

Abstract
Hepatocellular carcinoma, which is one of the most prevalent life-threatening human cancers, is showing an increased incidence worldwide. Recent evidence indicates that the development of hepatocellular carcinoma is associated with not only genetic alterations, but also with profound epigenetic changes. This review summarizes the current knowledge about epigenetic alterations during rodent hepatocarcinogenesis, considers the similarities and differences in epigenetic effects of genotoxic and non-genotoxic rodent liver carcinogens, and discusses the possible role of these effects in the causality of liver tumor development.
Keywords: epigenetics, hepatocarcinogenesis, rodents, genotoxic carcinogens, non-genotoxic carcinogens
INTRODUCTION
Hepatocellular carcinoma (HCC), which is one of the most prevalent life-threatening human cancers, is showing an increased incidence worldwide [Thorgeirsson and Grisham, 2002; Moradpour and Blum, 2005; McKillop et al., 2006]. HCC represents ∼85% of all liver cancers and is an aggressive disease [McKillop et al., 2006; Hussain et al., 2007]. The most prominent etiological factors associated with HCC are chronic viral hepatitis B and C infections, exposure to environmental chemicals and alcohol, and metabolic liver diseases [Moradpour and Blum, 2005; McKillop et al., 2006]; however, the molecular and cellular mechanisms of HCC pathogenesis are still poorly understood. Recent evidence indicates that HCC is associated with not only genetic alterations, such as DNA damage and chromosomal aberrations, but also with a substantial deregulation of the cellular epigenome, such as aberrant DNA methylation and histone modification [Shen et al., 1998; Lee et al., 2003; Pogribny et al., 2006a,b; Lehmann et al., 2007].
The development and progression of HCC in humans is a multistep, long-term process (more than 30 years) characterized by the progressive sequential evolution of morphologically distinct stages, such as chronic liver injury, necro-inflamation and regeneration, small cell dysplasia, low-grade and high-grade dysplastic nodules, and culminating in the formation of fully developed HCC [Thorgeirsson and Grisham, 2002; Libbrecht et al., 2005]. In humans, most of the research on HCC is conducted on patients who have already developed the disease. This limits the scope of the investigation to tumor biology and does not allow extensive inquiry into the mechanisms of disease progression. On the contrary, relevant rodent models of liver carcinogenesis provide a unique opportunity to understand the role of the etiological factors and mechanisms of tumor development [Lee et al., 2004].
In a broad sense, hepatocarcinogenesis may be induced through either genotoxic or non-genotoxic mechanisms. Environmental agents or chemicals are considered genotoxic if they, or the products of their metabolic activation, interact directly with DNA, causing mutations and leading to tumor formation [Shuker, 2002]. Non-genotoxic carcinogens are a diverse group of chemical compounds that are known to cause tumors by mechanisms rather than directly damaging DNA [Silva and Van der Laan, 2000]. Nonetheless, mounting evidence suggests that despite different mechanisms of action with regards to DNA reactivity both classes of agents were shown to lead to prominent epigenomic alterations in tissues that are targets for carcinogenesis as a result of exposure. This review considers the similarities and differences in the epigenetic effects of genotoxic and non-genotoxic rodent liver carcinogens and discusses the possible role of epigenetic changes in the causality of tumor development.
EPIGENETIC REGULATION AND LIVER CANCER
Classically, the development of cancer in humans has been viewed as a progressive multistep process involving the transformation of normal cells into malignant cells driven in part by genetic alterations that include mutations and deletions in tumor suppressor genes and oncogenes, and chromosomal abnormalities [Hanahan and Weinberg, 2000]. However, new data indicate the importance of epigenetic processes, knowledge that challenges our view on cancer as a disease dependent only on genetic changes [Jones and Baylin, 2007]. It is now clear now that cancer is a genetic and epigenetic disease, and both components cooperate at all stages of cancer development [Feinberg and Tycko, 2004; Jones and Baylin, 2007]. “Genetic” is defined as a heritable change in the DNA sequence (i.e., mutation), whereas “epigenetic” refers to heritable changes in gene expression that are not accompanied by changes in the DNA sequence. In normal cells, epigenetic information is hereditarily maintained to preserve cellular identity. In contrast, the epigenetic landscape of cancer cells is profoundly distorted, including a massive loss of global methylation throughout the genome accompanied by hypermethylation of certain promoters associated with gene silencing [Feinberg and Tycko, 2004; Jones and Baylin, 2007].
Global hypomethylation of DNA is one of the most common molecular alterations identified in human cancer cells [Feinberg and Tycko, 2004]. To understand and correctly recognize the importance of global DNA hypomethylation with respect to the carcinogenic process, it is necessary to consider the role and sites of DNA methylation in normal cell function. The majority of DNA methylation in mammalian cells occurs in repetitive DNA elements, and one of the primary functions of DNA methylation in normal cells is to silence foreign DNA sequences [Yoder et al., 1997; Goll and Bestor, 2005; Schulz et al., 2006]. It has been suggested that cancer-linked DNA hypomethylation largely affects the methylation status of repetitive elements [Yoder et al., 1997; Schulz et al., 2006]. Recent evidence has demonstrated decreased methylation of repetitive sequences (i.e., LINE1, LTR, SINE) during hepatocarcinogenesis [Asada et al., 2006; Pogribny et al., 2006a]. Consequently, hypomethylation of repetitive sequences compromises genomic integrity via chromatin decondensation and activation of repetitive DNA elements and proto-oncogenes, which results in a variety of genomic and chromosomal instability events, including cis- and trans-insertional mutagenesis, unequal homologous recombination, genomic rearrangements, and segmental duplications leading to deletions and duplications [Kazazian, 2004]. The causal role of these lesions in the etiology of cancer, including liver cancer, is now commonly accepted [Coleman and Tsongalis, 2006].
In addition to DNA hypomethylation, many key genes involved in metabolism and cell function, including APC, GSTP1, p16INK4A, SOCS1, and RASSF1, have been found to undergo DNA hypermethylation at early pre-cancerous stages of liver carcinogenesis [Lee et al., 2003; Yang et al., 2003]. Table I shows a selected list of the genes whose expression is associated with aberrant promoter methylation in HCC. Altered DNA methylation patterns in HCC are closely related to the disruption of the DNA methylation machinery. Several studies have demonstrated involvement of altered expression of the maintenance DNA methyltransferase 1, de novo DNA methyltransferases 3A and 3B, and methyl-CpG-binding proteins in the initiation, establishment, and maintenance of aberrant DNA methylation patterns during the development and progression of HCC [Saito et al., 2003; Park et al., 2006].
TABLE I. Selected List of the Genes Regulated by Epigenetic Mechanisms in HCC.
| Gene | Function | Consequences |
|---|---|---|
| Hypermethylated genes | ||
| RASSF1A | Ras effector homologue | Inhibition of cell cycle arrest |
| APC | Inhibitor of β-catenin | Activation of β-catenin pathway |
| p16INK4A | Cell cycle G1-to-S phase progression | Cell cycle alterations |
| CyclinD2 | Cell cycle G1-to-S phase progression | Cell cycle alterations |
| SOCS1/3 | Inhibitor of JAK/STAT pathway | Activation of JAK/STAT pathway |
| RB1 | Cell cycle G1-to-S phase progression | Cell cycle alterations |
| PTPRO | Protein tyrosine phosphatase receptor type O | Cell cycle alterations |
| PTEN | Regulation of PI3Ks | Activation of PI3K/Akt pathway |
| NORE1A/B | Ras effector homologue | Inhibition of cell cycle arrest |
| TIMP-3 | Inhibition of matrix metalloproteinases | Alteration in cytoskeletal organization, dissemination |
| Connexin 26 | Gap junctional intercellular communication | Alteration in cell-cell communication |
| E-cadherin | Cell adhesion | Dissemination |
| SYK | Immune and inflammatory responses, angiotensin II signaling pathway | Promotion of invasiveness and cell proliferation |
| GSTP1 | Xenobiotic metabolis, conjugation of glutathione | Accumulation of carcinogens and their metabolites |
| NQO1 | Xenobiotic metabolism | Accumulation of carcinogens and their metabolites |
| MGMT | DNA repair | Increased mutation rates |
| KLF6 | Zinc finger transcription factor | Abnormal cell proliferation |
| PROX1 | Homeobox gene | Misregulation of differentiation and cell proliferation |
| RIZ1 | Histone/protein methyltransferase | Alteration in heterochtomatin, aberrant gene expression |
| DLC-1 | Rho GTPase regulator | Misregulation of cell proliferation and cytoskeletal organization |
| MAT1A | Synthesis of S-adenosyl-L-methionine | Alteration in one carbon metabolism, misregulation of cell proliferation |
| Hypomethylated genes | ||
| MAT2A | Synthesis of S-adenosyl-L-methionine | Alteration in one carbon metabolism, misregulation of cell proliferation |
| uPA | Plasminogen activation system | Promotion of invasiveness and dissemination |
| Heparanase | Endoglycosidase | Promotion of invasiveness, invasiveness, and dissemination |
| SNCG | Member of synuclein proteins family | Promotion of cell proliferation, angiogenesis, and disseminationt |
| TFF3 | Member of trefoil peptides family | Increasing resistance to apoptosis |
| Maspin | Serine protease inhibitor | Promotion of invasiveness and dissemination |
| MAGE-A1 | Melanoma-associated antigen | Inhibition of apoptosis, promotion of cell proliferation |
| HKII | Glucose metabolism | Activation of glycolytic pathway |
EPIGENETIC ALTERATIONS DURING NON-GENOTOXIC HEPATOCARCINOGENESIS
The methyl-deficient model of endogenous liver carcinogenesis is one of the most extensively studied models of non-genotoxic rodent HCC [Nakae et al., 1992; James et al., 2003]. This model is unique because dietary omission of sources of methyl groups rather than xenobiotic addition leads to tumor formation [Nakae, 1999]. In addition, the sequence of pathological and molecular events is remarkably similar to the development of human HCC associated with viral hepatitis B and C infections, alcohol exposure, and metabolic liver diseases [Powel et al., 2005]. One of the earliest epigenetic alterations observed during hepatocarcinogenesis induced by methyl-deficiency is sustained global hypomethylation of liver DNA [Wainfan and Poirier, 1992; Christman, 2003; Pogribny et al., 2004]. Importantly, these changes are specific to liver tissue and do not occur in any other organs.
We have recently shown the importance of DNA hypomethylation as a promoting factor for the clonal expansion of initiated cells [Pogribny et al., 2006a,b]. Similar observations were reported with respect to other non-genotoxic liver carcinogens, specifically to one of the most extensively studied classes of non-genotoxic carcinogens—peroxisome proliferators. Treatment of mice with 4-chloro-6-(2,3-xylidino)-pyrimidinylthioacetic acid (WY-14,643), trichloroacetic acid, or dichloroacetic acid results in a rapid decrease in global DNA methylation as well as region-specific changes in DNA methylation [Tao et al., 2000; Ge et al. 2001; Pogribny et al., in press].
An altered pattern of DNA methylation was also observed in the livers of mice exposed to other non-genotoxic compounds, such as diethanolamine or phenobarbital, especially in the livers of the tumor-prone B6C3F1 and C3H mice [Bachman et al., 2006; Philips et al., 2007]. In mouse livers, treatment with these agents results in the rapid appearance of regions with altered DNA methylation, predominantly progressive accumulation of hypomethylated regions. Importantly, these changes were more pronounced in the livers of tumor-prone B6C3F1 mice as compared with the resistant C57BL/6 mice, and were also dependent on the availability of a functional Constitutive Androstane Receptor [Bachman et al., 2006; Philips et al., 2007]. This has led to the suggestion that sensitivity to hepatocarcinogenesis may be inversely related to the capacity to maintain normal patterns of DNA methylation [Goodman and Watson, 2002].
Another example demonstrating that hypomethylation of DNA is associated with malignant transformation and that this occurs at early stages of disease was obtained from studies on arsenic-induced hepatocarcinogenesis. Arsenic is a well-known human carcinogen that does not act through a classic genotoxic mechanism [Simeonova and Luster, 2000; Rossman, 2003]. In vitro exposure of the rat liver epithelial cell line TRL 1215 to arsenic produces malignant transformation concurrently with global DNA hypomethylation [Zhao et al., 1997]. The extent of DNA hypomethylation in these transformed cells was positively correlated with the tumorigenicity of the cells upon inoculation into nude mice, clearly indicating that DNA hypomethylation may be a causative factor in arsenic-induced malignancy [Zhao et al., 1997]. Additionally, long-term exposure of mice to arsenic induced global DNA and gene-specific hypomethylation in livers [Chen et al., 2004].
Hypomethylation of DNA is not the only mechanism involved in hepatocarcinogenesis. Several critical tumor suppressor genes, such as p16INK4A, PTPRO, E-cadherin, and Connexin26, exhibit DNA hypermethylation in liver at early precancerous stages of rodent liver carcinogenesis [Pogribny and James, 2002; Motiwala et al., 2003; Calvisi et al., 2004; Tsujiuchi et al., 2007]. The exact mechanisms causing aberrant DNA methylation in target organs during carcinogenesis, in general, and hepatocarcinogenesis, in particular, is currently unknown. However, one of the main factors that may cause this disruption is an alteration of DNA methylation machinery. Several lines of evidence indicate that altered activity and expression of DNA methyltransferases and methyl-CpG-binding proteins take place at early stages of liver carcinogenesis [James et al., 2003; Takiguchi et al., 2003; Li et al., 2006]. In addition, the presence of DNA and chromatin lesions, such as unrepaired DNA damage, several forms of cytosine damage products, and DNA-histone crosslink products, may alter the DNA methylation patterns [Voitkun and Zhitkovich, 1999; Valinluck and Sowers, 2007].
Epigenetic changes during liver carcinogenesis induced by non-genotoxic agents are not limited to altered DNA methylation patterns. Recently, using two different models of non-genotoxic hepatocarcinogenesis (methyl-deficient diets and WY-14,643), it was shown that marked alterations in the trimethylation of histone H3 lysine 9 (H3K9me3) and histone H4 lysine 20 (H4K20me3) occurred in the liver during carcinogenesis [Pogribny et al., 2006a; Pogribny et al., in press]. Specifically, early stages of hepatocarcinogenesis were characterized by a progressive decrease in H3K9me3 and H4K20me3. In contrast, a different trend in histone methylation changes was observed in full-fledged HCC, where there was a continuing decrease in H4K20me3 but an increase in H3K9me3. A decreased level of H4K20me3 has been observed in several forms of human cancer [Fraga et al., 2005] leading to the hypothesis that low levels of H4K20me3 may contribute to the etiology of cancer and can be used as an indicator and diagnostic marker for neoplastic transformation and tumor growth. The stage-dependent differences of H3K9me3 and H4K20me3 during carcinogenesis may by explained by their different functions in cells. One of the primary functions of H3K9me3 and H4K20me3 is in the formation of heterochromatin [Jenuwein, 2006]. Loss of H3K9me3 and H4K20me3 affects the stability of the genome by compromising the organisation of heterochromatin. Additionally, H4K20me3 plays an important role in damage checkpoint control [Sanders et al., 2004], and H3K9me3 is involved in terminal differentiation [Ait-Si-Ali et al., 2004]. Disturbances in any or all of these mechanisms induced by loss of H3K9me3 and H4K20me3 may promote the initial neoplastic cell transformation. In contrast, the increased level H3K9me3 in liver tumors may be a cellular defense mechanism safeguarding the viability of cancer cells and promoting tumor growth given the role of H3K9me3 in heterochromatin organisation and gene silencing.
EPIGENETIC ALTERATIONS DURING GENOTOXIC HEPATOCARCINOGENESIS
It is widely believed that genotoxic carcinogens, including hepatocarcinogens, cause tumor formation primarily through the direct induction of a variety of genotoxic DNA lesions. Although the presence of DNA adducts may be a necessary prerequisite, they are not sufficient for tumor formation, which results from a much broader alterations in cellular homeostasis, mainly from the inability of cells to properly maintain and control the expression of genetic information.
It has been shown that several genotoxic hepatocarcinogens (i.e., 1,2-dimethylhydrazine, N-nitrosodiethyamine, N-nitrosomorpholine) cause alterations in DNA methylation, in addition to exerting genotoxic effects [Rao et al., 1989; Münzel et al., 1991; Park et al., 2001]. Recently, it has been suggested that these epigenetic changes may play a leading causative role in carcinogenic process induced by genotoxic agents [Jaffe, 2003; Bombail et al., 2004; Karpinets and Foy, 2005]. Our studies on epigenetic mechanisms of tamoxifen-induced rat hepatocarcinogenesis support this suggestion. Feeding rats with a tamoxifen-containing diet resulted in an early and sustained loss of global DNA methylation, hypomethylation of repetitive DNA sequences, and an altered pattern of histone methylation [Tryndyak et al., 2007]. Importantly, the early appearance of epigenetic changes and the absence of the evident morphological abnormalities suggest that these alterations are directly related to the effect of carcinogen exposure. These changes were remarkably similar to alterations observed during non-genotoxic hepatocarcinogenesis indicating the significance of epigenetic alterations in the etiology of liver carcinogenesis induced by both genotoxic and non-genotoxic agents. In this context, monitoring epigenetic changes represents attractive molecular markers that can assist in molecular diagnostic and molecular classification of cancers, including HCC.
DISCUSSION AND CONCLUSION
Presently, it is becoming increasingly evident that epigenetic alterations are not only important features of cancer cells, but they also play a major role in the etiology of cancer [Jaffe, 2003; Feinberg, 2004; Jones and Baylin, 2007]. Results of recent studies on mechanisms of rodent hepatocarcinogenesis clearly show that the exposure of rats and mice to various genotoxic and non-genotoxic hepatocarcinogenic agents results in rapid alterations in the cellular epigenome. Loss of global and region-specific DNA hypomethylation, especially hypomethylation of repetitive DNA sequences, promoter hypermethylation of promoters in tumor suppressor genes, and progressive loss of histone H4 lysine 20 trimethylation accompanied by the dysbalance between cell proliferation and apoptosis leads to early disruption of cellular homeostasis in liver. This disruption, in turn, results in the emergence of epigenetically reprogrammed proliferating cells with a growth-advantage phenotype and a high potential for the activation of mutator pathways (Fig. 1). Recent evidence showing the importance of epigenetic changes in the establishment of mutator phenotype in human cancer cells supports this suggestion [Jacinto and Esteller, 2007]. The remarkable feature of epigenetic changes is their early appearance and correspondence to alterations in full-fledged HCC suggesting that these alterations may be used as biomarkers for the carcinogenic process. Lastly, the potential reversibility of epigenetic changes makes them promising targets for chemoprevention [Kopelovich et al., 2003].
Fig. 1.
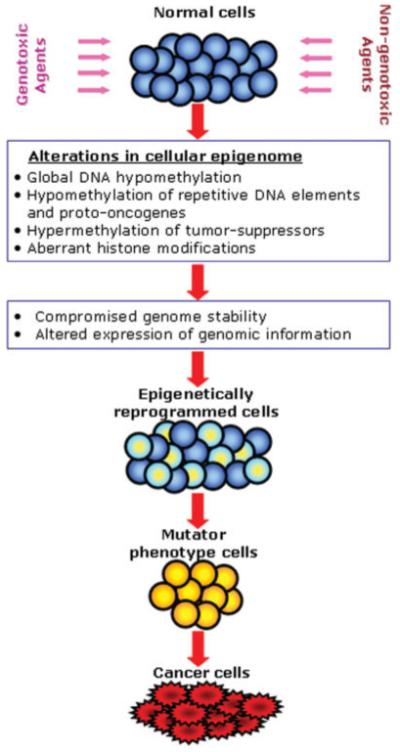
An integrated view of the role of epigenetic dysregulation in hepatocarcinogenesis. Genotoxic or non-genotoxic insults injure many liver cells triggering changes in the cellular epigenome. Alterations in epigenetic mechanisms lead to early disruption of homeostasis in liver cells characterized by a loss in the balance between cell proliferation and apoptosis, activation of DNA repetitive sequences in the genome, loss of genomic and chromosomal stability, and aberrant expression of genomic information. This results in the emergence of the population of epigenetically reprogrammed proliferating cells with a growth advantage and high potential for activation of a mutator phenotype and, consequently, leads to malignant cell transformation.
Footnotes
The views expressed in this article do not necessarily represent those of the U.S. Food and Drug Administration.
REFERENCES
- Ait-Si-Ali S, Guasconi V, Fritsch L, Yahi H, Sekhri R, Naguibneva I, Robin P, Cabon F, Polesskaya A, Harel-Bellan A. A Suv39h-dependent mechanism for silencing S-phase genes in differentiating but not in cycling cells. EMBO J. 2004;23:605–615. doi: 10.1038/sj.emboj.7600074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asada K, Kotake Y, Asada R, Saunders D, Broyles RH, Towner RA, Fukui H, Floyd RA. LINE-1 hypomethylation in a choline-deficiency-induced liver cancer in rats: Dependence on feeding period. J Biomed Biotechnol. 2006;1:17142. doi: 10.1155/JBB/2006/17142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman AN, Kamendulis LM, Goodman JI. Diethanolamine and Phenobarbital produce an altered pattern of methylation in GC-rich regions of DNA in B6C3F1 mouse hepatocytes similar to that resulting from choline deficiency. Toxicol Sci. 2006;90:317–325. doi: 10.1093/toxsci/kfj091. [DOI] [PubMed] [Google Scholar]
- Bombail V, Moggs JG, Orphanides G. Perturbation of epigenetic status by toxicants. Toxicol Lett. 2004;149:51–58. doi: 10.1016/j.toxlet.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Calvisi DF, Ladu S, Conner EA, Factor VM, Thorgeirsson SS. Disregulation of E-cadherin in transgenic mouse models of liver cancer. Lab Invest. 2004;84:1137–1147. doi: 10.1038/labinvest.3700147. [DOI] [PubMed] [Google Scholar]
- Chen H, Li SF, Liu J, Diwan BA, Barrett JC, Waalkes MP. Chronic inorganic arsenic exposure induces hepatic global and individual gene hypomethylation: Implications for arsenic hepatocarcinogenesis. Carcinogenesis. 2004;25:1779–1786. doi: 10.1093/carcin/bgh161. [DOI] [PubMed] [Google Scholar]
- Christman JK. Diet, DNA methylation and cancer. In: Daniel H, Zempleni J, editors. Molecular Nutrition. CABI Publishing; Oxon, UK: 2003. pp. 237–265. [Google Scholar]
- Coleman WB, Tsongalis GJ. Molecular mechanisms of human carcinogenesis. EXS. 2006;96:321–349. doi: 10.1007/3-7643-7378-4_14. [DOI] [PubMed] [Google Scholar]
- Feinberg AP. The epigenetics of cancer etiology. Semin Cancer Biol. 2004;14:427–432. doi: 10.1016/j.semcancer.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- Ge R, Wang W, Kramer PM, Yang S, Tao L, Pereira MA. Wy-14,643-induced hypomethylation of the c-myc gene in mouse liver. Toxicol Sci. 2001;62:28–35. doi: 10.1093/toxsci/62.1.28. [DOI] [PubMed] [Google Scholar]
- Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- Goodman JI, Watson RE. Altered DNA methylation: A secondary mechanism involved in carcinogenesis. Annu Rev Pharmacol Toxicol. 2002;42:501–525. doi: 10.1146/annurev.pharmtox.42.092001.141143. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 mutations and human hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene. 2007;26:2166–2176. doi: 10.1038/sj.onc.1210279. [DOI] [PubMed] [Google Scholar]
- Jacinto FV, Esteller M. Mutator pathways unleashed by epigenetic silencing in human cancer. Mutagenesis. 2007;22:247–253. doi: 10.1093/mutage/gem009. [DOI] [PubMed] [Google Scholar]
- Jaffe LF. Epigenetic theories of cancer initiation. Adv Cancer Res. 2003;30:209–230. doi: 10.1016/s0065-230x(03)90007-8. [DOI] [PubMed] [Google Scholar]
- James SJ, Pogribny IP, Pogribna M, Miller BJ, Jernigan S, Melnyk S. Mechanisms of DNA damage, DNA hypomethylation, and tumor progression in the folate/methyl-deficient rat model of hepatocarcinogenesis. J Nutr. 2003;133:3740S–3747S. doi: 10.1093/jn/133.11.3740S. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. The epigenetic magic of histone lysine methylation. FEBS J. 2006;273:3121–3135. doi: 10.1111/j.1742-4658.2006.05343.x. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpinets T, Foy BD. Tumorigenesis: The adaptation of mammalian cells to sustained stress environment by epigenetic alterations and succeeding matched mutations. Carcinogenesis. 2005;26:1323–1334. doi: 10.1093/carcin/bgi079. [DOI] [PubMed] [Google Scholar]
- Kazazian HH. Mobile elements: Drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- Kopelovich L, Crowell JA, Fay JR. The epigenome as a target for cancer chemoprevention. J Natl Cancer Inst. 2003;95:1747–1757. doi: 10.1093/jnci/dig109. [DOI] [PubMed] [Google Scholar]
- Lee S, Lee HJ, Kim JH, Lee HS, Jang JJ, Kang GH. Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am J Pathol. 2003;163:1371–1378. doi: 10.1016/S0002-9440(10)63495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Chu IS, Mikaelyan A, Calvisi DF, Heo J, Reddy JK, Thorgeirsson SS. Application of comparative functional genomics to identify best-fit mouse model to study human cancer. Nat Genet. 2004;36:1306–1311. doi: 10.1038/ng1481. [DOI] [PubMed] [Google Scholar]
- Lehmann U, Wingen LU, Brakensiek K, Wedemeyer H, Becker T, Heim A, Metzig K, Hasemeier B, Kreipe H, Flemming P. Epigenetic defects of hepatocellular carcinoma are already found in non-neoplastic liver cells from patients with hereditary haemochromatosis. Hum Mol Genet. 2007;16:1335–1342. doi: 10.1093/hmg/ddm082. [DOI] [PubMed] [Google Scholar]
- Li X, Ghoshal K, Datta J, Bai S, Pogribny I, Pogribny M, Huang Y, Young D, Jacob ST. Effect of folate and methyl deficient diet on expression of DNA methyltransferases and methyl CpG binding proteins involved in epigenetic gene silencing in livers of F344 rats. J Nutr. 2006;136:1522–1527. doi: 10.1093/jn/136.6.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libbrecht L, Desmet V, Roskams T. Preneoplastic lesions in human hepatocarcinogenesis. Liver Int. 2005;25:16–27. doi: 10.1111/j.1478-3231.2005.01016.x. [DOI] [PubMed] [Google Scholar]
- McKillop IH, Moran DM, Jin X, Koniaris LG. Molecular pathogenesis of hepatocellular carcinoma. J Surg Res. 2006;136:125–135. doi: 10.1016/j.jss.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Moradpour D, Blum HE. Pathogenesis of hepatocellular carcinoma. Eur J Gastroenterol Hepatol. 2005;17:477–483. doi: 10.1097/00042737-200505000-00002. [DOI] [PubMed] [Google Scholar]
- Motiwala T, Ghoshal K, Das A, Majumder S, Weichenhan D, Wu YZ, Holman K, James SJ, Jacob ST, Plass C. Suppression of the protein tyrosine phosphatase receptor type O gene (PTPRO) by methylation in hepatocellular carcinomas. Oncogene. 2003;22:6319–6331. doi: 10.1038/sj.onc.1206750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münzel PA, Pfohl-Leszkowicz A, Röhrdanz E, Keith G, Dirheimer G, Bock KW. Site-specific hypomethylation of c-myc protooncogene in liver nodules and inhibition of DNA methylation by N-nitrosomorpholine. Biochem Pharmacol. 1991;42:365–371. doi: 10.1016/0006-2952(91)90724-j. [DOI] [PubMed] [Google Scholar]
- Nakae D. Endogenous liver carcinogenesis in the rat. Pathol Int. 1999;49:1028–1042. doi: 10.1046/j.1440-1827.1999.00990.x. [DOI] [PubMed] [Google Scholar]
- Nakae D, Yoshiji H, Mizumoto Y, Horiguchi K, Shiraiwa K, Tamura K, Denda A, Konishi Y. High incidence of hepatocellular carcinomas induced by a choline deficient l-amino acid defined diet in rats. Cancer Res. 1992;52:5042–5045. [PubMed] [Google Scholar]
- Park TJ, Kim HS, Byun KH, Jang JJ, Lee YS, Lim IK. Sequential changes in hepatocarcinogenesis induced by diethylnitrosamine plus thioacetamide in Fisher 344 rats: Induction of gankyrin expression in liver fibrosis, pRB degradation in cirrhosis, and methylation of p16(INK4A) exon 1 in hepatocellular carcinoma. Mol Carcinogenesis. 2001;30:138–150. doi: 10.1002/mc.1022. [DOI] [PubMed] [Google Scholar]
- Park HJ, Yu E, Shim YH. DNA methyltransferase expression and DNA hypermethylation in human hepatocellular carcinoma. Cancer Lett. 2006;233:271–278. doi: 10.1016/j.canlet.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Phillips JM, Yamamoto Y, Negishi M, Maronpot RR, Goodman JI. Orphan nuclear receptor constitutive active/androstane receptor-mediated alterations in DNA methylation during phenobarbital promotion of liver tumorigenesis. Toxicol Sci. 2007;96:72–82. doi: 10.1093/toxsci/kfl188. [DOI] [PubMed] [Google Scholar]
- Pogribny IP, James SJ. De novo methylation of the p16INK4A gene in early preneoplastic liver tumors induced by folate/methyl deficiency in rats. Cancer Lett. 2002;187:69–75. doi: 10.1016/s0304-3835(02)00408-1. [DOI] [PubMed] [Google Scholar]
- Pogribny IP, James SJ, Jernigan S, Pogribna M. Genomic hypomethylation is specific for preneoplastic liver in folate/methyl deficient rats and does not occur in non-target tissues. Mutat Res. 2004;548:53–59. doi: 10.1016/j.mrfmmm.2003.12.014. [DOI] [PubMed] [Google Scholar]
- Pogribny IP, Ross SA, Tryndyak VP, Pogribna M, Poirier LA, Karpinets TV. Histone H3 lysine 9 and histone H4 lysine 20 trimethylation and the expression of Suv4-20h2 and Suv-39h1 histone methyltransferases in hepatocarcinogenesis induced by methyl deficiency in rats. Carcinogenesis. 2006a;27:1180–1186. doi: 10.1093/carcin/bgi364. [DOI] [PubMed] [Google Scholar]
- Pogribny IP, Ross SA, Wise C, Pogribna M, Jones EA, Tryndyak VP, James SJ, Dragan YP, Poirier LA. Irreversible global DNA hypomethylation as a key step in hepatocarcinogenesis induced by dietary methyl deficiency. Mutat Res. 2006b;593:80–87. doi: 10.1016/j.mrfmmm.2005.06.028. [DOI] [PubMed] [Google Scholar]
- Pogribny IP, Tryndyak VP, Woods CG, Witt SE, Rusyn I. Epigenetic effects of the continuous exposure to peroxisome proliferator WY-14,643 in mouse liver are dependent upon peroxisome proliferator activated receptor α. Mutat Res. doi: 10.1016/j.mrfmmm.2007.05.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powel CL, Kosyk O, Bradford BU, Parker JS, Lobenhofer EK, Denda A, Uematsu F, Nakae D, Rusyn I. Temporal correlation of pathology and DNA damage with gene expression in a choline-deficient model of rat liver injury. Hepatology. 2005;42:1137–1147. doi: 10.1002/hep.20910. [DOI] [PubMed] [Google Scholar]
- Rao PM, Antony A, Rajalakshmi, Sarma DSR. Studies on hypomethylation of liver DNA during early stages of chemical carcinogenesis in rat liver. Carcinogenesis. 1989;10:933–937. doi: 10.1093/carcin/10.5.933. [DOI] [PubMed] [Google Scholar]
- Rossman TG. Mechanism of arsenic carcinogenesis and integrated approach. Mutat Res. 2003;533:37–65. doi: 10.1016/j.mrfmmm.2003.07.009. [DOI] [PubMed] [Google Scholar]
- Saito Y, Kanai Y, Nakagawa T, Sakamoto M, Saito H, Ishii H, Hirohashi S. Increased expression of DNA methyltransferase (DNMT) 1 is significantly correlated with the malignant potential and poor prognosis of human hepatocellular carcinomas. Int J Cancer. 2003;105:527–532. doi: 10.1002/ijc.11127. [DOI] [PubMed] [Google Scholar]
- Sanders SL, Portoso M, Ata J, Bahler J, Allshire RC, Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–614. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Schulz WA, Steinhoff C, Florl AR. Methylation of endogenous human retroelements in health and disease. Curr Top Microbiol Immunol. 2006;310:211–250. doi: 10.1007/3-540-31181-5_11. [DOI] [PubMed] [Google Scholar]
- Shen L, Fang J, Qiu D, Zhang T, Yang J, Chen S, Xiao S. Correlation between DNA methylation and pathological changes in human hepatocellular carcinoma. Hepato-Gastroenter. 1998;45:1753–1759. [PubMed] [Google Scholar]
- Shuker DE. The enemy at the gates? DNA adducts as biomarkers of exposure to exogenous and endogenous genotoxic agents. Toxicol Lett. 2002;134:51–56. doi: 10.1016/s0378-4274(02)00162-5. [DOI] [PubMed] [Google Scholar]
- Silva LB, Van der Laan JW. Mechanisms of nongenotoxic carcinogenesis and assessment of the human hazard. Regul Toxicol Pharmacol. 2000;32:135–143. doi: 10.1006/rtph.2000.1427. [DOI] [PubMed] [Google Scholar]
- Simeonova PP, Luster MI. Mechanisms of arsenic carcinogenicity” genetic or epigenetic mechanisms? J Environ Pathol Toxicol Oncol. 2000;19:281–286. [PubMed] [Google Scholar]
- Takiguchi M, Achanzar WE, Qu W, Li G, Waalkes MP. Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp Cell Res. 2003;2886:355–365. doi: 10.1016/s0014-4827(03)00062-4. [DOI] [PubMed] [Google Scholar]
- Tao L, Yang S, Xie M, Kramer PM, Pereira MA. Effect of trichloroethylene and its metabolites, dichloroacetic acid and trichloroacetic acid, on the methylation and expression of c-Jun and c-Myc protooncogenes in mouse liver: Prevention by methionine. Toxicol Sci. 2000;54:399–407. doi: 10.1093/toxsci/54.2.399. [DOI] [PubMed] [Google Scholar]
- Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339–346. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- Tryndyak VP, Kovalchuk O, Muskhelishvili L, Montgomery B, Rodriguez-Juarez R, Melnyk S, Ross SA, Beland FA, Pogribny IP. Epigenetic reprogramming of liver cells in tamoxifen-induced rat hepatocarcinogenesis. Mol Carcinogenesis. 2007;46:187–197. doi: 10.1002/mc.20263. [DOI] [PubMed] [Google Scholar]
- Tsujiuchi T, Shimizu K, Itsuzaki Y, Onishi M, Sugata E, Fujii H, Honoki K. CpG site hypermethylation of E-cadherin and connexin26 genes in hepatocellular carcinomas induced by a choline-deficient L-amino acid-defined diet in rats. Mol Carcinogenesis. 2007;46:269–274. doi: 10.1002/mc.20268. [DOI] [PubMed] [Google Scholar]
- Valinluck V, Sowers LC. Inflammation-mediated cytosine damage: A mechanistic link between inflammation and the epigenetic alterations in human cancers. Cancer Res. 2007;67:5583–5586. doi: 10.1158/0008-5472.CAN-07-0846. [DOI] [PubMed] [Google Scholar]
- Voitkun V, Zhitkovich A. Analysis of DNA-protein crosslinking activity of malondialdehyde in vitro. Mutat Res. 1999;424:97–106. doi: 10.1016/s0027-5107(99)00011-1. [DOI] [PubMed] [Google Scholar]
- Wainfan E, Poirier LA. Methyl groups in carcinogenesis: Effects on DNA methylation and gene expression. Cancer Res. 1992;52:2071S–2077S. [PubMed] [Google Scholar]
- Yang B, Guo M, Herman JG, Clark DP. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am J Pathol. 2003;163:1101–1107. doi: 10.1016/S0002-9440(10)63469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:376–378. doi: 10.1016/s0168-9525(97)01181-5. [DOI] [PubMed] [Google Scholar]
- Zhao CQ, Young MR, Diwan BA, Coogan TP, Waalkes MP. Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc Natl Acad Sci USA. 1997;94:10907–10912. doi: 10.1073/pnas.94.20.10907. [DOI] [PMC free article] [PubMed] [Google Scholar]