

SUMMARY
NMR spectroscopy and X-ray crystallography are currently the two most widely applied methods for the determination of macromolecular structure at high resolution. More recently, significant advances have been made in algorithms for de novo prediction of protein structure, and in favorable cases, the predicted models agree extremely well with experimentally determined structures. Here we demonstrate a synergistic combination of NMR spectroscopy, de novo structure prediction, and X-ray crystallography in an effective overall strategy for rapidly determining the structure of the coat protein C-terminal domain from the Sulfolobus islandicus rod-shaped virus (SIRV). This approach takes advantage of the most accessible aspects of each structural technique and may be widely applicable for structure determination.
INTRODUCTION
One important goal in structural biology is the determination of representative structures for new folds in a rapid and cost-efficient manner. Nevertheless, the two major structural techniques that are widely employed can still suffer from roadblocks to structure determination. For instance, phase determination is often a rate-limiting step in X-ray crystallography and the assignment of side chain resonances and distance correlations in Nuclear Magnetic Resonance (NMR) spectra can be difficult and time-consuming. The identification of amenable protein constructs often hampers efforts in both techniques. Ab initio calculations from the primary sequence are an alternative means to obtain structures, but such structure predictions are dependent on locating the global minimum in a vast conformational space. A variety of techniques have been developed over the years to circumvent these various difficulties, but significant challenges in structure determination are still frequently encountered, both in structural genomics efforts and in focused studies from individual laboratories.
The combination of structure determination techniques has greatly aided in obtaining macromolecular structures. NMR provides information that may aid in the design of protein constructs that are more amenable for crystallization studies, and restraints that improve the models obtained from de novo structure prediction programs, such as Rosetta (Bowers et al., 2000; Rohl and Baker, 2002; Shen et al., 2008a). Structural models determined by NMR or computational techniques have had some limited success in serving as molecular replacement models for crystallography (Brünger et al., 1987; Chen and Clore, 2000; Chen et al., 2000; Qian et al., 2007; Ramelot et al., 2008), and several joint X-ray and NMR structure refinements have been reported (Chao and Williamson, 2004; Clore and Gronenborn, 1992; Hoffman et al., 1996; Raves et al., 2001; Schiffer et al., 1994; Shaanan et al., 1992). A variety of computational techniques have aided in the rapid automatic assignment of residues and determination of NMR structures, for instance ABACUS (Grishaev and Llinás, 2004; Lemak et al., 2008), ASCAN (Fiorito et al., 2008), ATNOS/CANDID (Herrmann et al., 2002a, b), and AutoStructure (Zheng et al., 2003).
Computational methods have a tremendous potential to bootstrap NMR or crystal structure calculations, potentially providing additional restraints or search models for molecular replacement. Historically, the success of molecular replacement has been limited when using crystal structures of proteins with low homology or moderate-resolution NMR models. Refinement of these structural models with the Rosetta algorithm, in one study, yielded successful molecular replacement models about 70% of the time (Qian et al., 2007). Both cases depend on the availability of a related structure, and such models do not always exist or can be difficult to obtain. The success of using Rosetta derived de novo models in molecular replacement, however, is minimal due to the quality of the structures generated by the algorithm (Qian et al., 2007; Rigden et al., 2008).
The success rate of using de novo models for molecular replacement will be enhanced through the inclusion of limited NMR-derived structural information that is straightforward to obtain. NMR chemical shift assignments or residual dipolar couplings, for instance, are sufficient to improve computationally generated de novo models (Bowers et al., 2000; Cavalli et al., 2007; Robustelli et al., 2008; Rohl and Baker, 2002; Shen et al., 2008a; Wishart et al., 2008). Most recently, CS-Rosetta was developed to include the backbone and Cβ chemical shifts, greatly improving the de novo structural models generated by the Rosetta algorithm (Shen et al., 2008a). Over 90% of the predicted structures had backbone RMSD values within 1.8 Å of the calculated NMR structure.
These advances point to a convergence of NMR, X-ray crystallography and computational techniques that may generally expedite the process of structure determination, and it is worthwhile considering an optimal global strategy that takes advantage of the strengths of each technique. Here, we report a synergistic approach to structure determination in the case of the major coat protein from Sulfolobus islandicus rod-shaped virus (SIRV-CP). The tactics used for solving the SIRV-CP structure are outlined in Figure 1, which illustrates the contributions of NMR, X-ray crystallography, and computation in the overall process. NMR was used to define the optimal construct for crystallization, and to obtain the chemical shifts that were used in CS-Rosetta to generate models of the structure. These models provided molecular replacement solutions that led to the determination of the X-ray crystal structure. This overall strategy may serve as a generally useful paradigm for the class of structural problems that are currently within the reach of all three techniques. This approach has the potential to accelerate the process of structure determination in cases where one method alone does not prevail.
Figure 1.
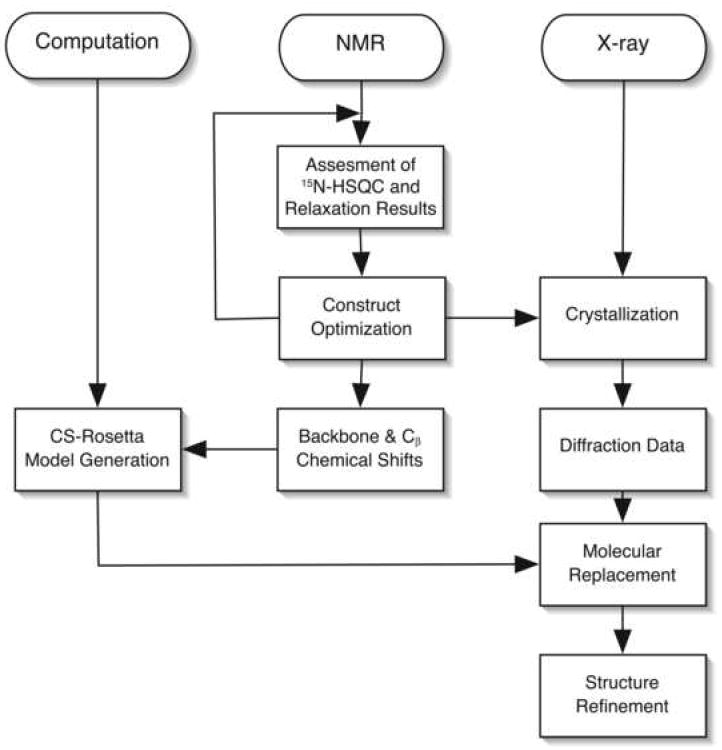
Synergetic Approach to Structure Determination. A flowchart describing the strategy for structure determination using a combination of computational techniques, NMR and X-ray Crystallography.
RESULTS
Defining the structured region of SIRV-CP
SIRV-YNP is a double-stranded DNA rod-shaped virus in the Rudiviridae family that infects strains of the thermophilic, acidophilic archaeon Sulfolobus and was isolated from hot springs in Yellowstone National Park (Rice et al., 2001). The viral rod of known Rudiviridae members is primarily composed of a single 145-residue coat protein that is thought to associate with the genomic DNA (Prangishvili et al., 1999; Steinmetz et al., 2008). Structural and biophysical characterization of this archaeal virus may provide a unique insight into the architecture, assembly and function of macromolecules under extreme conditions. The extraordinary stability of SIRV-2, even in organic solvents, makes it an attractive nano-building block, with potential uses in materials science and nanotechnologies (Steinmetz et al., 2008). Determination of the monomeric SIRV-CP structure is an important component in the effort to interpret cryo-electron microscopic reconstructions of the SIRV structure.
Despite extensive crystallization trials, no crystals of full-length SIRV-CP were obtained. NMR spectroscopy was applied to qualitatively diagnose regions of order and disorder in the protein. The 1H,15N-HSQC spectrum of the SIRV-CP is shown in Figure 2A, where each resonance originates from an individual backbone amide. The dispersion of resonances reveals that a portion of the protein is structured, but the concentration of sharp resonances in the unfolded region of the spectrum (1H: 7.8 - 8.5 ppm) suggested a significant portion of the protein is unstructured.
Figure 2.
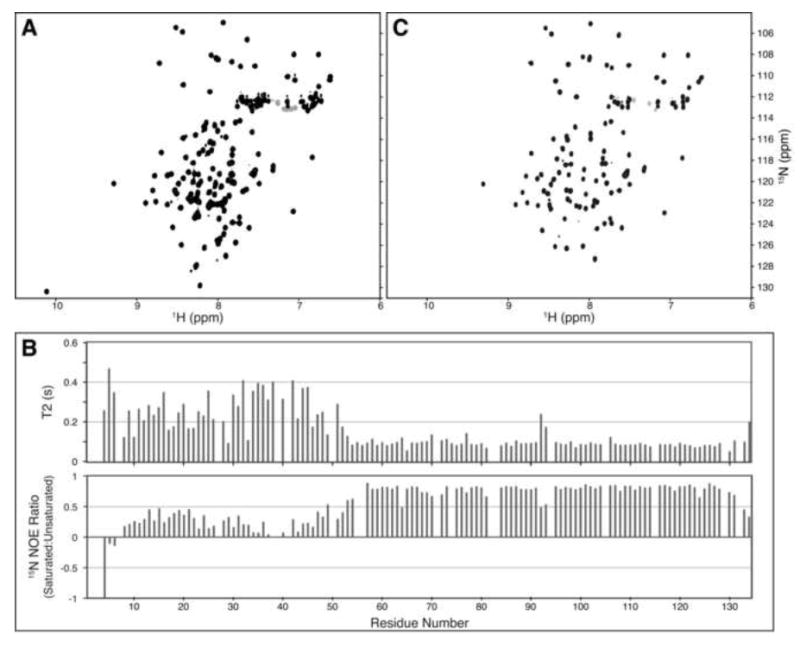
Identification of the C-terminal Structured Region in SIRV-CP by NMR Spectroscopy. 1H,15N-HSQC spectra for the full length SIRV-YNP coat protein (A) and the protein lacking the N-terminal 45 amino acids (C). The peaks that remain in the spectrum of the N-terminal deletion mutant correlate well with peaks in the full-length protein, suggesting that the remaining C-terminal residues are undisturbed by the deletion. (B) 15N-relaxation results suggest that the C-terminus is folded, while the N-terminus remains dynamic under these experimental conditions.
To determine the extent of the structured region, the assignments of the backbone (HN, N, C, Cα, Hα) and Cβ atoms of SIRV-CP were obtained from a 13C,15N-labeled sample using standard triple resonance NMR experiments (Sattler et al., 1999). The ordered region was defined by the 15N-relaxation values shown in Figure 2B. The N-terminal ~50 residues are dynamic relative to the C-terminus, as judged by the low values of the 15N-heteronuclear NOE ratios and the increased values of T2. Prediction of the secondary structure from the chemical shifts (Berjanskii et al., 2006; Wishart and Sykes, 1994) indicated that the C-terminal region consisted of four α-helical segments (Supplementary Figure 1, inset), but that the N-terminus was devoid of regular secondary structure elements. Based on these data, a shorter SIRV-CP(46-134) construct was prepared, resulting in a significantly improved 1H,15N-HSQC spectrum, as shown in Figure 2C. The strong correlation between the resonances in the full-length protein and N-terminal deletion mutant suggests that the C-terminal structure is retained in the absence of the N-terminus. Furthermore, crystallization trials using SIRV-CP(46-134) rapidly produced high quality crystals that diffracted to 1.67 Å.
Prediction of the SIRV-CP C-terminal domain structure using CS-Rosetta
The backbone (HN, N, C, Cα, Hα) and Cβ chemical shifts of resides 46-134, determined within the context of the full-length protein, were used as an input to CS-Rosetta for structure prediction. The CS-Rosetta models of SIRV-CP(46-134) did not appear to converge on a well-defined structure based on the range of Cα-RMSD values (Figure S1A), but each of the lowest energy models adopted the same four-helix bundle fold, with disordered N- and C-termini (Figure S2A). The average Cα-RMSD of the structured region (residues 51-128) of the 10 lowest energy structures is 1.2 ± 0.3 Å relative to the lowest energy structure. Indeed, when CS-Rosetta calculations were repeated with the residues 51-128, the newly generated models converged on the same four-helix bundle fold (Cα-RMSD = 0.72 Å). The RMSD values of most SIRV-CP(51-128) models cluster within 2Å of the lowest energy structure, and the average Cα-RMSD of the 10 lowest energy models is 1.1 ± 0.5 Å (Figure S1B).
Determination of the crystal structure by molecular replacement
The CS-Rosetta structure with the lowest energy score for SIRV-CP(46-134), shown in Figure 3A, was successfully used as a search model for molecular replacement with the 1.67 Å native data set obtained for SIRV-CP(46-134). Phase bias was removed using prime-and-switch in the program Resolve (Terwilliger, 2000, 2004) and the final crystallographic statistics for the structure are listed in Table 1. The relatively low Rfree and observation that some residues included in the molecular replacement search model are disordered in the refined structure indicate that the initial model did not bias the final structure. The final refined X-ray model of SIRV-CP(46-134), shown in Figure 3B, has a Cα-RMSD of 0.7 Å from the initial CS-Rosetta model (Figure 3C).
Figure 3.
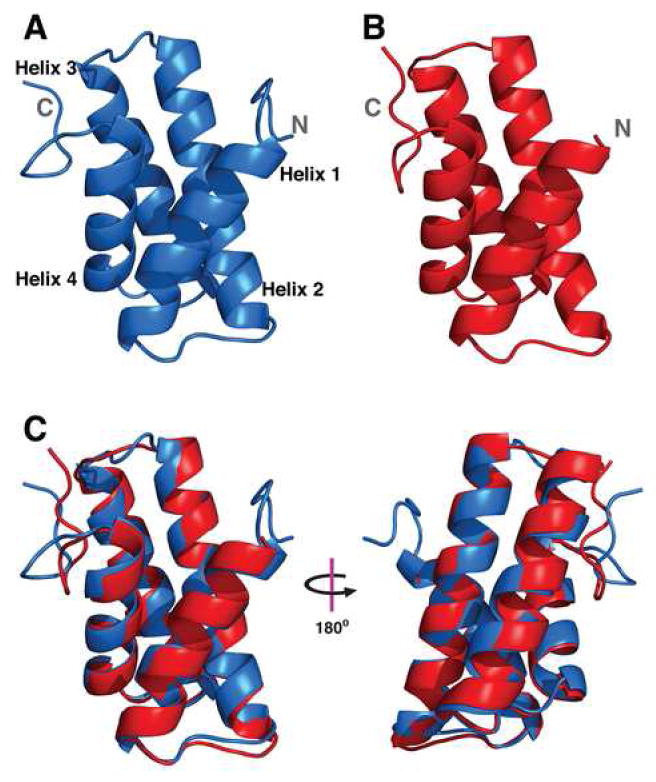
Structure of the SIRV-YNP Coat Protein C-terminal Domain. (A) Lowest energy model of SIRV-CP(46-134) determined by CS-Rosetta and used in molecular replacement. The N- and C-termini are indicated, and helices 1-4 are labeled at the N-terminal ends. (B) The refined X-ray crystal structure of SIRV-CP(46-134). The N-terminal five amino acids and the C-terminal His-tag are disordered in the electron density map. (C) Alignment of the two structures reveals they have a Cα-RMSD of 0.7 Å. The largest source of variance is within the unstructured region at the C-terminus of the helix bundle.
Table 1.
Crystallographic statistics
| Data Collection | |
|---|---|
| Space group | P43212 |
| Cell dimensions | |
| a, b, c (Å) | 54.41, 54.41, 77.79 |
| Resolution (Å) | 44.58 (1.70-1.67) |
| Rsym | 6.2 (37.1) |
| I/σI | 33.40 (8.27) |
| Completeness (%) | 99.1 (94.6) |
| Redundancy | 16.8 (14.5) |
| Refinement | |
| Resolution (Å) | 31.64-1.67 (1.8-1.67) |
| No. Reflections | 14,055 (2,675) |
| Rwork/Rfree | 18.4 (18.3) / 20.2 (23.1) |
| No. Atoms | |
| Protein | 627 |
| Water | 80 |
| B-factors | |
| Protein | 18.82 |
| Water | 31.59 |
| R.M.S. deviations | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 0.833 |
The domain adopts a four-helix bundle fold that is stabilized by an extensive hydrophobic core, with helices ranging from 11 to 19 amino acids in length. Helices 2 and 3 are almost parallel, while helices 4 and 1 are at increasingly larger angles relative to helix 2, giving the appearance of a helical twist. With the exception of the C-terminus, the loops are very short (4-5 residues), and only the five N-terminal amino acids and the C-terminal His-tag do not have an associated electron density. Although a search for proteins with a similar fold using the DALI database (Holm and Sander, 1993) did reveal weak structural homology to a subregion of the allophycocyanin protein [PDB: 1B33; Z score = 6.1] and other members of the phycobiliprotein family [Z-values > 5] (Betz, 1997; Reuter et al., 1999), the C-terminus of SIRV-CP does not resemble any known independently folded structure.
Validation of CS-Rosetta structures as molecular replacement models
To determine whether the chemical shift information was critical for providing a Rosetta model suitable for molecular replacement, a set of 10 control models was generated using Rosetta, without the chemical shift constraints, and compared to a test set of 10 models generated using CS-Rosetta (Figure S2). The molecular structures calculated using the Rosetta program, with fragments and input files derived from the Robetta website (Chivian et al., 2003; Kim et al., 2004), yielded models that did not converge on a single fold. The set of 10 lowest energy Rosetta structures for SIRV-CP(46-134) have an average Cα-RMSD of 6 ± 3 Å for residues 51-128, relative to the lowest energy structure determined using CS-Rosetta (Figure S2B). The two sets of 10 lowest energy Rosetta and CS-Rosetta models were then used as search models for molecular replacement. The EPMR (Kissinger et al., 1999) and Phaser (McCoy et al., 2007) programs gave virtually identical results, summarized in Table 2. The strong correlation in the plot of the correlation coefficient (CC) from EPMR versus the translation factor Z-score (TFZ) from Phaser (r2 = 0.81) indicates that the CS-Rosetta models give comparable results using these two methods (Figure S3). No such correlation is observed for the Rosetta models (r2 = 0.02), and none of these gave a molecular replacement solution, as judged by TFZ (Group 3, Figure 4). 6 of the top 10 CS-Rosetta models were effective in providing a molecular replacement solution, with TFZ-scores greater than 8 (Group 1, Figure 4), while only two of the models failed to provide a molecular replacement solution (Group 3, Figure 4). The remaining two CS-Rosetta models gave “borderline” TFZ-scores between 7 and 8 (Group 2, Figure 4). The electron density maps generated from these solutions were clean with clear solvent boundaries and side-chain density, and the R-values dropped in response to standard refinement procedures, indicating that the solutions were adequate. Inclusion of the chemical shift information as an experimental constraint in the generation of the Rosetta models using CS-Rosetta significantly improves the prediction of structures, and, in this case, the majority of structures predicted by CS-Rosetta were suitable for molecular replacement with X-ray diffraction data.
Table 2.
Results of molecular replacement tests with lowest energy Rosetta and CS-Rosetta models.
| Phaser Results | EPMR Results | ||||||
|---|---|---|---|---|---|---|---|
| Model number | Type | Cα-RMSDa | #Atoms aligned | TFZb | LLGc | CCd | Re |
| S_0177_4185* | CS-Rosetta | 0.723 | 72 | 7.0 | 157 | 0.339 | 0.598 |
| S_4043_8555 | CS-Rosetta | 0.672 | 74 | 10.5 | 136 | 0.365 | 0.583 |
| S_4086_5730 | CS-Rosetta | 0.727 | 78 | 12.4 | 165 | 0.404 | 0.562 |
| S_5184_4038 | CS-Rosetta | 0.807 | 73 | 8.7 | 127 | 0.357 | 0.597 |
| S_2061_3401 | CS-Rosetta | 0.860 | 74 | 9.4 | 117 | 0.325 | 0.595 |
| S_5130_9429 | CS-Rosetta | 0.871 | 76 | 9.3 | 128 | 0.349 | 0.592 |
| S_2068_3220 | CS-Rosetta | 0.874 | 73 | 9.5 | 113 | 0.355 | 0.593 |
| S_5151_9974 | CS-Rosetta | 0.901 | 70 | 3.9 | -2 | 0.242 | 0.646 |
| S_0121_7585 | CS-Rosetta | 1.246 | 74 | 4.1 | 6 | 0.245 | 0.647 |
| S_4093_7187 | CS-Rosetta | 1.356 | 76 | 7.0 | 81 | 0.304 | 0.620 |
| S_0051_7452 | CS-Rosetta | 1.496 | 80 | 7.5 | 40 | 0.257 | 0.654 |
| S_6086_2986 | Rosetta | 2.447 | 76 | 3.8 | 1 | 0.223 | 0.650 |
| S_4056_3063 | Rosetta | 3.343 | 78 | 3.2 | 0 | 0.207 | 0.673 |
| S_2018_1716 | Rosetta | 3.439 | 79 | 4.2 | -4 | 0.221 | 0.678 |
| S_2004_8972 | Rosetta | 4.426 | 76 | 4.8 | 4 | 0.208 | 0.644 |
| S_0144_4826 | Rosetta | 5.336 | 78 | 4.6 | -2 | 0.225 | 0.669 |
| S_2163_5218 | Rosetta | 5.844 | 77 | 3.8 | 2 | 0.236 | 0.675 |
| S_5001_5948 | Rosetta | 5.937 | 78 | 5.1 | 1 | 0.231 | 0.661 |
| S_3046_1450 | Rosetta | 7.471 | 74 | 3.9 | 0 | 0.237 | 0.687 |
| S_3083_2462 | Rosetta | 8.506 | 78 | 4.7 | 3 | 0.227 | 0.670 |
| S_6017_0972 | Rosetta | 11.088 | 80 | 4.2 | 3 | 0.210 | 0.670 |
Lowest energy CS-Rosetta model derived from an independent calculation of 800 structures, and used in initial molecular replacement to determine SIRV-CP(46-134) structure
RMSD from refined crystal structure, alignment performed in PyMOL
Translation function Z-score
Log Likelihood Gain, likelihood that the data would have been measured given the model
Correlation coefficient between the Fcalc of the potential solution and Fobs
R factor based on the molecular replacement solution
Figure 4.
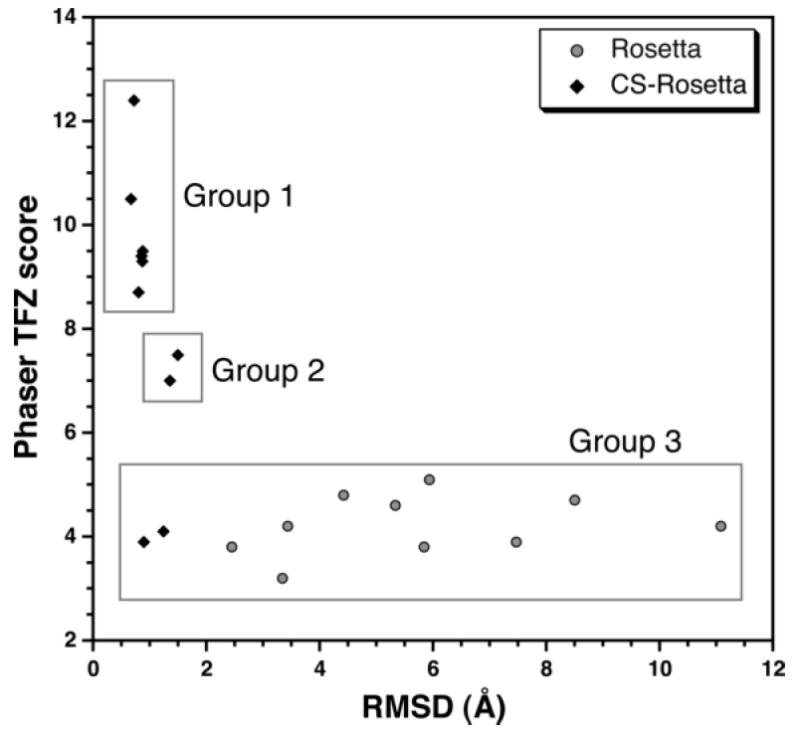
CS-Rosetta Provides Good Models for Molecular Replacement. Plot of the translation function Z-score (TFZ) versus the RMSD relative to the crystal structure of the SIRV-CP C-terminal domain. Most of the CS-Rosetta models fall in either Group 1 or 2, and may serve as good or decent molecular replacement models, respectively. All of the Rosetta models and two of the CS-Rosetta models (Group 3) failed to provide molecular replacement solutions.
Convergence of the SIRV-CP C-terminal domain structure was observed in a set of 1,000 models generated by CS-Rosetta. For the molecular replacement comparison, a similar number of models were generated using Rosetta alone. It is important to note, though, that the calculation of 20,000 – 30,000 de novo models is recommended in order to observe convergence of the Rosetta generated folds (Bradley et al., 2005). Subsequent calculation of 20,000 Rosetta models of SIRV-CP(46-134) still did not result in convergence, and did not yield the same four-helix bundle structure obtained using CS-Rosetta. The 10 lowest energy Rosetta models obtained from the calculation have an average Cα-RMSD of 6 ± 2 Å for residues 51-128, relative to the lowest energy structure determined using CS-Rosetta. Similar to the calculation of 1,000 Rosetta structures, it is expected that most of these models would not provide molecular replacement solutions.
DISCUSSION
There is considerable opportunity for synergy between NMR spectroscopy, X-ray crystallography, and computation. Structure determination using X-ray crystallography can be rapid, however crystallization and phasing may present major obstacles in this process. NMR and computation can be extremely powerful tools that help overcome these obstacles, as illustrated by the process carried out for the SIRV-CP(46-134) structure determination, shown in Figure 1. Provided soluble, mono-disperse samples of a suitable-sized protein are obtainable, the iterative evaluation of constructs for crystallization may be accomplished using simple 1H,15N-HSQC experiments. This assessment may be enhanced by 13C,15N-labeling the protein and assigning the backbone chemical shifts. Chemical shifts are useful in both the identification of regions within proteins, such as residues that interact with a ligand, and, in combination with 15N-relaxation data, the identification of flexible regions that may hamper crystallization efforts. Chemical shifts are also powerful when combined with the Rosetta tertiary structure prediction program (Bradley et al., 2005; Rohl et al., 2004; Shen et al., 2008a). The accuracy of the CS-Rosetta models is greatly improved relative to Rosetta alone.
One of the major challenges for the generation of suitable de novo structures is getting close enough to the correct location in conformational space such that the structural refinement procedures will identify the global minimum. The Rosetta program limits this search by selecting fragment conformations based upon secondary structure prediction from the protein sequence and its homology to other proteins. Although prediction tools have improved, the method still requires searching a large conformational space (Chivian and Baker, 2006).
The additional constraints provided by the backbone and Cβ chemical shifts in CS-Rosetta limits the range of conformational space that needs to be explored, increasing the rate of convergence of the structures. The SPARTA program (Shen and Bax, 2007; Shen et al., 2008a), which back-calculates chemical shifts from the generated model, is implemented at the end of the CS-Rosetta analysis to further assess the agreement of the model with the raw data, further improving the quality of the lowest energy models. The Rosetta algorithm, using only the secondary structure predictions obtained from Robetta, predicted the helical region of SIRV-CP, but the α-helixes were not as well defined and their orientation in the final predicted models deviated significantly from the crystal structure (Figure S2b). Nevertheless, 8 of the 10 lowest energy structures selected from only 1,000 CS-Rosetta models yielded molecular replacement solutions capable of determining the X-ray structure of the NMR-optimized construct, SIRV-CP(46-134). In cases where the protein domain studied is larger and the fold is more complicated, it may be necessary to generate the recommended 10,000 to 20,000 CS-Rosetta models in order to observe convergence of the fold (Shen et al., 2008a).
Molecular replacement is a quick and efficient means to obtain initial phases for structure determination, provided there is a model with high structural similarity. The use of previously determined, closely related structures is common in molecular replacement studies, but more time-consuming methods for determining the phases is necessary in the absence of such a model. Provided that the starting models are close enough in conformational space to the actual structure, the Rosetta program will generate models that are suitable for molecular replacement (Das and Baker, 2008; Qian et al., 2007; Ramelot et al., 2008). Weak homology models and NMR structures have a limited success rate with molecular replacement (Chen and Clore, 2000; Chen et al., 2000), but their ability to serve as suitable models is improved by the Rosetta algorithm. De novo models generated by Rosetta may also work in molecular replacement, but, due to the large conformational space that needs to be sampled, the inclusion of only secondary structure predictions has a limited success rate in generating suitable models (Qian et al., 2007; Rigden et al., 2008).
The application of CS-Rosetta on complete and incomplete chemical shift data sets reveals the robustness of the program and its ability to calculate accurate structural models (Shen et al., 2008a; Shen et al., 2008b). The original testing of the program on proteins with a variety of folds (all-α, all-β, mixed-α/β) demonstrated that almost all of the models have a backbone RMSD less than 1.8 Å relative to the actual NMR structure. Over half of the models have an RMSD less than 1.5 Å, the widely accepted rule-of-thumb limit for successful molecular replacement (Read, 2001; Rossman and Blow, 1963; Shen et al., 2008a). In addition, the CS-Rosetta structures were deemed to be higher in quality relative to NMR structures, based upon structure validation programs.
While molecular replacement can fail with search models that are highly similar to the crystal structure due to minor structural deviations, the failure of the Rosetta models to provide molecular replacement solutions in this study is not due to such effects. A comparison of the 10 lowest energy Rosetta and CS-Rosetta models clearly reveals that the Rosetta models did not converge to a uniform structure, indicating that the models were not near the global minimum (Figure S2). The eccentricities of molecular replacement may indeed be the reason that the two CS-Rosetta models in Group 3 failed to provide a solution, since they only deviate considerably from the crystal structure in their C-terminal loop conformations and the angle of helix 1. In this case, however, it is clear that Rosetta alone did not accurately predict the SIRV-CP fold, thus the models could not provide reasonable initial phases.
The combination of previous computational results with our successful application of CS-Rosetta to determine the crystal structure of the C-terminal domain of the SIRV-CP suggests this technique will likely be broadly applicable. This synergistic method has also been successfully applied in our laboratory to a dimeric protein complex with a different overall fold (data not shown). This approach will have its limitations, but the ability to include sparse, unambiguous long-range distance constraints in the CS-Rosetta program calculation will further improve the structures one can obtain and enhance the likelihood that the model will provide phases using molecular replacement. Other programs that implement chemical shifts in the prediction of tertiary structure, such as CHESHIRE (Cavalli et al., 2007; Robustelli et al., 2008) and CS23D (Wishart et al., 2008), may prove to be equally powerful in the generation of suitable molecular replacement models. This technique may also be invaluable in cases where phasing from heavy atoms is not possible.
The structure determination of SIRV-CP was driven by interest in virus structure, and an interdisciplinary approach was taken using a combination of NMR, X-ray crystallography and computation. The course taken proved to be extremely effective and has broader implications for efficient structure determination. Each of the three techniques has its strengths and limitations, and it may be a profitable strategy for both individual laboratories and structural genomics efforts to employ such a synergistic approach.
EXPERIMENTAL PROCEDURES
Gene cloning
The SIRV-YNP coat protein gene was amplified from an enrichment culture established from an acidic hot spring within the Rabbit Creek thermal area of Yellowstone National Park (83 C, pH = 3.1, 44°31.287’N, 110°48.647’W). PCR primers were designed based on a sequence alignment of the coat proteins from SIRV1 and SIRV2. The primers (DBP-F 5’-GATATTGACCAAAAATGGCAAAAGG-3’ and DBP-R 5’-GATATTGACCAAAAATGGCAAAAGG-3’) were used in a PCR amplification reaction and cloned into the pET 30a(+) expression vector (Novagen). The construct was confirmed by DNA sequencing.
Protein expression & purification
The full-length SIRV-YNP coat protein and N-terminal deletion mutant containing residues 46-134, SIRV-CP(46-134), were expressed from a pET30a-derived expression vector (Novagen). A C-terminal hexahistidine tag (GGSGHHHHHH) was included in the SIRV-CP(46-134) construct. The 15N,13C-labeled sample was expressed in E. coli BL21 (DE3) pLysS cells grown in standard M9 minimal media with 15NH4SO4 as the sole nitrogen source, 13C-glucose as the sole carbon source, and supplemental trace metals and vitamins. 15N-labeled samples were grown in media containing unlabeled glucose, while unlabeled samples were grown in Luria-Bertani media. Due to problems with proteolysis, even in the presence of PMSF and protease inhibitor cocktails, the cells were lysed by sonication in a denaturing buffer: 8 M urea, 100 mM sodium phosphate, 10 mM Tris•HCl (pH = 8.0). Cellular debris was pelleted at 20,000 rpm in a 50.2 Ti rotor using a Beckman Optima L-90k ultracentrifuge.
The supernatant for SIRV-CP was filtered and loaded onto a 5 mL HiTrap SP HP column (GE Healthcare). The column was washed with 30 column volumes of denaturing buffer (pH = 6.0), 30 column volumes of 50 mM sodium phosphate (pH = 6.0), and then eluted with a 50-column volume gradient to 50 mM sodium phosphate (pH = 6.0) with 1 M NaCl. The fractions containing the protein were dialyzed against 50 mM sodium phosphate (pH = 6.0).
The SIRV-CP(46-134) protein construct was purified using nickel affinity chromatography. The protein was bound in batch to 5 mL of Ni-NTA (Invitrogen), and was washed with 50 mL of 8 M urea, 100 mM sodium phosphate, 10 mM Tris•HCl (pH = 6.0) and then 50 mL of 100 mM sodium phosphate (pH = 6.0), 250 mM NaCl, and 20 mM imidazole. The protein was eluted in two steps with 100 mM sodium phosphate (pH = 6.0), 250 mM NaCl, 250 mM imidazole and 100 mM sodium phosphate, 250 mM NaCl, 500 mM imidazole. The fractions were dialyzed against 50 mM sodium phosphate (pH = 6.0), 0.5 M NaCl.
Samples were concentrated, and further purified by size exclusion on a Sephacryl S-200 column (26/60, GE Healthcare) in 50 mM sodium phosphate (pH = 6), 0.5 M NaCl. Isotope labeled samples were buffer exchanged into 20mM CD3COOH (pH = 4.5) containing 10% D2O/90% H2O, while unlabeled samples were dialyzed extensively against 20 mM MES (pH = 6) for crystallization. The final concentration of protein for the NMR samples was 0.8 mM.
NMR spectroscopy
Assignment of the coat protein backbone atoms was accomplished using classical triple resonance experiments. Three-dimensional HNCO (Grzesiek and Bax, 1992b; Kay et al., 1990), HNCA (Grzesiek and Bax, 1992b; Kay et al., 1990), HN(CO)CA (Bax and Ikura, 1991; Grzesiek and Bax, 1992b), HNCACB (Wittekind and Mueller, 1993), CBCA(CO)NH (Grzesiek and Bax, 1992a), and HBHA(CO)NH (Grzesiek and Bax, 1993a) experiments were collected on a 500 MHz Bruker Avance spectrometer equipped with a TXI 5mm probe. 1H,15N-HSQC experiments employing water flip-back pulses were also collected (Grzesiek and Bax, 1993b). For data processing and analysis, the NMRPipe program package (Delaglio et al., 1995) and CARA (Keller, 2005) programs were used. Doubling the number of points in the 15N and 13C dimensions by linear prediction increased the resolution of the spectra. NMR experiments were acquired at 35°C, and all chemical shifts are relative to 2,2-dimethyl-2-silapentane-5-sulfonate (Wishart et al., 1995).
Definition of the SIRV-YNP coat protein construct used for crystallography was based upon the Preditor and Chemical Shift Index (CSI) secondary structure prediction programs and 15N-relaxation data (Berjanskii et al., 2006; Farrow et al., 1994; Wishart and Sykes, 1994). Nitrogen T1, T2 and steady-state heteronuclear NOE relaxation experiments were collected for the full-length coat protein using sensitivity enhanced pulse programs (Farrow et al., 1994). The experiments were acquired at 24.6°C, which was calibrated using methanol, on a shielded 800 MHz Bruker Avance spectrometer equipped with a TXI 5mm probe. T1 and T2 relaxation delay times ranged from 10 ms to 3840 ms and 6 ms to 258 ms, respectively. Delay time points were collected in a random order to avoid any systematic errors, and fit using a 2-parameter formula in the CurveFit program (Mandel et al., 1995). The saturated and unsaturated heteronuclear NOE spectra were collected in an interleaved manner, and the ratio for each resonance was calculated using the signal intensities. Spectra were processed using NMRpipe (Delaglio et al., 1995) and analyzed using NMRVIEW (Johnson and Blevins, 1994).
Structure prediction
Structural models of the C-terminal structured region of SIRV-CP were obtained from both the CS-Rosetta and Rosetta programs (Bradley et al., 2005; Rohl et al., 2004; Shen et al., 2008a). The list of fragments used for the CS-Rosetta ab initio calculations were generated by inputting the chemical shifts for the backbone (HN, N, C, Cα, Hα) and Cβ atoms of residues 46-134 and 51-128 from the full-length protein into the CS-Rosetta program. Fragments for the Rosetta structural models were generated by submitting the protein sequence of SIRV-CP(46-134) to the Robetta server (Chivian et al., 2003; Kim et al., 2004). The generated fragments and associated files were downloaded and used as input to the Rosetta module within the CS-Rosetta program. CS-Rosetta calculations were run in parallel on a local 64-bit Linux computer cluster containing 3808 CPUs, to generate between 800-1200 structures for each of the fragment sets. On average the calculation times of the CS-Rosetta models were 15.6 min/model for SIRV(46-134), and 12.8 min/model for SIRV(51-128); calculation of the SIRV(46-134) Rosetta models required 14.3 min/model. The 500 lowest energy structural models were extracted to assess convergence, and the 10 lowest energy models were used in the molecular replacement trials.
X-ray crystallography
Needle clusters of His-tagged SIRV-CP(46-134) grew at 26°C in a 2 μl sitting drop. SIRV-CP(46-134) at 19 or 30 mg/mL was mixed 1:1 with a solution of 25% PEG 20,000, 0.1 M sodium citrate (pH = 3.6); inclusion of 4-9% sucrose in the PEG solution improved crystal morphology from needles to rods. Crystals were cryoprotected by soaking in a solution of 14% PEG 20,000, 50 mM sodium citrate (pH = 3.6), 15-25% sucrose; soak time did not appear to affect the diffraction. Diffraction was collected to 1.67 Å on a Rigaku FR-D generator with a MAR345 detector. The diffraction was indexed and processed using XDS (Kabsch, 1993), and molecular replacement was implemented with Phaser (McCoy et al., 2007). Prime-and-switch and statistical density modification were performed with Resolve (Terwilliger, 2000, 2004). CNS (Brünger, 2007; Brünger et al., 1998) was used for initial refinement, rigid body refinement and simulated annealing, with model-building in Coot (Emsley and Cowtan, 2004). PHENIX (Adams et al., 2002) was used in later rounds of refinement for coordinate minimization and adjustment of individual B-factors, with alternating steps of refitting and rebuilding in Coot. The coordinates were deposited in the Protein Data Bank under accession number 3F2E. Molecular replacement tests of Rosetta and CS-Rosetta models were implemented in both EPMR (Kissinger et al., 1999) and Phaser (McCoy et al., 2007).
Supplementary Material
Acknowledgments
We thank Gerard Kroon for helpful discussions, suggestions, and NMR support. We also thank Ian Wilson for the use of his X-ray generator. This work was supported by grants from the NIH to J.E.J. (GM-054076) and J.R.W. (GM-53320), a Ruth L. Kirschstein National Research Service Award to R.E.T. (GM-084476-01), and a CIHR fellowship to B.R.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Bax A, Ikura M. An efficient 3D NMR technique for correlating the proton and15N backbone amide resonances with the α-carbon of the preceding residue in uniformly15N/13C enriched proteins. J Biomol NMR. 1991;1:99–104. doi: 10.1007/BF01874573. [DOI] [PubMed] [Google Scholar]
- Berjanskii MV, Neal S, Wishart DS. PREDITOR: a web server for predicting protein torsion angle restraints. Nucleic Acids Res. 2006;34:W63–69. doi: 10.1093/nar/gkl341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz M. One century of protein crystallography: the phycobiliproteins. Biol Chem. 1997;378:167–176. [PubMed] [Google Scholar]
- Bowers PM, Strauss CE, Baker D. De novo protein structure determination using sparse NMR data. J Biomol NMR. 2000;18:311–318. doi: 10.1023/a:1026744431105. [DOI] [PubMed] [Google Scholar]
- Bradley P, Misura KM, Baker D. Science. Vol. 309. New York, N.Y: 2005. Toward high-resolution de novo structure prediction for small proteins; pp. 1868–1871. [DOI] [PubMed] [Google Scholar]
- Brünger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Brünger AT, Campbell RL, Clore GM, Gronenborn AM, Karplus M, Petsko GA, Teeter MM. Science. Vol. 235. New York, N.Y: 1987. Solution of a Protein Crystal Structure with a Model Obtained from NMR Interproton Distance Restraints; pp. 1049–1053. [DOI] [PubMed] [Google Scholar]
- Cavalli A, Salvatella X, Dobson CM, Vendruscolo M. Protein structure determination from NMR chemical shifts. Proc Natl Acad Sci USA. 2007;104:9615–9620. doi: 10.1073/pnas.0610313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao JA, Williamson JR. Joint X-ray and NMR refinement of the yeast L30e-mRNA complex. Structure. 2004;12:1165–1176. doi: 10.1016/j.str.2004.04.023. [DOI] [PubMed] [Google Scholar]
- Chen YW, Clore GM. A systematic case study on using NMR models for molecular replacement: p53 tetramerization domain revisited. Acta Crystallogr D Biol Crystallogr. 2000;56:1535–1540. doi: 10.1107/s0907444900012002. [DOI] [PubMed] [Google Scholar]
- Chen YW, Dodson EJ, Kleywegt GJ. Does NMR mean “not for molecular replacement”? Using NMR-based search models to solve protein crystal structures. Structure. 2000;8:R213–220. doi: 10.1016/s0969-2126(00)00524-4. [DOI] [PubMed] [Google Scholar]
- Chivian D, Baker D. Homology modeling using parametric alignment ensemble generation with consensus and energy-based model selection. Nucleic Acids Res. 2006;34:e112. doi: 10.1093/nar/gkl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivian D, Kim DE, Malmstrom L, Bradley P, Robertson T, Murphy P, Strauss CE, Bonneau R, Rohl CA, Baker D. Automated prediction of CASP-5 structures using the Robetta server. Proteins. 2003;53(Suppl 6):524–533. doi: 10.1002/prot.10529. [DOI] [PubMed] [Google Scholar]
- Clore GM, Gronenborn AM. NMR and X-ray analysis of the three-dimensional structure of interleukin-8. Cytokines. 1992;4:18–40. [PubMed] [Google Scholar]
- Das R, Baker D. Macromolecular modeling with Rosetta. Annu Rev Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- Fiorito F, Herrmann T, Damberger FF, Wüthrich K. Automated amino acid side-chain NMR assignment of proteins using (13)C- and (15)N-resolved 3D [(1)H, (1)H]-NOESY. J Biomol NMR. 2008;42:23–33. doi: 10.1007/s10858-008-9259-x. [DOI] [PubMed] [Google Scholar]
- Grishaev A, Llinás M. BACUS: A Bayesian protocol for the identification of protein NOESY spectra via unassigned spin systems. J Biomol NMR. 2004;28:1–10. doi: 10.1023/B:JNMR.0000012846.56763.f7. [DOI] [PubMed] [Google Scholar]
- Grzesiek S, Bax A. Correlating Backbone Amide and Side-Chain Resonances in Larger Proteins by Multiple Relayed Triple Resonance NMR. J Am Chem Soc. 1992a;114:6291–6293. [Google Scholar]
- Grzesiek S, Bax A. Improved 3D Triple-Resonance NMR Techniques Applied to a 31-kDa Protein. J Magn Reson. 1992b;96:432–440. [Google Scholar]
- Grzesiek S, Bax A. Amino-Acid Type Determination in the Sequential Assignment Procedure of Uniformly C-13/N-15-Enriched Proteins. J Biomol NMR. 1993a;3:185–204. doi: 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- Grzesiek S, Bax A. The Importance of Not Saturating H2O in Protein NMR - Application to Sensitivity Enhancement and NOE Measurements. J Am Chem Soc. 1993b;115:12593–12594. [Google Scholar]
- Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002a;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J Biomol NMR. 2002b;24:171–189. doi: 10.1023/a:1021614115432. [DOI] [PubMed] [Google Scholar]
- Hoffman DW, Cameron CS, Davies C, White SW, Ramakrishnan V. Ribosomal protein L9: a structure determination by the combined use of X-ray crystallography and NMR spectroscopy. J Mol Biol. 1996;264:1058–1071. doi: 10.1006/jmbi.1996.0696. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Blevins RA. NMR View: A Computer-Program for the Visualization and Analysis of NMR Data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Automatic Processing of Rotation Diffraction Data from Crystals of Initially Unknown Symmetry and Cell Constants. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- Kay LE, Ikura M, Tschudin R, Bax A. 3-Dimensional Triple-Resonancee NMR-Spectroscopy of Isotopically Enriched Proteins. J Magn Reson. 1990;89:496–514. doi: 10.1016/j.jmr.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Keller RLJ. ETH Zürich #15947. Zürich, Switzerland: ETH Zürich #15947; 2005. Optimizing the process of NMR spectrum analysis and computer aided resonance assignment. [Google Scholar]
- Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526–531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissinger CR, Gehlhaar DK, Fogel DB. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D Biol Crystallogr. 1999;55:484–491. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- Lemak A, Steren CA, Arrowsmith CH, Llinas M. Sequence specific resonance assignment via Multicanonical Monte Carlo search using an ABACUS approach. J Biomol NMR. 2008;41:29–41. doi: 10.1007/s10858-008-9238-2. [DOI] [PubMed] [Google Scholar]
- Mandel AM, Akke M, Palmer AG., 3rd Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J Mol Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prangishvili D, Arnold HP, Gotz D, Ziese U, Holz I, Kristjansson JK, Zillig W. A novel virus family, the Rudiviridae: Structure, virus-host interactions and genome variability of the sulfolobus viruses SIRV1 and SIRV2. Genetics. 1999;152:1387–1396. doi: 10.1093/genetics/152.4.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian B, Raman S, Das R, Bradley P, McCoy AJ, Read RJ, Baker D. High-resolution structure prediction and the crystallographic phase problem. Nature. 2007;450:259–264. doi: 10.1038/nature06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramelot TA, Raman S, Kuzin AP, Xiao R, Ma LC, Acton TB, Hunt JF, Montelione GT, Baker D, Kennedy MA. Improving NMR protein structure quality by Rosetta refinement: A molecular replacement study. Proteins. 2008 doi: 10.1002/prot.22229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raves ML, Doreleijer JF, Vis H, Vorgias CE, Wilson KS, Kaptei R. Joint refinement as a tool for thorough comparison between NMR and X-ray data and structures of HU protein. J Biomol NMR. 2001;21:235–248. doi: 10.1023/a:1012927325963. [DOI] [PubMed] [Google Scholar]
- Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D Biol Crystallogr. 2001;57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- Reuter W, Wiegand G, Huber R, Than ME. Structural analysis at 2.2 Å of orthorhombic crystals presents the asymmetry of the allophycocyanin-linker complex, AP.LC7.8, from phycobilisomes of Mastigocladus laminosus. Proc Natl Acad Sci USA. 1999;96:1363–1368. doi: 10.1073/pnas.96.4.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice G, Stedman K, Snyder J, Wiedenheft B, Willits D, Brumfield S, McDermott T, Young MJ. Viruses from extreme thermal environments. Proc Natl Acad Sci USA. 2001;98:13341–13345. doi: 10.1073/pnas.231170198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigden DJ, Keegan RM, Winn MD. Molecular replacement using ab initio polyalanine models generated with ROSETTA. Acta Crystallogr D Biol Crystallogr. 2008;64:1288–1291. doi: 10.1107/S0907444908033192. [DOI] [PubMed] [Google Scholar]
- Robustelli P, Cavalli A, Vendruscolo M. Determination of Protein Structures in the Solid State from NMR chemical shifts. Structure. 2008;16:1764–1769. doi: 10.1016/j.str.2008.10.016. [DOI] [PubMed] [Google Scholar]
- Rohl CA, Baker D. De novo determination of protein backbone structure from residual dipolar couplings using Rosetta. J Am Chem Soc. 2002;124:2723–2729. doi: 10.1021/ja016880e. [DOI] [PubMed] [Google Scholar]
- Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- Rossman MG, Blow DM. Determination of Phases by Conditions of Non-Crystallographic Symmetry. Acta Crystallogr. 1963;16:39–45. [Google Scholar]
- Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog Nucl Magn Reson Spectrosc. 1999;34:93–158. [Google Scholar]
- Schiffer CA, Huber R, Wüthrich K, van Gunsteren WF. Simultaneous refinement of the structure of BPTI against NMR data measured in solution and X-ray diffraction data measured in single crystals. J Mol Biol. 1994;241:588–599. doi: 10.1006/jmbi.1994.1533. [DOI] [PubMed] [Google Scholar]
- Shaanan B, Gronenborn AM, Cohen GH, Gilliland GL, Veerapandian B, Davies DR, Clore GM. Science. Vol. 257. New York, N.Y: 1992. Combining experimental information from crystal and solution studies: joint X-ray and NMR refinement; pp. 961–964. [DOI] [PubMed] [Google Scholar]
- Shen Y, Bax A. Protein backbone chemical shifts predicted from searching a database for torsion angle and sequence homology. J Biomol NMR. 2007;38:289–302. doi: 10.1007/s10858-007-9166-6. [DOI] [PubMed] [Google Scholar]
- Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, Eletsky A, Wu Y, Singarapu KK, Lemak A, et al. Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci USA. 2008a;105:4685–4690. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Vernon R, Baker D, Bax A. De novo protein structure generation from incomplete chemical shift assignments. Journal of biomolecular NMR. 2008b doi: 10.1007/s10858-008-9288-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz NF, Bize A, Findlay KC, Lomonossoff GP, Manchester M, Evans DJ, Prangishvili D. Site-specific and spatially controlled addressability of a new viral nanobuilding block: Sulfolobus islandicus rod-shaped virus. Adv Funct Mater. 2008;18:3478–3486. [Google Scholar]
- Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC. Using prime-and-switch phasing to reduce model bias in molecular replacement. Acta Crystallogr D Biol Crystallogr. 2004;60:2144–2149. doi: 10.1107/S0907444904019535. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Arndt D, Berjanskii M, Tang P, Zhou J, Lin G. CS23D: a web server for rapid protein structure generation using NMR chemical shifts and sequence data. Nucleic Acids Res. 2008;36:W496–502. doi: 10.1093/nar/gkn305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- Wittekind M, Mueller L. HNCACB, a High-Sensitivity 3D NMR Experiment to Correlate Amide-Proton and Nitrogen Resonances with the Alpha- and Beta-Carbon Resonances in Proteins. J Magn Reson B. 1993;101:201–205. [Google Scholar]
- Zheng D, Huang YJ, Moseley HN, Xiao R, Aramini J, Swapna GV, Montelione GT. Automated protein fold determination using a minimal NMR constraint strategy. Protein Sci. 2003;12:1232–1246. doi: 10.1110/ps.0300203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.