

Summary
Methylation of cytosine residues in CpG dinucleotides plays an important role in epigenetic regulation of gene expression and chromatin structure/stability in higher eukaryotes. DNA methylation patterns are established and maintained at CpG dinucleotides by DNA methyltransferases (Dnmt1 and Dnmts 3a and 3b). In mammals and many other eukaryotes, the CpG dinucleotide is underrepresented in the genome. This loss is postulated to be the result of unrepaired deamination of cytosine (C) and 5-methylcytosine (5mC) to uracil (U) and thymine (T), respectively. Two thymine glycosylases are believed to reduce the impact of 5mC deamination. G/T mismatch-specific thymine-DNA glycosylase (Tdg) and methyl-CpG binding domain protein 4 (Mbd4) can both excise uracil or thymine at U·G and T·G mismatches to initiate base excision repair. Here, we report the characterization of interactions between Dnmt3b and both Tdg and Mbd4. Our results demonstrate that both Tdg and Dnmt3b are colocalized to heterochromatin and also demonstrate reduction of T·G mismatch repair efficiency upon loss of DNA methyltransferase expression as well as a requirement for an RNA component for correct T·G mismatch repair.
Keywords: DNA methyltransferase, glycosylase, base excision repair, heterochromatin
Introduction
Genomic stability is required for proliferation of mitotic cells as well as for long-term viability of mitotic and post-mitotic cell populations. Repair of endogenous DNA damage and maintenance of chromatin structure are particularly important to cellular homeostasis. DNA methylation promotes genomic stability through repression of mitotic recombination, assurance of proper chromatid segregation and the maintenance of higher-order heterochromatin structure.1-3 In higher eukaryotes, DNA methylation occurs primarily at carbon 5 of the cytosine pyrimidine ring within CpG dinucleotides. Genomic methylation patterns are laid down in early embryogenesis prior to gastrulation by the de novo methyltransferases, Dnmt3a and Dnmt3b,4-6 and then propagated by the maintenance methyltransferase Dnmt1. A significant in vivo function of Dnmt3b is the establishment and maintenance of DNA methylation of centromeric and pericentromeric satellite repeats,4,7 chromosomal regions that are normally dense in methylated CpG sites. Genomic instability in the rare autosomal recessive disease, ICF syndrome has been attributed to reduction or loss of DNA methylation (hypomethylation) in pericentromeric heterochromatin.8,9 Except for a few documented cases, patients with ICF (Immunodeficiency, Centromeric instability and Facial anomalies) syndrome possess biallelic mutations in DNMT3B that ablate or drastically reduce its catalytic function leading to loss of CpG methylation of the satellite repeats in pericentromeric heterochromatin.10,11 Hypomethylation of these repeats results in chromatin decondensation and enhanced chromosomal rearrangement leading to chromosomal arm deletions and/or formation of multiradiate chromosomes.12 This points to the fact that proper DNA methylation patterns are crucial not only for regulation of gene expression but also for maintaining chromatin structure.
Repair of DNA damage is essential to ensure genomic integrity. A significant source of endogenous DNA damage is the deamination of cytosine (C) or 5-methylcytosine (5mC) residues which results in U·G or T·G mismatches, respectively.13 If these lesions are left unrepaired, mutagenic C·G to T·A transitions occur following DNA replication. It has been estimated that C·G to T·A transitions account for approximately 30% of all germline and somatic point mutations.14 Short-patch and long-patch base excision repair (BER) have evolved to repair this damage. Long-patch, or replicative, BER utilizes proteins involved in DNA replication (e.g. PCNA, FEN-1, RPA, LigI) and is physically and temporally associated with replication foci. On the other hand, short-patch BER, involves a different set of proteins (e.g. XRCC1, LigIIIα) and occurs throughout the cell cycle.15 It has been estimated that the short-patch repair pathway accounts for ~90% of BER activity.16 Thymine-DNA glycosylase (Tdg) initiates short-patch BER at T·G and U·G mismatches ultimately leading to reformation of the original C·G base pair.17-19 Tdg has also been shown to excise cytosine adducts that occur as a result of the metabolism of environmental pollutants such as vinyl chloride and by the natural metabolites of lipid peroxidation.20,21 However, when sequence-context effects are taken into account, the preferred substrate for Tdg is a T·G mismatch in a methylated or unmethylated CpG dinucleotide.19,22-24
Methyl-CpG binding domain protein 4 (Mbd4) is another DNA glycosylase implicated in the suppression of CpG mutation. MBD4 can bind methylated CpG sites through its N-terminal methyl-binding domain and has been demonstrated to excise thymine residues from T·G mismatches.25 However, its role in BER remains to be tested. A degree of functional redundancy between Tdg and Mbd4 seems likely as Mbd4 knockout mice exhibit a modest 2-3 fold increase in C→T transition mutations at CpG sites compared to wild-type mice.26
MBD4 has been shown to play a role in transcriptional repression and has been implicated in DNA repair at methylated promoters.27,28 While Tdg has also been implicated in transcriptional regulation,29-32 it remains the prime candidate for repair of T·G mismatches.
Here, we report that the DNA methyltransferase, Dnmt3b, interacts with both Tdg and Mbd4. We also identify the regions of Dnmt3b necessary for this interaction in vitro and in vivo and demonstrate that Dnmt3b and Tdg are targeted to and interact with heterochromatic regions of the genome that are heavily methylated. We provide evidence that DNA methyltransferases possess the ability to potentiate T·G mismatch repair and that proper T·G mismatch repair requires a previously unidentified RNA component.
Results
Dnmt3b and Tdg interact and localize to heterochromatin in vivo
In order to further elucidate the in vivo function of Dnmt3b, a yeast two-hybrid screen of a murine embryonic day 11 cDNA library (Clontech) was utilized to detect potential protein interactions with full-length murine Dnmt3b. Tdg was isolated as a positive interactor with Dnmt3b using the high stringency of reporter gene expression combined with several rounds of auxotrophic growth selection. The full-length Tdg cDNA isolated in the yeast two-hybrid screen was subcloned into a different Gal4(AD) vector and the interaction was confirmed by a forward yeast two-hybrid assay with Gal4(DBD)-Dnmt3b (data not shown).
To determine whether Dnmt3b and Tdg interact in vivo in mammalian cells, endogenous Dnmt3b in nuclear extracts from an asynchronously proliferating population of F9 embryonal carcinoma cells was immunoprecipitated with α-Dnmt3b antibody. The immunoprecipitated proteins were separated by SDS-PAGE and immunoblotted. This demonstrated that endogenous Tdg coimmunoprecipitates with Dnmt3b (Figure 1a).
Figure 1.
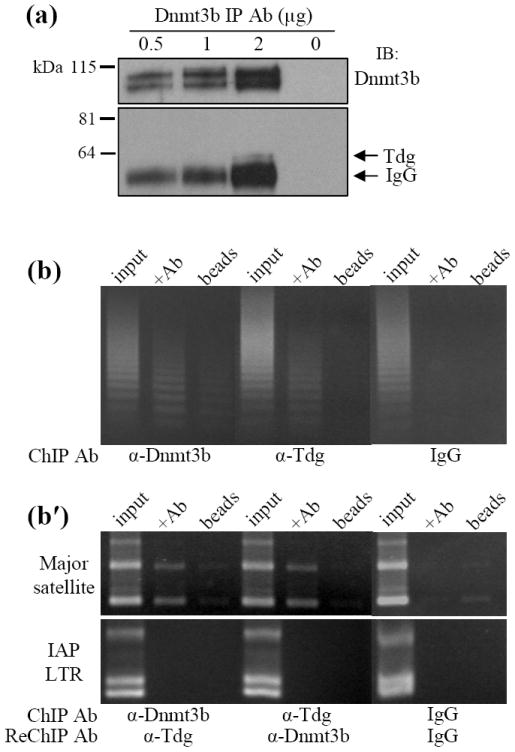
Interaction and genomic localization of Dnmt3b and Tdg in EC cells. (a) Endogenous Dnmt3b was immunoprecipitated from F9 EC cell nuclear extracts cells with increasing concentrations (0.5, 1.0, 2.0 μg) of α-Dnmt3b antibody. An immunoblot of the pull down product with α-Tdg antibody confirmed that Tdg coprecipitated with Dnmt3b. (b) Genomic localization of Dnmt3b and Tdg by chromatin immunoprecipitation (ChIP). Dnmt3b and Tdg were immunoprecipitated from crosslinked chromatin in P19 EC cells. PCR was performed using primers designed to amplify the centromeric minor satellite repeats. (b′) Dnmt3b and Tdg were immunoprecipitated from crosslinked chromatin. The complexes were purified by several wash steps and subjected to a second ChIP (i.e. ReChIP) with the antibody indicated above. PCR was performed using primers designed to amplify the pericentromeric major satellite repeats or the intracisternal A particle long terminal repeats (IAP LTR). Control PCR reactions utilized total input DNA, DNA isolated using species-matched normal IgG antibody and DNA isolated in the absence of immunoprecipitating antibody.
Dnmt3b is targeted to tandemly repeated minor and major satellite DNA that constitutes centromeric and pericentromeric heterochromatin, respectively.7,33 Chromatin immunoprecipitation (ChIP) was used to determine whether Dnmt3b and Tdg are localized to heterochromatin and ReChIP was used to determine whether Dnmt3b and Tdg associate with heterochromatic regions as part of a complex. P19 EC cells were treated with the crosslinking agent dithiobispropionimidate (DTBP) before crosslinking with formaldehyde. Dnmt3b and Tdg were separately immunoprecipitated from crosslinked chromatin and purifed DNA from the ChIP experiment was subjected to PCR using primers designed to amplify the centromeric minor satellite repeats. It was found that both Dnmt3b and Tdg are associated with minor satellite DNA (Figure 1b) and the major satellite repeats (data not shown). For the ReChIP experiments, Dnmt3b or Tdg was immunoprecipitated from formaldehyde-crosslinked chromatin (without prior DTBP treatment) in a primary ChIP experiment. The resulting complexes were purified and used in a second (ReChIP) experiment using α-Tdg or α-Dnmt3b antibody, respectively. Results obtained from the ReChIP experiments indicate that Dnmt3b and Tdg can be found complexed at pericentromeric heterochromatin (Figure 1b′, major satellites) but not at interspersed intracisternal A particle long terminal repeats (Figure 1b′, IAP LTR). This suggests that the Dnmt3b:Tdg complex may be targeted to specific regions of the genome.
Taken together, the data from the ChIP/ReChIP studies support the conclusion that Dnmt3b and Tdg are targeted to and interact with heterochromatic satellite repeats.
Identification of two separate Dnmt3b domains that interact with Tdg/Mbd4
Dnmt3b is composed of multiple domains (Figure 2a). The C-terminus of Dnmt3b contains the catalytic domain which harbors conserved C-5 DNA methyltransferase (MTase) motifs (depicted as solid black bars) that are necessary for catalyzing methyl transfer from the cofactor S-adenosyl-L-methionine (AdoMet) to the target cytosine. The N-terminus contains a PWWP domain and an ATRX-like domain; so called because it shares homology with the α-thalassemia/mental retardation syndrome, X-linked (ATRX) protein. The PWWP domain has been shown to bind DNA and target Dnmt3b to the major satellite repeats of pericentromeric heterochromatin.7,34 In contrast, the ATRX-like domain of Dnmt3b binds histone deacetylase 1 (HDAC1) allowing Dnmt3b to have a repressive effect on transcription that is independent of methyl transfer.35
Figure 2.
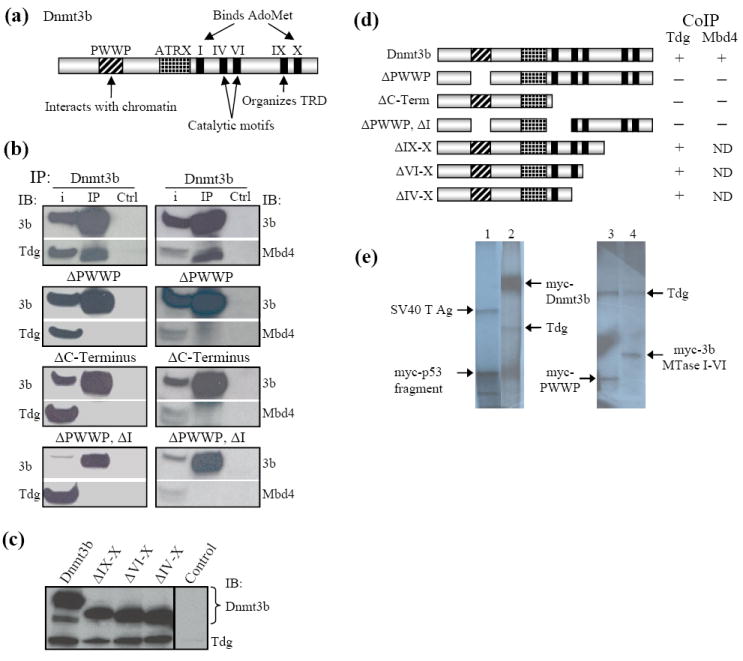
Tdg and Mbd4 make multiple interactions with Dnmt3b. (a) Schematic diagram of Dnmt3b showing the location of characterized domains and conserved MTase motifs of Dnmt3b. Refer to text for details. (b) Interaction between Tdg and Mbd4 and deletion mutants of Dnmt3b tested by coimmunoprecipitation and immunoblotting. When the PWWP domain and the region of the C-terminus that interacts with Tdg (identified in Figure 3c) were deleted from Dnmt3b, neither Tdg, nor Mbd4 precipitated with Dnmt3b. (c) Interaction between Tdg and Mbd4 and C-terminal truncation mutants of Dnmt3b tested by coimmunoprecipitation and immunoblotting. Note that while deletion of the complete C-terminal region of Dnmt3b led to loss of interaction, deletion of motifs IV-X did not reduce binding between Dnmt3b and either Tdg or Mbd4. This identifies a region containing the conserved MTase motif I as critical for interaction with both glycosylases. IB, immunoblot. (d) Summary of the results displayed in (b) and (c). (e) Tdg interacts with the PWWP and catalytic domains of Dnmt3b in vitro. 35S-Met-radiolabeled, myc-tagged Dnmt3b as well as myc-tagged Dnmt3b PWWP and MTase IVI domains and HA-tagged Tdg were generated by in vitro transcription/translation in rabbit reticulocyte lysates. Immunoprecipitations were performed with α-cMyc antibody. Lane 1, (positive control) cMyc-p53 immunoprecipitates the SV40 large T antigen. Lane 2, coimmunoprecipitation of HA-Tdg and cMyc-Dnmt3b. Lane 3, the isolated Dnmt3b PWWP domain (aa 229-301) effectively immunoprecipitates Tdg. Lane 4, a portion of the Dnmt3b C-terminus encompassing conserved DNA MTase motifs I-VI (aa 580-754) also interacts with Tdg.
In order to determine which domains of Dnmt3b interact with Tdg, the full-length cDNA encoding each enzyme was cloned into separate mammalian expression vectors to obtain pcDNA3.1-Dnmt3b and pCMV-Tdg. These plasmids express Xpress-Dnmt3b (Xpress epitope, Invitrogen) and cMyc-Tdg, respectively. Interaction between exogenously expressed Dnmt3b and Tdg was confirmed in HEK293T cells. Dnmt3b was immunoprecipitated with α-Xpress antibody. The pull-down product was washed in lysis buffer and subjected to SDS-PAGE. An immunoblot with an antibody directed against the cMyc tag of Tdg demonstrated that Tdg coimmunoprecipitates with Dnmt3b (Figure 2b left, Dnmt3b IP). As a control, lysate expressing Xpress-Dnmt3b and cMyc-Tdg was incubated with protein G Sepharose but without an immunoprecipitating antibody (Figure 2b, Ctrl). Pull-down with normal IgG was also negative. The other thymine-DNA glycosylase, Mbd4, was PCR amplified from P19 EC cell cDNA and cloned into the mammalian expression vector, pCMV-Tag 3 (Stratagene), to encode cMyc-Mbd4. Immunoprecipitation experiments similar to those described above identified an interaction between Dnmt3b and Mbd4 (Figure 2b right, Dnmt3b IP).
Although the Dnmt3b PWWP domain has been shown to bind DNA non-specifically,34 PWWP domains in general are thought to be involved in mediating protein-protein interactions.36 Therefore, Xpress-Dnmt3b lacking the PWWP domain (ΔPWWP) was tested for its ability to interact with cMyc-Tdg by coimmunoprecipitation. As shown in Figure 2b left, removal of the PWWP domain abolished the interaction of Dnmt3B with Tdg. A series of Xpress-tagged Dnmt3b C-terminal truncation mutants were designed to determine whether Tdg makes contacts with any region(s) of the Dnmt3b catalytic domain. Dnmt3b mutants possessing the conserved MTase motifs VI, IV or I were all capable of binding Tdg (Figure 2c). However, removal of the entire catalytic domain of Dnmt3b disrupted its interaction with Tdg (Figure 2b, ΔC-Terminus). These results strongly suggested that Dnmt3b possesses two regions capable of interacting with Tdg. To test this conclusion, both putative interaction regions were deleted from Dnmt3b (ΔPWWP, ΔI). Dnmt3b ΔPWWP, ΔI also failed to IP Tdg (Figure 2b). The same regions of Dnmt3b were also found to mediate the interaction with Mbd4 (Figure 2b, right panels). Together, the results summarized in Figure 2d support the conclusions that 1) both the PWWP domain and a region of the catalytic domain of Dnmt3b that contains conserved MTase motif I are responsible for Dnmt3b’s interaction with both Tdg and Mbd4 and 2) removal of either domain ablates interaction with Tdg or Mbd4. It should be noted that Li et al. identified an interaction between Tdg and the PWWP domain and the catalytic domain of Dnmt3a.37 Our results indicate that MTase motif I of the catalytic domain of Dnmt3b is sufficient for interaction with Tdg and Mbd4.
Interaction between Dnmt3b and Tdg was demonstrated by coimmunoprecipitation of full-length, 35S-Met-labeled, myc-Dnmt3b and HA-Tdg translated in vitro using rabbit reticulocyte lysates (Figure 2e, lane 2). A direct interaction between Tdg and the isolated PWWP and catalytic domains of Dnmt3b was demonstrated by coimmunoprecipitation [Figure 2e; lane 3, 3b PWWP (aa 229-301); lane 4, 3b catalytic domain (aa 580-754)]. This region of the catalytic domain spans the conserved MTase motifs I-VI and hence, includes the C-terminal Tdg interaction site identified above in Figure 2b, c.
Dnmt3b ICF mutations S277P, A609T and V612A do not affect association with Mbd4 or Tdg
The recessive genetic disorder, ICF syndrome, occurs as a result of DNMT3B mutation leading to decondensation of pericentromeric heterochromatin. Typically, the ICF mutations that have been characterized abolish the activity of DNMT3B by affecting its ability to bind cofactor, by disordering conserved motifs necessary for methyl-transfer38 or by affecting its ability to interact with chromatin.33 The same regions of Dnmt3b that interact with Tdg/Mbd4 have also be found to be important for targeting Dnmt3b to pericentromeric heterochromatin.7,33 While Tdg exhibits a rather diffuse nuclear distribution,31 Mbd4 is enriched at chromocenters when overexpressed in ES cells.39 To determine whether DNMT3B ICF mutations that lie within the Tdg/Mbd4 interacting regions (see Figure 3a) are capable of interfering with the interaction between Dnmt3b and Mbd4 or Tdg, mutations corresponding to the human ICF S270P, A603T and V606A mutations were individually introduced into the murine Dnmt3b cDNA (S277P, A609T and V612A, respectively). The resulting Dnmt3b ICF mutants were expressed in HEK293T cells and tested for their ability to interact with cMyc-Mbd4 by coimmunoprecipitation. Each of the Dnmt3b ICF mutant proteins was able to effectively pull down Mbd4 (Supplementary Figure 1) and Tdg (data not shown). These results indicate that the ICF mutations examined here, that lie within the regions of glycosylase association, do not affect the interaction between Dnmt3b and Mbd4 or Tdg.
Figure 3.
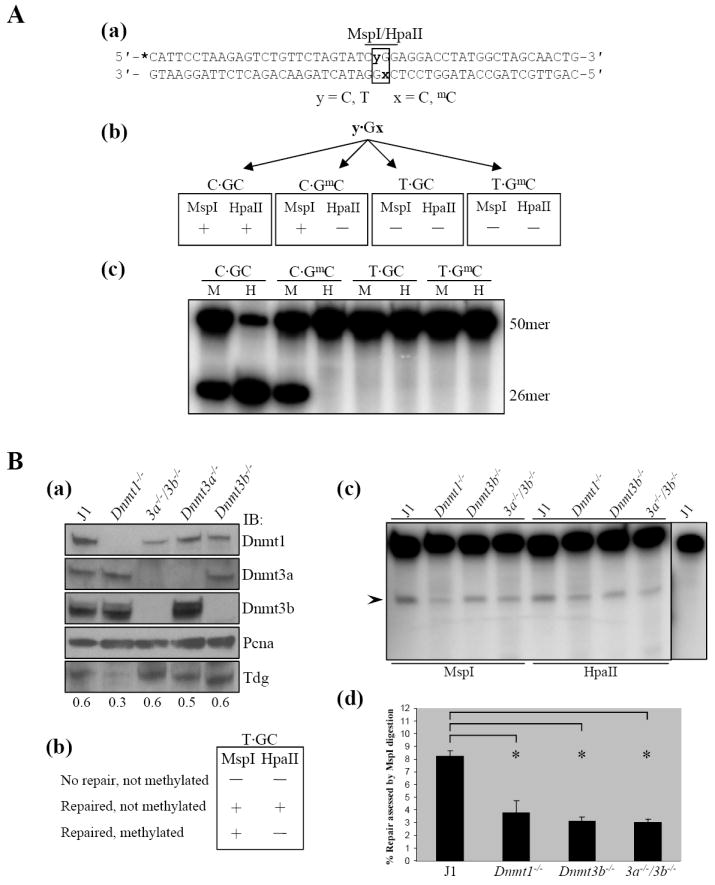
DNA methyltransferases stimulate base excision repair of T·G mismatches. A: Design and testing of oligodeoxyribonucleotides (ODNs) used in a mismatch repair assay. (a) Sequence of the double-stranded ODN. The sense strand contains either a normal CCGG site or a mutated CTGG site. The antisense strand contains either C or 5mC within an MspI/HpaII recognition site. Positions of variable nucleotides y and x are shown in bold. The MspI/HpaII recognition sequence is indicated with a bar. The asterisk indicates the position of the 32P label. (b) Predicted sensitivity to MspI or HpaII digestion of the control C·GC and C·GmC ODNs compared to the mismatched T·GC and T·GmC ODNs. (c) MspI digestion of the control ODNs results in the expected 26 nt long radiolabeled fragment from both controls, while a 26 nt long fragment is only observed after HpaII digestion of the unmethylated control. In contrast, T·G mismatch ODNs are refractory to digestion by either enzyme. M, MspI. H, HpaII. B: De novo DNA methyltrasnferases stimulate repair of T·G mismatches. (a) Evaluation of Dnmt and Tdg protein levels in J1 and Dnmt null ES cells. Nuclear extracts prepared from each cell line were immunoblotted using antibodies specific for Dnmt1, Dnmt3a, Dnmt3b and Tdg to verify the expression of Tdg and the absence of specific Dnmt expression in each cell line relative to wild-type J1. Immunoblot for Pcna served as a loading control. (b) Predicted T·GC ODN sensitivity to digestion by MspI or HpaII depending on extent of repair/DNA methylation. (c) Typical results from one of three independent experiments in which the T·GC ODN was incubated with wild-type J1 or Dnmt null ES cell nuclear extracts followed by digestion with either MspI or HpaII. The expected 26 nt repair product produced as a result of digestion is indicated by the arrowhead. In the absence of either Dnmt3b and/or Dnmt3a there is a reduction in repair efficiency when compared to wild-type nuclear extracts (refer to text for discussion of Dnmt1 null extracts). As a control (last lane), T·GC ODN was used in a repair assay but was not digested. Lack of a 26 nt fragment indicates the ODN is being repaired and not merely nicked 5′ to the mismatched thymine. (d) Quantitation of differences in the extent of mismatch repair by extracts from normal ES cells (J1) and ES cells nullizygous for the indicated Dnmts. Error bars represent the mean (± SD) of three independent experiments. [*] P < 0.005
De Novo DNA methyltransferase(s) potentiate T·G mismatch repair
It is well established that Tdg initiates base excision repair of T·G mismatches to restore the original C·G base pair in nuclear extracts.17,40 The majority of studies involving the functional aspects of Tdg have examined the glycosylase activity of the purified protein. In order to examine the role of DNA methyltransferase(s) in the entire process of T·G mismatch repair, we developed a T·G mismatch repair assay using nuclear extracts isolated from a series of mouse ES cell lines nullizygous for each of the DNA methyltransferases.4,41
A synthetic oligodeoxyribonucleotide (ODN) containing a single T·G mismatch embedded within the recognition sequence of the isoschizomers MspI and HpaII (5′-CTGG-3′) was designed to avoid any theromodynamically favorable secondary structure using the mfold algorithm.42 The sense strand was annealed to an antisense strand possessing either an unmethylated or a methylated cytosine within the MspI/HpaII target CpG sequence (5′-CCGG-3′). The sequence of the annealed, double-stranded ODN is shown in Figure 3A(a). Annealing the various sense and antisense ODN strands yielded the control unmethylated (C·GC) and hemimethylated (C·GmC) ODNs as well as an ODN containing a T·G mismatch within an unmethylated CpG site (T·GC) and a T·G mismatch within a hemimethylated CpG site (T·GmC). The expected MspI/HpaII digestion patterns of the T·GC and control ODNs are depicted in Figure 3A(b). The recognition sequence of MspI/HpaII cannot be cleaved by HpaII when the CpG site is methylated while MspI digestion is not inhibited by CpG methylation. Neither MspI nor HpaII is expected to cleave ODNs containing T·G mismatches because their recognition sequence is disrupted by the mispaired thymine. The data in Figure 3A(c) demonstrate that the annealed ODNs result in the expected digestion pattern to yield a 26 nucleotide (nt) long radiolabeled cleavage product when the purified ODN is analyzed on a denaturing polyacrylamide gel. As expected, neither endonuclease digests the T·G mismatch ODNs.
The panel of ES cell lines used for the repair assay included the wild-type parental J1 line,43 in addition to lines 36 (Dnmt1-/-), 6aa (Dnmt3a-/-), 8bb (Dnmt3b-/-) and 7aabb (Dnm3a-/-/Dnmt3b-/-).4,41 Immunoblot of nuclear extracts with antibodies specific for Dnmt1, Dnmt3a and Dnm3b confirmed the phenotype of wild-type J1 cells and the knockout phenotype of all four Dnmt-/- ES cell lines [Figure 3B(a)]. In addition, the expression of Tdg was confirmed in each of the ES cell lines [Figure 3B(a)]. The expected sensitivity of the T·GC ODN to digestion with MspI/HpaII depending on its repair/methylation status following incubation with nuclear extract are depicted in Figure 3B(b). Repair of the mispaired T to C restores the recognition sequence of both MspI and HpaII. Therefore, both will cleave the repaired, unmethylated ODN but only MspI will digest a T·GC ODN that has been repaired and methylated (mC·GC), thus allowing methylation of the repaired cytosine to be examined.
End-labeled T·GC ODN was incubated with nuclear extracts prepared from J1, Dnmt1-/-, Dnmt3b-/- or 3a-/-/3b-/- ES cells. The ODN was purified from the incubation mixture, divided into equal parts and digested with either MspI or HpaII. Digestion of T·GC ODN with either MspI or HpaII subsequent to incubation with J1 nuclear extracts produced the expected 26 nt digestion fragment as a result of repair of the T·G mismatch to C·G. As a control, ODN that had been incubated with J1 nuclear extract was incubated in the absence of restriction endonuclease to ensure that bona fide repair was occurring, not merely nicking of the DNA phosphodiester backbone (by apurinic/apyrimidinic endonuclease 1) 5′ to the excised thymine. As can be seen in Figure 3B(c) (last lane), T·GC ODN incubated with J1 nuclear extract but not digested fails to release a 26 nt fragment after denaturation. Furthermore, it can be concluded that the ODN was not methylated by Dnmts in the nuclear extract because HpaII digested the ODN to an extent equal to, or slightly greater than, MspI. Importantly, it was determined that repair of the T·GC ODN was reproducibly reduced ~50-60% in the absence of Dnmt3b or both Dnmt3a and 3b as assessed by MspI or HpaII digestion [Figures 3B(c), (d)]. Comparable results were obtained with Dnmt3a null nuclear extracts (see Supplementary Figure 2). A reduction in repair is also noted in Dnmt1 null ES cells [Figure 3B(c), (d)], although there is no evidence for a direct interaction between Dnmt1 and Tdg,37 (M.J. Boland and J.K. Christman, unpublished results). This result can be explained by greatly reduced levels of Tdg protein in the Dnmt1-/- ES cells compared to the other ES cell lines [Figure 3B(a), IB; Tdg]. In general, the absence of either or both of the de novo Dnmts results in ~50-60% reduction in T·G mismatch repair efficiency [Figure 3B(d)]. This indicates that Dnmt3a and Dnmt3b possess the ability to potentiate repair of T·G mismatches. These results are consistent with the findings of a recent study which demonstrated that purified Dnmt3a acted to stimulate the glycosylase activity of Tdg toward a T·G mismatch ~2-3 fold.37
Identification of a putative RNA component required for T·G mismatch repair
In vitro DNA methylation reactions using the C·GC and C·GmC ODNs as substrate together with J1, Dnmt1-/- and 3a-/-/3b-/- nuclear extracts as a source of Dnmts failed to transfer methyl groups to DNA when assayed under the same conditions used for DNA repair (data not shown). Predictably, this was the result of endogenous RNA in the extracts.44 Therefore, extracts were prepared from J1 and Dnmt null cells and treated with RNase A prior to use in a repair assay. First, an in vitro methylation assay confirmed that the RNase-treated nuclear extracts were capable of catalyzing DNA methylation by analyzing transfer of [3H]CH3 from cofactor S- adenosyl-L-methionine (AdoMet) to the ODN substrate. As expected the J1 and 3a-/-/3b-/- nuclear extracts were capable of methylating the hemimethylated C·GmC ODN whereas the Dnmt1-/- extracts failed to methylate this substrate (Supplementary Figure 3). This result is consistent with the higher specific activity of Dnmt1 as compared to Dnmt3a/3b, thereby reflecting the predominant maintenance methylation activity of Dnmt1.5 No de novo methylation activity (i.e. methylation of the C·GC ODN) was detected in any of the nuclear extracts.
In order to examine methylation of the repaired cytosine, the schema in Figure 4a was followed. A repair assay was performed using RNA-depleted nuclear extracts supplemented with fresh AdoMet and the T·GmC ODN. Repair would result in two of three possible outcomes: 1) repaired, fully methylated ODN, 2) repaired, hemimethylated ODN and 3) if no repair occurred; hemimethylated, mismatched ODN. Following the repair assay, the ODN was purified, heat denatured and annealed to an excess of unmethylated antisense GC strand. Depending on the methylation status of the repaired cytosine residue, this would result in either an unmethylated ODN that could be digested by MspI or HpaII or a hemimethylated ODN that could only be digested with MspI. However, an unexpected result was obtained, i.e., apparent degradation of the ODN (Figure 4b). The pattern of degradation was reproducible and consistent between nuclear extracts irrespective of methyltransferase expression. Since this result could have been due to contaminating DNase in the RNase A used to treat the nuclear extracts, repair assays were performed as above using nuclear extracts treated with DNase-free RNase A (Roche) or RNase A that had been heated at 100°C for 15 min to inactivate any DNase. In each case, a consistent but non-identical pattern of ODN “degradation” was obtained for both the T·GC and T·GmC ODNs when either the mismatch strand (shown) or the antisense strand (not shown) was end-labeled. Digestion of C·GC ODN after incubation with RNase-treated J1 nuclear extract yields the correct 26 nt fragment (Figure 4b, arrow).
Figure 4.
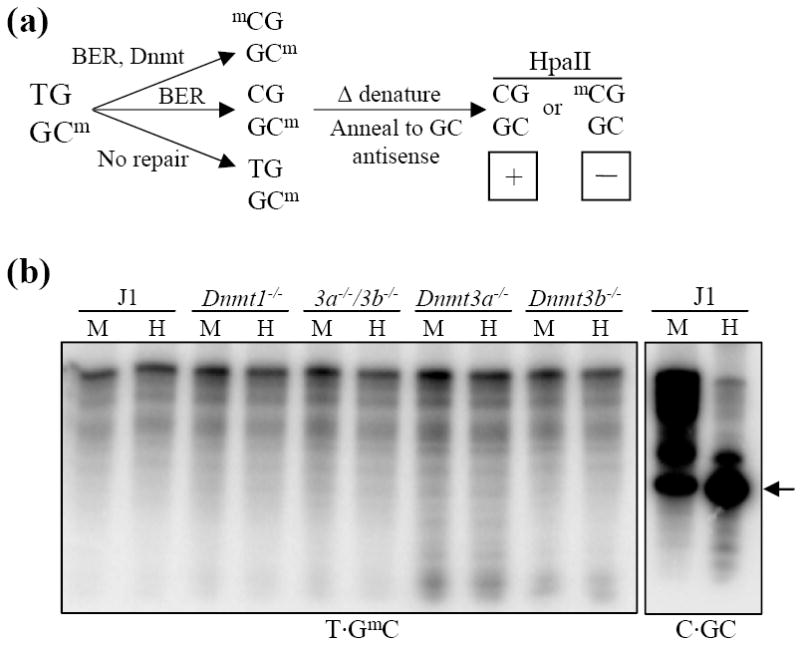
Treatment of nuclear extracts with RNase A leads to random cleavage of the T·GmC ODN. (a) Diagram depicting an experimental protocol devised to examine methylation of repaired T·GmC ODN in normal and Dnmt null ES cell lines. Following repair of the T·GmC ODN using RNase A-treated ES cell nuclear extracts, the ODN was purified and heat denatured. It was then annealed to a 5-fold excess of cold antisense CG strand. This will generate an unmethylated or hemimethylated ODN depending on the methylation status of the repaired cytosine. MspI will digest either ODN but HpaII digestion will be inhibited if the repaired cytosine is methylated. (b) Results of the experiment diagrammed in (a). The T·GmC ODN appears to be degraded by each RNase-treated nuclear extract in a consistent pattern irrespective of methyltransferase expression. As a control, the C·GC ODN was incubated with RNase-treated J1 nuclear extract. The expected 26 nt digestion product is indicated by an arrow.
These results suggest that an RNA component is necessary for the repair of T·G mismatches. If this hypothesis is true, cellular RNA should restore the repair capability of the nuclear extracts. Therefore, aliquots of J1 nuclear extracts were either 1) treated with DNase-free RNase A, 2) treated with DNase-free RNase A that was blocked with an excess of RNase A inhibitor or 3) treated with DNase-free RNase A for 30 min at which point an excess of RNase A inhibitor was added, the extract was incubated an additional 30 min and then supplemented with 10 μg of total J1 RNA. Each of these extracts was used in a T·GC mismatch repair assay. Representative results are displayed in Figure 5. As a control, naked T·GC ODN incubated with DNase-free RNase A was not detectably degraded (Figure 5a, lanes 1, 2). As before, J1 nuclear extract that had not been treated with RNase was competent for repair (lanes 3, 4). In contrast, incubation with nuclear extract treated with RNase resulted in degradation of the ODN (lanes 5, 6). As a control, nuclear extract treated with RNase A that had been blocked with RNase inhibitor was still able to perform repair (lanes 7, 8). More importantly, the J1 nuclear extract that had been first depleted of RNA and then supplemented with J1 cellular RNA regained the ability to repair the T·G mismatch ODN (lanes 9, 10). We note that RNA-depleted J1 nuclear extracts supplemented with tRNA or genomic DNA did not correctly repair the T·G mismatch ODN (Figure 5b). Addition of tRNA to RNA-depleted J1 extracts partially restored repair (Figure 5b, lane 5) but also resulted in non-specific nicking of the ODN phosphodiester backbone (lanes 5, 6). Furthermore, RNA-depleted J1 extracts supplemented with genomic DNA (lanes 7, 8) or poly(dA)·poly(dT) (data not shown) produced a single 28 nt non-specific product that resulted from backbone nicking (lane 8). These data indicate that an endogenously produced RNA species is required for base excision repair of T·G mismatches.
Figure 5.

Evidence for the existence of an endogenous RNA component essential to T·G mismatch repair. (a) T·GC ODN was treated with RNase A (Lanes 1, 2), J1 nuclear extract (Lanes 3, 4) or aliquots of a J1 nuclear extract were treated with DNase-free RNase A for 30 min (Lanes 5, 6), with RNase A blocked with an excess of RNase A inhibitor (Lanes 7, 8), or treated with RNase A for 30 min followed by addition of RNase A inhibitor for 30 min and then supplemented with 10 μg of J1 total RNA (Lanes 9, 10). The untreated or treated aliquots were used in T·GC repair assays. The expected 26 nt repair product is indicated by the arrowhead. Both the untreated extract and the extract treated with blocked RNase A were competent for repair. The extract treated with RNase A alone was not able to repair the ODN. However, when extracts treated with RNase A followed by RNase A inhibitor were supplemented with J1 total RNA (lanes 9, 10), they regained the ability to correctly repair the T·GC ODN. Naked ODN incubated with RNase A for 16 hs at 37°C was not detectably degraded (lanes 1, 2). M, MspI. H, HpaII. (b) J1 nuclear extracts treated sequentially with RNase A and RNase inhibitor were supplemented with J1 total RNA, E. coli tRNA or RNase-treated salmon sperm genomic DNA as indicated. Reactions were split in half and either digested with HpaII or not digested. Addition of tRNA or genomic DNA resulted in non-specific cleavage of the ODN phosphodiester backbone (lanes 6, 8 respectively). M, sizing markers. H, HpaII. Ø, no digestion.
DISCUSSION
The only known mammalian proteins that are able to excise mispaired thymine opposite guanidine are Tdg and Mbd4. Evolutionary forces have shaped the mammalian genome into regions of euchromatin and heterochromatin. The majority of 5mC in the mammalian genome is located in heterochromatic regions. As a consequence, the majority of T·G mismatches produced as a result of 5mC deamination are also likely to be localized in heterochromatin. DNA methyltransferases are responsible for the establishment and maintenance of methylation patterns at heterochromatin and hence, are targeted to heterochromatin. Our results suggest that interaction between Tdg and either Dnmt3b or Dnmt3a37 could serve to recruit/target Tdg to heterochromatin. Indeed, Tdg interacts with the regions of Dnmt3b that have been shown to target Dnmt3b to pericentromeric heterochromatin.7,33 Redundancy of the interaction with Dnmt3a and Dnmt3b might ensure that Tdg is targeted to spatially defined regions of heterochromatin (e.g. centromeric vs. pericentromeric heterochromatin).
Li et al. identified a stimulatory effect of Dnmt3a and Dnmt3b on the catalytic activity of Tdg37 whereas, here it was observed that T·G mismatch repair efficiency was reduced in the absence of DNA methyltransferases. Thus, the findings of these independent studies complement each other and indicate that DNA methyltransferases possess the ability to potentiate T·G mismatch repair. Furthermore, the involvement of Dnmt3a and 3b in BER is predicted to be replication-independent as Tdg is absent during S phase45 and there is no evidence of association of either Dnmt3a and 3b with replication foci.35,46
Although it cannot be ruled out that overexpression of proteins in HEK293T cells may mask ICF mutations that reduce the affinity of Tdg/Mbd4 interaction, none of the Dnmt3b ICF mutations that lie within the regions of Tdg/Mbd4 interaction seem to affect their association with Dnmt3b. Two separate explanations are plausible: 1) Disruption of the interaction has no phenotypic consequences on normal development or 2) Mutations that disrupt the interaction are lethal and therefore not observed. Mbd4 has already been shown to associate with heterochromatin,39 presumably through its methyl-CpG binding domain. If Dnmt3b is responsible for targeting Tdg to heterochromatin and for stimulating repair, loss of the interaction would result in greatly decreased repair efficiency at these genomic regions.
Recently, RNA molecules have been implicated in processes as diverse as repair of double-strand breaks in yeast47 and mediation of genomic rearrangement in ciliates.48 Jost and colleagues identified and characterized an RNA species that copurified with the chicken ortholog of Tdg. It was determined that the RNA stimulated the activity of Tdg against 5mC as a substrate but reduced the activity of Tdg when a T·G mismatch was used as substrate.49-51 We provide evidence suggesting that an RNA component is necessary for the proper repair of T·G mismatches in mammalian cells. While our experiments do not address the effect of RNA on the activity of Tdg, our data indicate that an RNA species acts downstream of Tdg in the BER pathway; possibly regulating or providing specificity to the endonucleolytic activity of APE1/HAP1 (Figure 5b). Additional experiments are in progress to further characterize the role of RNA in T·G mismatch repair.
A paucity of mutations among DNA glycosylases has led to the hypothesis that selective pressure to conserve glycosylases is necessary for long-term stability of the genome.15 Just as loss of methylation at CpG dinucleotides can lead to decondensation of pericentromeric DNA, unrepaired deamination of 5mC in satellite repeats over time could result in decondensation of pericentromeric heterochromatin leading to improper kinetochore formation, missegregation of sister chromatids and/or enhanced mitotic recombination; a cellular phenotype observed in many forms of cancer.52-55 The process of genetic repair initiated by Tdg concomitant with “epigenetic repair” by DNA methyltransferases provides a potential mechanism for preventing such mutagenesis.
Materials and Methods
Yeast two-hybrid screen/assay
The Matchmaker Two-Hybrid System 3 (BD Biosciences Clontech, Palo Alto, CA.) was utilized to screen a mouse embryonic day 11 cDNA library (Clontech) with full-length Dnmt3b as bait following the manufacturer’s instructions. Wild-type Dnmt3b was cloned into pGBKT7 to create a cMyc-tagged fusion protein with the Gal4-DBD. The library screen was performed under high stringency auxotrophic conditions on minimal medium lacking adenine and the amino acids histidine, leucine and tryptophan (SD-Ade/-His/-Leu/-Trp/X-α-Gal) where approximately 2.5 × 106 clones were screened. Plasmid DNA from positive clones was isolated, propagated in E. coli, and sequenced to identify clones.
Forward yeast two-hybrid assay
The full-length Tdg cDNA isolated in the two-hybrid screen was cloned into pGADT7 (Clontech) to create a Gal4(AD)-Tdg fusion protein. The plasmids encoding Gal4(DBD)-Dnmt3b and Gal4(AD)-Tdg were transformed into yeast strains AH109 (MATa, trp1-901, leu2-3, 112, ura3-52, his3-200, gal4Δ, gal80Δ, LYS2∷GAL1UAS-GAL1TATA-HIS3, GAL2UAS-GAL2TATA-ADE2, URA3∷MEL1UAS-MEL1TATA-lacZ, MEL1) and Y187 (MATα, ura3-52, his3-200, ade2-101, trp1-901, leu2-3, 112, gal4Δ, met−, gal80Δ, URA3∷GAL1UAS-GAL1TATA-lacZ, MEL1), respectively. Transformed yeast colonies were mated and the resultant diploids plated on QDO. Robust, blue colony growth on QDO medium after multiple rounds of selection indicated a positive interaction.
Cell culture
HEK293T cells were maintained in Dulbecco’s Modified Eagle’s Medium (4.5 g/L glucose) supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere containing 5% CO2. P19 embryonal carcinoma (EC) cells were maintained in α-Minimal Essential Medium supplemented with ribonucleosides and deoxyribonucleosides (7.5% fetal calf serum, 2.5% fetal bovine serum). J1 and Dnmt-/- embryonic stem (ES) cells were cultured on gelatinized dishes in DMEM (4.5 g/L glucose) containing 15% fetal bovine serum, 0.1 mM non-essential amino acids, 0.1 mM β-mercaptoethanol, 50U penicillin/ml, 50 μg streptomycin/ml and 1000U/ml leukemia inhibitory factor (ESGRO; Chemicon, Temecula, CA.).
Site-directed mutagenesis and cloning
The full-length Tdg cDNA isolated in the yeast two-hybrid screen was subcloned into pCMV-Tag 3 (Stratagene, La Jolla, CA.) and full-length Dnmt3b was cloned into pcDNA3.1 (Invitrogen, Carlsbad, CA.) as were all of the following Dnmt3b mutants. The PWWP domain was deleted from the Dnmt3b cDNA by overlap extension PCR.56 The primers designed to delete the Dnmt3b PWWP domain were A: 5’-ATCGGATCCCAATGAAGGGAGACAGCAGAC-3’, B: 5’-CCTATAAGA AACCAGTTTATCATCCTGAT-3’, C: 5’-ATCAGGATGATAAACTGGTTTCTTATA GG-3’, D: 5’-GCCTCTAGACTATTCACAGGCAAAGTAGTCC-3’. Dnmt3b C-terminal truncation mutants were created by PCR using the following antisense primers in combination with primer A (above): ΔIV-X 5’-CTAGCCCCACTTCTTCAATAT-3’, ΔVI-X 5’-CTAGTCGCCCTCCTTGGGGCGG-3’, ΔIX-X 5’-CTACCAGGAATCTTGA GATGTC-3’, ΔC-Terminus 5’-CTACAGGACTATAATGGGCCTCC-3’. Dnmt3b ΔPWWP was used as template to generate ΔPWWP, ΔI by overlap extension PCR using primers A and D (above) with B: 5’-TAAACCTTTGCGGGCCAGGACTCTAATGGG CCTCC-3’ and C: 5’-GCCCATTAGAGTCCTGGCCCGCAAAGGTTTATATGAGG-3’.
The QuikChange XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA.) was used to generate the Dnmt3b ICF mutants. Primer sequences to create the ICF Dnmt3b S277P are; sense 5’-GGTGATGGCAAGTTTCCCGAGATCTCTGCTG-3’/ antisense 5’-CAGCAGAGATCTAGGGAAACTTGCCATGACC-3’, A609T; sense 5’-GTGGAAAAGTACATTACCTCCGAAGTCTGTGC-3’, antisense 5’-GCACAGACTTCGGAGGTAATGTACTTTTCCAC-3’, V612A; sense 5’-CATTGCCTCCGAAGCCTGTGCAGAGTCCATCG-3’, antisense 5’-CGATGGACTCTGCACAGGCTTCGGAGGCAATG-3’. Underlined nucleotides correspond to the introduced mutations. Successful mutagenesis was verified by DNA sequencing.
Full-length Mbd4 was PCR amplified from P19 cDNA and cloned into pCMV-Tag 3 (Stratagene) using the following primers: sense 5’-GCAGGATCCATGG AGAGCCCAAACCTTGG-3’, antisense 5’-GCACTCGAGTCAAGATAGACTTAATTTTTCATG-3’.
Coimmunoprecipitation
In vitro: 35S-Methionine labeled proteins were prepared using the TnT T7 Coupled Reticulocyte Lysate System (Promega, Madison, WI.) according to the manufacturer’s protocol. Coimmunoprecipitation was performed using the Matchmacker Co-IP Kit (Clontech) as per the manufacturer’s instruction. In vivo: F9/P19 EC whole cell lysates were prepared from ~80% confluent 75 cm2 flasks as described below. Alternatively, HEK293T cells were transiently cotransfected with 5.0 μg of each plasmid per 75 cm2 flask by calcium phosphate precipitation. Whole cell lysates were prepared in 115 mM NaCl, 2.26 mM KCl, 8.4 mM Na2HPO4, 1.68 mM KH2PO4, 1.0% NP-40, 0.5% deoxycholate, 1.0% SDS containing protease inhibitors (12.5 μg/ml leupeptin, 12.5 μg/ml E-64, 12.5 μg/ml chymostatin, 12.5 μg/ml pepstatin A, 12.5 μg/ml antipain, 0.35 mM phenylmethylsulphonylfluoride). Lysates were precleared with protein G Sepharose (Sigma, St. Louis, MO.) and split in half before immunoprecipitation with 2.0 μg of the appropriate antibody (half of the lysate served as an IgG or no antibody control). Immunoprecipitates were collected by incubation with protein G Sepharose, washed twice, subjected to SDS-PAGE and analyzed by standard immunoblotting with appropriate antibodies: α-Xpress (Invitrogen), α-cMyc (Clontech), α-FLAG M2 (Stratagene), α-Dnmt3b (Imgenex, San Diego, CA.), α-TDG (Santa Cruz Biotechnology, Santa Cruz, CA.). Proteins were visualized by ECL(+) Western Blotting Detection System (Amersham Biosciences, Piscataway, N.J.).
Chromatin immunoprecipitation/ReChIP
P19 EC cells grown in 10 cm2 culture dishes were washed twice with cold 1X PBS and then incubated in freshly prepared 5 mM dithiobispropionimidate (DTBP; Pierce, Rockford, IL.) in 1X PBS pH 8.0 at 4°C for 30 min. Cells were washed twice with cold 1X PBS and incubated in Quenching buffer (100 mM Tris pH 8.0, 150 mM NaCl) for 10 min at 4°C. The DTBP and Quenching buffer treatments were omitted in the ReChIP experiments. Cells were washed in cold 1X PBS and incubated in 1% formaldeyde for 10 min at 24°C. Unreacted formaldehyde was quenched with 200 mM glycine and the cells were washed twice with cold 1X PBS. Cells were scraped into 1.5 ml centrifuge tubes in 1.0 ml 1X PBS containing Protease Inhibitor cocktail (PIN; 12.5 μg/ml leupeptin, 12.5 μg/ml E-64, 12.5 μg/ml chymostatin, 12.5 μg/ml pepstatin A, 12.5 μg/ml antipain, 0.35 mM phenylmethylsulphonylfluoride). Cells were pelleted by centrifugation at 1,000 × g for 5 min at 4°C. Cell pellets were suspended in Swelling buffer (25 M Hepes pH 7.8, 1.5 mM MgCl2, 10 mM KCl, 0.1% NP-40, 1 mM DTT, PIN) and incubated on ice for 10 min. During this incubation period cells were disrupted in a 1.5 ml tube with 10 strokes of a micropestle. This mixture was subjected to centrifugation at 2,000 × g for 5 min at 4°C to pellet nuclei. Nuclei were suspended in 100 μl / 2 × 106 cells of Lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris pH 8.1) containing PIN. Chromatin was sonicated on ice to fragment DNA to an average length of 500-600 bp using a Branson Digital Sonifier equipped with a microtip; 4 pulses (10 s/pulse) with a 20 s cooling period between each pulse. Length of sonicated chromatin was assessed by agarose gel electrophoresis. Samples were then centrifuged at 16,000 × g for 15 min at 4°C. The supernatant was placed in a clean tube and the samples were subjected to a second round of centrifugation at 16,000 × g for 15 min at 4°C. A 200 μl aliquot of the supernatant (corresponding to chromatin from 4 × 106 cells) per antibody treatment was diluted 10 fold in Dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris pH 8.1, 167 mM NaCl) and pre-cleared with 10 μl protein G Sepharose (Sigma) that had been pre-blocked with 25 mM Hepes, 150 mM NaCl, 1% BSA containing 2.0 μg sheared salmon sperm DNA for 1 h with rotation at 4°C. The mixture was centrifuged at 800 × g for 1 min at 4°C. A 20 μl aliquot was removed from the supernatant and saved as an Input control. The remainder was split in half; one half was treated with antibody and the other half served as a negative (beads only) control. Protein-specific antibody (α-Dnmt3b, Imgenex; α-TDG, Santa Cruz, 4.0 μg each) was added and samples were incubated at 4°C with rotation for 20 h. Immune-complexes were captured by adding 10 μl pre-blocked protein G Sepharose with gentle mixing for 1 h at 4°C followed by centrifugation at 800 × g for 1 min. Sepharose beads were washed sequentially with: a) one wash of Low Salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris pH 8.1, 150 mM NaCl); b) one wash of High Salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris pH 8.1, 500 mM NaCl); c) one wash of LiCl Wash Buffer (0.25% LiCl, 1% IGEPAL-CA630, 1% DOC, 1 mM EDTA, 10 mM Tris pH 8.1); two washes of TE (10 mM Tris, 1 mM EDTA, pH 8.0). (For ReChIP, beads were incubated with 25 μl of 10 mM DTT for 30 min at 37°C. Samples were centrifuged at 16,000 × g for 1 min. The supernatant was transferred to a new tube, diluted 40 fold with Dilution buffer and treated with the second antibody. Samples were then processed as follows). Each sample was eluted with 200 μl 1% SDS, 0.1 M NaHCO3 at 25°C for 30 min. Sepharose beads were pelleted by centrifugation at 800 × g for 1 min and the supernatant was placed in a new tube. Reversal of crosslinks was achieved by addition of 5 M NaCl (320 mM final concentration) and heating at 65°C for 16 h. Samples were treated with RNase A for 30 min at 37°C and then DNA was purified using the QIAquick PCR Purification kit (Qiagen, Valencia, CA.).
The minor/major satellites and IAP LTR were amplified by PCR using ChIP/ReChIP product as template and the following primers: Minor - sense, 5′-AAATCCCGTTTCAA CGAATGTG-3′; antisense, 5′-GTAGAACAGTGTATATCAATGAG-3′. Major - sense, 5′-GGACCTGGAATATGGCGAGAAA-3′; antisense, 5′-TTCAGTGTGCATTTCTCAT TTT-3′; IAP LTR - sense 5′-TTGATAGTTGTGTTTTAAGTGGTAAATAAA-3′, antisense, 5′-CCACAAACCAAAATCTCTAC-3′.
Immunoblot for DNA methyltransferases and Tdg
Nuclear extracts from all mouse ES cell lines were prepared as below (see mismatch repair assay). Equal amounts of each extract (12.0 μg) were loaded onto an 8% SDS-Tris glycine polyacrylamide gel and electrophoresed at 120 V for 90 min. Protein was transferred to PVDF membrane and analyzed by standard immunoblotting with protein specific antibodies: α-Dnmt1 (Santa Cruz, K-18), α-Dnmt3a (Imgenex), α-Dnmt3b (Imgenex), α-Tdg(37), α-PCNA (Santa Cruz, sc-56) with appropriate secondary antibodies conjugated to horse radish peroxidase (HRP). Proteins were visualized by ECL(+) Western Blotting Detection System (Amersham).
Mismatch repair assay
The oligodeoxyribonucleotide (ODN) substrate had the following sequence: sense 5’-CATTCCTAAGAGTCTGTTCTAGTATCyGGAGGACCTATGGC TAGCAACTG-3’, y = T or C; antisense 5’-CAGTTGCTAGCCATAGGTCCTCxGGAT ACTAGAACAGACTCTTAGGAATG-3’, x = C or 5mC. Sense strands were 5’-end labeled with [γ-32P]ATP using T4 Polynucleotide Kinase (New England Biolabs, Beverly, MA.) and annealed to a 2:1 molar excess of unlabeled antisense strand. Oligonucleotide sizing markers (Amersham) were 5′-end-labeled according to the manufacturers’ protocol. To prepare nuclear extracts; ~1.5 × 107 ES cells were harvested and washed with cold 1X phosphate buffered saline (PBS). Cells were suspended in isolation buffer (15 mM Tris-Cl pH 7.4, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, 1 mM DTT, 250 mM sucrose) containing protease inhibitors as above. An equal volume of isolation buffer containing 0.6% NP-40 was added and the mixture was incubated for 10 min on ice. Nuclei were pelleted at 2000 × g and washed in isolation buffer before suspension in extraction buffer (25 mM Tris-Cl pH 8.0, 250 mM NaCl, 1 mM EDTA, 10% glycerol, 0.2% NP-40). Extracts were subjected to centrifugation at 15,000 × g for 30 min at 4°C. Protein concentration was determined using a BCA Protein Assay Kit (Pierce, Rockford, IL.). Where appropriate, DNase-free RNase (10 μg/ml) was added to extract aliquots and incubated for 30 min at 37°C followed by addition of a 20% excess of RNase Block (Stratagene) to inhibit RNase A. As indicated, RNA-depleted nuclear extracts were supplemented with either 10 μg J1 total RNA, 10 μg Escherichia coli tRNA (Sigma), 10 μg RNase-treated salmon sperm genomic DNA (Sigma) or 5 μg poly(dA)·poly(dT) (Sigma). Repair reactions were performed using 5.0 μg ES cell nuclear extract in repair buffer (10X buffer: 250 mM HEPES-KOH, pH 7.9, 5 mM EDTA, 0.1 mM ZnCl2, 5 mM DTT) supplemented with 40 mM KCl, 20 mM creatine phosphate, 1 mM MgCl2, 50 μm ATP, 100 μM each dNTP17 and 10 μM S-adenosyl-L-methionine (AdoMet). Reactions were incubated for 16 hs at 37°C after which an additional 5.0 μg of nuclear extract and 10 μM AdoMet were added and incubated for 90 min. ODNs were purified using the QIAquick Nucleotide Removal Kit (Qiagen, Valencia, CA). To examine repair and methylation of the T·GC mismatch ODN, eluted ODN was divided in half and digested with 20 U MspI or 2 U HpaII (New England Biolabs). To examine repair and methylation of the T·GmC mismatch ODN following the repair/methylation reaction, purified ODNs were denatured at 95°C for 5 min and annealed to a 5 fold excess of unlabeled CG antisense strand before digestion as above. The radiolabel in an aliquot of each digested sample was determined by scintillation counting before addition of formamide loading buffer. An equal amount of ODN (determined by cpm) was loaded in each lane and electrophoresed on an 8 M urea/15% polyacrylamide gel for 90 min at 200V in 1X TBE. Gels were fixed in 15% methanol, 10% acetic acid, 5% glycerol before drying and exposure to a phosphorimaging screen. Images were acquired using a Typhoon 9410 Phosphorimager (Amersham). Signal intensity was quantified by densitometry using ImageQuant 5.2 software. Repaired ODN (i.e. digestion product) is expressed as a percentage of the total signal of ODN. % repair = (signal intensity of 26 nt fragment)/(signal intensity of uncleaved input DNA + signal intensity of 26 nt fragment) × 100. The control ODNs C·GC and C·GmC were processed identically to the T·GC ODN under repair assay conditions.
Supplementary Material
Acknowledgments
We would like to thank Drs. E. Li and T. Chen (Novartis Institutes for Biomedical Research, Cambridge, MA.) for providing the J1 and Dnmt null ES cell lines as well as Dr. G-L. Xu (Chinese Academy of Sciences, Shanghai, China) for providing the α-Tdg antibody. This work was supported in part by NIH-R21 CA91315 to J.K.C.
Footnotes
Note. While this manuscript was in preparation Li et al. published a study describing a similar interaction between Dnmt3a and Tdg.37
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- 2.Rizwana R, Hahn PJ. CpG methylation reduces genomic instability. J Cell Sci. 1999;112(Pt 24):4513–4519. doi: 10.1242/jcs.112.24.4513. [DOI] [PubMed] [Google Scholar]
- 3.Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- 4.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 5.Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 6.Hsieh CL. In vivo activity of murine de novo methyltransferases, Dnmt3a and Dnmt3b. Mol Cell Biol. 1999;19:8211–8218. doi: 10.1128/mcb.19.12.8211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen T, Tsujimoto N, Li E. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol Cell Biol. 2004;24:9048–9058. doi: 10.1128/MCB.24.20.9048-9058.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fryns JP, Azou M, Jaeken J, Eggermont E, Pedersen JC, Van den Berghe H. Centromeric instability of chromosomes 1, 9, and 16 associated with combined immunodeficiency. Hum Genet. 1981;57:108–110. doi: 10.1007/BF00271181. [DOI] [PubMed] [Google Scholar]
- 9.Tiepolo L, Maraschio P, Gimelli G, Cuoco C, Gargani GF, Romano C. Multibranched chromosomes 1, 9, and 16 in a patient with combined IgA and IgE deficiency. Hum Genet. 1979;51:127–137. doi: 10.1007/BF00287166. [DOI] [PubMed] [Google Scholar]
- 10.Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CM, Gartler SM. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci U S A. 1999;96:14412–14417. doi: 10.1073/pnas.96.25.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu GL, Bestor TH, Bourc’his D, Hsieh CL, Tommerup N, Bugge M, Hulten M, Qu X, Russo JJ, Viegas-Pequignot E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- 12.Tuck-Muller CM, Narayan A, Tsien F, Smeets DF, Sawyer J, Fiala ES, Sohn OS, Ehrlich M. DNA hypomethylation and unusual chromosome instability in cell lines from ICF syndrome patients. Cytogenet Cell Genet. 2000;89:121–128. doi: 10.1159/000015590. [DOI] [PubMed] [Google Scholar]
- 13.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 14.Shen J, Rideout W, 3rd, Jones P. The rate of hydrolytic deamination of 5-methylcytosine in double- stranded DNA. Nucl Acids Res. 1994;22:972–976. doi: 10.1093/nar/22.6.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nilsen H, Krokan HE. Base excision repair in a network of defence and tolerance. Carcinogenesis. 2001;22:987–998. doi: 10.1093/carcin/22.7.987. [DOI] [PubMed] [Google Scholar]
- 16.Bennett RA, Wilson DM, 3rd, Wong D, Demple B. Interaction of human apurinic endonuclease and DNA polymerase beta in the base excision repair pathway. Proc Natl Acad Sci U S A. 1997;94:7166–7169. doi: 10.1073/pnas.94.14.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiebauer K, Jiricny J. In vitro correction of G.T mispairs to G.C pairs in nuclear extracts from human cells. Nature. 1989;339:234–236. doi: 10.1038/339234a0. [DOI] [PubMed] [Google Scholar]
- 18.Neddermann P, Jiricny J. The purification of a mismatch-specific thymine-DNA glycosylase from HeLa cells. J Biol Chem. 1993;268:21218–21224. [PubMed] [Google Scholar]
- 19.Neddermann P, Gallinari P, Lettieri T, Schmid D, Truong O, Hsuan JJ, Wiebauer K, Jiricny J. Cloning and expression of human G/T mismatch-specific thymine-DNA glycosylase. J Biol Chem. 1996;271:12767–12774. doi: 10.1074/jbc.271.22.12767. [DOI] [PubMed] [Google Scholar]
- 20.Chung FL, Chen HJ, Nath RG. Lipid peroxidation as a potential endogenous source for the formation of exocyclic DNA adducts. Carcinogenesis. 1996;17:2105–2111. doi: 10.1093/carcin/17.10.2105. [DOI] [PubMed] [Google Scholar]
- 21.Saparbaev M, Laval J. 3,N4-ethenocytosine, a highly mutagenic adduct, is a primary substrate for Escherichia coli double-stranded uracil-DNA glycosylase and human mismatch-specific thymine-DNA glycosylase. Proc Natl Acad Sci U S A. 1998;95:8508–8513. doi: 10.1073/pnas.95.15.8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griffin S, Karran P. Incision at DNA G.T mispairs by extracts of mammalian cells occurs preferentially at cytosine methylation sites and is not targeted by a separate G.T binding reaction. Biochemistry. 1993;32:13032–13039. doi: 10.1021/bi00211a012. [DOI] [PubMed] [Google Scholar]
- 23.Sibghat U, Gallinari P, Xu YZ, Goodman MF, Bloom LB, Jiricny J, Day RS., 3rd Base analog and neighboring base effects on substrate specificity of recombinant human G:T mismatch-specific thymine DNA-glycosylase. Biochemistry. 1996;35:12926–12932. doi: 10.1021/bi961022u. [DOI] [PubMed] [Google Scholar]
- 24.Waters TR, Swann PF. Kinetics of the action of thymine DNA glycosylase. J Biol Chem. 1998;273:20007–20014. doi: 10.1074/jbc.273.32.20007. [DOI] [PubMed] [Google Scholar]
- 25.Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. 1999;401:301–304. doi: 10.1038/45843. [DOI] [PubMed] [Google Scholar]
- 26.Millar CB, Guy J, Sansom OJ, Selfridge J, MacDougall E, Hendrich B, Keightley PD, Bishop SM, Clarke AR, Bird A. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science. 2002;297:403–405. doi: 10.1126/science.1073354. [DOI] [PubMed] [Google Scholar]
- 27.Kondo E, Gu Z, Horii A, Fukushige S. The thymine DNA glycosylase MBD4 represses transcription and is associated with methylated p16(INK4a) and hMLH1 genes. Mol Cell Biol. 2005;25:4388–4396. doi: 10.1128/MCB.25.11.4388-4396.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watanabe S, Ichimura T, Fujita N, Tsuruzoe S, Ohki I, Shirakawa M, Kawasuji M, Nakao M. Methylated DNA-binding domain 1 and methylpurine-DNA glycosylase link transcriptional repression and DNA repair in chromatin. Proc Natl Acad Sci U S A. 2003;100:12859–12864. doi: 10.1073/pnas.2131819100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen D, Lucey MJ, Phoenix F, Lopez-Garcia J, Hart SM, Losson R, Buluwela L, Coombes RC, Chambon P, Schar P, et al. T:G mismatch-specific thymine-DNA glycosylase potentiates transcription of estrogen-regulated genes through direct interaction with estrogen receptor alpha. J Biol Chem. 2003;278:38586–38592. doi: 10.1074/jbc.M304286200. [DOI] [PubMed] [Google Scholar]
- 30.Lucey MJ, Chen D, Lopez-Garcia J, Hart SM, Phoenix F, Al-Jehani R, Alao JP, White R, Kindle KB, Losson R, et al. T:G mismatch-specific thymine-DNA glycosylase (TDG) as a coregulator of transcription interacts with SRC1 family members through a novel tyrosine repeat motif. Nucleic Acids Res. 2005;33:6393–6404. doi: 10.1093/nar/gki940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol Cell. 2002;9:265–277. doi: 10.1016/s1097-2765(02)00453-7. [DOI] [PubMed] [Google Scholar]
- 32.Missero C, Pirro MT, Simeone S, Pischetola M, Di Lauro R. The DNA glycosylase T:G mismatch-specific thymine DNA glycosylase represses thyroid transcription factor-1-activated transcription. J Biol Chem. 2001;276:33569–33575. doi: 10.1074/jbc.M104963200. [DOI] [PubMed] [Google Scholar]
- 33.Ge YZ, Pu MT, Gowher H, Wu HP, Ding JP, Jeltsch A, Xu GL. Chromatin Targeting of de Novo DNA Methyltransferases by the PWWP Domain. J Biol Chem. 2004;279:25447–25454. doi: 10.1074/jbc.M312296200. [DOI] [PubMed] [Google Scholar]
- 34.Qiu C, Sawada K, Zhang X, Cheng X. The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat Struct Biol. 2002;9:217–224. doi: 10.1038/nsb759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bachman KE, Rountree MR, Baylin SB. Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J Biol Chem. 2001;276:32282–32287. doi: 10.1074/jbc.M104661200. [DOI] [PubMed] [Google Scholar]
- 36.Stec I, Nagl SB, van Ommen GJ, den Dunnen JT. The PWWP domain: a potential protein-protein interaction domain in nuclear proteins influencing differentiation? FEBS Lett. 2000;473:1–5. doi: 10.1016/s0014-5793(00)01449-6. [DOI] [PubMed] [Google Scholar]
- 37.Li YQ, Zhou PZ, Zheng XD, Walsh CP, Xu GL. Association of Dnmt3a and thymine DNA glycosylase links DNA methylation with base-excision repair. Nucleic Acids Res. 2007;35:390–400. doi: 10.1093/nar/gkl1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lappalainen I, Vihinen M. Structural basis of ICF-causing mutations in the methyltransferase domain of DNMT3B. Protein Eng. 2002;15:1005–1014. doi: 10.1093/protein/15.12.1005. [DOI] [PubMed] [Google Scholar]
- 39.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiebauer K, Jiricny J. Mismatch-specific thymine DNA glycosylase and DNA polymerase beta mediate the correction of G.T mispairs in nuclear extracts from human cells. Proc Natl Acad Sci U S A. 1990;87:5842–5845. doi: 10.1073/pnas.87.15.5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol. 2003;23:5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucl Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 44.Bolden A, Ward C, Siedlecki JA, Weissbach A. DNA methylation. Inhibition of de novo and maintenance methylation in vitro by RNA and synthetic polynucleotides. J Biol Chem. 1984;259:12437–12443. [PubMed] [Google Scholar]
- 45.Hardeland U, Kunz C, Focke F, Szadkowski M, Schar P. Cell cycle regulation as a mechanism for functional separation of the apparently redundant uracil DNA glycosylases TDG and UNG2. Nucleic Acids Res. 2007 doi: 10.1093/nar/gkm337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Margot JB, Cardoso MC, Leonhardt H. Mammalian DNA methyltransferases show different subnuclear distributions. J Cell Biochem. 2001;83:373–379. doi: 10.1002/jcb.1236. [DOI] [PubMed] [Google Scholar]
- 47.Storici F, Bebenek K, Kunkel TA, Gordenin DA, Resnick MA. RNA-templated DNA repair. Nature. 2007;447:338. doi: 10.1038/nature05720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nowacki M, Vijayan V, Zhou Y, Schotanus K, Doak TG, Landweber LF. RNA-mediated epigenetic programming of a genome-rearrangement pathway. Nature. 2008;451:153. doi: 10.1038/nature06452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu B, Zheng Y, Hess D, Angliker H, Schwarz S, Siegmann M, Thiry S, Jost J-P. 5-Methylcytosine-DNA glycosylase activity is present in a cloned G/T mismatch DNA glycosylase associated with the chicken embryo DNA demethylation complex. PNAS. 2000;97:5135–5139. doi: 10.1073/pnas.100107597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jost JP, Fremont M, Siegmann M, Hofsteenge J. The RNA moiety of chick embryo 5-methylcytosine- DNA glycosylase targets DNA demethylation. Nucl Acids Res. 1997;25:4545–4550. doi: 10.1093/nar/25.22.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fremont M, Siegmann M, Gaulis S, Matthies R, Hess D, Jost JP. Demethylation of DNA by purified chick embryo 5-methylcytosine-DNA glycosylase requires both protein and RNA. Nucl Acids Res. 1997;25:2375–2380. doi: 10.1093/nar/25.12.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qu G, Dubeau L, Narayan A, Yu MC, Ehrlich M. Satellite DNA hypomethylation vs. overall genomic hypomethylation in ovarian epithelial tumors of different malignant potential. Mutat Res. 1999;423:91–101. doi: 10.1016/s0027-5107(98)00229-2. [DOI] [PubMed] [Google Scholar]
- 53.Qu GZ, Grundy PE, Narayan A, Ehrlich M. Frequent hypomethylation in Wilms tumors of pericentromeric DNA in chromosomes 1 and 16. Cancer Genet Cytogenet. 1999;109:34–39. doi: 10.1016/s0165-4608(98)00143-5. [DOI] [PubMed] [Google Scholar]
- 54.Narayan A, Ji W, Zhang XY, Marrogi A, Graff JR, Baylin SB, Ehrlich M. Hypomethylation of pericentromeric DNA in breast adenocarcinomas. Int J Cancer. 1998;77:833–838. doi: 10.1002/(sici)1097-0215(19980911)77:6<833::aid-ijc6>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 55.Tsuda H, Takarabe T, Kanai Y, Fukutomi T, Hirohashi S. Correlation of DNA hypomethylation at pericentromeric heterochromatin regions of chromosomes 16 and 1 with histological features and chromosomal abnormalities of human breast carcinomas. Am J Pathol. 2002;161:859–866. doi: 10.1016/S0002-9440(10)64246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.