

Abstract
Carbonic anhydrase, a zinc metalloenzyme, catalyzes the reversible hydration of carbon dioxide to bicarbonate. It is involved in processes connected with acid–base homeostasis, respiration, and photosynthesis. More than 100 distinct human carbonic anhydrase II (HCAII) 3D structures have been generated in last 3 decades [Liljas A, et al. (1972) Nat New Biol 235:131–137], but a structure of an HCAII in complex with CO2 or HCO3− has remained elusive. Here, we report previously undescribed structures of HCAII:CO2 and HCAII:HCO3− complexes, together with a 3D molecular film of the enzymatic reaction observed successively in the same crystal after extended exposure to X-ray. We demonstrate that the unexpected enzyme activation was caused in an X-ray dose-dependent manner. Although X-ray damage to macromolecular samples has long been recognized [Ravelli RB, Garman EF (2006) Curr Opin Struct Biol 16:624–629], the detailed structural analysis reports on X-ray-driven reactions have been very rare in literature to date. Here, we report on enzyme activation and the associated chemical reaction in a crystal at 100 K. We propose mechanisms based on water photoradiolysis and/or electron radiolysis as the main cause of enzyme activation.
Keywords: CO2 binding, crystal pressurization, radiation driven reaction, substrate–product complex structure
The HCAII enzyme is a functional 29-kDa monomer consisting of a 10-stranded, twisted β-sheet. The active site is located at the bottom of a 15-Å cone-shaped cavity that leads to the center of the protein (37). Key features of the active site (Fig. 1) include a zinc ion coordinated tetrahedrally by 3 histidine residues (His-94, His-96, and His-119) and a water molecule/hydroxide ion as a fourth ligand (Wat-263).
Fig. 1.

The active site of HCAII. The zinc ion is tetrahedrally coordinated by 3 histidines (His-94, His-96, and His-119) and catalytic water (Wat-263). The deep water (Wat-338) sits in a hydrophobic pocket lined by Leu-198, Trp-209, Val-143, and Val-121 at the bottom of the active site. Wat-318 is in a hydrophilic environment toward the mouth of the active site cone. The proton shuttle His-64, shown in both “in” and “out” positions, is linked via Wat-292 and Wat-318 to the catalytic water. Hydrogen bonds are depicted as dotted lines, and waters are labeled with numbers only. Numbering is according to PDB code 2CBA.
Thr-199, a key residue of the second coordination sphere, is important for enzyme activity; together with Thr-200, it is involved in a finely tuned network of hydrogen bonds leading toward the solvent-exposed His-64, which is located at the entrance of the active-site channel. Thr-199 forms a hydrogen bond to the zinc-bound water/hydroxide (Wat-263), thereby orienting the 2 lone hydroxide electron pairs toward the 2 neighboring water molecules (Wat-318 and Wat-338) that reside on potential substrate-binding sites. Although both positions are suitable for a nucleophilic attack of the zinc-bound hydroxide ion, their environments differ substantially.
Wat-318 is located in a hydrophilic environment on the way out of the active-site cone, whereas the “deep water” Wat-338 is located in a hydrophobic pocket that is lined by the following side chains: Leu-198, Trp-209, Val-143, and Val-121 (Fig. 1). A wealth of indirect evidence indicates that the deep water Wat-338 position serves as the primary substrate-binding site (1–12), although the atomic details of enzyme and substrate–product interaction have remained elusive until now.
The generally accepted catalytic mechanism of carbonic anhydrase (13) is described by a 3-step kinetic scheme: (i) a Zn-OH− moiety catalyzes the interconversion of CO2 to HCO3−, leaving a water molecule as the fourth zinc ligand (Eq. 1); (ii) a proton is then transferred from the zinc-bound water to the imidazole ring of His-64 (Eq. 2); and (iii) this proton then leaves His-64 for the surrounding solvent (Eq. 3).
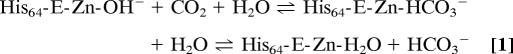 |
The pKa values for both the zinc-bound water and the proton shuttle (His-64) are close to 7.
Xenon under pressure was for the first time used in structural biology employing either solution NMR or single-crystal X-ray diffraction methods to probe cavities in protein structures (14, 15). The crystal pressurization with noble gases, such as xenon or krypton, was subsequently recognized as a successful heavy atom derivatization method to solve the phase problem in macromolecular crystallography (16). We used this method for functional studies of an enzyme–substrate complex, similarly to what has been reported for isopenicillin N synthase (17). Here, we exploit the pH decrease that is induced in the crystal by CO2, as well as the delivery of substrate at concentrations much greater than what is possible at atmospheric pressure.
Results and Discussion
The enzyme–substrate complex was generated by pressurizing a crystal of HCAII with CO2, followed by flash cryocooling at 77 K. Pressurization with CO2 has a dual effect: (i) it decreases the pH in the crystal via the spontaneous reaction of CO2 with water (CO2 + H2O ⇌ HCO3− + H+), and (ii) it supplies substrate to the enzyme at a high concentration. At pH levels lower than 6, the enzyme is in an inactive state (left part of Eq. 2), during which CO2 can bind the active site but cannot be converted to bicarbonate.
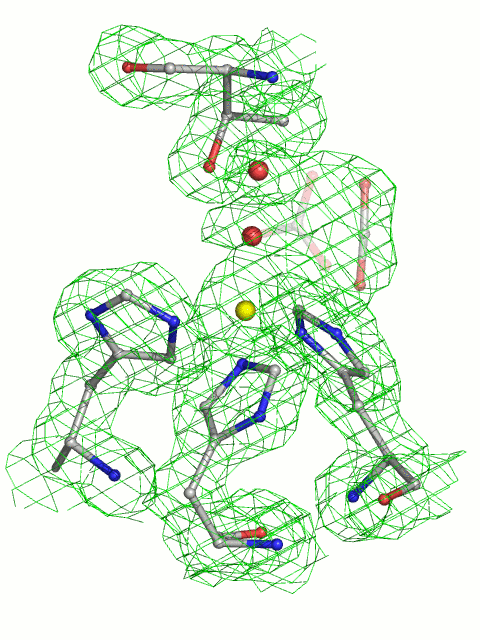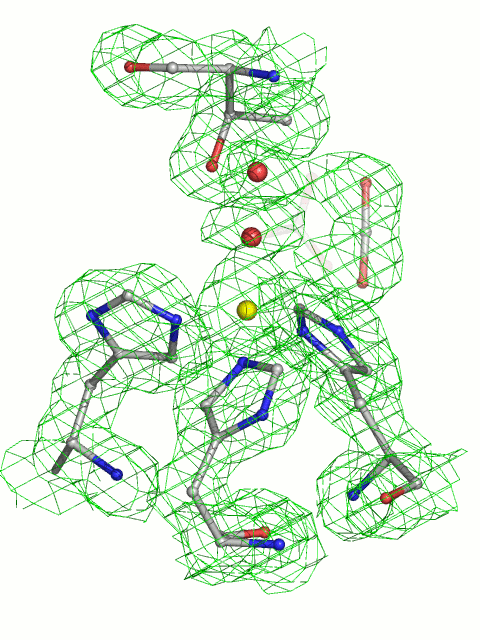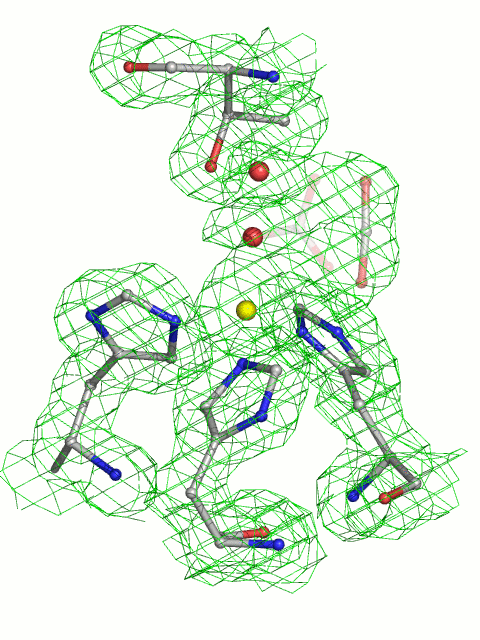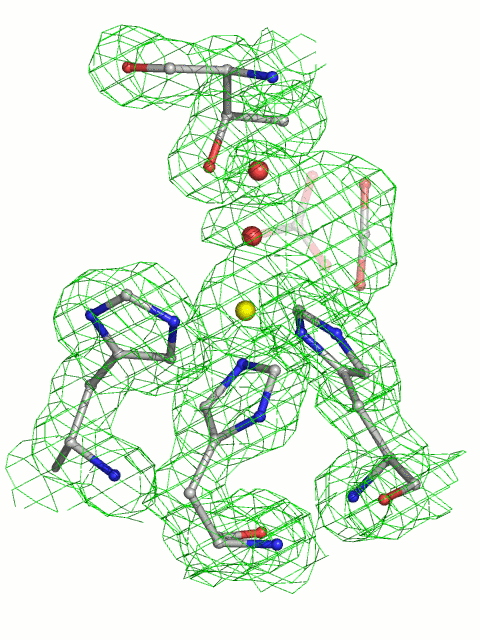
A structure obtained from diffraction data collected from a CO2-loaded HCAII crystal was refined to 1.56-Å resolution (Table 1) and displayed the CO2 located in the active site of the enzyme (Fig. 2A). The O2 oxygen atom of CO2 was at 0.5 Å from the position formerly occupied by the deep water Wat-338, which in turn was in a new location within hydrogen-bonding distance of Thr-199 N; Thr-200 N; Oγ; the catalytic waters Wat-263, Wat-318, and Wat-389 (the last not shown in Fig. 2); and the above-mentioned O2 oxygen of CO2.
Table 1.
Data collection and refinement statistics
| HCAII:CO2 | HCAII:HCO3− | |
|---|---|---|
| Data collection | ||
| Beamline | XRD1, Elettra | XRD1, Elettra |
| Wavelength, Å | 1.2 | 1.278 |
| Space group | P21 | P21 |
| Cell | a = 42.59 Å, b = 41.58 Å, c = 72.64 Å, β = 104.8° | a = 42.67 Å, b = 41.65 Å, c = 72.91 Å, β = 104.8° |
| Unique reflections | 35,235 | 29,567 |
| Resolution, Å | 28.90–1.56 (1.64–1.56) | 28.99–1.66 (1.73–1.66) |
| Redundancy | 4.7 (4.5) | 2.8 (2.6) |
| Completeness, % | 90.5 (93.7) | 87.7 (78.2) |
| Rmerge | 0.042 (0.108) | 0.068 (0.278) |
| Refinement | ||
| No. of reflections | 30,130 | 24,595 |
| R value | 0.153 | 0.168 |
| Rfree | 0.200 | 0.213 |
| rmsd from ideality, bonds, Å | 0.014 | 0.017 |
| rmsd from ideality, angles, ° | 1.530 | 1.587 |
| Overall B, Å2 | 13.6 | 18.4 |
| PDB entry | 2vva | 2vvb |
Numbers in parentheses refer to the last resolution shell. Rmerge = Σ|Ii − 〈I〉|/ΣIi, where Ii is the intensity of an individual reflection and 〈I〉 is the mean intensity of that reflection. R value = Σ|Fo − Fc|/ΣFo. Rfree is the cross-validation R value computed for a subset of reflections, omitted in the refinement process (5% of the total number of reflections).
Fig. 2.

Stereoview of HCAII in complex with carbon dioxide and bicarbonate. Distances listed below are indicated in the figures by dashed lines. (A) The 1σ level 2|Fo| − |Fc| electron density map and corresponding model for the CO2-loaded HCAII active site at 1.56-Å resolution. The carbon dioxide O2 atom is at a hydrogen-bonding distance of 3.4 Å from the main chain nitrogen of Thr-199, Wat-338 (3.1 Å), and Wat-263 (3.1 Å). The CO2 molecule also makes van der Waals contacts with all residues lining the hydrophobic pocket (Val-121, Val-143, Leu-198, and Trp-209) and the zinc ligands His-94 and His-119. (B) The 1σ level 2|Fo| – |Fc| electron density maps and the corresponding model for the complex of HCAII and bicarbonate at 1.66-Å resolution. The O3 oxygen of bicarbonate is bound to the zinc ion at 2.0 Å and is within hydrogen-bonding distance of Wat-318 (2.4 Å), Thr-199 OG1 (2.6 Å), and Wat-338 (2.9 Å). The bicarbonate O2 atom is within hydrogen-bonding distance of Wat-338 (2.7 Å) and Thr-199 N (3.1 Å). The O1 atom of bicarbonate is 2.9 Å away from the zinc ion. The HCO3− has van der Waals contacts with the same residues as CO2 (see A).
CO2 is sandwiched between the polar environment facing the active-site channel and the wall of the hydrophobic pocket. The position and orientation of the substrate are well-defined by several direct polar and van der Waals interactions, respectively (Fig. 2A). The binding mode of CO2 explains previous reports of drastically decreased enzymatic activity (10-fold to 105-fold compared with wild type) when the volume of the hydrophobic pocket is decreased by various residue mutations (Val-121, Val-143, Leu-198) with bulkier side chains, whereas enzymatic activity is only moderately decreased (2- to 3-fold) as a result of single-residue mutations that enlarge the pocket (4, 5, 9, 10, 18).
The fact that we observed the nonprocessed CO2 molecule bound at the active site clearly proves the Zn2+ ion is coordinated by the water molecule and not a hydroxide, which would immediately react with the CO2 to create bicarbonate. The distance of 2.0 Å between the catalytic water (Wat-263) and the Zn2+ ion is comparable to distances reported for other complexes mimicking the HCAII active site (1.97–2.04 Å) (19).
A second CO2 molecule was found in a hydrophobic cavity ≈12 Å from the zinc ion. This pocket was located in the hydrophobic core of the protein and was not surface-accessible. Such hydrophobic cavities are typical binding sites for noble gases (20), and indeed an Xe atom was observed in this position in an Xe-loaded HCAII crystal, as well as in the hydrophobic pocket of the relative active site.
There was a 2-month interval between the collection of the first and the second dataset from the same crystal, which had been stored in liquid nitrogen. Structural analysis showed that the CO2 bound in the active site was partially converted to bicarbonate (we detected a mixture of CO2/HCO3). This finding prompted the collection of a third dataset from the same crystal (Table 1), in which we observed only bicarbonate in the active site (Fig. 2B). In a control experiment, we loaded an HCAII crystal with CO2, flash-cooled it in liquid nitrogen, and collected a diffraction dataset from the crystal after storage for 2 months in liquid nitrogen. Structural analysis of this complex clearly showed only CO2 bound to the active site, with no evidence of product generation.
The 3 structures, in which the carbon dioxide in the active site was gradually transformed into bicarbonate as a function of the increasing absorbed X-ray dose, led to the design of an X-ray diffraction experiment to monitor and quantify structural changes associated with substrate–product interconversion and to determine the X-ray dose needed for enzyme activation. Diffraction data were collected at a 1.278-Å wavelength and processed in overlapping batches on an HCAII crystal freshly loaded with CO2. A total of 103 complete datasets staggered by 45° yielded 103 distinct refined structures (snapshots) that show consecutive average states of the enzyme in the crystal (Table S1).
The adopted experimental strategy resembles that of Berglund et al. (21) and is described in detail in Materials and Methods and in Movie S1, Movie S2, and Tables S1 and S2. We assembled the enzyme snapshots into a movie showing the conversion of CO2 to bicarbonate at the active site as a function of increasing applied X-ray dose (see Movie S1, Movie S2, and Tables S1 and S2). The movie starts in a state corresponding to the HCAII:CO2 complex, proceeds through decreasing substrate to product ratios, and ends in a state with 65% CO2 and 35% bicarbonate. At this point, the total dose absorbed by the crystal was 5.9 × 106 Gy (≈30% of the Henderson limit; ref. 22), corresponding to 1 absorbed photon in 37% of the unit cells of the crystal.
This indicates that 1 primary event of photon absorption leads to formation of approximately 2 molecules of bicarbonate; thus, the conversion from CO2 to HCO3− cannot be driven by the primary event of photon absorption alone, but must include secondary cascades resulting from the primary event.
Overall structures of the HCAII:CO2 complex are very similar to that of the wild-type enzyme [Protein Data Bank (PDB) code 2CBA] (3), with an rmsd of 0.36 Å after superposition of equivalent Cα atoms. Seemingly, the HCAII:HCO3− complex is highly preserved compared with the HCAII:CO2 (rmsd of 0.13 Å for equivalent Cα atoms). The second CO2 molecule, which we regard as an internal control, was unprocessed and bound in the hydrophobic cavity with a conserved occupancy of one.
The binding mode of bicarbonate was compared to that found in the HCAII T200H (12) mutant and human isoenzyme I (HCAI) (6): the O2 atom of bicarbonate is 0.7 Å from its former position in CO2 and 0.4 Å from the deep water Wat-338 position, whereas the O3 atom is coordinating the Zn2+ ion at a distance of 2.0 Å. The mode of binding of bicarbonate we reported is very similar to HCAI and HCAII T200H complexes; data clearly indicate the interaction we observed is genuine, and we were witnessing a reaction as it occurred in a native enzyme; hence, excluding X-ray radiation or crystal environment-induced artifacts.
Comparison of the active sites of CO2-loaded native enzyme and the HCAII:bicarbonate complex (Fig. 3) shows that active-site geometry remained unperturbed upon substrate–product conversion. The changes/differences only involve positions of critical water molecules and the binding of CO2/HCO3− along with their diverse environment interactions. HCO3− lies in approximately the same plane defined by CO2 and the catalytic water (Wat-263) within the HCAII:CO2 complex (Fig. 3). The short distance of 2.4 Å between Wat-318 and the zinc-bound oxygen O3 of HCO3− indicates this water will replace the bicarbonate O3 formerly bound to zinc, to become the catalytic water in the next enzymatic cycle. The position abandoned by Wat-318 will in the course of the catalytic cycle be filled by Wat-292, located at the entrance of the cone leading to the active site (Fig. 3).
Fig. 3.

Stereoview of the overlay of the resting enzyme with the substrate and product. Superposition of active sites of resting enzyme (red), enzyme with CO2 substrate (green), and enzyme with bound HCO3− (yellow). Arrows indicate potential pathways of the water molecules from the entrance of the active-site channel toward the catalytic site.
The unexpected recovery of enzyme activity during X-ray exposure may be caused by diverse radiation-induced events. For the reaction to proceed in the enzyme (inactive because of low pH), the zinc-bound water molecule must release a proton. This results in an active enzyme, in which a hydroxide ion reacts with the carbon dioxide to create bicarbonate.
We considered different, mutually nonexclusive mechanisms caused by the photoelectric effect, which is responsible for 84% of the photons absorbed by the matter at these energies (23): (i) a macroscopic increase in pH via radiolysis of water molecules in the bulk solvent caused by the primary and secondary electrons generated during X-ray–sample interaction (24, 25); and/or (ii) a shielding/masking effect by the produced electrons on the positively charged H+ species in the bulk, mimicking a pH increase; and/or (iii) a local event involving photoionization or electronic ionization of the zinc-bound water molecule, creating a hydroxyl radical that either recombines with an electron to yield the required hydroxyl ion or reacts with CO2 to create a bicarbonate radical that would, in turn, recombine with an electron or hydrogen radical from the neighborhood. The local photoionization or electronic ionization of water has a higher probability close to the zinc ion, which would result in generation of primary and secondary electrons in the vicinity of the metal ion because of its larger photoelectric cross-section.
Dubnovitsky et al. (26) observed a similar deprotonation event due to absorbed X-ray dose in crystals of phosphoserine aminotransferase, along with concomitant structural changes in a Schiff base, but the authors did not offer an explanation.
To attempt to distinguish between the possible mechanisms of HCAII activation by X-rays, we repeated the data collection at a 0.94-Å wavelength (Table S2), far from the zinc-absorption edge. At the same absorbed dose (30% of the Henderson limit), we found no significant difference in the substrate to product ratio with respect to the 1.278 Å dataset (70%:30% CO2/HCO3− at 0.94 Å compared with 65%:35% CO2/HCO3− at 1.278 Å). We additionally observe that the process does not depend on the dose rate: the 2 experiments were conducted by using beams that differed by more than one order of magnitude in flux (3.6 × 1010 at 1.278 Å and 7.2 × 1011 photons per second at 0.94 Å, respectively). The finding that the photon energy (at the zinc-absorption edge in the first case and far away from it in the second) does not influence the substrate to product ratios leads to the conclusion that the zinc ion and its interaction with X-rays do not play a pivotal role the process. This result does not clearly indicate the mechanism of the enzyme activation, but it does exclude a “local” effect affecting only the active-site zinc ion and bound water molecule, therefore promoting the hypothesis based on “bulk” effects resulting in a macroscopic increase of pH in the crystal.
Despite the high dissociation constant for CO2 (100 mM; ref. 5) and the very high enzyme turnover rate, we generated a stable complex and its X-ray structure by pressurizing crystals of HCAII with CO2, thus inactivating the enzyme by concomitantly lowering the pH. We show that CO2 has a distinct mode of binding in the hydrophobic pocket of HCAII, which explains many previous functional studies of active-site mutants. The enzyme reactivates in the crystal upon absorption of X-ray dose well below the Henderson limit, yielding an enzyme–product complex.
Radiation damage is a recognized phenomenon that damages and modifies macromolecular structure under crystallographic investigation (34). Here, we report a radiation-driven event occurring during X-ray data collection, eventually leading to enzyme activation where X-ray-activated carbonic anhydrase drives the conversion of CO2 to HCO3− in a single crystal kept at cryogenic temperature, and this is put forward as the most probable mechanism of activation radiolysis of water molecules in the bulk solvent caused by the primary and secondary electrons generated during X-ray–sample interaction.
Materials and Methods
Protein Production, Crystallization, and Pressurization.
The protein was expressed in Escherichia coli and purified as described (27). Crystals of HCAII were grown in a period of 3–4 days by using the hanging drop vapor diffusion method at 18.4 °C by mixing a 1:1 volume ratio of protein solution (20 mg/mL HCAII in 20 mM Tris, pH 7.5) with mother liquor [2.4 M (NH4)2SO4 in 100 mM Tris buffer, pH 8.5]. Before pressurization, the crystal was transferred to a drop of mother liquid supplemented with 20% glycerol. The drop was then overlaid with paraffin oil, transferred through the oil to the pressurization device (28), and pressurized for 10 min at 10 bar. After pressurization, the crystal was flash-cooled into liquid nitrogen within approximately 2 seconds from the release of pressure.
Structure Determination of Complexes.
X-ray diffraction data were collected at 100 K at the XRD1 beamline of the Elettra synchrotron source (Trieste, Italy) by using a Mar CCD detector. Data were processed by using Denzo (29) and programs from the CCP4 suite (30). The structure refinement was performed with Refmac5 (30) using PDB code 2CBA without waters as a starting model. Manual model building was carried out with Coot (31), and pictures were produced with Pymol (32).
Movie Data Collection and Data Treatment Strategy.
The data were collected at the European Molecular Biology Laboratory (EMBL) X11 beamline at the DORIS storage ring [Deutsches Elektronen Synchotron (DESY), Hamburg, Germany] by using a 1.278-Å wavelength, and at the European Synchotron Radiation Facility (ESRF, Grenoble, France) ID14-4 by using a 0.94-Å wavelength. In both cases, data collection strategy and data treatment were very similar.
For experiments at the EMBL/DESY X11 beamline, the flux of the X-ray beam was measured with a calibrated diode and found to be 3.6 × 1010 photons per second with a slit size of 0.3 × 0.3 mm2 and a current of 108 mA in the storage ring. The crystal used was chosen to be small enough to stay completely in the beam during the full data collection. A total of 4,770 diffraction images of 1° rotation each were collected from a single HCAII crystal pressurized with CO2 at 10 bars for 10 min. The size of the crystal was measured with a microscope to be 0.3 × 0.1 × 0.02 mm3, and the dose was calculated using RADDOSE (33).
At the ESRF ID14-4 beamline, the diffraction data were collected from a smaller crystal (0.1 × 0.14 × 0.01 mm3) and with smaller slits (0.15 × 0.1 mm2). The flux used for the dose calculations was taken as the average of the flux for each image (at ID14-4, the integrated counts are recorded for each image) of the particular dataset.
In both cases, the data were integrated with XDS (35) and scaled with Scala (30) in overlapping batches of 180° rotation. Each batch began at a phi-value that was 45° larger than the previous one, leading to a total number of 103 batches in case of the dataset collected at X11 at 1.278 Å. Because of the higher flux at ID14-4 compared with X11 (EMBL/DESY), only 11 batches of data were generated by using the same procedure for the dataset collected at 0.94 Å.
For both datasets, each structure was refined starting from the model derived from the data of the previous batch. The relative occupancy of CO2 and bicarbonate was determined by adjusting the occupancy of the CO2 (in steps of 5%) to keep its B factors approximately constant throughout all models. For the first batch, the model with the PDB code 2CBA (without water molecules) was used as a starting model. The movies present the refined models for the 103 and 11 data batches, respectively, together with the final 2|Fo| − |Fc| map contoured at 1σ level (Movie S1 and Movie S2).
Supplementary Material
Acknowledgments.
We thank D. Lamba, A. Cassetta, and L. Barba (Consiglio Nazionale delle Ricerche, Elettra, Sinchrotrone Trieste, Italy) for their help in collecting many of the diffraction datasets. We thank E. Garman (Laboratory for Molecular Biology, Oxford University, Oxford), D. Blaas (Max F. Perutz Laboratories, University of Vienna, Vienna), and C. Kratky (Department of Chemistry, University of Graz, Graz, Austria) for constructive discussions and critical reading of the manuscript. Many thanks to beamline scientists at EMBL Hamburg (Andrea Schmidt) and ESRF (Andrew McCarthy) for help with data collection setup. B.S. was a recipient of a postdoctoral fellowship funded by the European Commission (EC) 5th Framework Program Research Training Network CYTONET (contract HPRN-CT-2000-00096). Postdoctoral fellowship of M.P. was supported by the EC 5th Framework Program Research and Technological Development Project EXMAD (contract HPRI-CT-1999-50015) and by the EC 6th Framework Program Integrated Research project Biocrystallography (X) on a Highly Integrated Technology Platform for European Structural Genomics (BIOXHIT; contract LHSG-CT-2003-503420).
Note Added in Proof.
While this manuscript was under review, a crystallographic study of carbonic anhydrase in complex with substrate was published by Domsic et al. (36).
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2vva and 2vvb).
This article contains supporting information online at www.pnas.org/cgi/content/full/0904184106/DCSupplemental.
References
- 1.Alexander RS, Nair SK, Christianson DW. Engineering the hydrophobic pocket of carbonic anhydrase II. Biochemistry. 1991;30:11064–11072. doi: 10.1021/bi00110a008. [DOI] [PubMed] [Google Scholar]
- 2.Bottoni A, Lanza CZ, Miscione GP, Spinelli D. New model for a theoretical density functional theory investigation of the mechanism of the carbonic anhydrase: How does the internal bicarbonate rearrangement occur? J Am Chem Soc. 2004;126:1542–1550. doi: 10.1021/ja030336j. [DOI] [PubMed] [Google Scholar]
- 3.Håkansson K, Carlsson M, Svensson LA, Liljas A. Structure of native and apo carbonic anhydrase II and structure of some of its anion-ligand complexes. J Mol Biol. 1992;227:1192–1204. doi: 10.1016/0022-2836(92)90531-n. [DOI] [PubMed] [Google Scholar]
- 4.Huang S, Sjöblom B, Sauer-Eriksson AE, Jonsson BH. Organization of an efficient carbonic anhydrase: Implications for the mechanism based on structure-function studies of a T199P/C206S mutant. Biochemistry. 2002;41:7628–7635. doi: 10.1021/bi020053o. [DOI] [PubMed] [Google Scholar]
- 5.Krebs JF, Rana F, Dluhy RA, Fierke CA. Kinetic and spectroscopic studies of hydrophilic amino acid substitutions in the hydrophobic pocket of human carbonic anhydrase II. Biochemistry. 1993;32:4496–4505. doi: 10.1021/bi00068a004. [DOI] [PubMed] [Google Scholar]
- 6.Kumar V, Kannan KK. Enzyme-substrate interactions. Structure of human carbonic anhydrase I complexed with bicarbonate. J Mol Biol. 1994;241:226–232. doi: 10.1006/jmbi.1994.1491. [DOI] [PubMed] [Google Scholar]
- 7.Liang JY, Lipscomb WN. Binding of substrate CO2 to the active site of human carbonic anhydrase II: A molecular dynamics study. Proc Natl Acad Sci USA. 1990;87:3675–3679. doi: 10.1073/pnas.87.10.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mertz KM. CO2 binding to human carbonic anhydrase II. J Am Chem Soc. 1991;113:406–411. [Google Scholar]
- 9.Nair SK, Calderone TL, Christianson DW, Fierke CA. Altering the mouth of a hydrophobic pocket. Structure and kinetics of human carbonic anhydrase II mutants at residue Val-121. J Biol Chem. 1991;266:17320–17325. [PubMed] [Google Scholar]
- 10.Nair SK, Christianson DW. Structural consequences of hydrophilic amino acid substitutions in the hydrophobic pocket of human carbonic anhydrase II. Biochemistry. 1993;32:4506–4514. doi: 10.1021/bi00068a005. [DOI] [PubMed] [Google Scholar]
- 11.Xue Y, Liljas A, Jonsson BH, Lindskog S. Structural analysis of the zinc hydroxide-Thr-199-Glu-106 hydrogen-bond network in human carbonic anhydrase II. Proteins. 1993;17:93–106. doi: 10.1002/prot.340170112. [DOI] [PubMed] [Google Scholar]
- 12.Xue Y, et al. Crystallographic analysis of Thr-200–>His human carbonic anhydrase II and its complex with the substrate, HCO3. Proteins. 1993;15:80–87. doi: 10.1002/prot.340150110. [DOI] [PubMed] [Google Scholar]
- 13.Silverman DN, Lindskog S. The catalytic mechanism of carbonic anhydrase: Implications of a rate-limiting protolysis of water. Acc Chem Res. 1988;21:30–36. [Google Scholar]
- 14.Tilton RF, Jr, Kuntz ID., Jr Nuclear magnetic resonance studies of xenon-129 with myoglobin and hemoglobin. Biochemistry. 1982;21:6850–6857. doi: 10.1021/bi00269a035. [DOI] [PubMed] [Google Scholar]
- 15.Tilton RF, Jr, Kuntz ID, Jr, Petsko GA. Cavities in proteins: Structure of a metmyoglobin-xenon complex solved to 1.9 A. Biochemistry. 1984;23:2849–2857. doi: 10.1021/bi00308a002. [DOI] [PubMed] [Google Scholar]
- 16.Schiltz M, Prange T, Fourme R. On the preparation and X-ray data collection of isomorphous xenon derivatives. J Appl Cryst. 1994;27:950–960. [Google Scholar]
- 17.Burzlaff NI, et al. The reaction cycle of isopenicillin N synthase observed by X-ray diffraction. Nature. 1999;401:721–724. doi: 10.1038/44400. [DOI] [PubMed] [Google Scholar]
- 18.Fierke CA, Calderone TL, Krebs JF. Functional consequences of engineering the hydrophobic pocket of carbonic anhydrase II. Biochemistry. 1991;30:11054–11063. doi: 10.1021/bi00110a007. [DOI] [PubMed] [Google Scholar]
- 19.Harding MM. The geometry of metal-ligand interactions relevant to proteins. Acta Crystallogr D. 1999;55:1432–1443. doi: 10.1107/s0907444999007374. [DOI] [PubMed] [Google Scholar]
- 20.Gugganig M. Graz, Austria: Univ of Graz; 2001. Studies in protein crystallography: Atomic resolution studies of the hydroxynitrile lyase from Hevea Brasiliensis, Xenon as a heavy atom derivative and MAD with xenon at the LIII edge. PhD thesis. [Google Scholar]
- 21.Berglund GI, et al. The catalytic pathway of horseradish peroxidase at high resolution. Nature. 2002;417:463–468. doi: 10.1038/417463a. [DOI] [PubMed] [Google Scholar]
- 22.Henderson R. Cryo-protection of protein crystals against radiation damage in electron and X-ray diffraction. Proc Biol Sci. 1990;241:6–8. [Google Scholar]
- 23.Murray JW, et al. Parameters affecting the X-ray dose absorbed by macromolecular crystals. J Synchrotron Radiat. 2005;12:268–275. doi: 10.1107/S0909049505003262. [DOI] [PubMed] [Google Scholar]
- 24.Carugo O, Djinovic Carugo K. When X-rays modify the protein structure: Radiation damage at work. Trends Biochem Sci. 2005;30:213–219. doi: 10.1016/j.tibs.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 25.Southworth-Davies RJ, Garman EF. Radioprotectant screening for cryocrystallography. J Synchrotron Radiat. 2007;14:73–83. doi: 10.1107/S0909049506044177. [DOI] [PubMed] [Google Scholar]
- 26.Dubnovitsky AP, Ravelli RB, Popov AN, Papageorgiou AC. Strain relief at the active site of phosphoserine aminotransferase induced by radiation damage. Protein Sci. 2005;14:1498–1507. doi: 10.1110/ps.051397905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xue Y. Umeå, Sweden: Umeå University; 1992. Engineering of carbonic anhydrase: Structural and functional analyses of active-site mutants of human isoenzyme II. PhD thesis. [Google Scholar]
- 28.Djinovic Carugo K, Everitt P, Tucker P. A cell for producing xenon-derivatives crystals for cryocrystallographic analysis. J Appl Crystallogr. 1998;31:812–814. [Google Scholar]
- 29.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW, Sweet RM, editors. Methods in Enzymology. Vol 276. San Diego: Academic; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 30.CCP4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 31.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 32.DeLano WL. The PyMOL Molecular Graphics System. Palo Alto, CA: DeLano Scientific; 2000. Available at www.pymol.org. [Google Scholar]
- 33.Murray JW, Garman EF, Ravelli RBG. X-ray absorption by macromolecular crystals: The effects of wavelength and crystal composition on absorbed dose. J Appl Crystallogr. 2004;37:513–522. [Google Scholar]
- 34.Ravelli RB, Garman EF. Radiation damage in macromolecular cryocrystallography. Curr Opin Struct Biol. 2006;16:624–629. doi: 10.1016/j.sbi.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 35.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- 36.Domsic JF, et al. Entrapment of carbon dioxide in the active site of carbonic anhydrase. J Biol Chem. 2008;283:30766–30771. doi: 10.1074/jbc.M805353200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liljas A, et al. Crystal structure of human carbonic anhydrase C. Nat New Biol. 1972;235:131–137. doi: 10.1038/newbio235131a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}