

The structure of ribonuclease A in complex with uridine 5′-phosphate and uridine 5′-diphosphate has been determined at 1.4 Å resolution in order to facilitate the rational design of selective and potent inhibitors.
Keywords: ribnuclease A, uridine 5′-phosphate, uridine 5′-diphosphate, inhibitor design
Abstract
In the quest for the rational design of selective and potent inhibitors for members of the pancreatic ribonuclease A (RNase A) family of biomedical interest, the binding of uridine 5′-phosphate (U5P) and uridine 5′-diphosphate (UDP) to RNase A have been investigated using kinetic studies and X-ray crystallography. Both nucleotides are competitive inhibitors of the enzyme, with K i values of 4.0 and 0.65 mM, respectively. They bind to the active site of the enzyme by anchoring two molecules connected to each other by hydrogen bonds and van der Waals interactions. While the first of the inhibitor molecules binds with its nucleobase in the pyrimidinyl-binding subsite, the second is bound at the purine-preferring subsite. The unexpected binding of a pyrimidine at the purine-binding subsite has added new important elements to the rational design approach for the discovery of new potent inhibitors of the RNase A superfamily.
1. Introduction
Ribonucleases (RNases) are enzymes that catalyze the degradation of RNA. The most well studied RNase is the mammalian pancreatic ribonuclease A (RNase A; Raines, 1998 ▶). The RNA-binding site of RNase A is a deep groove in the molecular surface (Fig. 1 ▶) lined with positively charged residues that bind the phosphate groups of the substrate RNA, mainly by electrostatic interactions. Along this deep cleft, several subsites have been identified that accommodate the phosphate groups, the riboses and the bases of RNA. These subsites are denoted P0…Pn, R0…Rn and B0…Bn, respectively, where n indicates the position of the group with respect to the position at which phosphodiester-bond cleavage occurs (n = 1; Raines, 1998 ▶). RNase A has a preference for purines at subsite B2 and binds pyrimidines at subsite B1 (Raines, 1998 ▶).
Figure 1.

A schematic diagram of the RNase A molecule with U5P (yellow) and UDP (cyan) molecules superimposed bound at the active site. The molecular surface and secondary structure of the enzyme are also shown. Subsites P0 (Lys66), B2 (Asn67, Gln69, Asn71, Glu111, His119), P1 (Gln11, His12, Lys41, His119), B1 (Val43, Asn44, Thr45, Phe120, Ser123) and P2 (Lys7, Arg10) are labelled and marked on the molecular surface with different colours.
In recent years, the members of the RNase A superfamily have attracted considerable biomedical interest as targets for the discovery of new pharmaceuticals for the treatment of inflammatory disorders and cancer. The fact that the ribonucleolytic activity of these enzymes is a prerequisite for the pathological activities related to the proteins of this family has triggered a structure-assisted approach to the design of inhibitors, mainly for three human RNases: angiogenin (RNase 5; Ang), a potent inducer of neovascularization that manifests pathologically during tumour growth and metastasis, and two eosinophil RNases that have been implicated in inflammation and viral replication, eosinophil-derived neurotoxin (EDN) and eosinophil cationic protein (ECP) (Russo et al., 2001 ▶). Initial efforts have mostly targeted the parental protein RNase A, since it is more amenable to binding studies and its active site is conserved in all members of this superfamily. Several structures of RNase A in complex with phosphopurine nucleotide derivatives have been reported to date by X-ray crystallography or NMR [d(Ap)4 (McPherson et al., 1986 ▶); d(CpA) (Zegers et al., 1994 ▶; Toiron et al., 1996 ▶); UpcA (Richards & Wyckoff, 1973 ▶; Gilliland et al., 1994 ▶); 2′,5′-CpA (Wodak et al., 1977 ▶; Toiron et al., 1996 ▶); d(ApTpApA) (Fontecilla-Camps et al., 1994 ▶); ppA-3′-p, ppA-2′-p (Leonidas et al., 1997 ▶); 3′,5′-ADP, 2′,5′-ADP, 5′-ADP (Leonidas et al., 2003 ▶); dUppA-3′-p (Jardine et al., 2001 ▶); pdUppA-3′-p (Leonidas et al., 1999 ▶); AMP, IMP (Hatzopoulos et al., 2005 ▶) and d(GMP) (Larson et al., 2007 ▶)]. Most of these studies have focused on the purine-preferring subsite B2 and structure-based inhibitor-design efforts have been directed towards ligand molecules that contain various adenine derivatives, with the aim of exploiting the potential interactions offered by protein residues at this subsite. This approach has generated pdUppA-3′-p, the most potent low-molecular-weight inhibitor of RNase A reported to date (K i = 27 nM), which is also effective against two major nonpancreatic RNases, Ang and EDN (Russo et al., 2001 ▶; Leonidas et al., 1999 ▶).
In recent years, inhibitor-discovery efforts have shifted towards pyrimidine analogues that mostly explore the interactions with residues in subsite B1, generating several compounds which also inhibit angiogenin as well as RNase A (Maiti et al., 2006 ▶; Leonidas et al., 2006 ▶; Ghosh et al., 2008 ▶). However, the molecular recognition of pyrimidine derivatives by subsite B1 has only been studied in the complexes of RNase A with U-2′-p and U-3′-p (Leonidas et al., 2003 ▶); the structural mode of binding of 5′-phosphopyrimidines has not yet been analyzed. With the aim of studying the molecular recognition of 5′-phosphouridines by RNase A, we have determined the crystal structures of the U5P–RNase A and UDP–RNase A complexes and have studied the inhibitory potency of these ligands towards RNase A in solution.
2. Materials and methods
Bovine pancreatic RNase A (type XII-A), U5P, UDP, cytidine 2′,3′-cyclic phosphate (C>p) and other chemicals were obtained from Sigma–Aldrich (Athens, Greece). The enzymatic activity of RNase A was measured using a spectrophotometric method at 303 K in 0.1 M MES–NaOH buffer pH 6.0, 0.1 M NaCl with an enzyme concentration of 1 µM (Hatzopoulos et al., 2005 ▶). The inhibition constants (K i) were determined by the method of Dixon (1953 ▶) using nonlinear regression analysis with the program GraFit (Leatherbarrow, 2007 ▶).
Crystals of RNase A were grown at 289 K using the hanging-drop vapour-diffusion technique as described previously (Leonidas et al., 1997 ▶). Briefly, drops formed by mixing equal volumes of an RNase A solution (18 mg ml−1) in water and reservoir solution [20 mM sodium citrate buffer 5.5 and 25%(w/v) PEG 4000] were equilibrated against reservoirs containing 25%(w/v) PEG 4000 and 20 mM sodium citrate buffer pH 5.5. Single crystals (800 × 400 × 50 µm) appeared after 7–10 d at 289 K. Crystals of the inhibitor complexes were obtained by soaking RNase A crystals (Leonidas et al., 1997 ▶) in a solution of the crystallization medium [20 mM sodium citrate pH 5.5, 25%(w/v) PEG 4000] containing either 50 mM U5P for 45 h or 50 mM UDP for 2.5 h prior to data collection. Diffraction data to 1.4 Å resolution were collected on station PX10.1 (λ = 1.0448 Å), SRS Daresbury, England at 100 K [using a solution of 20 mM sodium citrate buffer pH 5.5, 25%(w/v) PEG 4000 and 20%(w/v) MPD as a cryoprotecting medium] on a MAR 225 CCD detector using the MAR CCD diffraction data-collection protocol. The exposure time was 10 s per image, the oscillation range was 0.8° and a total of 162 and 180 images were collected for the U5P and UDP complexes, respectively. Data were processed using the HKL package (Otwinowski & Minor, 1997 ▶) and the program TRUNCATE (French & Wilson, 1978 ▶). Phases were obtained using the structure of free RNase A (Leonidas et al., 2006 ▶) as a starting model. Alternate cycles of manual building with the program Coot (Emsley & Cowtan, 2004 ▶) and refinement using the maximum-likelihood target function and anisotropic temperature-factor refinement of all non-H atoms with the program REFMAC (Murshudov et al., 1997 ▶) improved the model. Inhibitor molecules were included in during the final stages of the refinement procedure using models from the REFMAC library. Details of data-processing and refinement statistics are provided in Table 1 ▶.
Table 1. Crystallographic statistics.
Values in parentheses are for the outermost shell.
| RNase A–U5P | RNase A–UDP | |
|---|---|---|
| Space group | C2 | C2 |
| Unit-cell parameters (Å, °) | a = 100.035, b = 32.299, c = 72.475, α = 90.00, β = 90.91, γ = 90.00 | a = 100.003, b = 32.337, c = 72.299, α = 90.00, β = 90.72, γ = 90.00 |
| Matthews coefficient (Å3 Da−1) | 2.10 | 2.09 |
| Resolution (Å) | 30.0–1.40 (1.42–1.40) | 30.0–1.40 (1.42–1.40) |
| Reflections measured | 414856 | 297437 |
| Unique reflections | 44340 (2291) | 45026 (2290) |
| Rmerge† | 0.106 (0.263) | 0.044 (0.111) |
| Completeness (%) | 95.3 (99.8) | 97.8 (100.0) |
| 〈I/σ(I)〉 | 34.5 (4.1) | 29.0 (8.2) |
| Rcryst‡ | 0.208 (0.217) | 0.188 (0.186) |
| Rfree§ | 0.254 (0.273) | 0.225 (0.270) |
| No. of solvent molecules | 358 | 367 |
| R.m.s. deviation from ideality | ||
| In bond lengths (Å) | 0.009 | 0.009 |
| In angles (°) | 1.4 | 1.4 |
| Average B factor (Å2) | ||
| Protein atoms (mol A/mol B) | 19.9/19.9 | 17.6/16.3 |
| Solvent molecules | 32.4 | 31.7 |
| Ligand atoms (mol A/mol B/mol C/mol D) | 24.2/20.9/24.7 | 26.5/22.7/24.1/28.2 |
R
merge = 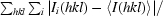
 , where I
i(hkl) and 〈I(hkl〉 are the ith and the mean measurements of the intensity of reflection hkl.
, where I
i(hkl) and 〈I(hkl〉 are the ith and the mean measurements of the intensity of reflection hkl.
R
cryst = 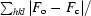
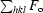 , where F
o and F
c are the observed and calculated structure-factor amplitudes of reflection hkl, respectively.
, where F
o and F
c are the observed and calculated structure-factor amplitudes of reflection hkl, respectively.
R free is the same as R cryst but for a randomly selected 5% subset of reflections not used in the refinement (Brünger, 1992 ▶).
The program PROCHECK (Laskowski et al., 1993 ▶) was used to assess the quality of the final structures. Analysis of the Ramachandran (ϕ–ψ) plot showed that all residues lay in the allowed regions. Solvent-accessible areas were calculated by the program NACCESS (Hubbard & Thornton, 1993 ▶). The atomic coordinates and structure factors of the two complexes have been deposited in the Protein Data Bank (http://www.pdb.org) with accession numbers 3dxg and 3dxh. Figures were prepared with the program PyMOL (DeLano, 2002 ▶).
3. Results and discussion
Both ligands are competitive inhibitors of the enzyme with respect to C>p. U5P is a moderate inhibitor (K i = 4.00 ± 0.41 mM), while UDP is more potent (K i = 0.65 ± 0.06 mM). The complex structures are very similar to that of the free RNase A (Leonidas et al., 2006 ▶) and the binding of the inhibitors did not cause any significant conformational change.
In the monoclinic crystals of RNase A there are two protein molecules in the crystallographic asymmetric unit (Leonidas et al., 2006 ▶). Two ligand molecules were found bound in the active site of the first protein molecule of the asymmetric unit, but only one was found in the second. This can be attributed, as previously (Leonidas et al., 2006 ▶; Hatzopoulos et al., 2005 ▶), to the impediments imposed by the crystal lattice that limit access to the active site of the second protein molecule in this crystal form. Our structural analysis was based on the ligand complex with the first RNase A molecule. All the atoms of U5P and UDP are well defined in the electron-density map of the protein complexes (Figs. 2 ▶ a and 2 ▶ b).
Figure 2.

The σA 2|F o| − |F c| electron-density map calculated from the RNase A model before incorporating the coordinates of U5P (a) or UDP (b) is contoured at the 1.0σ level and the refined structure of both the inhibitor molecules in the active site is shown. The numbering scheme used for each inhibitor molecule is also shown. Diagrams of the interactions between RNase A and U5P in mol A (c) and mol B (d) and UDP in mol A (e) and mol B (f) in the active site are shown. A standard colouring scheme is used (yellow for carbon, blue for nitrogen, red for oxygen and cyan for water molecules) and hydrogen-bond interactions are represented as dashed lines. C atoms in the inhibitor molecules are shown in grey.
Upon binding to RNase A, each inhibitor molecule adopts a different conformation. However, the glycosyl torsion angle χ′ in U5P and UDP adopts the frequently observed (Moodie & Thornton, 1993 ▶) anti conformation. The ribose of U5P adopts the two most preferred conformations for free and protein-bound nucleotides (Moodie & Thornton, 1993 ▶): C3′-endo and C2′-endo. The rest of the backbone torsion angles are in the common range for protein-bound pyrimidines (Moodie & Thornton, 1993 ▶). In UDP, the ribose of the three inhibitor molecules adopts the C1′-exo, C2′-endo and C3′-endo puckering, respectively, which together with the rest of the backbone and phosphate torsion angles are also in the preferred range for protein-bound pyrimidines (Moodie & Thornton, 1993 ▶).
The two inhibitors bind at the active site with one molecule in subsite B1 and the other in subsite B2 (referred to hereafter as mol A and mol B, respectively). In the U5P complex the uracil engages in hydrogen-bond interactions with Thr45 at subsite B1 (Table 2 ▶), the residue that is responsible for the pyrimidine specificity of this site (Raines, 1998 ▶), while the rest of the molecule is involved in van der Waals interactions, mainly with His12 and Phe120 (Fig. 2 ▶ c). The 5′-phosphate group moves away from subsite P1 towards P0 and the closest distance between the phosphate and the side chain of Lys66 (the sole component of subsite P0; Raines, 1998 ▶) is 4.9 Å. U5P mol B is bound with the uracil ring almost parallel to the side chain of His119 (Fig. 2 ▶ d) and is involved in hydrogen bonding and van der Waals interactions with all residues of subsite B2 (Table 2 ▶). The 5′-phosphate group binds at P1 and is hydrogen bonded to His119 (Fig. 2 ▶ d). In addition to the interactions between the ligands and the protein, the two ligands also interact with each other. Thus, the 5′-phosphate group of U5P mol B forms a hydrogen bond to the 3′-hydroxyl group of the ribose of U5P mol A, while two water molecules mediate polar interactions between the 5′-phosphate of U5P mol B and the 2′- and 3′-hydroxyl groups of the ribose of U5P mol A. Upon binding to RNase A, the two U5P molecules displace 13 water molecules from the active site of the unliganded enzyme (Leonidas et al., 2006 ▶). The two ligand molecules have a total solvent-accessible surface of 829 Å2, which shrinks to 326 Å2 upon binding to RNase A. Polar and nonpolar groups contribute almost equally to the buried surface (233 and 270 Å2, respectively).
Table 2. Potential hydrogen bonds of U5P and UDP with RNase A in the crystal.
Values in parentheses are distances in Å. Asterisks indicate residues from a symmetry-related protein molecule.
| RNase A–U5P complex | RNase A–UDP complex | ||||
|---|---|---|---|---|---|
| U5P/UDP atom | Mol A | Mol B | Mol A | Mol B | Mol C |
| O2 | Thr45 N (2.9) | Asn71 Nδ2 (2.9) | Thr45 N (2.9) | Asn71 Nδ2 (2.8) | Asn34 Nδ2 (2.7) |
| O2 | Glu111 O∊2 (3.2) | Gln69 N∊2 (3.3) | |||
| O4 | Water7 (3.2) | Asn67 Oδ1 (3.2) | Water35 (2.9) | Asn67 Oδ1 (3.1) | Water108 (2.7) |
| O4 | Water43 (2.8) | Asn67 Nδ2 (3.4) | Water49 (3.3) | Asn67 Nδ2 (3.1) | |
| O4 | Water15 (2.8) | Water10 (2.7) | |||
| N3 | Thr45 Oγ1 (2.8) | Gln69 O∊1 (3.4) | Thr45 Oγ1 (2.7) | Tyr76 Oη* (2.7) | |
| O2′ | His12 N∊2 (3.3) | His12 N∊2 (3.2) | Arg10 Nη1 (3.2) | ||
| O2′ | Water20 (3.1) | Lys41 Nζ (3.0) | |||
| O2′ | Water207 (3.0) | Water371 (3.0) | |||
| O2′ | Water215 (2.8) | ||||
| O3′ | Water207 (2.7) | Glu111 O∊1 (3.1) | |||
| O3′ | Water286 (2.7) | Water230 (3.0) | |||
| O3′ | Water364 (3.2) | ||||
| O4′ | Water38 (3.2) | Water20 (3.2) | Glu2 O∊1 (3.2) | ||
| O5′ | Water146 (3.3) | ||||
| O1A | Water146 (2.6) | Water263 (2.7) | Water355 (2.8) | ||
| O1P/O1B | Water175 (2.8) | Water286 (3.0) | Water221 (2.9) | ||
| O2P/O2B | Water236 (2.9) | Water207 (3.0) | His119 Nδ1 (2.9) | ||
| O2P/O2B | Water244 (2.8) | Water371 (2.7) | |||
| O3P/O3B | His119 Nδ1 (2.5) | Water221 (2.9) | |||
| O3P/O3B | Water120 (3.4) | Water20 (2.7) | |||
| O3P/O3B | Water236 (3.4) | Water207 (3.2) | |||
On binding, the two UDP molecules displace 15 water molecules from the active site of the free enzyme (Leonidas et al., 2006 ▶) and, like U5P, anchor one uracil to subsite B2 (Fig. 2 ▶ e) and another to B1 (Fig. 2 ▶ f). The β-phosphate rather than the α-phosphate group of the 5′-pyrophosphate group of UDP mol B binds to subsite P1, forming hydrogen-bond interactions with the side chain of His119 (Fig. 2 ▶ e). This phosphate group also engages in hydrogen bonds to the two hydroxyl groups of the ribose of UDP mol A, similarly to U5P (Table 2 ▶). The 5′-pyrophosphate of inhibitor mol A projects towards the solvent (the closest RNase A residue is Lys66, which is 5.6 Å away). Upon binding to RNase A, the UDP molecules become buried. Thus, the combined solvent-accessible surface of the two free ligand molecules is 899 Å2. When bound, this molecular surface shrinks to 361 Å2, indicating that 60% of the UDP surface becomes buried. The greatest contribution comes from the nonpolar groups, which contribute 381 Å2 (71%) of the surface that becomes inaccessible. The shape-correlation statistic Sc, which is used to quantify the shape complementarity of interfaces and gives an idea of the ‘goodness of fit’ between two surfaces (Lawrence & Colman, 1993 ▶), is 0.67 and 0.70 for the combined molecular surface of the two U5P molecules and the two UDP molecules, respectively.
A third UDP molecule was found to be bound in a location close to the N-terminus of RNase A (Fig. 3 ▶). There, it participates in a hydrogen-bond network of interactions with residues from both RNase A molecules of the asymmetric unit as well as with residues from a symmetry-related protein molecule (Table 2 ▶). X-ray diffraction data collected from RNase A crystals soaked with 20 mM UDP did not show any ligand binding at this location, indicating that this binding is nonspecific and can be attributed to the high concentration of UDP (50 mM) used for soaking the RNase A crystals.
Figure 3.

Stereo diagram of the interactions of UDP bound at the interface of the two RNase A molecules of the crystallographic asymmetric unit. Residues labelled with asterisks are from a symmetry-related molecule.
Crystallographic data from RNase A crystals soaked with 5 mM of either U5P or UDP for 1 h showed only one inhibitor molecule (mol A) bound at the active site. This indicated that the inhibitory effect of U5P and UDP in solution probably arises from the binding of mol A at the active site. Both uridylyl compounds bind similarly at the active site (Fig. 4 ▶ a). The differences in the potency of UDP and U5P might derive from a combination of positive factors associated with the addition of the extra phosphate group (an enthalpic gain arising from new van der Waals interactions between the β-phosphate and residues of the P1 subsite and the entropic advantage of the increased structural constraints on the α-phosphate) partially counterbalanced by enthalpic losses arising from this same conformational restriction of the α-phosphate, which would prevent this group from optimizing its interactions as in the U5P complex.
Figure 4.

Structural comparisons of the complexes. (a) RNase A–U5P (yellow) and RNase A–UDP (green). (b) RNase A–U-2′-p (green) and RNase A–U5P (white). (c) RNase A–U-3′-p (green) and RNase A–U5P (white). (d) RNase A–UDP (green) and RNase A pdUppA-3′-p (yellow).
U-2′-p and U-3′-p are potent inhibitors of RNase A, with K i values of 7 and 82 µM, respectively (Anderson et al., 1968 ▶). Superposition of the U5P and UDP complexes onto the U-2′-p or U-3′-p complexes (Leonidas et al., 2003 ▶) reveals that the uracil moieties bind similarly in all four complexes at subsite B1 (Fig. 4 ▶ b and 4 ▶ c). However, while in the U-2′-p and U-3′-p complexes the phosphate group binds at P1, in the U5P and UDP complexes it binds away from this site (Figs. 4 ▶ b and 4 ▶ c). Instead, it is the phosphate group of the U5P or UDP molecule that binds at subsite B2 that binds close to P1. Interestingly, although the structures of the RNase A complexes with either U-2′-p or U-3′-p were determined from protein crystals soaked in a solution containing 50 mM of each inhibitor for several hours (Leonidas et al., 2003 ▶), only one inhibitor molecule was found to be bound at the active site. Therefore, the question raised from the present structural study is why U-2′-p and U-3′-p are more potent inhibitors than U5P and UDP when two molecules of the latter instead of one molecule bind at the active site. The answer may lie in the number of interactions of the phosphate group at subsite P1. In the U-2′-p and U-3′-p complexes this group forms many more interactions than in the U5P and UDP complexes. Thus, while in the U-2′-p and U-3′-p complexes it forms hydrogen-bond interactions with Gln11, His12, Lys41, His119 and Phe120 (Leonidas et al., 2003 ▶), in the U5P and UDP complexes it only forms a hydrogen bond to His119. Therefore, it seems that these additional hydrogen-bond interactions in the U-2′-p and U-3′-p complexes counterbalance the interactions of the second uracil at subsite B2 in the U5P and UDP complexes. Differences in the potency between U-2′-p and U-3′-p or 2′-CMP and 3′-CMP (U-2′-p and 2′-CMP have a tenfold smaller K i for RNase A than U-3′-p and 3′-CMP, respectively; Anderson et al., 1968 ▶) have similarly been attributed to differences in the phosphate binding at subsite P1 (Leonidas et al., 2003 ▶; Howlin et al., 1987 ▶; Zegers et al., 1994 ▶).
A structural comparison of the UDP binding to that of pdUppA-3′-p (the most potent ribonucleolytic inhibitor found to date) reveals that the two ligands follow a similar binding pattern and the uracil ring of UDP at B2 superimposes onto the adenine ring of pdUppA-3′-p (Fig. 4 ▶ d). The pyrophosphate group of UDP does not bind at the same location as the analogous group of pdUppA-3′-p, but this is probably a consequence of steric impediments since it is not covalently bound to the second inhibitor molecule at the active site of the enzyme as is the case for pdUppA-3′-p. However, it is still the β-phosphate rather than the α-phosphate that binds at P1. Kinetic experiments have shown that pyrophosphate groups enhance binding by 300-fold, 130-fold and 790-fold compared with monophosphate in RNase A, EDN and RNase-4, respectively (Russo & Shapiro, 1999 ▶). The binding of the β-phosphate rather than the α-phosphate group at subsite P1 of a pyrophosphonucleotide bound at B2 has been observed with pdUppA-3′-p (Leonidas et al., 1999 ▶), dUppA-3′-p (Jardine et al., 2001 ▶), ppA-3′-p, ppA-2′-p (Leonidas et al., 1997 ▶) and 5′-ADP (Leonidas et al., 2003 ▶), showing that this structural feature confers higher affinity towards the enzyme. It is the first time that this has been shown for uridine nucleotides, although it has a smaller effect on the inhibition constants (UDP is sixfold more potent than U5P). Furthermore, the adenosine glycosyl bond torsion angle χ′ in the 5′-pyrophosphate nucleotides adopts the unusual syn conformation (Leonidas et al., 1997 ▶, 1999 ▶, 2003 ▶; Jardine et al., 2001 ▶), whereas in the uridine complexes it adopts the more energetically favoured anti conformation.
Since the two bound molecules interact with each other via hydrogen bonds, it is rational to propose that a new chemical entity that combines the two molecules with a suitable linker between the 5′-phosphate and one of the ribose hydroxyl groups might be more potent than these two together and we are currently pursuing its synthesis and study.
Supplementary Material
PDB reference: ribonuclease A, U5P complex, 3dxg, r3dxgsf
PDB reference: UDP complex, 3dxh, r3dxhsf
Acknowledgments
This work was supported by the Hellenic General Secretariat for Research and Technology (GSRT) and the Minister of Education, Youth and Sports of the Czech Republic through a Joint Research and Technology project between Greece and The Czech Republic (2006–2008) and the program ‘Excellence in Research Institutes (2nd Cycle)’. The support of the Commission of the European Communities under the FP7 ‘SP4-Capacities Coordination and Support Action, Support Actions’ EUROSTRUCT project (CSA-SA_FP7-REGPOT-2008-1 Grant Agreement No. 230146) is also acknowledged. This work was supported by grants from the European Community Research Infrastructure Action under the FP6 ‘Structuring the European Research Area’ Programme (through the Integrated Infrastructure Initiative ‘Integrating Activity on Synchrotron and Free Electron Laser Science’) for work at the Synchrotron Radiation Source, CCLRC, Daresbury, England, MAX-lab, Lund, Sweden and EMBL Hamburg Outstation, Germany.
References
- Anderson, D. G., Hammes, G. G. & Walz, F. G. (1968). Biochemistry, 7, 1637–1645. [DOI] [PubMed]
- Brünger, A. T. (1992). Nature (London), 355, 472–475. [DOI] [PubMed]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. http://www.pymol.org.
- Dixon, M. (1953). Biochem. J.55, 170–171. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fontecilla-Camps, J. C., de Llorens, R., le Du, M. H. & Cuchillo, C. M. (1994). J. Biol. Chem.269, 21526–21531. [DOI] [PubMed]
- French, S. & Wilson, K. (1978). Acta Cryst. A34, 517–525.
- Ghosh, K. S., Debnath, J., Dutta, P., Sahoo, B. K. & Dasgupta, S. (2008). Bioorg. Med. Chem.16, 2819–2828. [DOI] [PubMed]
- Gilliland, G. L., Dill, J., Pechik, I., Svensson, L. A. & Sjolin, L. (1994). Protein Pept. Lett.1, 60–65.
- Hatzopoulos, G. N., Leonidas, D. D., Kardakaris, R., Kobe, J. & Oikonomakos, N. G. (2005). FEBS J.272, 3988–4001. [DOI] [PubMed]
- Howlin, B., Harris, G. W., Moss, D. S. & Palmer, R. A. (1987). J. Mol. Biol.196, 159–164. [DOI] [PubMed]
- Hubbard, S. J. & Thornton, J. M. (1993). NACCESS Computer Program. Department of Biochemistry and Molecular Biology, University College London.
- Jardine, A. M., Leonidas, D. D., Jenkins, J. L., Park, C., Raines, R. T., Acharya, K. R. & Shapiro, R. (2001). Biochemistry, 40, 10262–10272. [DOI] [PubMed]
- Larson, S. B., Day, J. S., Cudney, R. & McPherson, A. (2007). Acta Cryst. F63, 728–733. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Lawrence, M. C. & Colman, P. M. (1993). J. Mol. Biol.234, 946–950. [DOI] [PubMed]
- Leatherbarrow, R. J. (2007). GraFit v.6. Erithacus Software Ltd, Horley, England.
- Leonidas, D. D., Chavali, G. B., Oikonomakos, N. G., Chrysina, E. D., Kosmopoulou, M. N., Vlassi, M., Frankling, C. & Acharya, K. R. (2003). Protein Sci.12, 2559–2574. [DOI] [PMC free article] [PubMed]
- Leonidas, D. D., Maiti, T. K., Samanta, A., Dasgupta, S., Pathak, T., Zographos, S. E. & Oikonomakos, N. G. (2006). Bioorg. Med. Chem.14, 6055–6064. [DOI] [PubMed]
- Leonidas, D. D., Shapiro, R., Irons, L. I., Russo, N. & Acharya, K. R. (1997). Biochemistry, 36, 5578–5588. [DOI] [PubMed]
- Leonidas, D. D., Shapiro, R., Irons, L. I., Russo, N. & Acharya, K. R. (1999). Biochemistry, 38, 10287–10297. [DOI] [PubMed]
- Maiti, T. K., Soumya, D., Dasgupta, S. & Pathak, T. (2006). Bioorg. Med. Chem.14, 1221–1228. [DOI] [PubMed]
- McPherson, A., Brayer, G. D. & Morrison, R. D. (1986). J. Mol. Biol.189, 305–327. [DOI] [PubMed]
- Moodie, S. L. & Thornton, J. M. (1993). Nucleic Acids Res.21, 1369–1380. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Raines, R. T. (1998). Chem. Rev.98, 1045–1065. [DOI] [PubMed]
- Richards, F. M. & Wyckoff, H. W. (1973). In Atlas of Molecular Structures in Biology, Vol. 1, Ribonuclease S, edited by D. C. Philips & F. M. Richards. Oxford: Clarendon Press.
- Russo, A., Acharya, K. R. & Shapiro, R. (2001). Methods Enzymol.341, 629–648. [DOI] [PubMed]
- Russo, N. & Shapiro, R. (1999). J. Biol. Chem.274, 14902–14908. [DOI] [PubMed]
- Toiron, C., Gonzalez, C., Bruix, M. & Rico, M. (1996). Protein Sci.5, 1633–1647. [DOI] [PMC free article] [PubMed]
- Wodak, S. Y., Liu, M. Y. & Wyckoff, H. W. (1977). J. Mol. Biol.116, 855–875.
- Zegers, I., Maes, D., Dao-Thi, M.-H., Poortmans, F., Palmer, R. & Wyns, L. (1994). Protein Sci.31, 2322–2339. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: ribonuclease A, U5P complex, 3dxg, r3dxgsf
PDB reference: UDP complex, 3dxh, r3dxhsf
PDB reference: ribonuclease A, U5P complex, 3dxg, r3dxgsf
PDB reference: UDP complex, 3dxh, r3dxhsf