B) from the URO-D enzymatic assay are separated by HPLC, the metabolites heptacarboxyl porphyrin (7), hexacarboxyl porphyrin (6), pentacarboxyl porphyrin (5) and coproporphyrin (4) are labeled with the corresponding isomer designation after the label. WT enzyme (pink), scURO-D (purple), scURO-D(FY) (green), scURO-D(YF) (blue) and scURO-D(FF) (red).
Table 3. Reaction products as a percent of total decarboxylation.
| 7-COOH | 6-COOH | 5-COOH | 4-COOH | |
|---|---|---|---|---|
| Isomer -I | % | % | % | % |
| URO-D | 23 | 7 | 42 | 27 |
| scURO-D | 27 | 8 | 40 | 26 |
| scURO-D(FY) | 34 | 10 | 39 | 17 |
| scURO-D(YY) | 77 | 15 | 8 | 00 |
| scURO-D(YF) | 35 | 10 | 39 | 16 |
| Isomer-III | ||||
| URO-D | 53 | 10 | 8 | 29 |
| scURO-D | 56 | 9 | 8 | 27 |
| scURO-D(FY) | 66 | 9 | 7 | 19 |
| scURO-D(YY) | 96 | 3 | 1 | 1 |
| scURO-D(YF) | 66 | 8 | 7 | 19 |
Reaction products expressed as a percentage of the total. Represented graphically in figure 4 (A) Isomer-I and (B) Isomer III.
Wild-type URO-D has a reported Km for uro'gen-I of 0.80 μM and a Km for uro'gen-III of 0.35 μM, suggesting that the binding is approximately 2 times tighter for the III isomer (11). An enzymatic assay, started using 5 μM uro'gen-I as the substrate, displayed formation of all the reaction intermediates (heptacarboxyl-porphyrin, one decarboxylation; hexacarboxyl-porphyrin, 2 decarboxylations; pentacarboxyl-porphyrin, 3 decarboxylations) and the product coproporphyrinogen (4-COOH) within five minutes (Figure 5 A-B). At 5 min, uro'gen-III was added at 45 μM and the continued accumulation of intermediates and products was monitored. Upon uro'gen III addition, the peak areas of the I-isomer reaction intermediates/products remained essentially constant, but the peak areas for the III-isomer intermediates/products continued to increase with time of incubation. The same is true if decarboxylation of uro'gen III is quenched after 5 minuets with uro'gen I (Figure 5 C-D). These observations indicate a mechanism in which the intermediates release and rebind to the enzyme for each successive decarboxylation. If the intermediates for the initial isomer present at the time of dilution were bound by URO-D and unable to exchange until completion of all decarboxylation reactions, the amount of coproporphyrin corresponding to the initial isomer would continue to accumulate after dilution with the second substrate. These data support the conclusion that reaction intermediates are released from the enzyme rather than shuttled to the partner active site of the dimer for the next step in formation of the ultimate product coproporphyrinogen.
Figure 5. Product profile of competition assays using wild-type URO-D. (A).
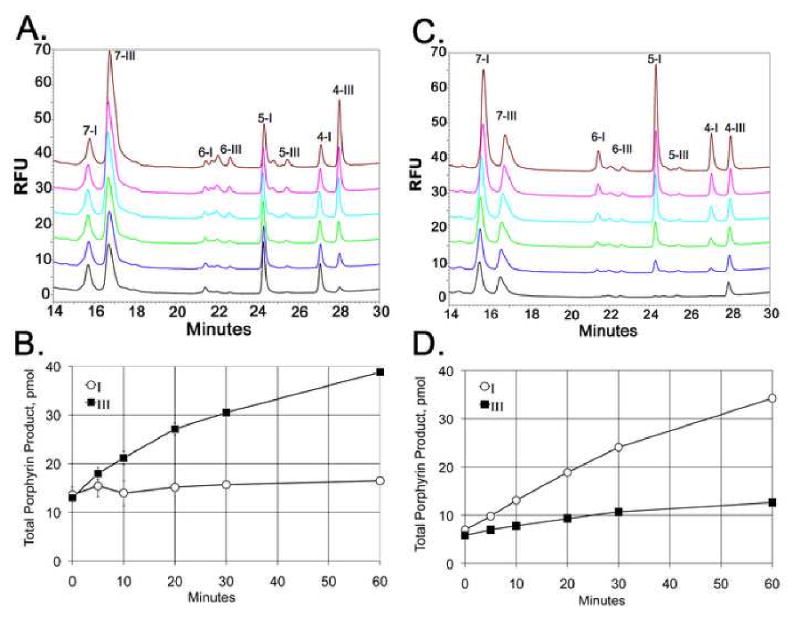
The URO-D reaction was started with 5 μM uro'gen-I, at 5 min 45 μM uro'gen-III was added and 1 min later as aliquot was removed and the product profile was measured by HPLC (black trace). The reaction was allowed to continue and samples were removed for analysis at 5 min (blue trace), 10 min (green trace) 20 min (light blue trace), 30 min (pink trace) and 60 min (brown trace). The metabolites heptacarboxyl-porphyrin (7), hexacarboxyl-porphyrin (6), pentacarboxyl-porphyrin (5) and coproporphyrin (4) are labeled with the corresponding isomer designation (I or III) are labeled. (B) The total reaction products, in pmol, for isomer I (blue) and isomer III (red) at time 1, 5, 10, 20, 30 and 60 min are shown, error bars represent 3 independent samples. (C and D) Data from an identical experiment where 5 μM uro'gen-III was used initially, followed by the addition of 45 μM uro'gen-I.
Because the dimeric structure of URO-D juxtaposes subunit active site clefts, shuttling between subunit active sites would provide an appealing mechanism to bind the physiologically relevant III isomer substrate and 7-COOH intermediate with equivalent geometry and without an intervening dissociation. Nevertheless, our observations with single chain variant URO-D proteins demonstrate that substrate shuttling between subunits is not required for catalysis and that intermediates are released, equilibrate with the bulk solvent and rebind. There are no data to support an obligate substrate shuttling mechanism where the substrate is transferred in a “ping-pong” fashion between active sites, as indicated by the competition experiments. The observed strict conservation of URO-D dimerization between species as diverse as human and the plant Nicotiana tabacum (13) may reflect the location of the dimer interface adjacent to the active site, with residues at the interface also lining the active site cleft. In this sense, dimerization might be viewed as essential to URO-D activity because it creates a cleft in a (β/α)8 protein that is large enough to accommodate a tetrapyrrole substrate.
Materials and Methods
Linking the C-terminus of one URO-D monomer to the N-terminus of a second URO-D monomer
A pET16b vector (Novagen, Madison, WI) containing a single copy of URO-D fused in frame behind a 21 residue, 10× histidine tag was used as a starting plasmid. An XhoI site was added to the 3′ end of the coding region, removing the stop codon. The QuikChange mutagenesis protocol (Stratagene, La Jolla, CA) was used for all plasmid mutagenesis. The NdeI to XhoI fragment of URO-D was cloned into pTYB1 (New England Biolabs, Beverly, MA) (Supplemental Figure 3). In a separate reaction the region 5′ to the 21 amino acid 10× histidine tag, between the XbaI site and the NcoI sites, was mutated in 2 additional rounds to produce a coding region in frame to the histidine tag. This changed the XbaI site into an XhoI site (Supplemental Figure 3). The URO-D inserts from both plasmids were sequenced to confirm the presence of all changes. The XhoI to BlpI fragment of the second plasmid was cloned into the first to create the initial URO-D fused to URO-D coding region, single-chain URO-D (scURO-D). Additional rounds of site directed mutagenesis, to add or remove residues, were performed using the QuikChange mutagenesis protocol (Stratagene, La Jolla, CA). All DNA modification enzymes were purchased from Invitrogen (Carlsbad, CA). Sequencing was performed in the University of Utah's Core Sequencing Laboratory. Mutations in the active site of URO-D were introduced into the parent plasmid and sequenced for confirmation. The fragments were sub-cloned into the scURO-D vector for protein expression.
Protein production
URO-D expression plasmids were transformed in BL-21 DE3 pLysS cells (Novagen, Madison WI). Proteins were expressed using the Overnight Express system (Novagen, Madison, WI) as described by the manufacturer. A 6 L culture was grown for each of the URO-D constructs. After overnight growth, samples were centrifuged at 2,500 × g for 15 min. The media were then decanted and the cell pellets frozen at -80 °C until used.
Cells were thawed in 30 ml lysis buffer (300 mM NaCl, 50 mM sodium phosphate, pH 6.8, 10% glycerol) and incubated for 30 min, at 4 °C, with 1.2 mL of 10 mg/mL lysozyme prior to sonication for 4 × 30 sec. Samples were returned to ice for 2 min between pulses. The membrane fraction was removed by centrifugation at 12,000 × g for 30 min. The supernatant was loaded onto a 1 mL Ni2+-NTA (Qiagen, Chatsworth, CA) column, at 4 °C. The column was washed with 40 mL buffer A (300 mM NaCl, 50 mM sodium phosphate pH 6.8, 10% glycerol, 1 mM β-mercaptoethanol (B-ME)), followed by elution of the protein in 30 mL buffer A with the addition of 250 mM imidazole. Fractions containing purified recombinant URO-D were dialyzed against 4 L of 20 mM Tris pH 7.0, 5% glycerol. The protein concentration was determined using BCA protein reagents, (Pierce, Rockford, IL). For crystallization, proteins were concentrated to 25 mg/mL using Centriprep concentrators (Amicon, Beverly, MA).
Analytical ultracentrifugation
Equilibrium sedimentation data were collected using a Beckman XL-A analytical ultracentrifuge. The sample buffer contained 50 mM Tris pH 7.5, 1 mM B-ME. The vbar and solvent density were calculated to be 0.7286 and 1.00379 respectively. The samples were scanned at 280 nm every 20 minutes until no changes were detected and the samples were determined to be at equilibrium. Five independent scans were collected. Data were merged and processed using the Heteroanalysis software (14).
URO-D assay
The assay for URO-D activity with uro'gen-I as substrate was adapted from the method described by Phillips and Kushner (15). The uro'gen-I substrate was enzymatically produced in a mixture containing 360 μM porphobilinogen (PBG), 75 mM Tris pH 7.7, 7.5 mM dithiothreitol (DTT), and 3-5 μg/mL of purified recombinant porphobilinogen deaminase (PBG-D). This reaction generates 30-35 nmol of uro'gen-I in 35 min at 37 °C in the dark. All subsequent steps were carried out either in the dark or under red darkroom light. To prepare uro'gen-III, the reaction mixture (described above) also contained 1 μg/mL of purified recombinant uroporphyrinogen-III-synthase. Following substrate generation the pH of the reaction was changed to pH 6.8, optimal for the URO-D reaction, by the addition of 20 μL/mL of 150 mM KH2PO4, pH 5.0.
A sample of 120-uL substrate was added to 80-μL of enzyme solution that contained 0.4 μg URO-D and 8 μg BSA in 50 mM potassium phosphate pH 6.8. The resulting 200 μL assay mixture was incubated at 37 °C for 30 min in the dark, and the reaction terminated by adding 200-μL 3M HCl. Porphyrinogens in the assay mixture were oxidized to porphyrins by exposure to UV light for 30 min or white light for approximately 2 h. The acidified sample was centrifuged at 14,000 × g for 10 min to remove precipitated proteins and products were analyzed by high performance liquid chromatography (HPLC) see below.
Isomer competition assay
Uro'gen-I and uro'gen-III were prepared as described (16). One microgram of URO-D was incubated with 5 μM of uro'gen-I for 5 min, whereupon a sample was removed for analysis and 45 μM of uro'gen-III was added to the reaction mixture, with additional samples removed for analysis at 1, 5, 10, 20, 30 and 60 min. Reaction products were separated by HPLC and peaks for each of the intermediate substrates were quantified. An identical experiment was run starting with 5 μM uro'gen-III and adding 45 μM uro'gen-I at 5 min, with intermediate time points as above.
HPLC of porphyrins
A 25 μL sample of the assay products was injected into a reverse phase HPLC system consisting of a Waters 2795 Separations module, a 3.9 × 300 mm μBondapak C18 column, and a Waters 474 scanning fluorescence detector that was set at 404 nm excitation and 618 nm emission to resolve the various porphyrins. The flow rate was set at 1.0 mL per min. Two solvents were used: Solvent A consisted of 50 mM sodium phosphate pH 4.5 in 50% v/v methanol and water, while Solvent B was 100% methanol. The milivolt signals detected for the various porphyrins were individually compared with those in a standard solution that contained 62.5 pmol each of uroporphyrin, heptacarboxyl-porphyrin, hexacarboxyl-porphyrin, pentacarboxyl-porphyrin, coproporphyrin and mesoporphyrin per 25-μL injection. Chromatograms were processed using the Waters Empower software.
Crystal growth and data collection
Single chain URO-D crystallized isomorphously to the native protein at room temperature (∼21 °C) under the same conditions as previously identified for wild type URO-D (6). The reservoir (500 μL) comprised 1.5 M sodium citrate, pH 6.5. The crystallization drop was prepared by mixing 5 μL of the URO-D protein solution (25 mg/mL) with 3 μL of the reservoir solution. Crystals typically grew in 3 to 10 days.
Prior to data collection, crystals were transferred to 1.7 M sodium citrate, pH 7.0, 5% glycerol for 2 min, suspended in a nylon loop, and plunged into liquid nitrogen. Crystals were maintained at 100 K during data collection. Data were integrated and scaled using DENZO and SCALEPACK (17). Data were phased directly using a model of the URO-D apo-enzyme monomer (pdb entry 1URO). The model was optimized by rigid-body minimization, positional and B-factor refinement using REFMAC (18, 19). Model building was performed using O (20). Figures were prepared using Pymol (21).
Supplementary Material
Acknowledgments
This work was supported by NIH grants RO1 DK20503, P30 DK072437, GM 56775. We thank Dr. Steve Alam for assistance in preparing samples and analyzing the data from analytical ultracentrifugation and Hector Bergonia for HPLC analysis of porphyrins. University of Utah Core Laboratories, supported in part by CA 42014, were used for sequencing of DNA and oligonucleotide production. The authors are grateful for assistance with data collection and crystal analysis provided by the use of synchrotron facilities at The National Synchrotron Light Source (Brookhaven National Laboratories, supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886) and The Stanford Synchrotron Radiation Lightsource (operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences).
Abbreviations
- URO-D
uroporphyrinogen decarboxylase
- PCT
porphyria cutanea tarda
- B-ME
β-mercaptoethanol
Footnotes
Accession numbers. Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers; scUROD - 3GVQ, scURO-D(GY) - 3GVR, scURO-D(FY) - 3GVV, scURO-D(YF) - 3GVW.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jackson AH, Sancovich HA, Ferrmolas AM, Evans N, Games DE, Matlin SA, Elder GH, Smith SG. Macrocyclic intermediates in the biosynthesis of porphyrins. Philos Trans R Soc Lond. 1976;273:191–206. doi: 10.1098/rstb.1976.0009. [DOI] [PubMed] [Google Scholar]
- 2.McLellan T, Pryor MA, Kushner JP, Eddy RL, Shows TB. Assignment of uroporphyrinogen decarboxylase (UROD) to the pter->p21 region of human chromosome 1. Cytogenet Cell Genet. 1985;39:224–227. doi: 10.1159/000132139. [DOI] [PubMed] [Google Scholar]
- 3.Phillips JD, Jackson LK, Bunting M, Franklin MR, Thomas KR, Levy JE, Andrews NC, Kushner JP. A mouse model of familial porphyria cutanea tarda. Proc Natl Acad Sci U S A. 2001;98:259–264. doi: 10.1073/pnas.011481398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of Heme Biosynthesis: X-linked Sideroblastic Anemia and the Porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th. McGraw-Hill; New York: 2001. pp. 2991–3062. [Google Scholar]
- 5.Phillips JD, Whitby FG, Kushner JP, Hill CP. Characterization and crystallization of human uroporphyrinogen decarboxylase. Protein Sci. 1997;6:1343–1346. doi: 10.1002/pro.5560060624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitby FG, Phillips JD, Kushner JP, Hill CP. Crystal structure of human uroporphyrinogen decarboxylase. EMBO J. 1998;17:2463–2471. doi: 10.1093/emboj/17.9.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillips JD, Whitby FG, Kushner JP, Hill CP. Structural basis for tetrapyrrole coordination by uroporphyrinogen decarboxylase. EMBO J. 2003;22:6225–6233. doi: 10.1093/emboj/cdg606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silva PJ, Ramos MJ. Density-functional study of mechanisms for the cofactor-free decarboxylation performed by uroporphyrinogen III decarboxylase. J Phys Chem B. 2005;109:18195–18200. doi: 10.1021/jp051792s. [DOI] [PubMed] [Google Scholar]
- 9.Nicholls CD, McLure KG, Shields MA, Lee PW. Biogenesis of p53 involves cotranslational dimerization of monomers and posttranslational dimerization of dimers. Implications on the dominant negative effect. J Biol Chem. 2002;277:12937–12945. doi: 10.1074/jbc.M108815200. [DOI] [PubMed] [Google Scholar]
- 10.Phillips JD, Parker TL, Schubert HL, Whitby FG, Hill CP, Kushner JP. Functional consequences of naturally occurring mutations in human uroporphyrinogen decarboxylase. Blood. 2001;98:3179–3185. doi: 10.1182/blood.v98.12.3179. [DOI] [PubMed] [Google Scholar]
- 11.de Verneuil H, Grandchamp B, Nordmann Y. Some kinetic properties of human red cell uroporphyrinogen decarboxylase. Biochim Biophys Acta. 1980;611:174–186. doi: 10.1016/0005-2744(80)90053-4. [DOI] [PubMed] [Google Scholar]
- 12.de Verneuil H, Sassa S, Kappas A. Purification and properties of uroporphyrinogen decarboxylase from human erythrocytes. A single enzyme catalyzing the four sequential decarboxylations of uroporphyrinogens I and III. J Biol Chem. 1983;258:2454–2460. [PubMed] [Google Scholar]
- 13.Martins BM, Grimm B, Mock HP, Huber R, Messerschmidt A. Crystal structure and substrate binding modeling of the uroporphyrinogen-III decarboxylase from Nicotiana tabacum. Implications for the catalytic mechanism. J Biol Chem. 2001;276:44108–44116. doi: 10.1074/jbc.M104759200. [DOI] [PubMed] [Google Scholar]
- 14.Cole JL. Analysis of heterogeneous interactions. Methods Enzymol. 2004;384:212–232. doi: 10.1016/S0076-6879(04)84013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phillips JD, Kushner JP. Measurement of Uroporphyrinogen Decarboxylase Activity. In: Maines MD, Costa LG, Reed DJ, Sassa S, Sipes IG, editors. Current Protocols in Toxicology. John Wiley & Sons, Inc; New York: 1999. pp. 8.4.1–8.4.13. [DOI] [PubMed] [Google Scholar]
- 16.Bergonia HA, Phillips JD, Kushner JP. Reduction of porphyrins to porphyrinogens with palladium on carbon. Anal Biochem. 2009;384:74–78. doi: 10.1016/j.ab.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Otwinowski Z, Minor W. Processing X-ray diffraction data in oscillation mode. In: Carter C, Sweet R, editors. Methods in Enzymology. Vol. 276. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 18.Murshudov GN. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 19.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 20.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 21.DeLano WL. DeLano Scientific; San Carlos, CA, USA: 2002. The PyMOL Molecular Graphics System. http://www.pymol.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.