

Summary
In fission yeast, assembly of centromeric heterochromatin requires the RITS complex, which consists of Ago1, Tas3, Chp1, and siRNAs derived from centromeric repeats. Recruitment of RITS to centromeres has been proposed to depend on siRNA-dependent targeting of Ago1 to centromeric sequences. Previously, we demonstrated that methylated lysine 9 of histone H3 (H3K9me) acts upstream of siRNAs during heterochromatin establishment. Our crystal structure of Chp1's chromodomain in complex with a trimethylated lysine 9 H3 peptide reveals extensive sites of contact that contribute to Chp1's high affinity binding. We found that this high affinity binding is critical for the efficient establishment of centromeric heterochromatin, but preassembled heterochromatin can be maintained when Chp1's affinity for H3K9me is greatly reduced.
Introduction
The eukaryotic genome is structurally divided into dense, heterochromatic regions with low transcriptional activity, and more permissive, transcriptionally active euchromatic regions. Heterochromatin is critical for genomic integrity and is localized to centromeres and telomeres, as well as other repetitive sequences that often contain transposons and other parasitic genomic elements. In the fission yeast S. pombe, assembly of specialized chromatin at the centromere allows faithful segregation of chromosomes. The centromere consists of a central region on which the kinetochore assembles, flanked by pericentromeric regions consisting of repeat sequences that are packaged into heterochromatin (Allshire et al. 1995; Partridge et al. 2000; Takahashi et al. 2000). Pericentromeric heterochromatin is characterized by high densities of histone H3 lysine 9 (H3K9) methylation and binding of the S. pombe HP1 homologue, Swi6. Its formation relies on an intricate interplay between transcription by RNA polymerase II, the RNAi machinery and chromatin modifying enzymes (Buhler et al. 2007). Centromeres are transcribed at low levels in G1/S phase (Chen et al. 2008; Kloc et al. 2008), and these transcripts provide a source for the production of double-stranded RNAs that are diced to form siRNAs (Volpe et al. 2002; Reinhart et al. 2002; Cam et al. 2005). The siRNAs are loaded into the argonaute protein Ago1, a component of the RNAi effector complex RITS (Verdel et al. 2004). In addition to Ago1 the RITS complex contains Chp1, a chromodomain protein that binds methylated H3K9 (Partridge et al. 2002) and a GW-repeat protein, Tas3 (Verdel et al. 2004; Partridge et al. 2007; Till et al. 2007). siRNA-loaded RITS targets and slices nascent transcripts (Irvine et al. 2006; Buker et al. 2007), thereby amplifying the siRNA response by providing templates for the RNA dependent RNA polymerase (Motamedi et al. 2004; Sugiyama et al. 2005). Recruitment of RITS to centromeres promotes the association of the Clr4 containing complex CLRC to the centromere in a positive feedback loop (Partridge et al. 2002; Verdel et al. 2004; Zhang et al. 2008). Methylation of nucleosomes on histone H3K9 by Clr4, the Su(var)3-9 homologue, facilitates the robust assembly of centromeric heterochromatin by promoting recruitment of Swi6 and cohesin for proper centromere function (Bannister et al. 2001; Nakayama et al. 2001; Bernard et al. 2001; Nonaka et al. 2002).
Chromodomains, originally identified in heterochromatin-associated factors (Paro et al. 1991), are recognized as modules used to target proteins to specific chromosomal loci (Brehm et al. 2004). The chromodomain family display a range of activities, including methyl lysine recognition (Lachner et al. 2001; Bannister et al. 2001; Jacobs et al. 2002), and RNA and DNA binding (Akhtar et al. 2000; Bouazoune et al. 2002). As shown for HP1 and Polycomb, chromodomains can discriminate between different methyl marks in almost identical sequence contexts (Fischle et al. 2008; Min et al. 2003). In S. pombe, many of the key factors for the assembly of centromeric heterochromatin have a chromodomain. The chromodomain of Chp1 is crucial for tethering the RITS complex to centromeric regions (Petrie et al. 2005). The chromodomain of Clr4 links the deposition of methyl marks to their readout and thereby provides a feed forward mechanism for amplification and spreading of the initial signal (Zhang et al. 2008). Swi6 uses its chromodomain to bind to methylated H3K9, and since it is dimeric it could tether adjacent nucleosomes thereby inducing a specialized chromatin structure (Cowieson et al. 2000; Bannister et al. 2001; Nakayama et al. 2001). The second HP1 homologue in S. pombe, Chp2, is similarly localized to regions of heterochromatin, and has recently been found to be part of the SHREC2 complex (Sadaie et al. 2008; Motamedi et al. 2008).
The RITS complex is critical for the assembly of heterochromatin, thus determination of the mechanism by which RITS is recruited to centromeres is of great interest (Verdel et al. 2004; Partridge et al. 2007). Earlier models proposed that siRNAs bound by Ago1 in the RITS complex targeted RITS to either nascent transcripts or centromeric DNA, and that once RITS facilitated recruitment of Clr4 activity to centromeres, di- or trimethylated H3K9 (H3K9me) stabilized association of RITS by providing a binding site for the chromodomain of Chp1 (Verdel et al. 2004). However, strains lacking components of the RNAi pathway maintain low levels of H3K9 methylation (Volpe et al. 2002; Sadaie et al. 2004; Partridge et al. 2007) that might initially target Chp1 to the centromere. Indeed, we previously demonstrated that H3K9 methylation by Clr4 acts upstream of siRNA production by Dcr1 during the establishment of heterochromatin, suggesting that Chp1's binding to H3K9me might play a crucial role in the initial targeting of RITS to centromeric sequences (Partridge et al. 2007).
Here, we demonstrate that heterochromatin establishment depends on Chp1's high affinity for H3K9me. Our crystal structure of the Chp1 chromodomain bound to an H3K9me3 peptide highlights residues that may contribute to this high affinity interaction. Mutation of these sites reduced Chp1's binding affinity 2 to 500-fold and in vivo studies demonstrate that a 5 fold reduction in Chp1's affinity for H3K9me is sufficient to abolish the establishment of centromeric heterochromatin. Interestingly, these mutants express robust levels of centromeric siRNAs, which cannot promote heterochromatin establishment when Chp1's H3K9me binding affinity is reduced. Together these results reveal that Chp1's high affinity binding to H3K9me is critical for the establishment of heterochromatin.
Results
Chp1 chromodomain binds H3K9me with high affinity
To investigate the role of Chp1's chromodomain in targeting RITS to centromeres, we determined the binding affinity of Chp1, Swi6 and Clr4 for H3K9 methylated peptides by fluorescence polarization (Fig. 1A, B). All proteins bound specifically to the di- or trimethylated peptides but not to unmethylated peptide. Interestingly, the Chp1 chromodomain bound both H3K9me2 and H3K9me3 peptides with significantly higher affinity than either Clr4 or Swi6 (Table 1), and all proteins bound H3K9me3 more tightly than H3K9me2.
Figure 1. Binding and structural analysis of the Chp1 chromodomain.
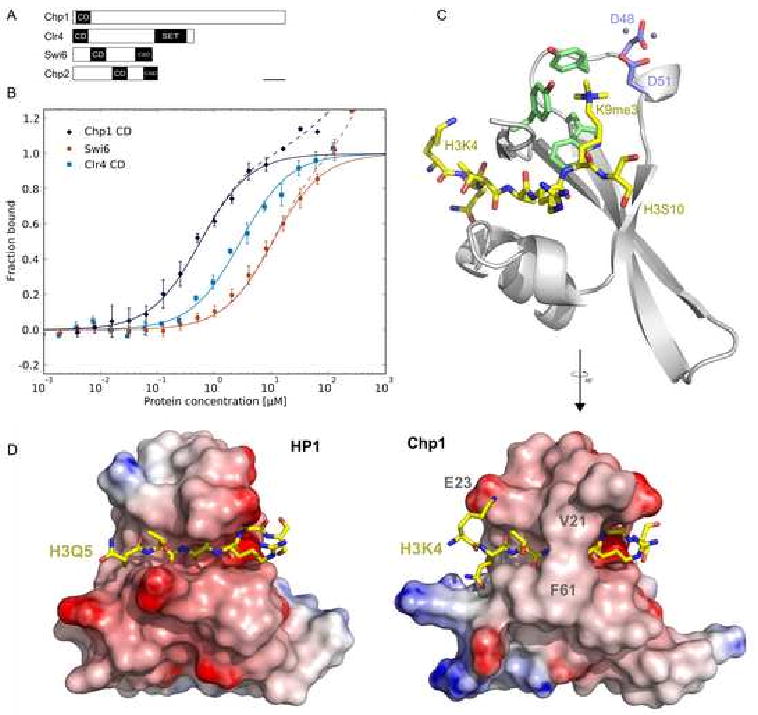
A. Domain architecture of Chp1, Clr4, Swi6 and Chp2 (CD:chromodomain, CSD: chromo shadow domain, SET: Suppressor of variegation-enhancer of zeste-trithorax domain).
B. Affinity measurements for binding of chromodomains to fluorescein labeled H3K9me2 peptides by fluorescence polarization. Error bars indicate standard deviation (SD) of 3 or more measurements. Solid lines represent fit for specific binding, dashed lines include fit for non-specific binding.
C. Crystal structure of the Chp1 chromodomain bound to H3K9me3 peptide. Cartoon representation of the Chp1 chromodomain is shown in gray, the H3 peptide in gold stick representation with oxygens shown in red and nitrogens in blue. The “aromatic cage” residues are shown in green, zinc binding aspartates in blue. Zinc ions are shown as gray spheres.
D. Side-by-side comparison of HP1 and Chp1 co-crystal structures with H3K9me3 peptides. Electrostatic surfaces computed by DelPhi (Rocchia et al. 2001) are shown for both chromodomains. H3K9me3 peptide is shown in gold.
Table 1.
Binding constants (Kd) to di- and trimethylated H3 peptides in μM for the constructs used in this study
| Construct | Kd for H3K9me2 [μM] | Kd for H3K9me3 [μM] |
|---|---|---|
| Chp1 CD (15-76) | 0.55 ± 0.09 | 0.19 ± 0.02 |
| Chp1 F61A | 1.02 ± 0.28 | 0.54 ± 0.20 |
| Chp1 V21A | 3.38 ± 0.60 | 1.46 ± 0.40 |
| Chp1 E23V | 4.10 ± 0.37 | 3.16 ± 0.32 |
| Chp1 N59A | 6.22 ± 1.12 | 5.95 ± 1.83 |
| Chp1 V24M | 9.58 ± 0.55 | 5.00 ± 0.45 |
| Chp1 E23V V24M | 22.13 ± 1.70 | 14.67 ± NA |
| Chp1 V24R | >500 | >500 |
| Swi6 | 10.28 ± 1.69 | 3.34 ± 0.55 |
| Swi6 V82E | 2.19 ± 0.13 | 1.04 ± 0.10 |
| Swi6 E80V V82E | 1.07 ± 0.03 | 0.61 ± 0.12 |
| Clr4 CD (1-70) | 2.86 ± 0.27 | 0.60 ± 0.08 |
We further characterized the binding of Chp1 and Swi6 to H3K9me peptides by isothermal titration calorimetry (ITC) experiments (Fig. S1, Table S1). This analysis showed Chp1 binding H3K9me3 with at least 5 fold higher affinity than Swi6, and confirmed the unusually tight association of Chp1 CD to H3K9me.
Structure of the Chp1 chromodomain H3K9me3 complex
To identify the source of Chp1's increased affinity for H3K9me, we solved the crystal structure of the Chp1 chromodomain (CD) in complex with an H3K9me3 peptide (Fig. 1C, Table 2). The Chp1 CD structure shows the typical chromodomain architecture with a three-stranded β-sheet supporting the peptide binding groove and a C-terminal α-helix packing against the β-sheet. Forming a β-sheet with the N-terminus of the chromodomain, the H3 peptide sits across the chromodomain with the methylated lysine bound by the “aromatic cage” residues. When compared with the co-crystal structure of the Swi6 homolog HP1 (Jacobs et al. 2002) (Fig. 1D), it is evident that Chp1 interacts with the H3 peptide more tightly than HP1. This is reflected in the larger interface between the chromodomain and the peptide for the Chp1 complex (625 Å2 vs. 521 Å2 for HP1). Most of the difference between the two complexes can be attributed to a better ordered N-terminus of the H3 peptide when bound to Chp1. We clearly observe K4 of H3 engaging in a salt bridge and van der Waals contacts with E23 of Chp1, whereas K4 is disordered when bound to HP1. E23 is not conserved in the chromodomains of the HP1 class, in which the corresponding residue, valine or alanine, cannot form such a salt bridge (Fig. 2A). Additionally, the amide oxygen of H3Q5 is pointing into the N-terminal end of Chp1's C-terminal helix, interacting with the positive end of the helix dipole. A smaller contribution that increases Chp1's binding interface compared to HP1 originates from an increased contact interface with H3R8 and a bridge over H3A7 formed by a van der Waals contact between V21 and F61 (Fig. 1D).
Table 2. Data collection and refinement statistics.
| Chp1 CD | |
|---|---|
| Data collection | |
| Space group | P43212 |
| Cell dimensions | |
| a, b, c (Å) | 41.56, 41.56, 87.38 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Resolution (Å) | 19.3-2.2 (2.4-2.2)* |
| Rmerge (%) | 4.7 (31.4) |
| I /σ(I) | 14.9 (3.5) |
| Completeness (%) | 97.9 (99.8) |
| Redundancy | 3.4 (3.3) |
| Refinement | |
| Resolution (Å) | 19.3-2.2 |
| No. reflections | 7255 |
| Rwork / Rfree (%) | 19.58 / 23.68 |
| No. atoms** | 576 |
| Protein | 540 |
| Ligand/ion | 4 |
| Water | 29 |
| <B-factors> (Å2) *** | |
| Protein | 46.80 |
| Peptide | 51.75 |
| Water | 53.29 |
| r.m.s. deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 0.798 |
Data collected on one crystal; numbers in parentheses are for highest resolution shell.
Non hydrogen atoms. Riding hydrogens were used in refinement and are included in the deposited structure.
Only non hydrogen atoms included.
Figure 2. Effect of mutations in the Chp1 chromodomain on binding affinity.
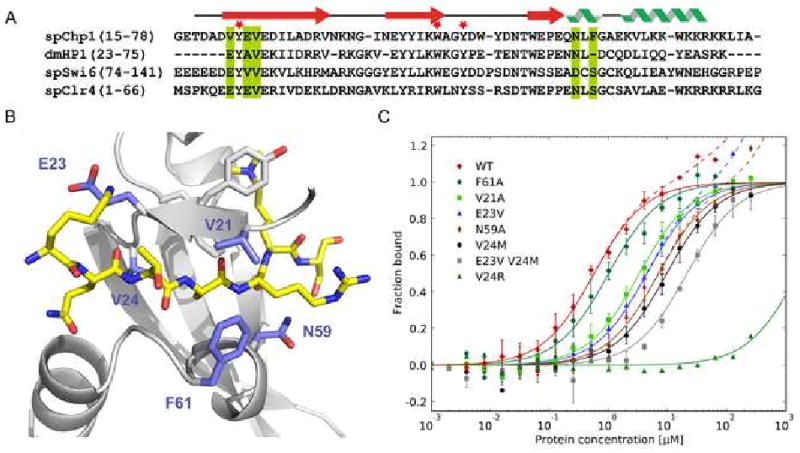
A. Sequence alignment of the chromodomains examined. The secondary structure diagram corresponds to the secondary structure determined in the Chp1 structure. Green bars indicate the mutated residues shown on the structure in panel B and red asterisks mark the position of the aromatic cage residues.
B. Residues mutated in Chp1. Close up view of the Chp1 structure (gray) in complex with the trimethylated H3 peptide (gold). Chp1 residues shown in blue were mutated to investigate their contribution to the Chp1 affinity.
C. Fluorescence polarization data for each Chp1 chromodomain mutant binding to fluorescein labeled H3K9me2 peptide. Error bars indicate SD of ≥3 measurements. Solid lines represent fit for specific binding, dashed lines include fit for non-specific binding. Chp1 wild type data from Figure 1B is shown for comparison.
Structure based mutants decrease affinity of Chp1 chromodomain for H3K9me in vitro
To determine which residues of Chp1's CD contribute to its binding affinity, mutations were generated to specifically perturb interactions identified within the co-crystal structure. The binding affinity of these recombinant mutants was assessed by fluorescence polarization binding assays (Fig. 2C, Table 1). The observed affinities ranged from close to wild type to complete loss of specific binding. The F61A mutant showed little reduction in binding affinity compared with wild type Chp1. A second class of mutants showed a 5 to 17-fold reduction in binding affinity for H3K9me2 compared with the wild type Chp1 chromodomain (V21A, E23V, N59A, V24M). A third class showed a more profound reduction in binding affinity: the E23V,V24M mutant reduced binding affinity ∼40 fold, and the V24R mutant abolished the specificity of the chromodomain interaction for K9 methylated peptides (Kd > 500 μM).
The observation that disruption of the salt bridge between E23 of Chp1 and K4 of histone H3 (in the E23V mutant) reduced Chp1's binding affinity for H3K9me peptides was intriguing, since the residue at this position is frequently valine in other chromodomain proteins. To directly assess the importance of this salt bridge for high affinity binding, we replaced the corresponding valine in the Swi6 chromodomain with glutamate (V82E). The Swi6 V82E mutant bound H3K9me2 with 5 fold higher affinity than wild type Swi6 (Table 1, Fig. S2A), indicating that the presence of glutamate at this position is an important determinant of high-affinity H3K9me association. Introduction of an E80V mutation, corresponding to V21 of Chp1, into Swi6V82E, further increased Swi6's affinity by 2 fold (Table 1, Fig. S2A). Importantly, these Swi6 mutants showed no increased affinity for the unmethylated histone H3 peptide, indicating that the salt bridge between K4 and a correctly positioned glutamate depends on the specific interaction of the positively charged methyllysine with the aromatic cage residues.
In our crystal structure, the side chain of K4 lies across the aliphatic portion of E23's side chain and displays an average B-factor significantly higher than the average of the peptide (83.7 vs. 51.8 Å2), indicating increased flexibility of this residue. Given the importance of H3K4 methylation in regulation of transcription (Martin et al. 2005), it is possible that methylation could act as a modulator of Chp1's affinity for the H3 peptide. However, because of the open binding site and flexibility of H3K4, we think it unlikely that methylation of H3K4 would have a strong effect on binding affinity.
It is interesting that although valine is widespread amongst chromodomains at the position corresponding to Chp1 E23, both Chp1 and Clr4 possess a glutamate at this position. We predict that the increased affinity of Clr4 compared with Swi6 for binding H3K9me may be, at least in part, due to glutamates' role in formation of the salt bridge with K4 of the H3K9me peptide. Indeed, a recent report has shown high affinity binding for the chromodomain Y chromosome (CDY) family of proteins (Fischle et al. 2008), which also have a glutamate in this position.
S10 of histone H3 is phosphorylated during mitosis, and displaces HP1 proteins (including Swi6) from chromatin (Fischle et al. 2005; Hirota et al. 2005; Yamada et al. 2005). We investigated whether binding of Swi6 and Chp1 to H3K9me peptides was affected by S10 phosphorylation, and found a strong reduction in both Chp1 and Swi6 binding affinity (Table S2, Fig. S2B), making it unlikely that Chp1 persists on chromatin during mitosis.
A 40 fold reduction in Chp1's H3K9me2 binding affinity does not affect the maintenance of centromeric heterochromatin
To test the physiological consequences of lowering Chp1's binding affinity, mutations were engineered into the chromodomain of the fully functional chp1-6xmyc allele at the normal chromosomal locus (Partridge et al. 2000). One set of mutants (V24M, V24R and N59A) was designed to mimic previously identified HP1 mutants that affect heterochromatic gene silencing in Drosophila by destabilization of the H3 binding interface (Platero et al. 1995; Jacobs et al. 2001). We further generated mutants that block the salt bridge between E23 and K4 of the peptide (E23Vchp1), disrupt the V21-F61 “bridge” over the peptide (V21Achp1 and F61Achp1), or block the coordination of zinc by D48 and D51 (D48S,D51Schp1) (see supplemental information). Additionally, a replacement of the entire chromodomain of Chp1 with that of Swi6 (chp1Swi6CD) was included. Western blot analysis with an anti-myc antibody showed that all the mutant proteins were stably expressed in fission yeast (Fig. S3).
In wild type cells (chp1+) or cells bearing wild type chp1-6xmyc, centromeric heterochromatin silences a transgene (cen∷ura4+) inserted at the dg region of the outer repeats in centromere 1, allowing growth on media containing FOA, a drug toxic to ura4+ expressing cells (Allshire et al. 1995; Partridge et al. 2000). In contrast, cells deficient for Chp1 (chp1Δ) cannot assemble centromeric heterochromatin, and exhibit robust expression of the cen∷ura4+ reporter and a failure to grow on FOA (Partridge et al. 2000). Interestingly, cells expressing most of the mutant alleles of Chp1 showed no defect in heterochromatin assembly as measured in this assay (Fig. 3A), with the exception of the double mutant E23V,V24M and the V24R Chp1 mutant, which did show silencing defects.
Figure 3. Mutants of Chp1 that reduce H3K9me binding activity by 40- fold show little defect in maintenance of centromeric heterochromatin.
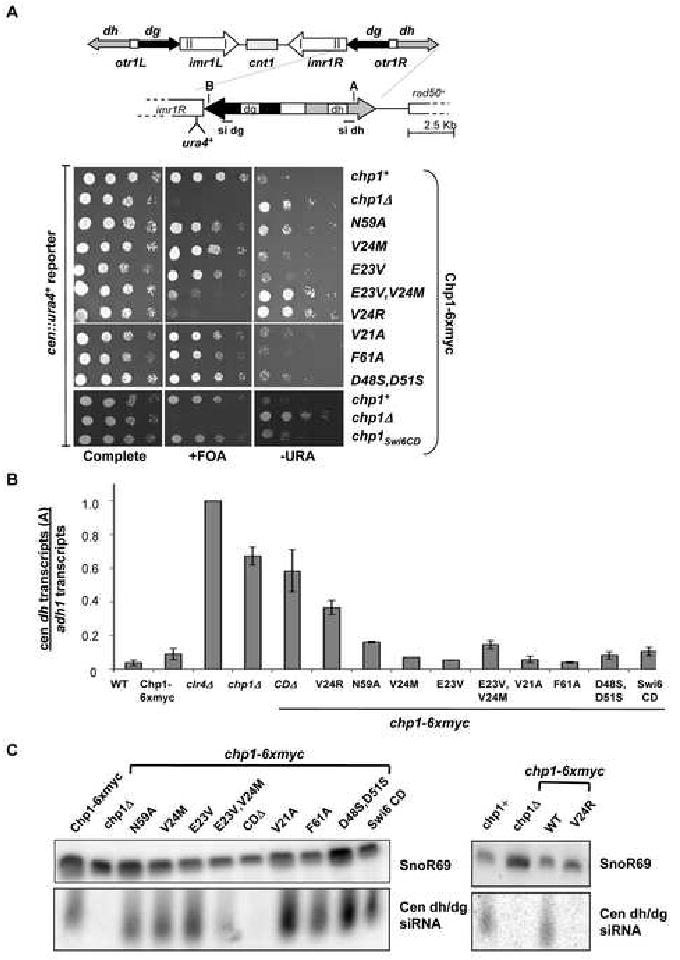
A. Schematic of centromere 1 illustrating the site of insertion of the ura4+ transgene in cen∷ura4+ strains [otr1R(dg-glu)Sph1∷ura4+], sites monitored in real time PCR assays (A in dh and B in dg) and the position of probes used for siRNA analyses. Comparative growth assay of serially diluted strains bearing the cen∷ura4+ reporter which are wild type (chp1+) or mutant at the chp1-6xmyc genomic locus, compared with strains lacking chp1 (chp1Δ). Strains were assessed for growth on PMG complete medium, PMG medium lacking uracil (-URA) or PMG medium supplemented with 5-FOA (+FOA).
B. Real time PCR analysis of centromeric transcripts (site A in dh) relative to adh1 in cDNA derived from chp1+, chp1Δ, clr4Δ, or chp1-6xmyc mutant strains. Data are represented as mean +/- SEM, after normalization to transcript levels in clr4Δ cells.
C. Small RNAs purified from the indicated strains were hybridized with centromeric probes (dg siRNA, dh siRNA) to reveal centromeric siRNAs and to snoR69 as a loading control.
High levels of centromeric transcripts accumulate in chp1 null strains or strains lacking the chromodomain of Chp1; both situations in which RITS is delocalized (Verdel et al. 2004; Petrie et al. 2005). In contrast, centromeric transcripts were not significantly elevated in any of the chp1 mutant strains, with the exception of V24R (Fig. 3B and Fig. S4).
Defects in centromeric heterochromatin can lead to disruption of Rad21 recruitment (or cohesin) at centromeres, which results in premature sister chromatid separation during mitosis (Bernard et al. 2001; Nonaka et al. 2002). The efficiency of chromosome segregation was monitored in asynchronous cell populations. Unlike chp1 null cells, which showed ∼23% of mitotic cells undergoing chromosome missegregation, the chromodomain point mutated strains, with the exception of V24Rchp1, showed few cells undergoing aberrant mitoses (Table S3). Together, these data suggest that dropping the affinity of Chp1's H3K9me interaction 40 fold (eg E23V,V24M) has little impact on heterochromatin maintenance, whereas loss of H3K9me binding affinity (eg V24R or chp1CDΔ) results in accumulation of transcripts and chromosome loss.
siRNA production is lost in Chp1 mutants with >40 fold reduced binding affinity
Mutants that lack the Chp1 component of RITS, or which cannot recruit Chp1 to centromeres are devoid of centromeric siRNAs (Verdel et al. 2004; Motamedi et al. 2004; Noma et al. 2004; Iida et al. 2008). To determine whether the Chp1 chromodomain mutants can generate siRNAs, we purified small RNA populations from these mutants (Fig. 3C). While chp1Δ and chp1CDΔ cells lack centromeric siRNAs, they were present in all other mutants, with the exception of V24R. These results correlate with our observation that centromeric transcripts, which are the precursors for siRNA production, accumulate only in the V24R and chp1CDΔ mutants.
Chp1 mutants show reduced centromeric association, but normal levels of Swi6 and H3K9me2 at centromeres
Given the mild effect on chromosome segregation, we anticipated that a 40 fold reduction of Chp1's H3K9me binding affinity would not perturb Chp1's localization to centromeres. Association of the mutant Chp1 proteins with the centromeric outer repeats was monitored in ChIP assays. Surprisingly, in contrast to robust association of wild type Chp1 at the centromeric outer repeat sites, we found only very low levels of many of the mutant Chp1 proteins at centromeres under our standard ChIP conditions (Fig. 4A and Fig. S5A). We obtained similar results when we improved crosslinking by using multiple fixation steps (Fig. S6A, B). Thus Chp1 mutants with more than 5-fold reduced affinity for H3K9me in vitro, showed little association with centromeres (eg V21Achp1). Immunofluorescent localization experiments confirmed that only the F61Achp1 mutant (with a ∼2 fold reduced affinity) supported near-normal localization of Chp1 to chromatin (Fig. S7).
Figure 4. Reduction of Chp1 binding affinity causes defective Chp1 localization, but heterochromatin is maintained at centromeres.
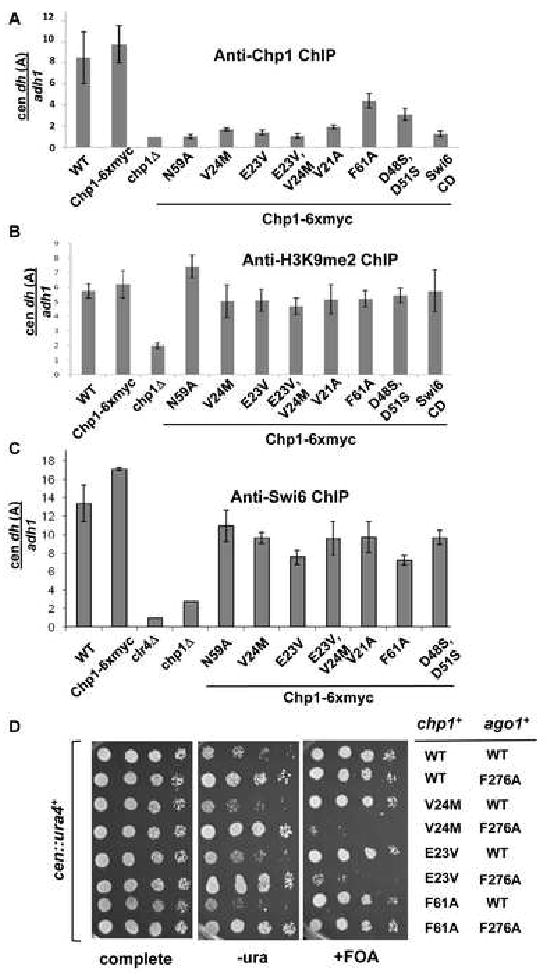
A. ChIP analysis of Chp1 association with dh (site A) centromeric repeat sequences in strains bearing chp1 chromodomain mutants, normalized to the euchromatic adh1 sequence, and to the signal obtained from chp1Δ cells (set at 1). Data were obtained by real-time PCR and are represented as mean +/- SEM
B. ChIP analysis of H3K9me2 association with centromeric dh sequences relative to adh1, measured by real-time PCR. Data was normalized to clr4Δ which lacks H3K9me2 and is represented as mean +/- SEM.
C. ChIP analysis of Swi6 association with centromeric sequences from dh (site A), relative to adh1, measured by real-time PCR. Values were normalized to clr4Δ and presented as mean +/- SEM.
D. Comparative growth assay of serially diluted strains bearing the cen∷ura4+ reporter which are wild type (chp1+) or mutant at the chp1 genomic locus, and wild type or mutant (F276A) at the ago1 locus. Strains were assessed for growth on PMG complete medium, PMG medium lacking uracil (-URA) or PMG medium supplemented with 5-FOA (+FOA).
We also tested whether Tas3 localization was affected by mutation of the Chp1 chromodomain, and found that its association with centromeres mirrored that of Chp1 in the mutants tested (Fig. S6C).
We next assessed if there were other changes in the centromeric chromatin of mutants with reduced affinity Chp1. ChIP experiments demonstrated that unlike chp1 null cells, there was no significant decrease in centromeric H3K9me2 or Swi6 association in any of the chp1 mutants tested (Fig. 4B, 4C and Fig. S5B).
We measured Swi6 and Chp1 levels in wild type cells and found that Swi6 is at least 100 fold more abundant than Chp1 (data not shown). The reduced association of Chp1 with centromeres upon reduction of affinity strongly suggests that proper localization of Chp1 depends mainly on H3K9me interaction, and that high affinity prevents Chp1 from being competed away from chromatin by the high levels of Swi6.
Reducing Ago1-siRNA interaction or displacing Ago1 from RITS in Chp1 chromodomain mutant strains causes loss of heterochromatin
Chp1 mutants with 40 fold reduced affinity for H3K9me can thus maintain centromeric heterochromatin, although Chp1 and Tas3 are present at low levels at centromeres in such mutants. This implies that low levels of the RITS complex are still recruited to centromeres in these mutant strains, and that this is sufficient to maintain centromeric silencing.
To test this hypothesis, we investigated to what extent the RITS complex could be weakened in the Chp1 mutant background. We first looked at a mutant Ago1 protein, in which siRNA binding by the PAZ domain is compromised (F276Aago1) (Partridge et al. 2007; Buker et al. 2007; Song et al. 2003). Under wild type conditions, this mutant does not show defects in centromeric heterochromatin. We found, however, that introduction of F276Aago1 into either the E23Vchp1 or V24Mchp1 mutants resulted in loss of silencing of cen∷ura4+. In contrast, the F61Achp1, F276Aago1 mutant strain showed no defect in the silencing of the reporter (Fig. 4D). These results suggest that full Ago1 activity is required when Chp1's association with centromeres is reduced. Next we tested whether the integrity of the RITS complex was essential in the Chp1 chromodomain mutant cells. A mutant Tas3 protein, Tas3WG, which separates Chp1-Tas3 from Ago1 (Partridge et al. 2007) was introduced into the chromodomain mutant strains, and loss of maintenance of heterochromatin was seen in cells expressing both Tas3WG and E23Vchp1, unlike cells expressing either single mutant (Fig. S8). This result suggests that the integrity of the RITS complex is required in the chromodomain mutant cells, further providing evidence that severely reduced levels of RITS at the centromeres are tolerated, but that a limiting amount of intact RITS with a fully active Ago1 is required to maintain centromeric heterochromatin.
Chp1 chromodomain mutants prevent establishment of de-novo centromeric heterochromatin
Next we asked whether centromeric heterochromatin establishment was affected in the chp1 mutants. First, we ablated heterochromatin in cells by removal of clr4+. clr4Δ cells lack H3K9me and centromeric siRNAs, but on reintroduction of clr4+ into the normal genomic locus, cells re- establish heterochromatin efficiently (Sadaie et al. 2004; Partridge et al. 2007). Reintegration of clr4+ into cells bearing the chp1 chromodomain mutants showed a striking separation of phenotypes- with some mutants unable to re-establish centromeric heterochromatin (eg E23Vchp1clr4Δ to clr4+, V24Mchp1clr4Δ to clr4+, N59Achp1clr4Δ to clr4+) and others showing efficient re-establishment (eg F61Achp1clr4Δ to clr4+), as assessed by silencing of the cen∷ura4+ transgene (Fig. 5A). These phenotypes correlated with binding affinity, such that mutants with >5-fold reduced H3K9me binding affinity failed to re-establish centromeric heterochromatin on reintroduction of clr4+. This defect in centromeric heterochromatin establishment was not a transient phenotype, since it persisted for more than 100 generations after clr4+ reintroduction (data not shown). These results reveal that high-affinity binding of Chp1 to H3K9me at centromeres is critical for heterochromatin establishment.
Figure 5. Chp1 mutants with 5 fold reduced H3K9me binding affinity cannot establish centromeric heterochromatin, but accumulate siRNAs to normal levels.
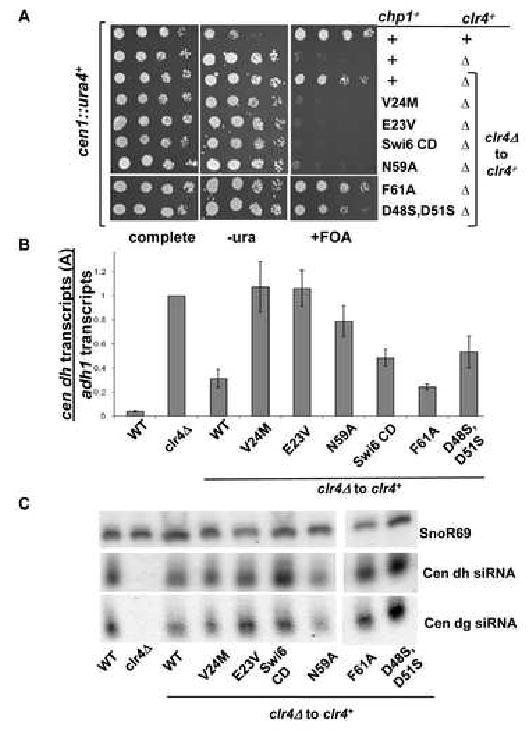
A. Comparative growth assay of serially diluted cells bearing the cen∷ura4 transgene and plated on media as outlined for Fig. 4D. Cells were wild type for clr4+ or were null for clr4 (clr4Δ). clr4+ was reintroduced by integration into the clr4 locus (clr4Δ to clr4+).
B. Real-time PCR analysis of centromeric transcripts from the dh region (A) relative to euchromatic adh1 transcripts in cDNA. Data are normalized to the values obtained for clr4Δ and represent the mean +/- SEM.
C. Small RNAs were hybridized with centromeric probes to reveal dg and dh siRNAs and to a probe for the small snoR69 RNA as a loading control.
High levels of centromeric transcripts also accumulated in these establishment-defective clr4+ reintroduction strains E23Vchp1clr4Δ to clr4+, V24Mchp1clr4Δ to clr4+, and N59Achp1clr4Δ to clr4+, but not in the establishment-competent F61Achp1clr4Δ to clr4+ cells (Fig. 5B and Fig. S9A).
The efficiency of chromosome segregation was also monitored in clr4+ reintroduction chp1 mutant cells (Table S4), and closely correlated with Chp1's binding efficiency. Mutants with > 5 fold reduction in H3K9me binding efficiency that cannot establish centromeric heterochromatin exhibited elevated rates of chromosome missegregation.
Centromeric siRNAs are present in establishment–defective chp1 mutant strains
Surprisingly, when we monitored the presence of centromeric siRNAs in these clr4+ reintroduction strains, we found that even mutants that were defective for establishment of centromeric heterochromatin efficiently synthesized siRNAs derived from both the dg and dh centromeric repeats (Fig. 5C). Thus, in contrast to other strains which are defective in heterochromatin establishment (such as chp1Δ), establishment defective Chp1 mutants with lowered affinity for H3K9me can efficiently generate siRNAs following reintroduction of clr4+. This suggests that RDRC can act, in conjunction with Dcr1, on the abundant centromeric transcripts produced in these mutants to generate centromeric siRNAs. However, these siRNAs are not sufficient to establish heterochromatin when Chp1's binding to H3K9me is impaired.
H3K9me2 is not enriched at centromeres in establishment-defective chp1 mutant strains
Chp1's association with centromeres was monitored in these clr4+ reintroduction cells by ChIP (Fig. 6A and Fig. S9B). Results for Chp1 localization were similar to that seen in maintenance strains with mutants such as E23Vchp1 clr4Δ to clr4+ and V24Mchp1clr4Δ to clr4+ showing little association with centromeres, whereas F61Achp1clr4Δ to clr4+ was enriched at levels close to wild type Chp1clr4Δ to clr4+. These results were confirmed by using double fixation (Fig. S10). However, in contrast to the maintenance situation, centromeric H3K9me2 levels were low in many of the chp1 chromodomain clr4+ reintroduction strains, and closely mirrored the levels of Chp1 recruitment in the various mutant backgrounds (Fig. 6B). This suggests therefore that mutants with weakened Chp1 association with chromatin exhibit a profound defect in recruitment of Clr4 to centromeres when clr4+ is reintegrated into these backgrounds.
Figure 6. Establishment defective Chp1 mutants are deficient for H3K9me2 at centromeres, but silencing defect can be suppressed by multi-copy clr4+.
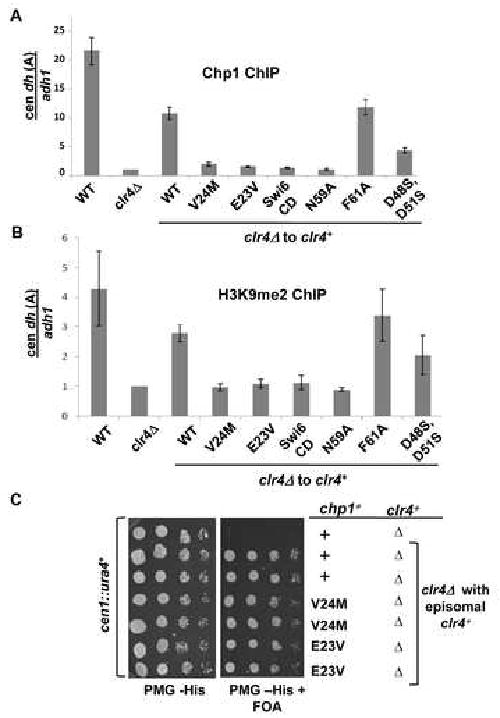
A. ChIP analysis of Chp1 association with centromeric repeat sequences (dh-site A) relative to adh1, in strains in which clr4 has been reintegrated (clr4Δ to clr4+). Data are normalized to clr4Δ, and represent mean +/- SEM of real-time PCR analyses.
B. ChIP analysis of H3K9me2 association with centromeric dh sequences (at site A) relative to the adh1 control, measured by real-time PCR. Values represent mean +/-SEM, and are normalized to clr4Δ.
C. Comparative growth assay of serially diluted cells transformed with empty vector, or an episomal genomic clone of clr4+.
Establishment defect of Chp1 chromodomain mutants can be compensated by increased dosage of clr4+
Our results suggest that high affinity interaction of Chp1 with chromatin is required for efficient establishment of heterochromatin. We hypothesize that the failure of Chp1 chromodomain mutants with reduced affinity for H3K9me to efficiently establish heterochromatin following reintroduction of clr4+ may be due to the low levels of H3K9me that can accumulate at centromeres in the absence of high affinity Chp1-H3K9me association. We predicted that increasing levels of centromeric H3K9me may compensate for the low binding affinity of Chp1 mutants and suppress their establishment defect. To test this idea, clr4 null cells bearing chp1 point mutants were transformed with an episomal plasmid carrying the clr4+ gene, and the effect on heterochromatin establishment was monitored by assessing silencing of a cen∷ura4+ transgene. High copy expression of clr4+ in the E23Vchp1clr4Δ and in the V24Mchp1clr4Δ cells allowed efficient establishment of centromeric heterochromatin (Fig. 6C). Thus the establishment defect of chp1 mutants with reduced H3K9me binding affinity can be compensated by an increased dosage of clr4+.
Discussion
Here, we demonstrate a critical requirement for the high affinity interaction of the Chp1 chromodomain with H3K9me for the establishment of centromeric heterochromatin. Mutants that reduce the H3K9me binding affinity of Chp1 in vitro by just 5 fold fail to establish centromeric heterochromatin following removal and reintroduction of the Clr4 histone methyltransferase. These findings suggest that Chp1 is a key sensor of the abundance of the methyl mark at centromeres. Thus, during the initial establishment of heterochromatin, when levels of H3K9me are low, Chp1's high affinity for H3K9me is critical for association of Chp1 (and RITS) with centromeres. In wild-type cells, following productive association of RITS, a positive feedback loop is triggered resulting in recruitment of robust Clr4 activity and increased H3K9me. Once high levels of H3K9me are achieved, high affinity binding by Chp1 is no longer critical for the maintenance of heterochromatin. The decreased dependence on RITS as centromeric H3K9me increases possibly reflects a requirement for RITS to either directly or indirectly facilitate Clr4 recruitment or activity during the initial stages of heterochromatin formation (Fig. S11). This is supported by our finding that the establishment defect of mutants with reduced H3K9me-binding affinity can be compensated by increased gene dosage of Clr4. Such a model fits well with theoretical models for heterochromatin assembly (Dodd et al. 2007).
There has been a long standing debate over how RITS is initially recruited to centromeres. Original models proposed that siRNAs target the RITS complex to homologous centromeric sequences or nascent transcripts. Once recruited, RITS triggers Clr4 recruitment and methylation of histone H3 on K9, enhancing binding of RITS to centromeric chromatin by association of Chp1's chromodomain with the methyl-marked chromatin (Verdel et al. 2004). However, low levels of H3K9me persist at centromeres in RNAi-deficient backgrounds (Volpe et al. 2002; Sadaie et al. 2004; Partridge et al. 2007). Evidence that these low-level H3K9me marks play an important role in the initial steps of heterochromatin assembly was provided by a Tas3 mutant that separates Ago1 from the rest of the RITS complex. This mutant can assemble heterochromatin in cells transiently depleted for siRNAs, but not following transient depletion of both siRNAs and H3K9me (Partridge et al. 2007). Our observation that Chp1 mutants with just 5 fold-reduced H3K9me binding affinity efficiently synthesize centromeric siRNAs after reintroduction of Clr4, but fail to establish heterochromatin, strongly supports the idea that establishment of heterochromatin depends crucially on recruitment of RITS to H3K9me, and that the presence of siRNAs is not sufficient to trigger heterochromatin establishment.
Several possible models could explain the production of siRNAs in low affinity Chp1 mutants. One possibility is that low levels of components of the RNAi pathway are recruited to the centromeres in the Chp1 mutants and are able to produce functional siRNAs. However heterochromatin assembly may still be defective because of the inability of poorly localized RITS to promote efficient recruitment of Clr4, or because of compromised co-transcriptional or transcriptional gene silencing in these mutants.
A second possibility is that in the absence of high affinity Chp1 binding, the processing in cis of centromeric transcripts into siRNAs by RDRC and Dcr1 is lost (Noma et al., 2004, Motamedi et al., 2004, Iida et al., 2008). Thus, silencing is blocked because the siRNAs that are produced are mislocalized and therefore unable to elicit efficient silencing. Precedent for this second model comes from the demonstration that siRNAs produced from synthetic ura4 hairpin RNAs can enter the RITS complex, but only heterochromatic target sites, or sites likely to have transient H3K9me marks are silenced by these siRNAs (Iida et al. 2008; Gullerova et al. 2008). Together, these findings suggest that it is the coupling of siRNA production with tethering of RITS and RDRC to chromatin via H3K9me that leads to efficient assembly of heterochromatin.
In mammals, artificial generation of siRNAs can lead to chromatin modification at select promoters (Morris 2008). However, it remains to be determined whether endogenous small RNAs (such as piRNAs that silence retrotransposons) elicit transcriptional gene silencing in mammals. It is important to note that several retrotransposons are targeted to integrate within regions of heterochromatin by a chromodomain motif within the integrase protein (Kordis 2005; Hizi et al. 2005; Gao et al. 2008). It has recently been demonstrated in flies that Piwi, which binds piRNAs to mediate transposon silencing, interacts directly with the chromodomain protein HP1a (Brower-Toland et al. 2007), and that this interaction is required for transcriptional gene silencing. Thus it is feasible that chromodomain-mediated targeting of RNAi machinery to heterochromatic domains via chromodomain-mediated interaction with H3K9me may be a widespread theme in RNAi-based transcriptional silencing mechanisms.
Experimental Procedures
Plasmid construction
GST fusion proteins: Chp1 chromodomain (amino acids 9-76) was cloned into pGEX-GK to yield constructs for expression of Chp1 CD mutants fused to GST. Integrity of cloned DNA was confirmed by sequence analysis. Mutant chromodomain sequences were generated by PCR.
Plasmids for overexpression or reintegration of Clr4 (JP-1078, JP1084) have previously been described (Partridge et al. 2007).
Expression and purification of proteins
Chp1 CD (residues 15-75) for crystallization and full length Swi6 were expressed as Sumo fusion proteins (Zuo et al. 2005) in E. coli BL21 (DE3) RIPL cells, purified by affinity chromatography on Ni-NTA resin and then treated with ULP-1 to remove the Sumo tag. The untagged chromodomain was collected from Ni-NTA flow through and purified by gel filtration. Chp1 CD proteins (residues 9-76) for binding studies were expressed as GST fusion constructs in E. coli BL21 RIPL cells. Proteins were purified by glutathione affinity chromatography, treated with thrombin to remove the GST tag and further purified by gel filtration. Clr4 CD (residues 1-70) was expressed as a Sumo fusion protein and purified by affinity chromatography, Mono Q ion exchange and gel filtration.
Crystallization and structure determination of Chp1 chromodomain/H3K9me3 complex
The complex of H3K9me3 peptide (ARTKQTARKme3STGGKAY, Peptide Protein Research Ltd, UK) with purified Chp1 CD was prepared by mixing the two components at a ratio of 1.5:1 (H3K9me3:Chp1CD) and purification by gel filtration. Crystals were grown in 50 mM zinc acetate and 20% PEG 3,350, by sitting drop vapor diffusion at 17°C. Data were collected under cryo conditions at the National Synchrotron Light Source beam line X29. The structure was solved by molecular replacement using the HP1 chromodomain structure (Jacobs et al. 2002) as a search model. Further details are in the supplementary data. Structure coordinates and data are accessible through PDB code 3G7L.
Peptide binding assays
Details are in the supplementary data.
Strain generation
Strains used in this study are listed in Table S5 and construction details are in the supplementary data.
Transcript and siRNA analyses
Transcript and siRNA analyses were performed essentially as described (Partridge et al. 2007; DeBeauchamp et al. 2008). RNA was extracted from 3 × 108 cells and following Peg 8000 precipitation, large RNA was used for cDNA synthesis and analysis. Small RNAs were recovered by ethanol precipitation of the supernatant , and were electrophoresed on an 8% polyacrylamide urea gel, blotted and hybridized with cen dg and dh probes and a probe to detect snoR69 as a loading control. Duplicate RNA samples were assessed for each experiment.
Cell growth analyses
5-fold serial dilutions of strains bearing a centromeric ura4+ marker were plated on PMG complete, or PMG medium lacking uracil, or PMG medium supplemented with 2g FOA per liter as described previously (DeBeauchamp et al. 2008), and incubated for 5 days at 25°C. To assess silencing in strains transformed with plasmids, cells were grown in PMG-his medium and were plated on minimal medium lacking histidine (PMG-his), or lacking histidine and supplemented with 2g FOA per liter (PMG-his+FOA) and incubated for 5 days at 25°C.
Chromatin immunoprecipitation
ChIP assays were performed with anti-Chp1 antibody (Abcam), anti dimethyl H3K9 antibody (Upstate), or anti-Swi6 antibody (Abcam) as described previously (Partridge et al. 2007). Results represent mean of three independent experiments.
Chromosome Segregation assays
Rates of chromosome missegregation were obtained by scoring lagging chromosomes (stained with DAPI) in late anaphase cells (exhibiting a long anaphase spindle in anti-TAT1 stained cells) as previously described (Petrie et al. 2005).
Supplementary Material
Acknowledgments
Thanks to J. Wuitschick, A. Kosinski, J.DeBeauchamp, T. Izard and D. Myszka for early contributions to this work. Thanks to K. Gull and R. Allshire for antibodies, and to M. Bix, P. Brindle, J. Ihle, S. Whitehall and Joshua-Tor and Partridge lab members for useful discussions. Thanks to H. Schindelin and G. Zhao for support with ITC measurements, to H. Robinson for help at the National Synchotron Light Source which is supported by Department of Energy, Office of Basic Energy Sciences. T.S. was supported by an EMBO fellowship and is a Human Frontiers in Research Program postdoctoral fellow. L.J. is an investigator of the Howard Hughes Medical Institute. This work was supported by NIH grants 1R01-GM084045 (JFP), GM072659 (LJ), by Cancer Center support grants CCSG 2 P30 CA 21765-25 and -26 (JFP) and CA21765 (St. Jude), and the American Lebanese Syrian Associated Charities (ALSAC) of St. Jude Children's Research Hospital.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhtar A, Zink D, Becker PB. Chromodomains are protein-RNA interaction modules. Nature. 2000;407:405–409. doi: 10.1038/35030169. [DOI] [PubMed] [Google Scholar]
- Allshire RC, Nimmo ER, Ekwall K, Javerzat JP, Cranston G. Mutations derepressing silent centromeric domains in fission yeast disrupt chromosome segregation. Genes Dev. 1995;9:218–233. doi: 10.1101/gad.9.2.218. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- Bernard P, Maure J, Partridge JF, Genier S, Javerzat J, Allshire RC. Requirement of Heterochromatin for Cohesion at Centromeres. Science. 2001;294:2539–2542. doi: 10.1126/science.1064027. [DOI] [PubMed] [Google Scholar]
- Bouazoune K, Mitterweger A, Längst G, Imhof A, Akhtar A, Becker PB, Brehm A. The dMi-2 chromodomains are DNA binding modules important for ATP-dependent nucleosome mobilization. EMBO J. 2002;21:2430–2440. doi: 10.1093/emboj/21.10.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm A, Tufteland KR, Aasland R, Becker PB. The many colours of chromodomains. BioEssays. 2004;26:133–140. doi: 10.1002/bies.10392. [DOI] [PubMed] [Google Scholar]
- Brower-Toland B, Findley SD, Jiang L, Liu L, Yin H, Dus M, Zhou P, Elgin SC, Lin H. Drosophila PIWI associates with chromatin and interacts directly with HP1a. Genes Dev. 2007;21:2300–2311. doi: 10.1101/gad.1564307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler M, Moazed D. Transcription and RNAi in heterochromatic gene silencing. Nat Struct Mol Biol. 2007;14:1041–1048. doi: 10.1038/nsmb1315. [DOI] [PubMed] [Google Scholar]
- Buker SM, Iida T, Buhler M, Villen J, Gygi SP, Nakayama J, Moazed D. Two different Argonaute complexes are required for siRNA generation and heterochromatin assembly in fission yeast. Nat Struct Mol Biol. 2007;14:200–207. doi: 10.1038/nsmb1211. [DOI] [PubMed] [Google Scholar]
- Cam HP, Sugiyama T, Chen ES, Chen X, FitzGerald PC, Grewal SIS. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat Genet. 2005;37:809–819. doi: 10.1038/ng1602. [DOI] [PubMed] [Google Scholar]
- Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, Grewal SIS. Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature. 2008;451:734–737. doi: 10.1038/nature06561. [DOI] [PubMed] [Google Scholar]
- Cowieson NP, Partridge JF, Allshire RC, McLaughlin PJ. Dimerisation of a chromo shadow domain and distinctions from the chromodomain as revealed by structural analysis. Current Biology. 2000;10:517–525. doi: 10.1016/s0960-9822(00)00467-x. [DOI] [PubMed] [Google Scholar]
- DeBeauchamp JL, Moses A, Noffsinger VJP, Ulrich DL, Job G, Kosinski AM, Partridge JF. Chp1-Tas3 Interaction Is Required To Recruit RITS to Fission Yeast Centromeres and for Maintenance of Centromeric Heterochromatin. Mol Cell Biol. 2008;28:2154–2166. doi: 10.1128/MCB.01637-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd IB, Micheelsen MA, Sneppen K, Thon G. Theoretical Analysis of Epigenetic Cell Memory by Nucleosome Modification. Cell. 2007;129:813–822. doi: 10.1016/j.cell.2007.02.053. [DOI] [PubMed] [Google Scholar]
- Fischle W, Franz H, Jacobs SA, Allis CD, Khorasanizadeh S. Specificity of the Chromodomain Y Chromosome Family of Chromodomains for Lysine-methylated ARK(S/T) Motifs. J Biol Chem. 2008;283:19626–19635. doi: 10.1074/jbc.M802655200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438:1116–1122. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- Gao X, Hou Y, Ebina H, Levin HL, Voytas DF. Chromodomains direct integration of retrotransposons to heterochromatin. Genome Res. 2008;18:359–369. doi: 10.1101/gr.7146408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullerova M, Proudfoot NJ. Cohesin Complex Promotes Transcriptional Termination between Convergent Genes in S. pombe. Cell. 2008;132:983–995. doi: 10.1016/j.cell.2008.02.040. [DOI] [PubMed] [Google Scholar]
- Hirota T, Lipp JJ, Toh B, Peters J. Histone H3 serine[thinsp]10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005;438:1176–1180. doi: 10.1038/nature04254. [DOI] [PubMed] [Google Scholar]
- Hizi A, Levin HL. The Integrase of the Long Terminal Repeat-Retrotransposon Tf1 Has a Chromodomain That Modulates Integrase Activities. J Biol Chem. 2005;280:39086–39094. doi: 10.1074/jbc.M506363200. [DOI] [PubMed] [Google Scholar]
- Iida T, Nakayama J, Moazed D. siRNA-Mediated Heterochromatin Establishment Requires HP1 and Is Associated with Antisense Transcription. Mol Cell. 2008;31:178–189. doi: 10.1016/j.molcel.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine DV, Zaratiegui M, Tolia NH, Goto DB, Chitwood DH, Vaughn MW, Joshua-Tor L, Martienssen RA. Argonaute Slicing Is Required for Heterochromatic Silencing and Spreading. Science. 2006;313:1134–1137. doi: 10.1126/science.1128813. [DOI] [PubMed] [Google Scholar]
- Jacobs SA, Khorasanizadeh S. Structure of HP1 Chromodomain Bound to a Lysine 9-Methylated Histone H3 Tail. Science. 2002;295:2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- Jacobs SA, Taverna SD, Zhang Y, Briggs SD, Li J, Eissenberg JC, Allis C, Khorasanizadeh S. Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J. 2001;20:5232–5241. doi: 10.1093/emboj/20.18.5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloc A, Zaratiegui M, Nora E, Martienssen R. RNA Interference Guides Histone Modification during the S Phase of Chromosomal Replication. Current Biology. 2008;18:490–495. doi: 10.1016/j.cub.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordis D. A genomic perspective on the chromodomain-containing retrotransposons: Chromoviruses. Gene. 2005;347:161–173. doi: 10.1016/j.gene.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- Min J, Zhang Y, Xu R. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003;17:1823–1828. doi: 10.1101/gad.269603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris KV. RNA Interference Current Topics in Microbiology and Immunology. Springer; 2008. RNA-Mediated Transcriptional Gene Silencing in Human Cells; pp. 211–224. [DOI] [PubMed] [Google Scholar]
- Motamedi MR, Hong EE, Li X, Gerber S, Denison C, Gygi S, Moazed D. HP1 Proteins Form Distinct Complexes and Mediate Heterochromatic Gene Silencing by Nonoverlapping Mechanisms. Mol Cell. 2008;32:778–790. doi: 10.1016/j.molcel.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motamedi MR, Verdel A, Colmenares SU, Gerber SA, Gygi SP, Moazed D. Two RNAi Complexes, RITS and RDRC, Physically Interact and Localize to Noncoding Centromeric RNAs. Cell. 2004;119:789–802. doi: 10.1016/j.cell.2004.11.034. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SIS. Role of Histone H3 Lysine 9 Methylation in Epigenetic Control of Heterochromatin Assembly. Science. 2001;292:110–113. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- Noma K, Sugiyama T, Cam H, Verdel A, Zofall M, Jia S, Moazed D, Grewal SIS. RITS acts in cis to promote RNA interference-mediated transcriptional and post-transcriptional silencing. Nat Genet. 2004;36:1174–1180. doi: 10.1038/ng1452. [DOI] [PubMed] [Google Scholar]
- Nonaka N, Kitajima T, Yokobayashi S, Xiao G, Yamamoto M, Grewal SIS, Watanabe Y. Recruitment of cohesin to heterochromatic regions by Swi6/HP1 in fission yeast. Nat Cell Biol. 2002;4:89–93. doi: 10.1038/ncb739. [DOI] [PubMed] [Google Scholar]
- Paro R, Hogness DS. The Polycomb protein shares a homologous domain with a heterochromatin-associated protein of Drosophila. Proc Natl Acad Sci USA. 1991;88:263–267. doi: 10.1073/pnas.88.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge JF, Borgstrom B, Allshire RC. Distinct protein interaction domains and protein spreading in a complex centromere. Genes Dev. 2000;14:783–791. [PMC free article] [PubMed] [Google Scholar]
- Partridge JF, DeBeauchamp JL, Kosinski AM, Ulrich DL, Hadler MJ, Noffsinger VJ. Functional Separation of the Requirements for Establishment and Maintenance of Centromeric Heterochromatin. Mol Cell. 2007;26:593–602. doi: 10.1016/j.molcel.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Partridge JF, Scott KSC, Bannister AJ, Kouzarides T, Allshire RC. cis-Acting DNA from Fission Yeast Centromeres Mediates Histone H3 Methylation and Recruitment of Silencing Factors and Cohesin to an Ectopic Site. Current Biology. 2002;12:1652–1660. doi: 10.1016/s0960-9822(02)01177-6. [DOI] [PubMed] [Google Scholar]
- Petrie VJ, Wuitschick JD, Givens CD, Kosinski AM, Partridge JF. RNA Interference (RNAi)-Dependent and RNAi-Independent Association of the Chp1 Chromodomain Protein with Distinct Heterochromatic Loci in Fission Yeast. Mol Cell Biol. 2005;25:2331–2346. doi: 10.1128/MCB.25.6.2331-2346.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platero JS, Hartnett T, Eissenberg JC. Functional analysis of the chromo domain of HP1. EMBO J. 1995;14:3977–3986. doi: 10.1002/j.1460-2075.1995.tb00069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart BJ, Bartel DP. Small RNAs Correspond to Centromere Heterochromatic Repeats. Science. 2002;297:1831. doi: 10.1126/science.1077183. [DOI] [PubMed] [Google Scholar]
- Rocchia W, Alexov E, Honig B. Extending the Applicability of the Nonlinear Poisson-Boltzmann Equation: Multiple Dielectric Constants and Multivalent Ions. J Phys Chem B. 2001;105:6507–6514. [Google Scholar]
- Sadaie M, Iida T, Urano T, Nakayama J. A chromodomain protein, Chp1, is required for the establishment of heterochromatin in fission yeast. EMBO J. 2004;23:3825–3835. doi: 10.1038/sj.emboj.7600401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadaie M, Kawaguchi R, Ohtani Y, Arisaka F, Tanaka K, Shirahige K, Nakayama J. Balance between Distinct HP1 Family Proteins Controls Heterochromatin Assembly in Fission Yeast. Mol Cell Biol. 2008;28:6973–6988. doi: 10.1128/MCB.00791-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Liu J, Tolia NH, Schneiderman J, Smith SK, Martienssen RA, Hannon GJ, Joshua-Tor L. The crystal structure of the Argonaute2 PAZ domain reveals an RNA binding motif in RNAi effector complexes. Nat Struct Mol Biol. 2003;10:1026–1032. doi: 10.1038/nsb1016. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Cam H, Verdel A, Moazed D, Grewal SIS. RNA-dependent RNA polymerase is an essential component of a self-enforcing loop coupling heterochromatin assembly to siRNA production. Proc Natl Acad Sci USA. 2005;102:152–157. doi: 10.1073/pnas.0407641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Chen ES, Yanagida M. Requirement of Mis6 Centromere Connector for Localizing a CENP-A-Like Protein in Fission Yeast. Science. 2000;288:2215–2219. doi: 10.1126/science.288.5474.2215. [DOI] [PubMed] [Google Scholar]
- Till S, Lejeune E, Thermann R, Bortfeld M, Hothorn M, Enderle D, Heinrich C, Hentze MW, Ladurner AG. A conserved motif in Argonaute-interacting proteins mediates functional interactions through the Argonaute PIWI domain. Nat Struct Mol Biol. 2007;14:897–903. doi: 10.1038/nsmb1302. [DOI] [PubMed] [Google Scholar]
- Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SIS, Moazed D. RNAi-Mediated Targeting of Heterochromatin by the RITS Complex. Science. 2004;303:672–676. doi: 10.1126/science.1093686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe TA, Kidner C, Hall IM, Teng G, Grewal SIS, Martienssen RA. Regulation of Heterochromatic Silencing and Histone H3 Lysine-9 Methylation by RNAi. Science. 2002;297:1833–1837. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- Yamada T, Fischle W, Sugiyama T, Allis CD, Grewal SI. The Nucleation and Maintenance of Heterochromatin by a Histone Deacetylase in Fission Yeast. Mol Cell. 2005;20:173–185. doi: 10.1016/j.molcel.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Zhang K, Mosch K, Fischle W, Grewal SIS. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat Struct Mol Biol. 2008;15:381–388. doi: 10.1038/nsmb.1406. [DOI] [PubMed] [Google Scholar]
- Zuo X, Li S, Hall J, Mattern M, Tran H, Shoo J, Tan R, Weiss S, Butt T. Enhanced Expression and Purification of Membrane Proteins by SUMO Fusion in Escherichia coli. J Struct Funct Genomics. 2005;6:103–111. doi: 10.1007/s10969-005-2664-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.