

Abstract
Cyclic amines can be encapsulated in a water-soluble self-assembled supramolecular host upon protonation. The hydrogen-bonding ability of the cyclic amines, as well as the reduced degrees of rotational freedom, allows for the formation of proton-bound homodimers inside of the assembly that are otherwise not observable in aqueous solution. The generality of homodimer formation was explored with small N-alkyl aziridines, azetidines, pyrrolidines, and piperidines. Proton-bound homodimer formation is observed for N-alkylaziridines (R = methyl, isopropyl, tert-butyl), N-alkylazetidines (R = isopropyl, tert-butyl), and N-methylpyrrolidine. At high concentration, formation of a proton-bound homotrimer is observed in the case of N-methylaziridine. The homodimers stay intact inside the assembly over a large concentration range, thereby suggesting cooperative encapsulation. Both G3(MP2)B3 and G3B3 calculations of the proton-bound homodimers were used to investigate the enthalpy of the hydrogen bond in the proton-bound homodimers and suggest that the enthalpic gain upon formation of the proton-bound homodimers may drive guest encapsulation.
Keywords: host–guest chemistry, molecular recognition, guest encapsulation, amine protonation
Hydrogen bonding and protonation play vital roles in both chemical and biological phenomena as one of the most prevalent intermolecular forces in nature (1–4). Although hydrogen bonding is often viewed as a weak interaction, the strength of a special class of strong hydrogen bonds, often referred to as ionic hydrogen bonds, can range from 5 to 35 kcal/mol (5). These strong hydrogen bonds are important in nucleation, self-assembly, protein folding, reactivity of enzyme active sites, formation of membranes in biological systems, and biomolecular recognition (5–7). Because of the importance of hydrogen bonding in so many aspects of chemical and biological systems, a number of synthetic and theoretical studies have examined their magnitude and origin (8, 9). Many of the computational studies have focused on simple proton-bound homodimers and heterodimers, where 2 neutral bases are bound by 1 proton (Fig. 1), because experimental energies for these complexes can be obtained from gas phase thermochemical studies such as variable-temperature high-pressure mass spectrometry (5).
Fig. 1.
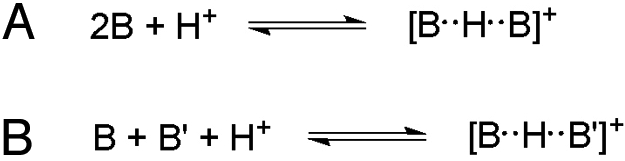
Formation of proton bound homodimers (A) and heterodimers (B).
In simple hydrogen-bonded complexes such as those shown in Fig. 1, the hydrogen bond strength is maximized when the proton donor and the conjugate base of the proton acceptor have similar proton affinities. Therefore, the strongest hydrogen bonds are formed when the 2 basic components, minus the proton, are identical (5). Although the proton-bound homodimers and heterodimers can be observed in the gas phase, solution studies of these types of complexes remain rare. To the best of our knowledge, proton-bound homodimers or heterodimers predominantly form in aprotic organic solvents, rather than protic hydrogen-bonding solvents (5, 10).
Previous work in the K.N.R. group has explored the formation, guest exchange dynamics, and mediated reactivity of the self-assembled supramolecular host molecule [Ga4L6]12− (1, L = N,N′-bis(2,3-dihydroxybenzoyl)-1,5-diaminonaphthalene) (Fig. 2) (11–14). The water-soluble 1 maintains a hydrophobic interior cavity, ranging from ≈250 to 430 Å3 depending on the encapsulated guest (15), which is isolated from bulk solution. Although both monocationic and neutral guests are encapsulated in aqueous solution (16), we have previously shown that monocations are preferentially encapsulated. In our exploration of encapsulated protonated guests, we have shown that protonation can allow for encapsulation of amine guests and also that chelating amines are able to intramolecularly share 1 proton to allow for observation of the nitrogen inversion bond rotation process (NIR) inside of the assembly (17). We have used the thermodynamic stabilization of protonated guests in 1 to demonstrate that acid-catalyzed hydrolyses of acetals and orthoformates can be carried out in basic solution inside of 1 (18–21). Herein, we expand on the ability of 1 to encapsulate protonated substrates in the simultaneous encapsulation of multiple cyclic amines as proton-bound homodimers and also present an example of heterodimer and homotrimer formation.
Fig. 2.

Graphical representation of 1. (Left) A schematic representation of 1 with only 1 ligand shown for clarity. (Right) A space-filling model of 1 as viewed down the 2-fold axis defined by the naphthalene-based ligand.
Results and Discussion
After our previous observations that chelating amines are able to intramolecularly bind a proton when encapsulated in 1, we surmised that 2 separate amines may be able to simultaneously coordinate to 1 proton in 1. Furthermore, synthetic host molecules have been shown to stabilize proton-bound amines, such as N2H7+, in the solid state (22). To test this hypothesis, a number of cyclic amines were investigated as potential guests because of their strong hydrogen-bonding ability (23). Furthermore, the reduced rotational degrees of freedom in the cyclic amines should attenuate the entropic penalty for simultaneously encapsulating multiple guest molecules. For example, in the study of encapsulated neutral guests in 1, we have observed that multiple substituted arenes with minimal degrees of rotational freedom can be simultaneously encapsulated in 1 (24).
When an excess of N-methylpyrrolidine was added to an aqueous solution of 1, the 1H NMR integration of the encapsulated guest region showed that 2 equivalents of the amine were encapsulated (Fig. 3). When only 1 equivalent of N-methylpyrrolidine was added to 1, half of an equivalent of 1 remained empty while the other half of an equivalent of 1 encapsulated 2 N-methylpyrrolidine molecules. Encapsulation of the second equivalent of amine also minimizes the contacts between the polar N H of the guest with the hydrophobic cavity of 1 while simultaneously maximizing the favorable hydrophobic interactions between host and guest. To probe the structure of the encapsulated guest, the importance of protonation was investigated. Because neutral guests are encapsulated in only 1 in aqueous solution (16), the encapsulation of N-methylpyrrolidine was investigated in both d4-MeOD and d6-DMSO to eliminate the possibility that the neutral amines were being encapsulated. The host–guest complex containing 2 equivalents of N-methylpyrrolidine was cleanly formed in both d4-MeOD and d6-DMSO, thereby eliminating the possibility of neutral substrate encapsulation.
H of the guest with the hydrophobic cavity of 1 while simultaneously maximizing the favorable hydrophobic interactions between host and guest. To probe the structure of the encapsulated guest, the importance of protonation was investigated. Because neutral guests are encapsulated in only 1 in aqueous solution (16), the encapsulation of N-methylpyrrolidine was investigated in both d4-MeOD and d6-DMSO to eliminate the possibility that the neutral amines were being encapsulated. The host–guest complex containing 2 equivalents of N-methylpyrrolidine was cleanly formed in both d4-MeOD and d6-DMSO, thereby eliminating the possibility of neutral substrate encapsulation.
Fig. 3.

1H NMR spectrum of 2 equivalents of N-methylpyrrolidine encapsulated in 1 in D2O. The resonances corresponding to the assembly (*), external N-methylpyrrolidine (○) and encapsulated N-methylpyrrolidine (■) are labeled for clarity.
To understand the structure of the encapsulated species, variable-temperature 1H NMR experiments were performed with the intention of freezing out 1 conformation of the encapsulated complex. However, experiments ranging from 0 °C to 80 °C in water and from −60 °C to 50 °C in methanol did not change the 1H NMR spectrum of the encapsulated guest. A likely explanation for the invariant 1H NMR spectrum is that the structure of any of the conformations of the encapsulated proton-bound homodimer is C2 symmetric. For example, the 2 extreme rotational conformations of the proton-bound homodimer have C2v and C2h symmetries, respectively, but encapsulation in the T-symmetric host reduces the effective symmetry of both conformations to C2 [see supporting information (SI) Fig. S1]. We hoped to gain further information about the simultaneous encapsulation through the use of natural-abundance 14N and 15N NMR experiments; however, neither of these methods was amenable to this system. Methylphospholane, the phosphorus analog of 2, was also prepared in hopes that 31P NMR experiments could be used to investigate protonation. However, methylphospholane is encapsulated as a protonated monomer rather than a proton-bound homodimer.
Expanding the Homodimer Scope.
After the observation of encapsulation of 2 equivalents of N-methylpyrrolidine, we sought to determine whether the simultaneous encapsulation was independent or cooperative. To address this question, the concentrations of N-methylpyrrolidine and 1 were varied by the same factor from 22 to 0.50 mM. If the encapsulation of each molecule of N-methylpyrrolidine is independent, then new 1H NMR resonances corresponding to the encapsulated protonated monomer should appear as the concentration of the solution is lowered. However, if encapsulation of the 2 molecules of N-methylpyrrolidine is strongly cooperative, then the 1H NMR resonances of the encapsulated complex should not change during the course of the dilution experiment. In the dilution experiment, the observed 1H NMR resonances at each concentration are identical, suggesting that encapsulation of both molecules of N-methylpyrrolidine is cooperative (Fig. 4).
Fig. 4.

Dilution 1H NMR experiments of N-methylpyrrolidine and 1. The resonances corresponding to the assembly (*), external N-methylpyrrolidine (○) and encapsulated N-methylpyrrolidine (■) are labeled for clarity. The guest region of the spectra (3 to −3 ppm) has been enlarged 4 times for clarity. The full dilution experiment is shown in Fig. S2.
To test the generality of simultaneous encapsulation of multiple guests in 1, a variety of N-alkyl aziridines (2–5), azetidines (6–10), pyrrolidines (11–14), and piperidines (15–18) were prepared and screened as potential guests in 1 (Fig. 5). [N-ethylazetidine (7) were excluded from the guest screening because attempts to prepare 7 have proved unsuccessful.] Although the toxicity and difficulty of preparation of low-molecular-mass aziridines and azetidines have previously limited the number of synthetic studies, both synthetic and computational efforts have investigated the hydrogen-bonding ability of these cyclic amines. Based on infrared studies of the N-methyl derivatives of the cyclic amines, it has been shown that the hydrogen-bonding ability of these complexes parallels the basicity with 2 ≪ 14 < 6 < 10 (23).
Fig. 5.

The scope of cyclic amines probed in 1.
In screening the encapsulation of the cyclic amines shown in Fig. 5, 5 equivalents of the desired amine were added to a 20 mM solution of 1 in D2O. Under these conditions, 2 equivalents of N-methylaziridine (2) were observed to be encapsulated. However, when >50 equivalents of 2 were added to 1, 3 equivalents of 2 were found to be encapsulated. Although the 1H NMR spectrum of N-ethylaziridine (3) indicated only monomeric guest encapsulation, the 1H NMR spectrum was broad, suggesting either fast guest exchange or interconversion of different conformations of the guest on the NMR time scale. Increasing the concentration of either 1 or 3 did not result in more than a single guest being encapsulated. Variable-temperature 1H NMR experiments from −70 °C to 50 °C did not result in sharper guest resonances or a change in the number of encapsulated molecules. For the 2 most sterically demanding aziridines investigated, N-isopropylaziridine (4) and N-tert-butylaziridine (5), clean dimer encapsulation was observed in both cases regardless of the number of equivalents of the cyclic amine added. In the azetidine family, N-methylazetidine (6) is only encapsulated as a monomer whereas N-isopropylazetidine (8) and N-tert-butylazetidine (9) were encapsulated as dimers. Attempts to encapsulate higher-order oligomers of 6 under higher concentrations of either 1 or 6 proved unsuccessful. Similar encapsulation experiments in buffered solution provided equivalent results, thereby suggesting that the pH of the solution did not account for the difference in amine encapsulation. Of the pyrrolidines investigated, only N-methylpyrrolidine (10) was encapsulated as a proton-bound homodimer, and none of the other pyrrolidines formed clean host–guest complexes with 1. Similarly, N-methylpiperidine (14) was the only pyrrolidine investigated that is encapsulated in 1, and it was encapsulated as a monomer (Fig. 6). It is likely that as the N-alkyl substitution is increased, the size of the pyrrolidines and piperidines becomes prohibitively large for formation and encapsulation of the proton-bound homodimers in 1. The calculated volumes of the [B··H··OH2]+ and [B··H··B]+ complexes are tabulated in Table S1. However, as we have previously shown, 1 is able to distort to accommodate variously sized guests, so that gauging the selectivity of amine encapsulation solely on the volume of the guest is difficult (15). For all of the host–guest complexes containing encapsulated protonated amines, encapsulation of the amines into 1 was fast, and stable host–guest complexes were formed within 10 s of addition.
Fig. 6.

Scope and results of encapsulation studies in 1.
Observation of a Proton-Bound Heterodimer.
After the investigation of encapsulation of the proton-bound homodimers, the possibility of encapsulating heterodimers was explored. Binary combinations of the cyclic amines in Fig. 6 were added to a solution of 1 by adding 1, 2, 5, 10, or 20 equivalents of each amine. In all but 1 case, a mixture of encapsulated homodimers was observed. However, when 4 and 10 were mixed, in either equal or unequal proportions, encapsulation of a species that did not correspond to either of the homodimers was observed (Fig. 7). Based on 1H NMR analysis, this encapsulated species contained 1 molecule of 4 and 1 molecule of 10, suggesting the formation of a proton-bound heterodimer. Although the origin of this selectivity is difficult to explain, the encapsulation of the proton-bound heterodimer does highlight the ability of 1 to selectively recognize the properties of the heterodimer over either of the possible homodimers.
Fig. 7.

1H NMR spectra of a solution of 10 with 1 (A), 4 with 1 (B), and an equimolar combination of 4 and 10 with 1 (C).
Computational Studies of Proton-Bound Homodimers.
In hopes of further understanding the driving force for proton-bound amine homodimer formation in 1, calculations were carried out by using Gaussian 03 (25) to investigate both the geometry of the complex and the enthalpic strength of the hydrogen bond in the proton-bound homodimers. One potential problem with calculations of this type is that standard methods of DFT single-point energy calculations often do not accurately describe hydrogen-bonded compounds (26). To overcome this problem, single-point energy calculations were performed by using the compound G3(MP2)B3 and G3B3 methods. These 2 methods combine multiple levels of theory using MP4 methods for G3B3 and the less computationally demanding MP2 methods for G3(MP2)B3. Both types of G3B3 calculations have been shown to be accurate for small-molecule systems, with most calculated energies being within 1 kcal of experimentally determined energies for simple systems (27, 28). (For a comparison of calculated and experimental hydrogen bond enthalpies of model systems, see SI Text and Tables S1–S49; please see also Figs. S1–S10.)
All of the compounds 2–10 and 14 were optimized at the B3LYP/6–31++G(d,p) level of theory with subsequent energy calculations using the B3LYP/6–31++G(d,p), G3(MP2)B3, and G3B3 methods. The calculated proton affinities for each of the cyclic amines are shown in Table 1. Although experimental proton affinities are known for only 2 (221.6 kcal/mol), 10 (227.9 kcal/mol), and 11 (228.9 kcal/mol), they agree well with the calculated values (29, 30). [Experimentally determined pKa values are known for only 2 (7.86), 6 (10.40), 10 (10.46), and 14 (10.08).] From the proton affinity calculations, 2 main trends are observed: (i) For a given ring size, the proton affinity increases as the size of the N-alkyl substitution increases, and (ii) as the ring size increases the proton affinity also increases.
Table 1.
Calculated proton affinities for N-alkyl cyclic amines
| Substrate | Proton affinity, kcal/mol |
||
|---|---|---|---|
| B3LYP | G3MP2 | G3 | |
| 2 | 222.8 | 221.6 | 221.8 |
| 3 | 225.2 | 223.9 | 224.0 |
| 4 | 227.0 | 225.5 | 224.0 |
| 5 | 230.3 | 228.8 | 228.9 |
| 6 | 228.0 | 227.6 | 227.6 |
| 7 | 228.3 | 227.8 | 227.7 |
| 8 | 232.3 | 231.4 | 231.3 |
| 9 | 234.6 | 233.8 | 233.7 |
| 10 | 229.0 | 228.8 | 228.8 |
| 14 | 227.8 | 230.6 | 230.3 |
Having demonstrated the validity of these computational methods in accurately calculating the proton affinity of the model compounds, the enthalpy of the hydrogen bond formed between the protonated amine and a second amine molecule ([B··H··B]+) was calculated. We have previously shown through kinetic analysis that water, in either its neutral or protonated form, can enter 1, so the enthalpy of the hydrogen bond formed between a protonated amine and water molecule ([B··H··OH2]+) was also calculated (21, 31). The geometry optimized structures for the [B··H··B]+ and [B··H··OH2]+ complexes are shown in Fig. 8. For each calculated structure, the single-point energy was calculated at the B3LYP, G3(MP2)B3, and G3B3 level of theory (Table 2). As expected, based on the difference between the proton affinity of the amines and water, the enthalpy of the hydrogen bond formed in [B··H··OH2]+ complexes is lower than that for the [B··H··B]+ complexes. For the [B··H··OH2]+ complexes, all 3 levels of theory gave similar results although the B3LYP energy calculations began to deviate from the G3(MP2)B3 and G3B3 calculations as the size of the molecule increased. For the [B··H··B]+ complexes, the B3LYP calculations greatly underestimated the enthalpy of the hydrogen bond when compared with the G3(MP2)B3 and G3B3 methods.
Fig. 8.

Calculated geometries for [B··H··OH2]+ and [B··H··B]+ complexes of 2–14. Atoms are color coded for clarity: carbon (blue), nitrogen (purple), oxygen (red), hydrogen (gray).
Table 2.
Calculated hydrogen-bond enthalpies for the [B··H··OH2]+ and [B··H··B]+ complexes
| Substrate | ΔH [B··H··OH2]+, kcal/mol |
ΔH [B··H··B]+, kcal/mol |
||||
|---|---|---|---|---|---|---|
| B3LYP | G3MP2 | G3 | B3LYP | G3MP2 | G3 | |
| 2 | 15.8 | 15.4 | 15.9 | 23.1 | 24.9 | 25.5 |
| 3 | 14.9 | 14.8 | 15.3 | 21.6 | 25.2 | 25.8 |
| 4 | 14.2 | 14.4 | 15.0 | 19.5 | 25.2 | 25.7 |
| 5 | 13.6 | 14.1 | 14.7 | 17.0 | 24.4 | — |
| 6 | 14.5 | 14.4 | 15.0 | 20.1 | 24.3 | 24.8 |
| 7 | 15.8 | 15.9 | 16.4 | 18.7 | 25.1 | 25.4 |
| 8 | 11.1 | 11.5 | 12.0 | 11.9 | 19.8 | — |
| 9 | 12.3 | 13.1 | 13.7 | 11.5 | 21.3 | — |
| 10 | 13.8 | 14.1 | 14.6 | 17.7 | 24.4 | 24.9 |
| 14 | 15.9 | 13.6 | 14.0 | 16.5 | — | — |
Based on the enthalpic difference between the [B··H··OH2]+ and [B··H··B]+ complexes, formation of the proton-bound homodimer is highly enthalpically favorable when compared with the formation of the water adduct, which may help to explain the observation of the encapsulation of proton-bound homodimers in 1 (Table 3). In the case of the observed proton-bound heterodimer [4··H··10]+, the enthalpy of the hydrogen bond was calculated (G3MP2) to be 24.3 kcal/mol, suggesting that the shape of this heterodimer is preferentially recognized by 1 over either of the homodimers. Although we did not pursue entropy calculations of proton-bound homodimer formation in the gas phase, the interpretation of entropy for the experimental solution studies with 1 are likely more complicated. Changes in solvation entropy and enthalpy in the desolvation of the amines upon encapsulation, release of solvent from the interior of 1, and the change in solvation of 1 when changing from a 12- to an 11- complex upon guest encapsulation prohibit definitive deconvolution of the different entropic components (32).
Table 3.
Comparison of the enthalpies of the hydrogen bond formation in the [B··H··OH2]+ and [B··H··B]+ complexes
| Substrate | ΔΔH*, kcal/mol |
||
|---|---|---|---|
| B3LYP | G3MP2 | G3 | |
| 2 | 7.3 | 9.5 | 9.6 |
| 3 | 6.7 | 10.4 | 10.5 |
| 4 | 5.2 | 10.8 | 10.7 |
| 5 | 3.4 | 10.3 | — |
| 6 | 5.7 | 9.8 | 9.8 |
| 7 | 2.9 | 9.2 | 8.9 |
| 8 | 0.8 | 8.3 | — |
| 9 | −0.8 | 8.2 | — |
| 10 | 3.9 | 10.3 | 10.3 |
| 14 | 0.6 | — | — |
*ΔΔH = ΔH([B··H··B]+) + ΔH(H2O) − ΔH([B··H··OH2]+) − ΔH(B).
Conclusions
In conclusion, we have demonstrated the formation and encapsulation of hydrogen-bonded homodimers in the interior of a water-soluble supramolecular assembly. To the best of our knowledge, the systems described in this study are among the largest molecules that have been subjected to G3(MP2)B3 or G3B3 calculations to date. The calculations suggest that formation of the proton-bound amine homodimers is highly enthalpically favorable when compared with the solvent adducts of the protonated amines.
Materials and Methods
General Procedures.
All NMR spectra were obtained by using an AV 500-MHz spectrometer. The temperature of all variable-temperature NMR experiments was calibrated with methanol or ethylene glycol standards. All reagents were obtained from commercial suppliers and used without purification unless otherwise noted. The host assembly K12[Ga4L6] was prepared as described in the literature (33). The N-alkylaziridines 2–5 (34), N-alkylazetidines 6 (35) and 9 (36), N-alkylpyrrolidines 11–13 (37), and N-alkylpiperidines 15–17 (38) were prepared as described in the literature. Because of a large number of overlapping peaks prohibiting unambiguous assignment of the 1H NMR resonances, the 1H NMR spectra have been included in SI Text rather than being numerically tabulated.
Synthetic Procedures.
N-isopropylaminopropanol.
This procedure is modified from a method described for the preparation of β-aminoalcohols (39). In a 5-L 3-neck flask equipped with a magnetic stir bar, nitrogen inlet and oil bubbler were added to absolute ethanol (2 L), 3-amino-1-propanol (75 g, 1.0 mol), and acetone (110 mL, 1.5 mol). After stirring overnight at room temperature, the reaction mixture was cooled to 0 °C in a salt/ice bath and NaBH4 (56.6 g, 1.5 mol) was added slowly under a gentle flow of nitrogen over the course of 3 h, keeping the temperature of the reaction mixture at 5–10 °C. The reaction mixture was allowed to stir at 5 °C for 2 h before the careful addition of 135 mL of cold H2O, dilution with 1.5 L of methylene chloride, and filtration of the reaction mixture. The solvents were removed by rotary evaporator, and the resultant oil was extracted with ether (3 × 200 mL) and dried over MgSO4. After removal of the solvents, the residual oil was distilled under reduced pressure (bp 40 °C at 2 mmHg) (40) to afford a clear liquid that solidified after standing in the collection flask (61 g, 52% yield). 1H NMR (500 MHz, CDCl3) δ: 3.52 (t, J = 7.5 Hz, 2H, CH2), 3.01 (sept, J = 6.5 Hz, 1H, CH), 2.61 (t, J = 7.5 Hz, 2H, CH2), 2.02 (bs, 2H, NH/OH), 1.52 (pent, J = 7.0 Hz, 2H, CH2), 1.03 (d, J = 7.5 Hz, 6H, 2 × CH3), 13C{1H} NMR (125 MHz, CDCl3) δ: 64.2, 52.1, 42.8, 23.6, 22.1.
N-isopropylaminoazetidine.
To a flame-dried 2-L flask was added dry ether (1.5 L) and N-isopropylaminopropanol (61 g, 0.52 mol). Dry HCl was bubbled through the solution that resulted in the formation and the precipitation of the HCl salt of the amine. The ammonium salt was filtered and washed with dry ether under a stream of nitrogen, and the residual ether was removed overnight under vacuum. The N-isopropylaminopropanol HCl salt (70.5 g, 0.459 mol) was added to a 250-mL Schlenk flask under N2. The flask was cooled to 0 °C in an ice bath, and chlorosulfonic acid (46 mL, 0.69 mol) was added dropwise with an addition funnel over the course of 1 h. The reaction mixture was heated to 50 °C for 1 h, to 150 °C for 2 h, and then heated an additional 1 h at 150 °C under moderate vacuum (≈15 mmHg). The reaction mixture was cooled to room temperature and 75 mL of water was added. The resultant solution was stirred slowly for 3 h until most of the solid had dissolved, cooled to 0 °C, and adjusted to pH 11 with KOH pellets. The solution was extracted with Et2O (3 × 150 mL), and the extract was dried over magnesium sulfate. Filtration, followed by removal of the solvent under vacuum and subsequent distillation of the resultant oil under atmospheric pressure (bp 117 °C) (41), yielded the desired product (2.8 g, 6% yield). 1H NMR (500 MHz, CDCl3) δ: 3.07 (t, J = 8.0 Hz, 4H, 2 × CH2), 2.17 (sept, J = 6.5 Hz, 1H, CH), 1.97 (pent, J = 7.0 Hz, 2H, CH2), 0.83 (d, J = 7.5 Hz, 6H, 2 × CH3), 13C{1H} NMR (125 MHz, CDCl3) δ: 58.6, 53.2, 19.2, 18.4.
Computational Methods.
All calculations were performed by using the Gaussian 03 software package with the GaussView graphical user interface. Geometry optimizations and unscaled frequency calculations were carried out at the B3LYP/6–31++G(d,p) level of theory. Frequency calculations were performed on all converged structures to confirm that they corresponded to local minima on their respective potential-energy surfaces. These structures and frequencies were then used as input in the G3(MP2)B3 or G3B3 zero-point corrected enthalpy calculations. Some of the [B··H··B]+ calculations were omitted because of large size of the molecules and the corresponding time required for calculation.
Supplementary Material
Acknowledgments.
We thank Drs. Jamin Krinsky and Kathleen Durkin for assistance with calculations. This work was supported by the Director, Office of Science, Office of Advanced Scientific Computing Research, Office of Basic Energy Sciences (U.S. Department of Energy) under contract DE-AC02-05CH11231 and National Science Foundation Predoctoral Fellowships (to M.D.P. and J.S.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0809806106/DCSupplemental.
References
- 1.Bachovchin WW. Contributions of NMR spectroscopy to the study of hydrogen bonds in serine protease active sites. Magn Res Chem. 2001;39:S199–S213. [Google Scholar]
- 2.Frey PA. Strong hydrogen bonding in molecules and enzymatic complexes. Magn Res Chem. 2001;39:S190–S198. [Google Scholar]
- 3.Nadassy K, Wodak SJ, Janin J. Structural features of protein-nucleic acid recognition sites. Biochemistry. 1999;38:1999–2017. doi: 10.1021/bi982362d. [DOI] [PubMed] [Google Scholar]
- 4.Wiest O, Houk KN. Stabilization of the transition state of the chorismate-prephenate rearrangement: An ab-initio study of enzyme and antibody catalysis. J Am Chem Soc. 1995;117:11628–11639. [Google Scholar]
- 5.Mautner M. The ionic hydrogen bond. Chem Rev. 2005;105:213–284. doi: 10.1021/cr9411785. [DOI] [PubMed] [Google Scholar]
- 6.Chang CJ, Chang MCY, Damrauer NH, Nocera DG. Proton-coupled electron transfer. A Unifying mechanism for biological charge transport, amino acid radical initiation and propagation, and bond making/breaking reactions of water and oxygen. Biochim Biophys Acta Bioenerg. 2004;1655:13–28. doi: 10.1016/j.bbabio.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 7.Kearley GJ, Fillaux F, Baron M-H, Bennington S, Tomkinson J. A new look at proton transfer dynamics along the hydrogen bonds in amides and peptides. Science. 1994;264:1285–1289. doi: 10.1126/science.264.5163.1285. [DOI] [PubMed] [Google Scholar]
- 8.Jeffrey GA. An Introduction to Hydrogen Bonding. New York: Oxford Univ Press; 1997. [Google Scholar]
- 9.Scheiner S. Hydrogen Bonding. A Theoretical Perspective. New York: Oxford Univ Press; 1997. [Google Scholar]
- 10.Graton J, Berthelot M, Besseau F, Laurence C. An enthalpic scale of hydrogen-bond basicity. 3. Ammonia, primary, secondary, and tertiary amines. J Org Chem. 2005;70:7892–7901. doi: 10.1021/jo050535g. [DOI] [PubMed] [Google Scholar]
- 11.Caulder DL, Raymond KN. Supermolecules by design. Acc Chem Res. 1999;32:975–982. [Google Scholar]
- 12.Davis AV, et al. Guest exchange dynamics in an M4L6 tetrahedral host. J Am Chem Soc. 2006;128:1324–1333. doi: 10.1021/ja056556+. [DOI] [PubMed] [Google Scholar]
- 13.Davis AV, Raymond KN. The big squeeze: Guest exchange in an M4L6 supramolecular host. J Am Chem Soc. 2005;127:7912–7919. doi: 10.1021/ja051037s. [DOI] [PubMed] [Google Scholar]
- 14.Fiedler D, Leung DH, Bergman RG, Raymond KN. Selective molecular recognition, C-H bond activation, and catalysis in nanoscale reaction vessels. Acc Chem Res. 2005;38:349–358. doi: 10.1021/ar040152p. [DOI] [PubMed] [Google Scholar]
- 15.Pluth MD, et al. Structural consequences of anionic host–cationic guest interactions in a supramolecular assembly. Inorg Chem. 2009;48:111–120. doi: 10.1021/ic8012848. [DOI] [PubMed] [Google Scholar]
- 16.Biros SM, Bergman RG, Raymond KN. The hydrophobic effect drives the recognition of hydrocarbons by an anionic metal–ligand cluster. J Am Chem Soc. 2007;129:12094–12095. doi: 10.1021/ja075236i. [DOI] [PubMed] [Google Scholar]
- 17.Pluth MD, Bergman RG, Raymond KN. Encapsulation of protonated diamines in a water-soluble, chiral, supramolecular assembly allows for measurement of hydrogen-bond breaking followed by nitrogen inversion/rotation. J Am Chem Soc. 2008;130:6362–6366. doi: 10.1021/ja076691h. [DOI] [PubMed] [Google Scholar]
- 18.Pluth MD, Bergman RG, Raymond KN. Acid catalysis in basic solution: A supramolecular host promotes orthoformate hydrolysis. Science. 2007;316:85–88. doi: 10.1126/science.1138748. [DOI] [PubMed] [Google Scholar]
- 19.Pluth MD, Bergman RG, Raymond KN. Catalytic deprotection of acetals in basic solution with a self-assembled supramolecular “nanozyme”. Angew Chem Int Ed. 2007;46:8587–8589. doi: 10.1002/anie.200703371. [DOI] [PubMed] [Google Scholar]
- 20.Pluth MD, Bergman RG, Raymond KN. Making amines strong bases: thermodynamic stabilization of protonated guests in a highly-charged supramolecular host. J Am Chem Soc. 2007;129:11459–11467. doi: 10.1021/ja072654e. [DOI] [PubMed] [Google Scholar]
- 21.Pluth MD, Bergman RG, Raymond KN. Supramolecular catalysis of orthoformate hydrolysis in basic solution: An enzyme-like mechanism. J Am Chem Soc. 2008;130:11423–11429. doi: 10.1021/ja802839v. [DOI] [PubMed] [Google Scholar]
- 22.Atwood JL, Barbour LJ, Jerga A. Supramolecular stabilization of N2H7+ J Am Chem Soc. 2002;124:2122–2123. doi: 10.1021/ja017722q. [DOI] [PubMed] [Google Scholar]
- 23.Searles S, Tamres M, Block F, Quarterman LA. Hydrogen bonding and basicity of cyclic imines. J Am Chem Soc. 1956;78:4917–4920. [Google Scholar]
- 24.Hastings CJ, Pluth MD, Biros SM, Bergman RG, Raymond KN. Simultaneously bound guests and chiral recognition: A chiral self-assembled supramolecular host encapsulates hydrophobic guests. Tetrahedron. 2008;64:8362–8367. [Google Scholar]
- 25.Frisch MJ, et al. Wallingford, CT: Gaussian Inc.; 2004. Gaussian 03 Revision C.02. [Google Scholar]
- 26.Pudzianowski AT. A systematic appraisal of density functional methodologies for hydrogen bonding in binary ionic complexes. J Phys Chem A. 1996;100:4781–4789. [Google Scholar]
- 27.Anantharaman B, Melius CF. Bond additivity corrections for G3B3 and G3MP2B3 quantum chemistry methods. J Phys Chem A. 2005;109:1734–1747. doi: 10.1021/jp045883l. [DOI] [PubMed] [Google Scholar]
- 28.Redfern PC, Zapol P, Curtiss LA, Raghavachari K. Assessment of Gaussian-3 and density functional theories for enthalpies of formation of C1-C16 alkanes. J Phys Chem A. 2000;104:5850–5854. [Google Scholar]
- 29.Hunter EPL, Lias SG. Evaluated gas-phase basicities and proton affinities of molecules: An update. J Phys Chem Ref Data. 1998;27:413–656. [Google Scholar]
- 30.Ohwada T, Hirao H, Ogawa A. Theoretical analysis of Lewis basicity based on local electron-donating ability. Origin of basic strength of cyclic amines. J Org Chem. 2004;69:7486–7494. doi: 10.1021/jo0486589. [DOI] [PubMed] [Google Scholar]
- 31.Pluth MD, Bergman RG, Raymond KN. The acid hydrolysis mechanism of acetals catalyzed by a supramolecular assembly in basic solution. J Org Chem. 2009;74:58–63. doi: 10.1021/jo802131v. [DOI] [PubMed] [Google Scholar]
- 32.Leung DH, Bergman RG, Raymond KN. Enthalpy–entropy compensation reveals solvent reorganization as a driving force for supramolecular encapsulation in water. J Am Chem Soc. 2008;130:2798–2805. doi: 10.1021/ja075975z. [DOI] [PubMed] [Google Scholar]
- 33.Caulder DL, Powers RE, Parac TN, Raymond KN. The self-assembly of a predesigned tetrahedral M4L6 supramolecular cluster. Angew Chem Int Ed. 1998;37:1840–1843. [Google Scholar]
- 34.Elderfield RC, Hageman HA. The von Braun cyanogen bromide reaction I. Application to pyrrolidines and ethyleneimines. J Org Chem. 1949;14:605–637. [Google Scholar]
- 35.Lambert JB, Oliver WL, Jr, Packard BS. Nitrogen inversion in cyclic N-chloroamines and N-methylamines. J Am Chem Soc. 1971;93:933–937. [Google Scholar]
- 36.Bottini AT, Roberts JD. Nuclear magnetic resonance spectra. Nitrogen inversion rates of N-substituted aziridines (ethylenimines) J Am Chem Soc. 1958;80:5203–5208. [Google Scholar]
- 37.Aitken DJ, et al. Theoretical and model studies on the chemoselectivity of a Grignard reagent's reaction with a combined aminonitrile–oxazolidine system. Tetrahedron. 2002;58:5933–5940. [Google Scholar]
- 38.Hayat S, et al. N-Alkylation of anilines, carboxamides and several nitrogen heterocycles using CsF–Celite/alkyl halides/CH3CN combination. Tetrahedron. 2001;57:9951–9958. [Google Scholar]
- 39.Saavedra JE. Reductive alkylation of β-alkanolamines with carbonyl compounds and sodium borohydride. J Org Chem. 1985;50:2271–2273. [Google Scholar]
- 40.Katritzky AR, Baker VJ, Brito-Palma FMS. Conformational analysis of saturated heterocycles. Part 100. 1-Oxa-3-azacyclohexanes. J Chem Soc Perkin Trans. 1980;2:1739–1745. [Google Scholar]
- 41.Crimaldi K, Lichter RL. Nitrogen-15 nuclear magnetic resonance spectroscopy. Natural-abundance nitrogen-15 chemical shifts of aziridines and azetidines. J Org Chem. 1980;45:1277–1281. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.