

Abstract
Inappropriate accumulation of the misfolded Z variant of alpha1-antitrypsin in the hepatocyte endoplasmic reticulum is a risk factor for the development of end-stage liver disease. However, the genetic and environmental factors that contribute to its etiology are poorly understood. ER mannosidase I (ERManI) is a quality control factor that plays a critical role in the sorting and targeting of misfolded glycoproteins for proteasome-mediated degradation. In this study, we tested whether genetic variations in the human ERManI gene influence the age at onset of end-stage liver disease in ZZ patients. We sequenced all 13 exons in a group of unrelated Caucasian ZZ transplant recipients with different age at onset of the end-stage liver disease. Homozygosity for the minor A allele at 2484G/A (rs4567) in the 3'-untranslated region was prevalent in the infant ZZ patients. Functional studies indicated that rs4567(A), but not rs4567(G), suppresses ERManI translation under ER stress conditions. These findings suggest that the identified SNP can accelerate the onset of the end-stage liver disease associated with alpha1-antitrypsin deficiency and underscore the contribution of biosynthetic quality control as a modifier of genetic disease.
Introduction
Alpha1-antitrypsin (AAT) is the predominant circulating protease inhibitor (Pi) in human plasma and protects lung elastin fibers from excess proteolytic destruction (Sifers et al. 1992; Lomas 2002). The Z variant of alpha1-antitrypsin (Pi Z), which bears a single amino acid substitution (G342K) (Carrell et al. 1982), is defective in secretion from primary and transfected cells in culture due to its inappropriately folded conformation (Sifers et al. 1989; Graham et al. 1990). The decreased concentration of circulating protease inhibitor leads to the hydrolytic destruction of lung elastin as an etiologic factor for panlobular emphysema (Eriksson 1965). The vast majority of retained Pi Z becomes a substrate for intracellular elimination (Le et al. 1990; Teckman and Perlmutter 1996; Teckman et al. 2001) by a process known as endoplasmic reticulum (ER)-associated degradation (ERAD) (McCracken and Brodsky 1996). However, the polymerizationprone conformation of variant Pi Z leads to the accumulation of loop-sheet polymers in the hepatocyte endoplasmic reticulum (ER) (Lomas et al. 1992). Liver injury, as the primary gain-of-toxic-function disorder, is associated with the intracellular accumulation of Pi Z polymers and can progress to end-stage pathology requiring liver transplantation (Perlmutter 2000; Perlmutter 2006). In recent years, AAT deficiency has emerged as a paradigm to investigate the pathologic variability associated with conformational diseases, all of which are caused by cytotoxicity associated with the inappropriate accumulation of a structurally aberrant protein (Kopito and Ron 2000).
Based on the most comprehensive study by Sveger and colleagues who screened 200,000 newborns in Sweden in the 1970s and the follow-up studies in the 1990s, about 1 in 1700 among mixed North America and European populations are homozygous for the Z allele (referred to as “PIZZ” individuals). However, only about 17% of the ZZ newborns exhibited clinically significant liver disease in infancy and less than 3% of those progressed to life-threatening end-stage disease as infants (Sveger 1976; Sveger and Eriksson 1995). The high variation in the severity of the liver disease in PIZZ individuals suggests that genetic modifiers or environmental factors might contribute to the development of end-stage pathology. Understanding the mechanistic principles that underlie the variability of liver disease progression among ZZ homozygotes is predicted to aid in the identification of biomarkers for improved prognosis, and have far-reaching consequences with regard to the development of effective treatment strategies (Balch et al. 2008).
Accumulation of variant Pi Z in the hepatocyte ER lumen, rather than in the cytosol where proteasomal degradation takes place, implies that an early step in the intracellular disposal process might exist. In support of this notion, a lag in degradation of transduced Pi Z in skin fibroblasts from a subgroup of “susceptible” ZZ homozygotes (Wu et al. 2003) led to the realization that a subtle defect in the protein disposal machinery likely contribute to the etiology of the disorder. For misfolded asparaginelinked secretory glycoproteins like alpha1-antitrypsin, the removal of mannose units from N-linked glycans represents a crucial early event in the disposal system (Cabral et al. 2001; Lederkremer and Glickman 2005). Studies from several groups including ours have demonstrated that ERManI, a putative ER mannosidase, plays a stochastic and rate-limiting role in both distinguishing and targeting misfolded glycoproteins for ER-associated degradation (ERAD) (Liu et al. 1999; Wu et al. 2007; Avezov et al. 2008). In the present study, a candidate gene approach was performed to determine whether variations in the corresponding gene (MAN1B1) might influence the age at onset of the end-stage liver disease.
Patients and Methods
Study subjects
We obtained from the Liver Tissue Cell Distribution System (LTCDS, University of Minnesota and University of Pittsburgh, #N01-DK-7-0004 / HHSN267200700004C) all available excised livers (n=30) from unrelated PIZZ individuals who had undergone orthotopic liver transplantation for end-stage liver disease. No additional confounding factors were identified. All ZZ patients were Caucasian, and included 10 females, and twenty males (Figure 2A). The age at which each patient was listed for liver transplantation was used to indicate the onset of end-stage of liver disease.
Figure 2.

Percentage of identified SNPs in different groups. (A) Percentage of allelic genotypes of each SNP in the indicated groups. P values are calculated with the χ2 statistic with 2 df. (B) Frequency of the rs4567 genotypes in the Caucasian population and in 200 ZZ individuals without liver disease.
We also acquired liver samples from 32 liver transplant donors (wildtype for AAT) from LTCDS. These are unrelated Caucasian individuals that had deceased from diseases other than liver disease. In addition, we obtained genomic DNA samples from 200 adult Caucasian ZZ patients (via the Alpha-1 Research Program at the University of Florida), none of which ever exhibited a history of clinically diagnosed liver disease. The entire study was approved by members of the appropriate institutional review boards.
Genotyping
Genomic DNA was extracted from each of the liver samples using Easy DNA kit (Invitrogen, Carlsbad, CA) following the manufacture's instructions. The 13 exons, plus intron/exon junctions, of the ERManI gene (GeneBank accession number NM_016219) were amplified by the polymerase chain reaction (PCR). Amplification primers were designed to target regions incorporating 5' ends complementary to forward and reverse sequence primers and are available upon request. PCR targets were amplified using the HotStarTaq Master Mix kit (Qiagen #203446) according to the manufacturer's instructions with some modifications. PCR products were generated using 10 ng of DNA for both the experimental and cell line reference samples, 2.8 μl of the 2X Hot Star Taq Master Mix, 3.2 pmol of each primer and sterile H2O for a total reaction volume of 8 μl. Reactions were cycled following the Qiagen protocol with annealing and extension times of 45 sec for each amplification cycle. Excess primers and dNTPs were removed from the PCR reaction by treatment with 5 μl of a 1:10 dilution of Exosap-IT (USB #78202). The PCR products were incubated with Exosap-IT at 37°C for 15 min and then inactivated by heating at 80°C for 15 min. Samples were then diluted with 22 μl of 1 × TE (10-mM Tris, pH 8.0; 0.2-mM EDTA) to a concentration of approximately 20-40 ng/μl in preparation for cycle sequencing. Sanger reactions were generated using Applied Biosystems BigDye Terminator v3.1 at 1/64th dilution, 4 pmol primer, 40 ng of PCR product and standard cycling conditions. Reactions were purified by ethanol precipitation and dried under a vacuum. The reactions were resuspended in 20 μl of 0.1-mM EDTA and sequenced with an Applied Biosystems 3730xl DNA Analyzer using the RapidSeq36 run module. Base calls were determined using the 3XX basecaller software provided by Applied Biosystems.
Statistical analysis
Genotype frequencies for SNPs identified between the early-onset group and late-onset groups were compared to their distribution in the general Caucasian population using the χ2 test. P values < 0.01 were considered statistically significant.
Plasmid construction
The cDNA for ERManI containing the 3'UTR was amplified using I.M.A.G.E. Clone 3533651 purchased from ATCC as a template. The following primers were used: ataagcttgcctgggtggcgaattc and agcggccgcatagatgcctcgag, with HindIII and NotI restriction endonuclease sites incorporated into 5' and 3' ends, respectively. The amplified fragment was inserted into the pMH vector and the directionality was confirmed by DNA sequencing.
Cell culture, transient transfection and Western blotting
HeLa cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Mediatech, Inc.) supplemented with 10% fetal bovine serum (Gemeni Bio-Products) and 1% ampicillin/streptomycin (Invitrogen). The day prior to transfection, cells were plated into 6-well dishes and allowed to reach 80% confluence by the time of transfection. 4μg of the plasmids were transfected into each well with lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. 24 hr post-transfection, cells were lysed and immunoblotted for ERManI following the protocol described previously (Wu et al. 2003).
Metabolic radiolabeling and immunoprecipitation
24 hr post-transfection, cells were starved in methionine- & cysteine-free medium for 1hr, and then subjected to metabolic pulse-radiolabeling with [35S]methionine for 20min. In selected experiments, the pre-starvation step was eliminated and 10% fetal bovine serum (Invitrogen) was included during the pulse. For chase experiments, pulse medium was replaced by fresh medium containing unlabelled methionine, and cells were incubated at 37°C for the indicated time. At each time point, cells were then incubated on ice in lysis buffer containing 50-mM TrisHCl, 150-mM NaCl, 0.5% NP-40, 2-mM PMSF and protease inhibitors (Sigma). Following centrifugation at 4°C for 30min, the supernatant of each sample was collected and mixed with polyclonal anti-ERManI antibody (Wu et al. 2003) and protein G agarose beads (Calbiochem). The mixtures were rotated at 4°C for overnight. After stringent washes with lysis buffer and lysis buffer containing 500-mM NaCl, the immunoprecipitates were eluted with SDS sample buffer and separated by SDS-PAGE. Radiolabeled proteins were detected by fluorography and quantified by either densitometric scanning using the National Institutes of Health Image Program, or by PhosphoImager analysis.
Results
Identification of a SNP associated with early-onset end-stage liver disease in ZZ patients
Genotyping of the unrelated Caucasian ZZ transplant recipients led to the identification of six single nucleotide polymorphisms (SNPs): one in exon 1 (176G/A), one in exon 4 (590C/T), and four in exon 13 (2046C/T; 2484G/A; 2542G/T; 2587C/CC) (Figure 1A). Two of these, 176G/A and 2484G/A have been reported by the dbSNP database as rs968733 and rs4567, respectively. The remaining four polymorphisms were novel. Both 176G/A and 590C/T represent non-synonymous SNPs of S59N and P197L, respectively, while 2046C/T generates a synonymous change (D681D). Nucleotide polymorphisms 2484A/G, 2542G/T, and 2587C/CC were located in the 3'-untranslated region (UTR). Of all the identified SNPs, 2484G/A (rs4567) was the only one that exhibited a clustered distribution with age. As shown in Figure 1B, 6 out of 10 patients who develop end-stage liver disease at less than 2 years of age were homozygous for the A allele at rs4567, but only 3 out of 20 in the remaining patients displayed the same genotype. The distribution of rs4567, but not any of the other five identified SNPs, was significantly different between these two groups based on Chi-square analysis (Figure 2A). The distribution of 2484A/G in the infant ZZ transplant patients, which was 60% (AA), 40% (A/G), and 0% (G/G) (Figure 2A), is also significantly different from the reported distribution in the Caucasian population with a p value < 0.01 (Figure 2B). To further confirm this finding, we extracted genomic DNA from 32 unrelated Caucasian livers carrying wildtype AAT and determined their genotypes at rs4567. The results were: 18% (AA), 69% (A/G), and 13% (G/G), which is significantly different from that of the infant ZZ transplant patients (p<0.0004). The prevalence of homozygosity for the A allele at rs4567 among the PIZZ infant group strongly suggests its contribution to an accelerated onset of the end-stage liver disease.
Figure 1.

Identified SNPs and distribution of rs4567. (A) Illustration of the positions of the six SNPs identified in the ERManI gene. (B) Genotypes of rs4567 among PIZZ individuals that developed end-stage liver disease, requiring transplantation at different ages. The age at which each patient was placed on the transplant list (age at listing) was used as an indicator for age of onset. TX, transplant.
Because a few PIZZ patients over 2 years of age are also homozygous for the rs4567(A) allele (Figure 1B), it is unlikely that this genotype is solely responsible for the development of end-stage liver disease. To provide further evidence, we acquired (from the Alpha-1 Research Program at the University of Florida) genomic DNA samples from 200 adult Caucasian ZZ patients without a history of clinically diagnosed liver disease. Homozygosity for rs4567(A) among these individuals was nearly identical to the general Caucasian population (Figure 2B), excluding the possibility that the SNP plays a causative role in end-stage liver disease. Rather, the data imply that rs4567(A) functions as a modifier of the disorder, accelerating the time at onset.
Functional studies
Previous studies have indicated that the ERManI concentration is primarily regulated at translational and/or post-translational levels (Hosokawa et al. 2001; Wu et al. 2003). Therefore, we asked whether the rs4567 genotype might influence the protein expression level for ERManI. To test this hypothesis, we generated mammalian expression constructs containing either nucleotide A or G at rs4567 in the 3'UTR of the human ERManI cDNA, which were designated ERManI(A) and ERManI(G), respectively. Equal amounts of the cDNA expression constructs were separately transfected into the human cervical carcinoma cell line HeLa, and the synthesis of the recombinant ERManI was monitored by metabolic radiolabeling. Based on our sequence data, HeLa cells are heterozygous (A/G) at rs4567. Therefore overexpression of ERManI(A) or ERManI(G) should have similar effects on A and G allele, respectively. No differences in the translational efficiency of the encoded products were detected under basal conditions. However, when cells were co-transfected with the Z variant of alpha1-antitrypsin, ERManI(A) exhibited a 50% decrease in synthesis as compared to rs4567(G) (Figure 3A). Consistently, the steady-state concentrations of the corresponding proteins were diminished to a similar extent, providing additional support for the validity of the observed phenomenon (Figure 3B). The doublet bands detected during steady-state conditions were not unexpected, as the post-translationally modification of the protein has been previously reported (Wu et al. 2007). Also, these distinctions did not reflect differences at either the transcriptional or post-translational levels because the respective mRNA levels were similar by Northern blotting (Figure 4A), as were the intracellular stabilities of the translated proteins (pulse-chase radiolabeling) (Figure 4B), indicating that rs4567(A) influences ERManI at the translational level. Moreover, the pre-starvation step used in our routine metabolic method does not influence the effect of rs4567 (A) on the translational suppression of ERManI, since an identical result was obtained in the absence of serum starvation. (Figure 4C).
Figure 3.

rs4567(A) suppresses the translation of transfected ERManI in HeLa cells. (A) The translational efficiency of transfected ERManI (during a 20 minute pulse with 35S-methionine) in HeLa cells, with or without co-transfected Pi Z, were evaluated by a combination of immunoprecipitation, SDS-PAGE, autoradiography and quantified by PhosphoImager analysis. The error bars represent standard error of the mean. (B) The concentration of transfected ERManI (from above) under steady-state conditions was evaluated by Western blotting of cell lysates. The relative ERManI concentrations, as compared with control, were quantified in five separate experiments. The error bars represent a standard error of the mean.
Figure 4.

rs4567 does not affect transcription or turnover rate of ERManI and its expression is not affected by serum. (A) HeLa cells were either not transfected [C], or cotransfected with ERManI(A) or ERManI(G) plus empty vector or Pi Z. 24hr after transfection. Cells were lysed and total mRNA was extracted and separated on agarose gels, and then subjected to Northern Blotting with ERManI-or Actin-specific probes. The bands were quantified using NIH Image J, and the ratio between ERManI and Actin was calculated. (B) HeLa cells co-transfected with ERManI(A) or ERManI(G) plus empty vector or Pi Z. 48 hr after transfection, cells were metabolically labeled with 35S-methionine and chased for 0.5, 1, and 2 hr. At each time point, cells were lysed and subjected to immunoprecipitation of ERManI. The immunoprecipitates were resolved by SDS-PAGE and detected by autoradiography. After quantification by PhosphoImager analysis, the percentage of ERManI left at each chase time point was calculated and plotted. Error bars represent standard deviation from three independent experiments. (C) HeLa cells were co-transfected with ERManI(A) plus empty vector or Pi Z , and then metabolically labeled with 35S-methionine in the presence 10% fetal bovine serum for 20 min. Cells were then lysed followed by the immunoprecipitation of ERManI. The immunoprecipitates were resolved by SDS-PAGE and detected by autoradiography. Quantification was performed by PhosphoImager analysis. The percentage of ERManI level in the presence of Z versus in the absence of Z was calculated with error bars representing the standard error of the mean.
We next asked whether rs4567(A)-mediated translational suppression of ERManI occurs only in the presence of Pi Z. To address this question, the ERManI constructs were co-transfected with wildtype alpha1-antitrypsin and the expression levels of ERManI(A) and ERManI(G) were examined by metabolic radiolabeling. A minor, but significant difference was also observed under this condition (Figure 5). This result suggests that the rs4567(A)-mediated translational suppression of ERManI is associated with certain ER stress conditions.
Figure 5.
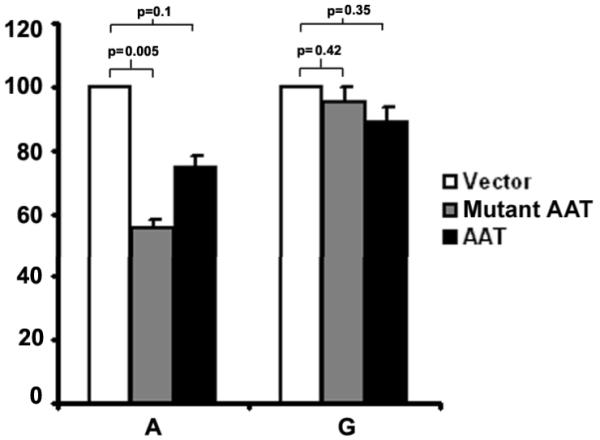
Overexpression of mutant or wild type AAT induces rs4567(A)-mediated translational suppression of ERManI. The translational efficiency of ERManI (A) or ERManI (G) in the presence of co-transfected Z or wildtype AAT were evaluated by metabolic radiolabeling and quantified by PhosphoImager analysis. The p-value was calculated by the Student's t-test.
Discussion
Due to its pathological variation and rare occurrence, the development of end-stage liver disease is difficult to predict in ZZ individuals. Although SNPs were recently implicated in the etiology of the PI Z-associated lung and liver diseases (Chappell et al. 2008; Demeo et al. 2008), findings from the present study represent a functionally-defined genetic modifier of the end-stage pathology. Based on the results of this study, we predict that lower levels of ERManI in individuals homozygous for the A allele at rs4567 generate a conditional hypomorphic allele for ERManI that impairs the liver's capacity to deal with the accumulation of misfolded alpha1-antitrypsin, likely accelerating the rate at which a tolerable threshold is surpassed, resulting in the liver failure. However, it is worth noting that the rs4567(A)-mediated translational suppression of ERManI itself could not be the only modifier, because several older ZZ individuals homozygous for rs4567(A) did not exhibit an accelerated onset of end-stage liver disease. Moreover, not all the affected ZZ infants exhibited this genotype. Importantly, our conclusions need confirmation with carefully collected liver tissue from the analysis of a larger group of affected ZZ patients before the role of ERManI can be substantiated.
The suppression of ERManI occurs when Pi Z or AAT are over-expressed, suggesting that cellular stress is likely required. Previous studies have shown that overexpression of Pi Z does not activate the unfolded response (UPR), but rather augments the NFκB and autophagy pathways (Teckman and Perlmutter 2000; Teckman et al. 2002; Hidvegi et al. 2005). It is possible that certain components of either pathway are actually responsible for the suppression of ERManI translation. The exact mechanism for the above suppression is unknown. However, the in silico analysis (MicroInspector web server (http://www.imbb.forth.gr/microinspector)) of sequences flanking rs4567, has identified alternative microRNA binding sites with constitution of the A or G allele (Figure 6). The possibility that certain microRNAs are responsible for the rs4567(A)-mediated translational suppression of ERManI is currently under investigation.
Figure 6.
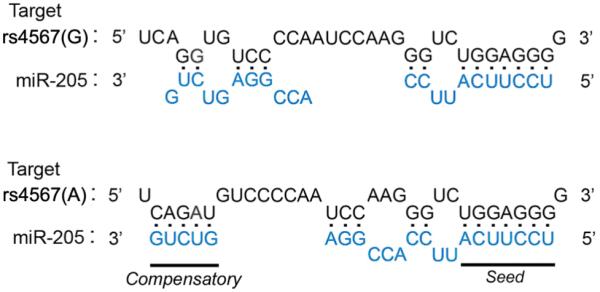
Predicted binding of miR-205 to rs4567 in ERManI. The predicted binding pattern between mRNA sequences of ERManI is shown. The nucleotide at rs4567 (red) and flanking sequences (black) are shown, as is the sequence for mature human miR-205 (blue). The seed region and predicted 3' compensatory sequences that contribute to hybridization are underlined.
The frequency of homozygous A allele at rs4567 is ~ 28% in the general Caucasian population. Therefore, under stress conditions similar to Pi Z accumulation, these individuals may not be able to cope with ER stress as efficiently as those homozygous for the G allele, due to the suppression of ERManI translation. This may eventually lead to other pathological conditions, and it will therefore be interesting to determine whether the genotype is associated with other conformational diseases.
Although present within the 3'UTR of ERManI, we cannot dismiss the possibility that rs4567 might also reside in a regulatory region of another gene involved in glycoprotein quality control. It will be interesting to eventually determine whether the genes upstream or downstream of ERManI are also affected by the nucleotide at rs4567.
The present study demonstrates the utility of functional studies to validate the contribution of a SNP in disease pathogenesis, and introduces a novel paradigm in which a subtle defect in the multilevel regulation of gene expression can modify a classical gain-of-toxic-function disorder. Whether a similar modality might undermine pathologies associated with other conformational disorders is still unknown. However, the present findings provide an exciting prospect for early prognosis and hold considerable promise for tapping a new avenue for therapeutic intervention.
Acknowledgements
This study was supported by NIH grant DK064232 (R.N.S.), Fernandez Liver Initiative research grant FO3-12 from the Alpha-1 Foundation (R.N.S.), and a post-doctoral research grant (S.P.) from the Alpha-1 Foundation. Also, we acknowledge the Liver Tissue Cell Distribution System (LTCDS) (#N01-DK-7-0004 / HHSN267200700004C) and the Alpha1-Foundation-University of Florida DNA and Tissue Bank. Finally, we thank Marion Namenwirth's (LTCDS) contribution toward the collection of data, and Sandra McGill for scientific editing.
References
- Avezov E, Frenkel Z, Ehrlich M, Herscovics A, Lederkremer GZ. Endoplasmic Reticulum (ER) Mannosidase I Is Compartmentalized and Required for N-Glycan Trimming to Man5 6GlcNAc2 in Glycoprotein ER-associated Degradation. Mol Biol Cell. 2008;19:216–225. doi: 10.1091/mbc.E07-05-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Cabral CM, Liu Y, Sifers RN. Dissecting glycoprotein quality control in the secretory pathway. Trends Biochem Sci. 2001;26:619–624. doi: 10.1016/s0968-0004(01)01942-9. [DOI] [PubMed] [Google Scholar]
- Carrell RW, Jeppsson JO, Laurell CB, Brennan SO, Owen MC, Vaughan L, Boswell DR. Structure and variation of human alpha 1-antitrypsin. Nature. 1982;298:329–334. doi: 10.1038/298329a0. [DOI] [PubMed] [Google Scholar]
- Chappell S, Hadzic N, Stockley R, Guetta-Baranes T, Morgan K, Kalsheker N. A polymorphism of the alpha1-antitrypsin gene represents a risk factor for liver disease. Hepatology. 2008;47:127–132. doi: 10.1002/hep.21979. [DOI] [PubMed] [Google Scholar]
- Demeo DL, Campbell EJ, Barker AF, Brantly ML, Eden E, McElvaney NG, Rennard SI, Sandhaus RA, Stocks JM, Stoller JK, Strange C, Turino G, Silverman EK. IL10 polymorphisms are associated with airflow obstruction in severe alpha1-antitrypsin deficiency. Am J Respir Cell Mol Biol. 2008;38:114–120. doi: 10.1165/rcmb.2007-0107OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson S. Studies in alpha 1-antitrypsin deficiency. Acta Med Scand Suppl. 1965;432:1–85. [PubMed] [Google Scholar]
- Graham KS, Le A, Sifers RN. Accumulation of the insoluble PiZ variant of human alpha 1-antitrypsin within the hepatic endoplasmic reticulum does not elevate the steady-state level of grp78/BiP. J Biol Chem. 1990;265:20463–20468. [PubMed] [Google Scholar]
- Hidvegi T, Schmidt BZ, Hale P, Perlmutter DH. Accumulation of mutant alpha1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and BAP31 but not the unfolded protein response. J Biol Chem. 2005;280:39002–39015. doi: 10.1074/jbc.M508652200. [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Wada I, Hasegawa K, Yorihuzi T, Tremblay LO, Herscovics A, Nagata K. A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2001;2:415–422. doi: 10.1093/embo-reports/kve084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito RR, Ron D. Conformational disease. Nat Cell Biol. 2000;2:E207–209. doi: 10.1038/35041139. [DOI] [PubMed] [Google Scholar]
- Le A, Graham KS, Sifers RN. Intracellular degradation of the transport-impaired human PiZ alpha 1-antitrypsin variant. Biochemical mapping of the degradative event among compartments of the secretory pathway. J Biol Chem. 1990;265:14001–14007. [PubMed] [Google Scholar]
- Lederkremer GZ, Glickman MH. A window of opportunity: timing protein degradation by trimming of sugars and ubiquitins. Trends Biochem Sci. 2005;30:297–303. doi: 10.1016/j.tibs.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Liu Y, Choudhury P, Cabral CM, Sifers RN. Oligosaccharide modification in the early secretory pathway directs the selection of a misfolded glycoprotein for degradation by the proteasome. J Biol Chem. 1999;274:5861–5867. doi: 10.1074/jbc.274.9.5861. [DOI] [PubMed] [Google Scholar]
- Lomas DA. Chronic obstructive pulmonary disease. Introduction. Thorax. 2002;57:735. doi: 10.1136/thorax.57.8.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature. 1992;357:605–607. doi: 10.1038/357605a0. [DOI] [PubMed] [Google Scholar]
- McCracken AA, Brodsky JL. Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J Cell Biol. 1996;132:291–298. doi: 10.1083/jcb.132.3.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter DH. Liver injury in alpha 1-antitrypsin deficiency. Clin Liver Dis. 2000;4:387–408. vi. doi: 10.1016/s1089-3261(05)70115-x. [DOI] [PubMed] [Google Scholar]
- Perlmutter DH. Pathogenesis of chronic liver injury and hepatocellular carcinoma in alpha-1-antitrypsin deficiency. Pediatr Res. 2006;60:233–238. doi: 10.1203/01.pdr.0000228350.61496.90. [DOI] [PubMed] [Google Scholar]
- Qu D, Teckman JH, Perlmutter DH. Review: alpha 1-antitrypsin deficiency associated liver disease. J Gastroenterol Hepatol. 1997;12:404–416. doi: 10.1111/j.1440-1746.1997.tb00451.x. [DOI] [PubMed] [Google Scholar]
- Sifers RN, Finegold MJ, Woo SL. Alpha-1-antitrypsin deficiency: accumulation or degradation of mutant variants within the hepatic endoplasmic reticulum. Am J Respir Cell Mol Biol. 1989;1:341–345. doi: 10.1165/ajrcmb/1.5.341. [DOI] [PubMed] [Google Scholar]
- Sifers RN, Finegold MJ, Woo SL. Molecular biology and genetics of alpha 1-antitrypsin deficiency. Semin Liver Dis. 1992;12:301–310. doi: 10.1055/s-2008-1040399. [DOI] [PubMed] [Google Scholar]
- Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 1976;294:1316–21. doi: 10.1056/NEJM197606102942404. [DOI] [PubMed] [Google Scholar]
- Sveger T, Eriksson S. The liver in adolescents with alpha 1-antitrypsin deficiency. Hepatology. 1995;22:514–7. doi: 10.1002/hep.1840220221. [DOI] [PubMed] [Google Scholar]
- Teckman JH. Alpha1-antitrypsin deficiency in childhood. Semin Liver Dis. 2007;27:274–281. doi: 10.1055/s-2007-985072. [DOI] [PubMed] [Google Scholar]
- Teckman JH, An JK, Loethen S, Perlmutter DH. Fasting in alpha1-antitrypsin deficient liver: constitutive [correction of consultative] activation of autophagy. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1156–1165. doi: 10.1152/ajpgi.00041.2002. [DOI] [PubMed] [Google Scholar]
- Teckman JH, Burrows J, Hidvegi T, Schmidt B, Hale PD, Perlmutter DH. The proteasome participates in degradation of mutant alpha 1-antitrypsin Z in the endoplasmic reticulum of hepatoma-derived hepatocytes. J Biol Chem. 2001;276:44865–44872. doi: 10.1074/jbc.M103703200. [DOI] [PubMed] [Google Scholar]
- Teckman JH, Perlmutter DH. The endoplasmic reticulum degradation pathway for mutant secretory proteins alpha1-antitrypsin Z and S is distinct from that for an unassembled membrane protein. J Biol Chem. 1996;271:13215–13220. doi: 10.1074/jbc.271.22.13215. [DOI] [PubMed] [Google Scholar]
- Teckman JH, Perlmutter DH. Retention of mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physiol Gastrointest Liver Physiol. 2000;279:G961–974. doi: 10.1152/ajpgi.2000.279.5.G961. [DOI] [PubMed] [Google Scholar]
- Wu Y, Swulius MT, Moremen KW, Sifers RN. Elucidation of the molecular logic by which misfolded alpha 1-antitrypsin is preferentially selected for degradation. Proc Natl Acad Sci U S A. 2003;100:8229–8234. doi: 10.1073/pnas.1430537100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Termine DJ, Swulius MT, Moremen KW, Sifers RN. Human endoplasmic reticulum mannosidase I is subject to regulated proteolysis. J Biol Chem. 2007;282:4841–4849. doi: 10.1074/jbc.M607156200. [DOI] [PMC free article] [PubMed] [Google Scholar]