

Abstract
This opinion identifies inconsistencies in the generally-accepted surface biophysics involved in contact activation of blood-plasma coagulation, reviews recent experimental work aimed at resolving inconsistencies, and concludes that this standard paradigm requires substantial revision to accommodate new experimental observations. Foremost among these new findings is that surface-catalyzed conversion of the blood zymogen factor XII (FXII, Hageman factor) to the enzyme FXIIa ( , a.k.a. autoactivation) is not specific for anionic surfaces, as proposed by the standard paradigm. Furthermore, it is found that surface activation is moderated by the protein composition of the fluid phase in which FXII autoactivation occurs by what appears to be a protein adsorption-competition effect. Both of these findings argue against the standard view that contact activation of plasma coagulation is potentiated by assembly of activation-complex proteins (FXII, FXI, prekallikrein, and high-molecular-weight kininogen) directly onto activating surfaces (procoagulants) through specific protein/surface interactions. These new findings supplement the observation that adsorption behavior of FXII and FXIIa is not remarkably different from a wide variety of other blood proteins surveyed. Similarity in adsorption properties further undermines the idea that FXII and/or FXIIa are distinguished from other blood proteins by unusual adsorption properties resulting in chemically-specific interactions with activating anionic surfaces.
Keywords: Blood coagulation, FXII, Hageman factor, contact activation, autoactivation, protein adsorption
1. Introduction
The need for improved cardiovascular healthcare worldwide is large and growing. Some statistics give a sense of scale. According to the Centers for Disease Control, 23 million were diagnosed with heart problems in 2001 [1, 2], driving use of nearly 2 million stents and implantation of more than 200,000 heart valves [3]. In that same year, 700,142 people in the United States died from cardiovascular disease, making it the leading cause of death. The estimated 2005 United States market for cardiovascular-related medical products exceeded $14.7 billion. Medical devices such as in-dwelling pumps, stents, valves, and ventricular assists are an important part of the cardiovascular-healthcare strategy aimed at servicing this demand. Biomaterials exhibiting good hemocompatibility are an essential enabling technology for this strategy. And yet, even after decades of focused research, thrombosis remains the significant barrier to development and implementation of advanced blood-contacting medical devices [4, 5]. Clearly, the field of biomaterials requires an improved understanding of the events leading to thrombus formation on biomaterials that will pave new bioengineering routes to hemocompatibility.
Thrombus that forms on all known biomaterials is partly due to platelet-mediated reactions and partly due to coagulation of blood plasma itself, in proportions that presumably depend on the surface chemistry of the biomaterial and characteristics of the blood flow in which the biomaterial is immersed [6, 7]. This Leading Opinion is focused only on blood-plasma coagulation as it occurs in vitro using plasma that has been substantially, but not wholly, depleted of platelets by centrifugation as a test vehicle (platelet-poor plasma). This approach simplifies the whole-blood coagulation problem by dividing it into plasma- and platelet-mediated regimes and eliminates pulsatile flow under blood pressure as a variable [8]. Whereas conclusions drawn from this work must therefore be applied to whole-blood coagulation and in vivo conditions with considerable caution, the simplification permits close examination of the protein reactions involved in thrombosis that might otherwise be obscured or greatly complicated by platelet contributions.
Research into plasma-phase coagulation has focused on plasma protein/surface interactions that activate the so-called plasma-coagulation cascade. The idea that plasma-protein adsorption is somehow responsible for plasma coagulation, now standard biomaterials-science thinking, can be traced at least as far back as Johlin’s 1929 work [9, 10] showing that “…clotting can be induced by the contact of the plasma with substances which produce an adsorbing interface…”. As this Leading Opinion will attempt to show, activation of the plasma coagulation cascade is apparently catalyzed by contact of certain blood factors with surfaces. But this contact does not necessarily require adsorption of these factors, at least not as protein adsorption is formally understood as an interfacial concentration of protein caused by chemically-specific and/or physical processes. This latter finding alone is a significant motivation to revise the currently-accepted mechanism of contact-activated plasma coagulation outlined below.
1.1 The Plasma Coagulation Cascade
Plasma coagulation is thought to occur through a series of interconnected self-amplifying, zymogen-enzyme conversions (Fig. 1, see ref. [11] for more discussion) that penultimately produces thrombin (FIIa), a powerful serine protease [6, 12-14]. In the final step of the plasma coagulation cascade, FIIa hydrolyses fibrinogen into fibrin units that oligomerize into a fine mesh which, in turn, causes plasma to gel or clot. Biochemistry of the plasma coagulation cascade represented in Fig. 1 was assembled from the highly-original work of a number of investigators starting as early as the 1930’s (see ref. [15] and citations therein) and continuing through the 1970’s. In particular, the pioneering work of Oscar Ratnoff discovering the role of Hageman factor (FXII) in contact activation and proposal of the waterfall or cascade mechanism of coagulation deserves special mention (see refs. [14, 16-19] and citations therein for historical reviews). Collectively, these efforts identified the important blood factors involved in coagulation that circulate in normal blood as inactive zymogens (Table 1).
Figure 1.
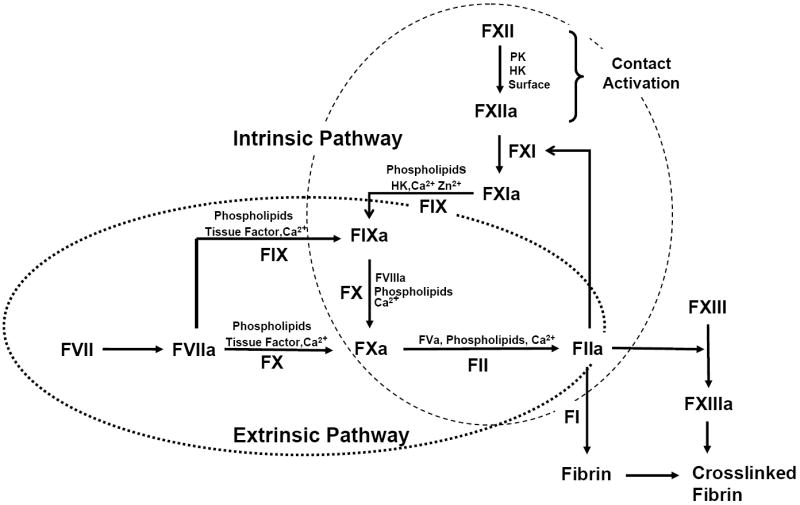
Simplified line diagram of the plasma-coagulation cascade showing intersection of the intrinsic and extrinsic pathways (many mediators and cofactors involved in hemostasis are not shown and the interaction with platelets has been ignored for simplicity). Contact-activation reactions that are proposed to be surface mediated by the consensus mechanism are expanded in Fig. 2. Blood zymogens are listed in Table 1 and activated forms are denoted by an “a” suffix. Calcium-dependent reactions are suspended in the presence of calcium-chelating anticoagulants such as citrate used to prepare platelet-poor plasma for coagulation studies. Activation of the intrinsic pathway by contact activation of FXII (upper-right, Intrinsic Cascade) is the focus of this Leading Opinion.
Table 1.
Plasma Coagulation Factors [19]
| Factor | Common Names | Molecular Weight (kDa) | Plasma Concentration (mg/dl) |
|---|---|---|---|
| FI | Fibrinogen | 340 | 200-400 |
| FII | Prothrombin | 72 | 12 |
| FIII | Tissue Thromboplastin* | - | - |
| FIV | Divalent Calcium Ion** | - | 2.2-2.5 mEq/l |
| FV | Proaccelerin (Labile Factor) | 330 | 0.4-1.4 |
| FVI | Not assigned*** | - | - |
| Factor VII | Proconvertin (Stable Factor) | 48 | 0.05-0.06 |
| Factor VIII | Antihemophilic Factor | 1000-12000 | 0.5-1 |
| Factor IX | Christmas Factor | 57 | 0.4-0.5 |
| Factor X | Stuart Prower Factor | 59 | 0.7-1.2 |
| Factor XI | Plasma Thromboplastin Antecedent | 160 | 0.4-0.6 |
| Factor XII | Hageman Factor | 80 | 1.5-4.5 |
| Factor XIII | Fibrin Stabilizing Factor | 320 | 1-2 |
| Plasma prekallikrein | Fletcher factor | 88 | 3.5-4.5 |
| High Molecular Weight Kininogen | Fitzgerald, Williams, or Flaujeac factor | 120 | 8-9 |
| Plasminogen | - | 92 | 20 |
Notes:
A lipoprotein complex principally located on cell membranes with 50<MW<330 kDa.
Total plasma calcium = 8.5-10.5 mg/dl.
FVI is no longer considered to be a coagulation following the discovery that FVI is actually an activated form of FV [110].
The plasma-coagulation cascade is usually divided into two branches for convenience of discussion and coagulopathy testing. The intrinsic and extrinsic branches can be separately potentiated but merge into a common pathway leading to FIIa. The extrinsic pathway is responsible for hemostatic control and response to vascular injury. The normal physiological function of the intrinsic pathway is not well understood [17, 20, 21], but has been implicated as an important cause for poor hemocompatibility of cadiovascular biomaterials because it is this branch that is triggered by contacting blood with artificial materials (referred to as procoagulants herein) [12, 14, 19, 22-25]. Recent work suggests that these two pathways are interconnected [18].
The coagulation cascade is sometimes referred to as an “enzyme amplifier” system for which kinetics based on coupled Michaelis-Menten type reactions was worked out shortly after elucidation of the biochemical pathways involved [26-28]. Modeling kinetics of a series of interconnected zymogen-enzyme conversions in which the product of a preceding reaction is the enzyme of a subsequent reaction is a rather complicated affair, especially when self-amplifying loops are taken into account. Early efforts have led to informative computational models of the extrinsic pathway [29-42], but much less work has been dedicated to modeling the intrinsic pathway that is activated by contact with material surfaces [43, 44]. No doubt a limitation has been the lack of fundamental information relating activator properties (surface area, chemistry, energy, etc.) to the extent that the intrinsic pathway is potentiated and the manner by which this activation is propagated down the cascade [45-48] (see Fig. 1). Evidence reviewed in this Leading Opinion suggests that contact activation does not follow coupled Michaelis-Menten type reactions (Section 5).
1.2 Contact Activation of Plasma Coagulation and the Chemical-Specificity Idea
The initiating step of the intrinsic pathway outlined above is surface-contact activation of the blood zymogen FXII (Hageman factor) into an active enzyme form FXIIa ( ), sometimes referred to as “autoactivation” in the hematology literature [17, 20]. As diagrammed in Fig. 2, binding of FXII with activating surfaces is thought to lead to the assembly of an activation complex involving FXIIa and the allosteric proteins prekallikrein (PK, Fletcher factor), high-molecular-weight kininogen (HK, Williams-Fitzgerald factor, frequently referred to as HMWK in the literature; see ref. [49] for standardized nomenclature), and FXI (thromboplastin antecedent) [19, 20]. Surface-bound HK is thought to bring all factors and co-factors into reactive proximity [24, 50] in a way that can multiply the amount of FXIIa produced by autoactivation through “reciprocal amplification” [20, 25] (see further Section 6).
Figure 2.
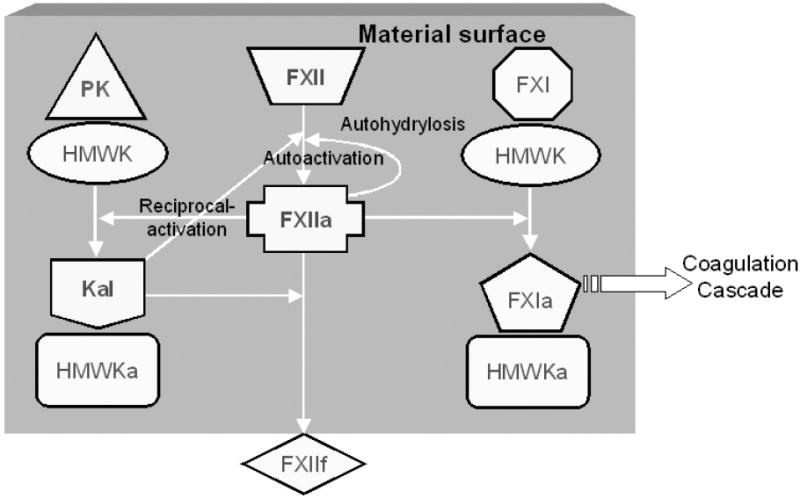
Surface-mediated interactions in contact activation of plasma coagulation according to the consensus mechanism involving the participating proteins: prekallikrein (PK), high molecular weight kininogen (HK), factor XII (FXII), factor XI (FXI), kallikrein (Kal). Suffixes “a” and “f” represent activated and fragmented forms, respectively. According to this traditional biochemical theory, FXII “binds” to a surface (represented by the grey box) bearing negatively-charged functional groups through domains rich in positively-charged lysine residues [100, 101]. Binding purportedly induces a conformational change in FXII, ultimately leading to a transformation into an active-enzyme form FXIIa through a process known as autoactivation [102, 103]. Structural changes of FXII upon binding is thought to sharply increase susceptibility to proteolytic cleavage by Kal [104]. FXIIa generated at the surface can, in turn, cleave PK bound to the surface as a complex with HK [105]. This mutual activation of PK and FXII at the surface is referred to as reciprocal-amplification. FXIIa apparently can also hydrolyze FXII by an “autohydrolysis” reaction, sometimes referred to as self-amplification [94, 106]. FXIIa is also implicated in an “autoinhibition” reaction whereby FXIIa itself inhibits production of FXIIa [8, 52]. Ultimately, FXIIa activates FXI bound at the surface as a complex with HK to generate FXIa, leading to propagation of subsequent coagulation cascade reactions diagrammed in Fig. 1 [107, 108]. The exact chemistry of putative autoactivation, autohydrolysis, and autoinhibition reactions is unknown.
The exact nature of the FXII adsorption/binding/contact step is a matter of continued investigation in biomaterials surface science [46, 47] and the central subject of this Leading Opinion. But experiment clearly shows that plasma coagulation is most efficiently activated in contact with anionic [19, 51, 52] or hydrophilic (water wettable) solid surfaces [45-47] (see Section 1.4 for a brief discussion of terminology). In particular, studies aimed at elucidating quantitative relationships among procoagulant surface chemistry, energy, and the propensity to activate the intact plasma coagulation cascade [45-48, 53-55] show that contact activation is all-but-specific for fully-water-wettable surfaces (water contact angle θ = 0°). Activation by less-water-wettable surfaces (θ > 0°) is sharply reduced. These studies are completely consistent with ordinary hematology-lab experience that blood clots much faster in glass (wettable or hydrophilic) tubes than in silanized-glass or plastic (poorly wettable or hydrophobic) tubes [56-59]. Indeed, it this simple observation that led to discovery of contact activation of blood-plasma coagulation [51] and the connection to anionic surface chemistries.
The perceived relationship between contact activation and anionic surfaces led directly to the chemical-specificity idea that confers special surface-interaction properties onto FXII that other blood proteins do not share. Without preferential attraction of FXII to anionic surfaces, FXII would be diluted out by a plethora of other blood proteins (about 490 of them at concentrations varying over six decades [60, 61]). Indeed, high-concentration blood proteins such as albumin, fibrinogen, or IgG would surely swamp assemblage of activation-complex proteins at procoagulant surfaces that are at considerably lower blood concentrations (referred to herein as an “adsorption-dilution” effect; see Section 7 for further discussion). This eventuality would sharply reduce frequency of FXII/surface interactions and render an anionic surface nearly inert to coagulation. But this is not observed to occur, leading to the idea that there is some sort of chemically-specific binding to anionic surfaces. By logical extension, this chemical-specificity idea must also mean that proteins unrelated to contact activation do not adsorb to activating surfaces, otherwise inviting the adsorption-dilution effect.
FXII specificity for anionic surfaces is proposed to be due to a chemical-binding event between positive charges (cluster of lysines) on the solvent-exposed surface of FXII molecules and negatively-charged moieties on the anionic surface [14, 62, 63]. Accordingly, FXII and/or FXIIa must be expected to adsorb to hydrophilic surfaces, at least those bearing anionic surface functionalities that react with water through formation of hydrogen bonds. It is hydrogen bonding of water to surfaces that drives the water-wetting phenomenon and gives rise to hydrophilicity [57, 59, 64]. Indeed, putative FXII adsorption from aqueous solution to hydrophilic surfaces would be in competition with water adsorption. Again, logical extension of the chemical-specificity idea implies that blood proteins unrelated to contact activation do not adsorb to hydrophilic surfaces. In sharp contrast to these expectations, recent evidence reviewed in Sections 2 and 7 strongly suggests that FXII and FXIIa do not, in fact, exhibit special anionic-surface-binding properties and cannot successfully compete with water for adsorption to hydrophilic surfaces. Also, it appears that the adsorption-dilution effect dominates activation properties of hydrophobic surfaces immersed in plasma, rendering hydrophobic materials inefficient contact activators of FXII.
1.3 Focus on Terminology
Terminology used in discussing contact activation is more than a bit vague from a purely surface-science perspective. Binding, assembly, and adsorption are used interchangeably in reference to activation-complex proteins. But it is not at all clear that is what is intended by different investigators. For the purposes of this Opinion, the term adsorption is meant to subsume all physicochemical events leading to an excess accumulation of solute or solvent at the interface between a solid procoagulant and aqueous solution [64, 65]. This excess may be positive or negative relative to bulk concentration. If solute (e.g. FXII) is at positive excess then solvent (water) is necessarily at negative excess since two objects cannot occupy the same space at the same time. Thus, proteins accumulating at a surface by adsorption from aqueous solution must displace interfacial water and compete with water adsorption at that surface, as mentioned in the preceding section. Descriptors such as binding, charge interactions, directed assembly, ion-exchange, and the like are nothing more than specific ways surface-active solutes such as proteins might adsorb to a surface (depending on surface chemistry) [66] and are not different processes than adsorption [59]. Adsorption can be detected and quantified by a great number of different sensitive techniques [59, 64, 67], perhaps the simplest of which are contact angle and wetting methods that are capable of detecting minute traces of organic substances adsorbed to the fully-water-wettable procoagulants [46, 64] that are the most potent activators of plasma coagulation.
The literature frequently refers to surfaces as either hydrophilic or hydrophobic with no general agreement as to what these relative terms quantitatively mean (see ref. [68] and citations therein). As a matter of practical convenience, we define hydrophilic as all solid surfaces that support water contact angles θ < 60° and hydrophobic as solid surfaces supporting θ > 60° [57]. Plasma coagulation by so-defined hydrophobic surfaces is so much less efficient than by an equal area of hydrophilic surface that materials fall into two groups; efficiently-activating hydrophilic procoagulants and inefficiently-activating hydrophobic procoagulants [45, 58, 59]. This is perhaps the most profound example of hydrophilic/hydrophobic contrast in the biological response to materials. We purposely exclude hydrogels from the surfaces under consideration herein because of the ambiguity in rating water-swollen materials on any sensible water-wetting (surface-energy) scale. These special materials probably require separate treatment altogether, but activation biochemistry occurring at ordinary ceramic and polymeric materials to which we generally refer (glass, silanized glass, polystyrene, gas-discharge-treated polymers and the like) is probably not different at hydrogel surfaces; except perhaps to the extent that large amounts of ABsorbed water influences this biochemistry.
The surface chemistry giving rise to hydrophilicity is sometimes referred to as “anionic” or “cationic” in the literature, presumably referring to a preponderance of negatively- or positively-charged surface-functional groups, respectively. These designations are, by themselves, incomplete specification of Lewis acid/base strength that dominates the interaction of the surface with water and solutes [57, 64]. There is sparse information in the literature that cationic surfaces are not strong activators of plasma coagulation in comparison to anionic surfaces [45, 69], which is quite consistent with the chemical-specificity idea (although we believe this to be entirely fortuitous). We refer to activating procoagulants as “anionic hydrophilic” so that there is no confusion with cationic-hydrophilic surfaces that represent a rather special subset of surfaces.
Extensive studies from our laboratories show that anionic-hydrophilic surfaces (as narrowly defined above) bearing relatively weak Lewis-base functional groups (e.g. oxidized functionalities such as hydroxyl, carbonyl; conjugate bases such as ionized carboxyl, etc.) resist protein adsorption by hydrogen-bonding to water so strongly that protein cannot displace interfacial water and become adsorbed (see, for examples, refs. [46, 70-72] and citations therein). Nevertheless, protein adsorption is without doubt a controversial subject in biomaterials surface science and many practitioners hold that protein adsorbs to all materials, including hydrophilic materials. In our opinion, both claims – all and hydrophilic – are too broad to be seriously considered in view of the facts that not “all” have been, or will ever be, fully tested and the notorious lack of a quantitative rating scale for hydrophobic/hydrophilic terminology [57, 68, 73]. Furthermore, there is need for systematic categorization of hydrophilic materials according to class (hydrogel, Lewis acid/base strength, etc.). It is of interest at this juncture to point out that acceptance of the idea that blood proteins adsorb to all materials leads to a logical conflict with the chemical-specificity idea because wholesale protein adsorption to anionic-hydrophilic surfaces would surely lead to the adsorption-dilution effect mentioned above in Section 1.2.
Theories and expectations aside, we find experimentally that that the fully-water-wettable surfaces that most efficiently activate coagulation do not measurably adsorb (as defined herein) blood proteins from solution [46, 58, 59, 72]. This finding, which is specific for the group of materials for which we have measured both protein-adsorption and coagulation-activation properties (typically, but not exclusively, borosilicate/lime glass and silanized analogs), not only conflicts with those who claim blood protein adsorbs to hydrophilic materials (however this may be defined) but also is in sharp contrast to expectations of the chemical-specificity idea.
In the continued context of Lewis acid/base strength of surface functionalities giving rise to hydrophilicity and resistance to protein adsorption, we further note that hydrophilic surfaces bearing strong Lewis acid/base functional groups exhibiting authentic ion-exchange properties in the conventional-chemistry sense of the term (such as sulfopropyl, carboxymethyl, diethylaminoethyl, or quarternary ammonium, etc.) are efficient protein adsorbents relative to hydrophobic analogs [74]. Surfaces bearing such functionalities can adsorb proteins through an ion-exchange mechanism that is unavailable to the hydrophilic surfaces bearing only relatively weak Lewis acid/base groups mentioned above [74]. This finding underscores the unmet need to scale surface properties on a more comprehensive basis that measures both surface density and acid/base properties of surface functional groups [57]. We know of no studies measuring contact activation by ion-exchange materials and it is not clear from the literature if non-activating cationic-hydrophilic surfaces exhibit authentic ion-exchange properties.
1.4 Troubling Open Questions
A number of troubling questions remain unanswered by the consensus mechanism of contact activation discussed above (numbered here for easy reference in subsequent sections). A first group of questions is related to the strong hydrophilic/hydrophobic contrast noted in blood plasma coagulation [58, 59]; namely, that coagulation activation by anionic-hydrophilic surfaces is very much more efficient than by hydrophobic analogs [45, 47]. A second group of questions is related to the unique biochemistry, not shared with all other blood proteins, proposed by the consensus mechanism to give activation-complex proteins (especially FXII) chemically-specific interactions with anionic-hydrophilic procoagulants.
Group 1 Questions
Exactly how does the strong hydrophilic/hydrophobic contrast in activation of plasma coagulation arise by adsorption? After all, it is well known that proteins adsorb more efficiently to hydrophobic surfaces than hydrophilic surfaces (except perhaps ion-exchange surfaces mentioned in Section 1.3). On this basis, hydrophobic surfaces might be expected to be activating; arguably more activating than hydrophilic surfaces, not less as observed. If adsorption is to be more specifically interpreted as assembly of activation-complex proteins that can only occur on anionic-hydrophilic surfaces, what direct surface-science evidence supports such a proposition? Or should this proposition be regarded as an unproven assertion of convenience that seems to explain the hydrophilic/hydrophobic contrast in contact activation?
Does inefficient activation by hydrophobic surfaces imply that activation-complex proteins exhibit little-or-no affinity for hydrophobic surfaces, in addition to having special affinity for hydrophilic surfaces?
Is inefficient activation by hydrophobic surfaces due to an adsorption-dilution effect by which adsorption of a plethora of proteins unrelated to coagulation dilutes FXII contact with procoagulant surfaces?
If an adsorption-dilution effect dominates hydrophobic surfaces immersed in plasma, would hydrophobic surfaces activate FXII in neat-buffer solution, and if so, what does that mean for the chemical-specificity idea?
If anionic-hydrophilic surfaces are essential for contact activation of coagulation, why does plasma slowly coagulate in hydrophobic tubes, rather than not coagulate at all? Background activation of plasma and platelets by blood collection and processing is sometimes offered as an explanation [45, 56], but without more data, background activation must be regarded as another unproven assertion of convenience.
Group 2 Questions
-
(6)
What combination of the 20 familiar proteins of the mammalian proteome could possibly be responsible for the (presumably Lewis acid-base type) interactions with anionic-hydrophilic procoagulants that are strong enough to displace bound interfacial water and adsorb to (or assemble on, bind to…) these activating surfaces? After all, even cationic surfactants such as cetyl diethylmethyl ammonium bromide (CDAB) with a quarternary-amine head group bearing a permanent positive charge (strong Lewis acid) do not adsorb to anionic-hydrophilic surfaces (Lewis bases such as clean glass) from aqueous solution because displacement of surface-bound water is energetically unfavorable [75-77]. Hence it would seem that the lysine chemistry that putatively supports the chemical-specificity idea [14, 62, 63] would not only be unique to protein biochemistry, but also broadly unique to surfactant science. Indeed, proteins do not detectably adsorb or bind to, or assemble on, anionic-hydrophilic surfaces from aqueous solution (see, for examples, refs. [46, 70-72] and citations therein), including aqueous mixtures such as serum and plasma [46, 71].
-
(7)
Ignoring that protein adsorption from plasma to anionic hydrophilic surfaces cannot be detected by sensitive techniques such as tensiometry [70, 71, 78, 79] (see also section 7) and going forward with the chemical-specificity idea for the sake of continued discussion, one is led to ask how it is that the same proteins that adsorb to anionic-hydrophilic surfaces with sufficient strength to displace bound water can also readily desorb into bulk solution to account for solution-phase reactions necessary to propagate the coagulation cascade through the common pathway (see Figs. 1,2)?
These questions, and the experimental evidence reviewed in the following sections, provoke serious concern that the consensus mechanism of contact activation that has so long served as an explanation of how biomaterials activate blood-plasma coagulation and as a rationale for poor hemocompatibility is an inadequate basis for surface engineering of advanced cardiovascular biomaterials.
2. FXII Activation by Anionic-Hydrophilic and Hydrophobic Surfaces
Given the criticality of the chemical-specificity idea to the consensus mechanism of contact activation, it is somewhat surprising that quantitative comparison of FXII autoactivation in neat-buffer solution by anionic-hydrophilic and hydrophobic surfaces seems to have only recently been performed [80]. Perhaps the apparent success of the standard paradigm in accounting for available experimental evidence neutered curiosity about such matters. Perhaps it was anticipated that interfacial behavior of purified-plasma proteins would not illuminate the network behavior of the plasma coagulation cascade [45]. Or perhaps it is because commercial availability of purified blood factors is a relatively recent development. Whatever the reasons, experiments performed with FXII in neat-buffer solution show quite conclusively that FXII autoactivation is not, in fact, specific to anionic-hydrophilic surfaces. This finding undermines a critical cornerstone of the consensus mechanism of contact activation.
By measuring rate-and-yield of FXIIa by activation of FXII in neat-buffer solution (with fixed anion/cation composition) by anionic-hydrophilic (fully-water-wettable clean glass) or hydrophobic (poorly-water-wettable silanized glass) procoagulant particles, Zhuo et al [80] showed that was effectively instantaneous for either activator type. FXIIa enzyme activity (measured by a standard plasma-coagulation-time assay) quickly rose to a fixed level that did not vary within experimentally-accessible reaction time, even though FXII was in continuous contact with activator particles. We return to this self-limiting feature of FXII activation in Section 3. In fact, Zhuo found that FXIIa yield at hydrophobic procoagulants was slightly greater than at an equivalent surface area of anionic-hydrophilic procoagulants. In sharp contrast to these results, both activation rate-and-yield was found to be significantly attenuated at hydrophobic surfaces immersed in plasma. It was concluded that was not, in fact, specific for anionic-hydrophilic procoagulants. This, in combination with the contrasting activation in neat buffer and plasma, led to the speculation that FXII activation in the presence of plasma proteins leads only to an apparent specificity for anionic-hydrophilic surfaces that is actually due to a relative diminution of the reaction at hydrophobic surfaces. Diminution of hydrophobic-procoagulant activity in the presence of plasma proteins is thus not only an alternative to the consensus mechanism but also an explanation that is consistent with activation properties measured in neat buffer solutions of FXII.
2.1 The Adsorption-Dilution Effect on Contact Activation
Zhuo pursued initial studies of FXII activation in neat buffer solutions [80] with FXII dissolved in buffer cocktails prepared from proteins unrelated to the coagulation cascade [8]. Protein cocktails were intended as chemically-defined surrogates for plasma in the sense that plasma is a complex mixture of proteins with many constituents at physiologic concentrations much higher than FXII. Activation studies in cocktails compared to neat buffer showed that added proteins reduced yield of FXIIa at hydrophobic surfaces by a mechanism unrelated to enzyme inhibitors putatively present in authentic plasma.
Although Zhuo’s studies involved only a single cocktail formulation of arbitrarily-chosen proteins and concentrations, results strongly supported earlier speculation [80] that diminution of hydrophobic-procoagulant activity in the presence of plasma proteins was, at least in part, due to the adsorption-dilution effect discussed in Section 2. This adsorption-dilution effect in plasma might well be amplified by the adsorption of activation moderators such as α-macroglobulin, C1-INH, and anti-thrombin III at hydrophobic procoagulant surfaces. More surprisingly, perhaps, it was found that added proteins increased yield of FXIIa produced at hydrophilic procoagulants compared to neat buffer solutions of FXII. Thus, it is apparent that hydrophobic surfaces appear to be relatively inert relative to hydrophilic surfaces not only because of an adsorption-dilution effect occurring at hydrophobic surfaces but also because of an enhancement of autoactivation at hydrophilic surfaces, as further discussed in Section 3.1.
2.2 Retrospective on the Chemical-Specificity Idea
It is of interest at this juncture to consider Zhuo’s surprising discovery of nearly equal autoactivation at hydrophobic and anionic-hydrophilic surfaces in light of copious literature evidence that autoactivation is specific to anionic chemistries. How could activating properties of hydrophobic materials be overlooked for so long? A primary answer appears to be that slow-to-negligible activation of plasma coagulation in hydrophobic tubes reinforced an implicit conviction that only anionic materials had contact-activation properties. This conviction apparently swayed the experimental focus away from hydrophobic materials. After all, why pursue hydrophobic test materials that appeared to be all but inert?
Following discovery of FXII by Ratnoff (see Section 1.1), many different proteases and materials were found to promote autoactivation; the latter including kaolin clay, dextran sulfates, and various phospholipid vesicles. Extensive mechanistic work by Greip et al. ([51] summarizes work up to 1986) showed that “…virtually any negatively charged surface could promote contact activation…” and that “…this is a general surface phenomenon rather than biological recognition of certain functional groups.” Griep also showed that autoactivation exhibited unusual thermal behavior, as well as specific-ion effects on autoactivation rates. Subsequent work (see, for examples, refs. [19, 52, 81]), especially using lipid vesicles as test procoagulants in plasma, served to concretize Griep’s basic conclusion that autoactivation was indeed a surface-mediated reaction peculiar to hydrophilic materials. In this latter regard, work of Zhuo et al. outlined in the preceding sections is but one more piece of corroborating evidence that anionic-hydrophilic surfaces activate FXII, even if obtained using solid materials somewhat more relevant to biomaterials than lipid vesicles.
A second factor contributing to the disregard of hydrophobic materials in past contact-activation studies is that many hematology researchers (including the authors) have a strong predilection to use plasma, if not whole blood, as a test vehicle. Clearly, purified-protein solutions cannot possibly embrace the manifold of interactions and mutual dependencies that characterize whole plasma reactions [45]. However, in the case of autoactivation (and probably other similar protein-adsorption-mediated reactions at surfaces such as complement activation), this predilection proves not to be the best course of scientific investigation. Here, complexities of protein-adsorption competition lead only to the appearance that hydrophobic surfaces are inert when, in fact, these surfaces exhibit substantial autoactivation properties. Zhuo’s inclusion of hydrophobic test procoagulants resolved an understandable oversight in prior work as part of a larger program measure FXII autoactivation by materials drawn from the full range of observable surface chemistries. This objective yet remains to be accomplished because of the labor intensity of such studies. As pointed out by Griep in 1986 “…thorough, detailed analysis of each surface with respect to solution and temperature conditions for each combination of contact activation proteins would require a prohibitive number of assays…”. Yet, a thorough understanding of contact activation may well depend on working through these combinations.
2.3 Answers to Troubling Questions
The work of Zhuo et al reviewed in the preceding sections provides a new insight into contact activation suggesting unanticipated, yet simple, answers to questions posed in Section 1.4:
Q1: Exactly how does the strong hydrophilic/hydrophobic contrast in activation of plasma coagulation arise by adsorption?
FXII activates upon contact with either hydrophobic or anionic-hydrophilic surfaces and requires no “assembly” of activation-complex proteins, as shown by neat-buffer experiments (see further Section 6). Competitive-protein adsorption occurring at hydrophobic (activating) surfaces greatly minimizes frequency of FXII contact with these surfaces when immersed in multi-component protein solutions wherein FXII is relatively dilute. Protein does not adsorb or bind to (or assemble on) anionic-hydrophilic surfaces, no adsorption competition occurs, and FXII/surface contacts are proportional to “collisions” with hydrated, anionic-hydrophilic surfaces.
Q2: Does FXII exhibit little-or-no affinity for hydrophobic surfaces?
FXII adsorbs to hydrophobic surfaces in a manner similar to any other blood protein. Interfacial energetics of adsorption suggest there is nothing unique about FXII or FXIIa in comparison to a broad range of other blood proteins (see further Section 7).
Q3: Is lack of activation by hydrophobic surfaces due to the adsorption-dilution effect?
FXII competes with other blood proteins in solution on the basis of solution concentration and characteristic adsorption affinity as further described in answer to Q1 above.
Q4: Would hydrophobic surfaces activate FXII in neat-buffer solution, and if so, what does that mean for the chemical-specificity idea?
Hydrophobic surfaces are at least as efficient FXII activators as hydrophilic surfaces in neat-buffer solution, rendering the chemical-specificity idea untenable.
Q5: Why does plasma slowly coagulate in hydrophobic tubes?
Even in the face of significant adsorption competition with a host of other plasma proteins, FXII molecules eventually contact a hydrophobic surface and activate to FXIIa. This FXIIa subsequently desorbs into plasma solution to propagate the coagulation cascade. The result is sluggish blood coagulation relative to that observed in hydrophilic tubes in which no adsorption competition occurs and FXIIa produced by autoactivation is readily available in bulk solution to propagate the coagulation cascade.
Q6: What combination of the 20 familiar amino acids comprising the mammalian proteome could possibly be responsible for the (presumably Lewis acid-base type) interactions with anionic-hydrophilic procoagulants that are strong enough to displace bound interfacial water?
No such chemistry exists, except perhaps between proteins and surfaces bearing very strong Lewis acid/base sites, such as that occurring on ion-exchange resins [74].
Q7: How it is that the same proteins that adsorb to hydrophilic surfaces with sufficient strength to displace bound water can also readily desorb into bulk solution to account for solution-phase reactions necessary to propagate the coagulation cascade through the common pathway (see Figs. 1,2)?
Proteins do not adsorb to anionic-hydrophilic surfaces. Protein adsorption to hydrophobic surfaces is, at least in part, reversible (see citations of Section 1.3). FXII and FXIIa adsorb/desorb from activating surfaces and propagate coagulation within the plasma solution phase as the next section further elaborates.
3. Reactions of FXII at Procoagulant Surfaces
Results highlighted in Section 2 do more to erode confidence in a comfortable paradigm than to replace it with and equally understandable mechanism that is consistent with new experimental results. There are at least two factors that greatly complicate assembly of a replacement mechanism, especially one that semi-quantitatively accounts for measured rate-and-yield data discussed in Section 2. First, it is evident that competitive-protein adsorption plays a significant role in mediating/moderating FXII activation. Second, the self-limiting yield of FXIIa in neat buffer solution implicates some sort of autoinhibition reaction that prevents continuous activation of FXII in the continuous presence of procoagulant surfaces. This latter feature is apparently operative in whole plasma as well and possibly controls how a “procoagulant dose” propagates through the coagulation cascade (see Section 5).
Discussing these two complicating factors briefly here for the purpose of this Leading Opinion, suffice it to repeat that protein adsorption is one of the most controversial subjects in biomaterials surface science. There is little agreement among investigators about the physical chemistry of adsorption of single proteins from solution, let alone competition among two-or-more proteins from a mixture (see, for example, ref. [82] and citations therein). Likewise kinetics of protein adsorption seems to be an unsettled matter (see, for example, ref. [83] and citations therein and Section 7). Effectively then, there is no way to predict concentrations or identities of proteins adsorbed from solution in a way that would help understand plasma coagulation - or any other biological response we wish to contemplate for that matter. Clearly, if the number and kind of proteins adsorbed to a surface are not known, then evidence-based biochemical mechanisms of the biological response to materials cannot be responsibly proposed.
And then there is the matter of autoactivation and autoinhibition for which, to our knowledge, no definitive biochemical mechanisms, or even stoichiometries, have been proposed. There is no known biochemistry that explains how (presumably) non-enzymatic cleavage of the FXII Arg353-Val354 bond involved zymogen activation [20] occurs with nearly equal efficiency at either hydrophobic or anionic-hydrophilic surfaces (see ref. [8] for more discussion). Worse, there is no known reaction that might be termed “autoinhibition” wherein FXIIa curiously inhibits production from FXII. For example, we find no experimental evidence for enzyme activity loss upon storage of FXIIa in either glass or silanized-glass vials that would suggest that FXIIa hydrolysis of FXIIa (leading to loss of FXIIa activity) is a facile reaction. And yet there must be some biochemistry that prevents continuous FXIIa production in the continuous presence of activating surfaces; especially in neat-buffer solution wherein antagonists such as C1, α-macroglobulin, and ATIII can be definitively ruled out. Simply put, the presumptive reactions termed “auto…” used to explain contact activation of plasma coagulation are themselves assertions of convenience to fit experimental observations. Clearly, direct spectroscopic (such as in refs. [25, 84]) or crystallographic evidence for reactive intermediates to auto… reactions is required to supplement inferential measurements of rate-and-yield.
Finally, it deserves mention that autoactivation is sensitive to ion composition, as already briefly discussed in Section 2.1. This factor might conspire with preferential ion adsorption at hydrophilic and hydrophobic surfaces [85] in a way that might enhance or inhibit FXII activation at different surface types [59]. Until more specific information on the exact mechanism(s) involved in autoactivation is available, such speculation must remain in that category.
3.1 Zhuo Model of Contact Activation
In full view of the uncertainties mentioned above, Zhuo et al proposed an ad hoc model of contact activation in buffer and protein cocktails [8] based on the reaction scheme of Fig. 3. Application of this minimal model to activation in plasma must be made with due caution since it does not directly consider a host of complexities, not the least of which are reciprocal amplification and/or moderation by inhibitors mentioned in Section 2.2. Essential to this scheme is the concept of an aqueous “interphase” that surrounds an activating particle and separates the physical procoagulant surface from bulk solution (the interphase concept [59] has been discussed elsewhere in the context of protein adsorption [86-93]). Proteins (minimally FXII and FXIIa in neat buffer solution) “partition” in-and-out of this interphase with a characteristic affinity measured by a partition coefficient ; where WI and WB represent interphase (I) and bulk-solution (B) concentrations (mg/mL), respectively. The only difference between anionic-hydrophilic (“philic”) and hydrophobic (“phobic”) particles recognized by Zhuo’s scheme is that Pphobic ≫ Pphilic = 1, so that WI > WB for hydrophobic surfaces (by about 100X); but WI = WB for anionic-hydrophilic surfaces (no concentration effect due to adsorption). Once in the interphase, FXII and FXIIa (FXII·S and FXIIa·S in Fig. 3) participate in autoactivation and/or autoinhibition reactions. In this way, Zhuo et al couple adsorption/desorption (partitioning) to interphase concentrations that presumably control the putative reactions autoactivation and autoinhibition.
Figure 3.

Zhuo model of FXII activation (FXII → FXIIa) at a hypothetical procoagulant particle [8] (compare to Fig. 2). All biochemical reactions are proposed to occur within a vicinal “interphase” region that surrounds a procoagulant particle immersed in aqueous FXII solutions (grossly out of scale). Adsorption/desorption is viewed as a partitioning of species between interphase and bulk-solution phases indicated by the double-headed arrows labeled “FXII Partition” and “FXIIa Partition”. Partitioning establishes the interphase concentrations FXII·S and FXIIa·S that ultimately control rate and yield of FXIIa production in solution. FXII activation in buffer solution occurs by autoactivation ( ) and autohydrolysis ( ) whereas FXII activation in plasma occurs by autoactivation and reciprocal activation (kallikrein mediated hydrolysis, not indicated in diagram; see Fig. 2) [48, 109]. FXII activation competes with an autoinhibition reaction indicated by the double-headed curved arrow in which FXIIa itself inhibits FXII→FXIIa [8, 52]. Partitioning concentrates protein within the interphase of hydrophobic procoagulant particles but not within the interphase surrounding hydrophilic procoagulants. As a consequence, surface contact activation of FXII is moderated by the protein composition of the fluid phase in which FXII is dissolved (buffer, protein cocktail, or plasma). The exact chemistry of putative autoactivation, autohydrolysis, and autoinhibition reactions is unknown.
General trends observed in FXII activation are qualitatively explained according to Zhuo’s model as follows. Adsorption at hydrophobic surfaces increases WF12·S 100-fold over that at hydrophilic surfaces thereby accelerating the reaction. This enhancement is nearly counter balanced by the combined opposing effects of increased autoinhibition (also increasing with increasing WF12·S) and enhanced FXIIa adsorption affinity at hydrophobic surfaces that resists FXIIa desorption into solution. Accordingly, FXII activation at hydrophobic surfaces would be much higher than at hydrophilic surfaces were it not for these latter compensating effects (see Question 4 of Sections 1 and 2). The model further explains that FXIIa yields are decreased at hydrophobic surfaces immersed in plasma or protein cocktail (relative to the neat-buffer solution) because these added proteins (at relatively high concentrations) effectively compete with FXII for adsorption to hydrophobic procoagulant surfaces (adsorption-dilution effect). FXII adsorption to hydrophobic procoagulant surfaces is thus significantly blocked by competing proteins, efficiency of FXII contacts are sharply diminished, and FXIIa production is commensurately reduced (see Question 3 of Sections 1 and 2). The overarching proposition of Zhuo’s model is that total FXIIa yield at a particular procoagulant surface depends on a balance between activation and autoinhibition reactions which are, in turn, moderated by the total protein composition and concentration of the fluid phase in which FXII is dissolved.
Zhuo et al. quantitatively tested the scheme of Fig. 3 by expressing core ideas in mathematical terms of a kinetic model that could be statistically fit to measured rate data. And, by taking the long-time limit of a master rate equation, Zhuo also tested kinetic formulations against steady-state activation data. The model was able to reproduce general trends observed in the data and provided semi-quantitative (±20%) estimates of parameters measuring (hypothetical) autoactivation and autoinhibition rate constants. Adequate fit of the model to experimental data suggests that the qualitative mechanism expressed above is feasible, but by no means implies that the model captures the full essence of contact activation. For a model is only as good as its weakest assumptions and, as mentioned above, there is no secure biochemistry known for either autoactivation and autoinhibition. Hence, Zhuo’s model is more reliable than the consensus mechanism only in the sense that it accommodates equal autoactivation by hydrophilic and hydrophobic surfaces and competitive-protein-adsorption moderation of FXIIa yields at hydrophobic procoagulants (adsorption-dilution effect).
3.2 The Value of Modeling Hypotheticals
Zhuo’s model, like the consensus mechanism of contact activation, is based on purely hypothetical biochemical reactions inferred from experiment but not validated by standard methods of chemistry and enzymology. It is useful, therefore, to consider the value of such speculation in light of the importance of an improved understanding of hemocompatibility mentioned in Section 1. Three benefits come to mind in this regard. First, proposition of an alternative model underscores inadequacy of the consensus mechanism that hinges on the chemical-specificity idea which experiment shows to be incorrect (Section 2). Hopefully, this will reinvigorate research into blood compatibility and the related problem of protein adsorption, both of which have suffered a degree of neglect over the last decade or so. Second, proposition of an alternative model helps clarify testable hypotheses that can lead to more definitive experiments. Third, and perhaps most significantly, Zhuo’s model gives a glimpse of factors that a more comprehensive, reaction-mechanism-based model based on secure biochemistry might be required to embrace:
Two or more surface catalyzed/mediated reactions of FXII and derivative proteins (FXIIa, FXIIf, etc.), possibly including autohydrolysis ( ; see further Section 4) [8, 80, 94].
Two or (many) more proteins engaged in adsorption competition at the same procoagulant surface, especially including activation-complex proteins.
Reciprocal (kallikrein-mediated) amplification of FXIIa produced by autoactivation in plasma.
Surface energy effects on protein adsorption.
In full view of the complexities subsumed by this brief list, there can be little doubt that the challenge facing biomaterial surface scientists hoping to quantitatively understand contact activation of blood plasma is great and will require substantial advances in understanding mechanisms of both zymogen activation at surfaces and protein adsorption from mixtures.
4. Is Autoactivation Really Surface-Mediated Autohydrolysis?
Enzyme-like specificity involved in the surface-catalyzed reaction is an enigma made even more curious by Zhuo’s finding that this reaction occurs with nearly equal efficiency at either anionic-hydrophilic or hydrophobic surfaces. Given the substantial surface-chemical differences between fully-water-wettable (anionic) glass and silanized glass used by Zhuo, one is led to suppose that cleavage of the FXII Arg353-Val354 bond involved zymogen activation [20] is generic and completely unrelated to surface chemistry. That is to say, FXII autoactivation readily occurs in contact with any artificial surface regardless of surface-chemistry type (contact is meant here to mean that a protein partitions into the interphase region surrounding the procoagulant particle; i.e. not necessarily on the physical surface but rather in the interphase region; see Fig. 3). Whereas this proposition is consistent with the observation that thrombus eventually forms on all known biomaterials, it would be somewhat dismal news for biomaterials scientists because it suggests that there is no purely surface-engineering route to improved hemocompatibility.
Zhuo et al. has speculated that autoactivation involves surface-mediated covalent-bond breakage similar to that observed to occur in polymer-brush adsorption to various liquid and solid surfaces [95]. Accordingly, cleavage of the Arg353-Val354 bond occurs only when FXII within the interphase region (FXII·S in Fig. 3) finds itself in a reaction-conducive proximity and orientation. Alternatively, it may be proposed that many random polypeptide-bond scissions occur upon entering the interphase but only cleavage of the Arg353-Val354 bond leads to FXIIa with enzyme activity. Evaluation of these and other alternatives is made difficult by the autohydrolysis reaction [80, 94] which appears to be facile in neat-buffer solution but insignificant in plasma [48, 94]. Hence, a hypothesis that cannot be rigorously ruled out with existing data suggests that autoactivation is really surface-catalyzed autohydrolysis due to the inevitable presence of vanishingly-small quantities FXIIa. Indeed, the lack of surface-chemical specificity observed in FXII activation in neat-buffer solutions would seem, on the face of it, to support a trace-FXIIa argument. And logically speaking, one cannot rigorously prove that something does not exist.
However, Zhuo et al. point out that FXII stocks used in their work failed to induce coagulation of Factor XII-depleted plasma, implying that putative FXIIa contamination must indeed be vanishingly small. Furthermore, Zhuo et al. were unable to rationalize all experimental observations by simply invoking FXIIa contamination; especially the adsorption-dilution effect and kinetic saturation of FXIIa yield in both plasma and buffer solution. Zhuo found that a minimum of two competing reactions, autoactivation and autohydrolysis, was required to accommodate all experimental outcomes. In face of the overwhelming complexity of contact activation, it is perhaps adequate to regard the reaction we term “autoactivation” as a composite reaction that includes both physical activation ( ) and enzymatic activation ( ), with both leading to FXIIa that can be subsequently amplified by reciprocal activation in plasma.
5. Procoagulant Stimulus Processing by the Intrinsic Pathway
One of the more fascinating outcomes of the work reviewed in Section 3 is the self-limiting yield of FXIIa obtained by activation of FXII in the continuous presence of procoagulant particles suspended in either neat-buffer solution or plasma. After considering various alternative options that might lead to self-limiting yield of FXIIa, Zhuo et al. concluded that autoinhibition was the best explanation of kinetic and steady-state activation data. Autoinhibition implies that the intrinsic pathway cannot be modeled as series of Michaelis-Menten type reactions that link the activation complex with the common pathway (see Fig. 1) because, according to Zhuo’s model, there is a surface-mediated interaction between FXII and FXIIa that inhibits FXIIa production; thus falling outside the scope of soluble-enzyme kinetics. Modeling contact activation may well require alternative methods [45, 47, 48] to supplement approaches that have been successfully used to model the extrinsic pathway of coagulation mentioned in Section 1.
It is of interest to speculate that autoinhibition might account, at least in part, for the discontinuous activation of plasma coagulation that leads to bolus production of FIIa in concentration proportion to the intensity of contact activation, as measured by procoagulant surface area or surface energy [47]. Bolus FIIa production suggests that the cascade somehow ‘turns off’ in the continuous presence of activating surfaces. Otherwise, continuous production of FIIa would be anticipated, not the punctuated release either inferred from experiment [47] or directly measured (ref. [96] and Fig. 4). Contact activation appears to be one likely point of control [52] and Zhuo’s work further suggests that autoinhibition is the specific limiting step involved in the process of activation that caps the total amount of FXIIa that can be produced. Even so, autoinhibition cannot alone be responsible for the discontinuous activation of the blood–plasma coagulation cascade. For some as-yet unknown reason(s), the continuous presence of enzymatically active FXIIa in plasma does not lead to continuous activation of subsequent steps of the plasma coagulation cascade. Apparently, other reactions of the plasma coagulation cascade downstream of autoactivation are subject to inhibition that limits zymogen-enzyme conversion in a peculiar proportionality to activation stimulus that affects not only the time-to-appearance of the FIIa bolus but also the magnitude and shape of the thrombin-release profile (Fig. 4).
Figure 4.

Kinetics of thrombin (FIIa) production by contact activation of recalcified plasma using glass procoagulant particles (following activation protocol outlined in refs. [45, 47]). Panel A collects results obtained with water-wettable particles (water contact angle θ < 10°) at three different surface areas: 353 mm2 (solid line), 78 mm2 (dotted line), and 15 mm2 (dashed line). Panel B collects results obtained with water-wettable glass particles (solid line), aminopropyltriethoxysilane-treated glass particles (θ ~ 70°, dotted line), or octadecyltrichlorosilane-treated glass particles (θ ~ 110°, dashed line) at either 353 mm2 (upper series) or 78 mm2 (bottom series) procoagulant surface area (see annotations). Ordinate plots fluorescence intensity (FIIa concentration) at various time points following procoagulant-plasma contact. FIIa kinetics exhibit dependence on both procoagulant surface area and surface energy, with higher surface area of any particular procoagulant yielding an earlier onset of FIIa production and increased magnitude of FIIa bolus produced (Panel A). Plasma coagulation was coincident with peak FIIa output, after which FIIa concentration decayed. Decreasing hydrophilicity (decreasing procoagulant efficiency, see Section 1.2) correlated with decreasing FIIa bolus and delayed onset of FIIa production (Panel B). FIIa concentration was measured using a fluorogenic assay that employed Z-Gly-Gly-Arg-(7-amido-4-methylcoumarin) as a substrate (Bachem Biosciences; 20 μl of Hepes Buffer and 10 μl of a 2.5mM stock solution of the fluorogenic substrate were added to a polystyrene culture dish well containing a 40 μl aliquot of plasma. After 15 min incubation at 37°C, 50ml of 20% glacial acetic acid was used to quench the reaction. Fluorescence intensity was measured in sterile Corning black-polystyrene clear-bottom 96-well plates in an excitation wavelength of 390 nm and an emission wavelength of 460 nm).
6. Autoactivation and Reciprocal Amplification
FXIIa produced by autoactivation apparently can be amplified through kallikrein-mediated reactions of the reciprocal loop [18] diagrammed in Fig. 2. By comparing activation of prekallikrein-deficient plasma and prekallikrein-reconstituted deficient plasma by hydrophobic and anionic-hydrophilic procoagulant particles, Chatterjee et al. recently determined that autoactivation can account for no more than approximately 25% of the total FXIIa produced by the intrinsic pathway [97]. Interestingly, Chatterjee found that autoactivation and reciprocal amplification increase in the same proportion with procoagulant surface energy (water-wettability), while total amount of FXIIa produced per-unit-area procoagulant remains roughly constant for any particular procoagulant. Chatterjee et al. thus corroborate Zhuo in that procoagulant surfaces are found to initiate the intrinsic cascade by producing a bolus of FXIIa in proportion to activation stimulus (measured in terms of surface energy or surface area) but activating surfaces apparently play no additional role in subsequent molecular events in the cascade. Results further suggest that the amount of enzyme produced by reciprocal-amplification is proportional to the amount of FXIIa produced by the initiating autoactivation step. In this way, it appears that the total output of the activation complex is in proportion to the autoactivation stimulus which, in plasma, is determined by both procoagulant surface chemistry (hydrophilic vs. hydrophobic, anionic vs. cationic) and surface area.
7. Adsorption of FXII from Blood
Krishnan et al. carried out a systematic comparison of blood-protein adsorption using time-and-concentration-dependent tensiometry that sensitively measures either solution/air (liquid-vapor, LV) interfacial tensions or solution/surface (solid-liquid, SL) contact angles. Adsorption energetics of selected purified blood proteins spanning 3 decades of molecular weight (including FXII and FXIIa) measured by tensiometry were compared to protein adsorption from citrated plasma and serum (bovine, ovine, equine, and human). Adsorption to the molecularly-smooth, maximally hydrophobic, LV interface [86-89] was used as a standard of comparison for adsorption to SL adsorption measured at surfaces selected from the full range of observable water-wettability [78, 90-92]. One of the compelling observations arising from this survey relevant to this Leading Opinion was that adsorption energetics of diverse proteins from purified solution or complex mixtures was strikingly similar when scaled on a weight/volume concentration basis. Krishnan et al. concluded that the structural variability among diverse blood proteins that confers profoundly different bioactivity does not greatly affect interfacial energetics in aqueous solution that drives adsorption. Rather, solution concentration, not diversity in molecular structure, was proposed to be the significant energetic driver of adsorption.
However interpreted, Krishnan’s results show that FXII and FXIIa certainly adsorb to hydrophobic surfaces and do not exhibit remarkable properties that distinguish these proteins from any other blood constituents, including failure to adsorb to fully-water-wettable surfaces (see Question 2 and 6 of Section 1.4). Furthermore, FXII was found to be only weakly surface active at physiological concentrations (about 40 μg/mL) compared to, for example, albumin (with about 45 mg/mL plasma concentrations) or IgG (with about 25 mg/mL plasma concentrations) [60, 61]. This observation raises an important question that asks how FXII can selectively adsorb from blood in the overwhelming presence of other blood proteins (see Question 3 Section 1.4). The work of Zhuo et al. reviewed in Section 3 indicates that FXII does not, in fact, effectively engage in adsorption competition with other proteins at such high concentration.
7.1 Adsorption and Coagulation Kinetics
Krishnan also observed that change in interfacial energetics due to FXII adsorption to hydrophobic surfaces was, like that for all other blood proteins tested, far slower (typically 30-60 minutes to steady state) than the plasma-coagulation process (typically minutes in the presence of hydrophilic surfaces) [86, 91]. Slow change in interfacial energetics has been taken as evidence in the past that protein adsorption is also quite slow to come to steady state. Slow protein adsorption compared to rapid coagulation is inconsistent with the consensus mechanism of activation that requires rapid adsorption/assembly of activation-complex proteins onto procoagulant surfaces. Slow protein adsorption is also inconsistent with Zhuo’s model discussed in Section 3 that requires rapid adsorption/desorption to/from procoagulant surfaces (hydrophilic or hydrophobic) at physiological concentrations to accommodate experimentally-measured autoactivation rates.
Recent investigations of protein-adsorption kinetics may have resolved this apparent inconsistency by showing that the rate-of-protein-mass accumulation at an adsorbing surface is significantly faster than the rate-of-change in interfacial energetics due to adsorption [83, 98]. In other words, protein molecules arrive at, and adsorb within, the surface region (interphase) much faster than interfacial tensions respond. Thus, the widely-held inference drawn from interfacial energetics that protein molecules slowly arrive at a surface and then undergo rate-limiting adsorption is not only incorrect, but also very misleading. A more correct view seems to be that proteins rapidly diffuse from concentrated solution within milliseconds and inflate the interphase (see further ref. [83] and citations therein). This interphase then undergoes slow decrease in volume due to efflux of interfacial water that, in turn, increases interphase protein concentration; an effect that causes decrease in measured interfacial tensions (LV or SL). Accordingly, Barnthip et al. speculate that the concentration profile of initially-adsorbed protein resembles that of the contacting solution phase [83] so that “…in mixtures such as blood, the proteins would be adsorbed simply in proportion to their surface collision frequency or concentrations…”; as argued by Brash and Lyman in the early 1960’s [99] and supported by more recent studies of protein-adsorption competition [82].
Although these latter findings do not rehabilitate the consensus mechanism of contact activation, rapid diffusion of protein molecules into an interphase surrounding a procoagulant particle does help explain rapid production of FXIIa from neat-buffer solutions of FXII measured by Zhuo et al. in a way that supports an autoactivation model like that illustrated in Fig. 3. Clearly, the problem of contact activation of blood plasma is inexorably bound to the problem of protein adsorption.
8. Conclusions
Close scrutiny of the generally-accepted surface biophysics of contact activation of blood-plasma coagulation reveals serious inconsistencies that cannot be resolved by minor modification. Furthermore, this standard paradigm cannot explain recent experimental work showing that the (autoactivation) reaction is not, in fact, specific for anionic-hydrophilic surfaces and that autoactivation is moderated by the protein composition of the solution phase in which autoactivation is carried out. We conclude that the consensus biochemical mechanism of contact activation that has so long served as a rationale for poor hemocompatibility is an inadequate basis for surface engineering of advanced cardiovascular biomaterials. Development of a replacement paradigm that can account for all experimental observations remains one of the most challenging problems of modern biomaterials surface science. Resolution of an improved reaction scheme for contact activation will require solution to vexing problems of protein adsorption and protein-adsorption competition, as well as a greatly improved understanding of the biochemistry involved in surface activation of zymogens such as blood factor XII.
Acknowledgments
This work was supported by the National Institute of Health grant PHS 2R01HL069965. Authors appreciate additional support from the Materials Research Institute and Department of Materials Science and Engineering, Penn State University. Careful reading and editorial assistance by Dr. Avantika Golas is sincerely appreciated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Citations
- 1.Anderson RN, Minino AM, Hoyert DL, Rosenberg HM. National Vital Statistics Reports. 2001;49:1–32. [PubMed] [Google Scholar]
- 2.Guzzo M. Medical Device Firms Focus on Cardiac, Respiratory Care. Pittsburgh Business Times. 2004 [Google Scholar]
- 3.Lysaght MJ, O’Loughlin JA. Demographic Scope and Economic Magnitude of Contemporary Organ Replacement Therapies. ASAIO Journal. 2000;46(5):515–521. doi: 10.1097/00002480-200009000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Rose EA, Gelijns AC, Moskowitz A, Heitjan DF, Stevenson LW, Dembitsky W, et al. Long-term Use of a Left Ventricular Assist Device for End-Stage Heart Failure. N Engl J Med. 2001;345(20):1435–1443. doi: 10.1056/NEJMoa012175. [DOI] [PubMed] [Google Scholar]
- 5.Lavine M, Roberts M, Smith O. Bodybuilding: The Bionic Human. Science. 2002;295:995–1032. [Google Scholar]
- 6.Hanson SR, Harker LA. Blood Coagulation and Blood-Materials Interactions. In: Ratner BD, Hoffman AS, Schoen FJ, Lemons JE, editors. Biomaterials Science: An Introduction to Materials in Medicine. 2. San Diego: Elsevier Academic Press; 1996. pp. 193–199. [Google Scholar]
- 7.Hanson SR, Ratner BD. Evaluation of Blood-Materials Interactions. In: Ratner B, Hoffman A, editors. Biomaterials Science: An Introduction to Materials in Medicine. 2. San Diego: Elsevier Academic Press; 2004. pp. 367–379. [Google Scholar]
- 8.Zhuo R, Siedlecki CA, Vogler EA. Competitive-Protein Adsorption in Contact Activation of Blood Factor XII. Biomaterials. 2007;28:4355–4369. doi: 10.1016/j.biomaterials.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johlin JM. Interfacial Adsorption as a Function of the Concentration of Colloidal Solutions. J Biol Chem. 1929;84:543–551. [Google Scholar]
- 10.Johlin JM. Interfacial Adsorption as a Factor in the Clotting of Blood Plasma. J Biol Chem. 1929;81:99–113. [Google Scholar]
- 11.Jenny NS, Mann KG. Coagulation Cascade: An Overview. In: Loscalzo J, Schafer A, editors. Thrombosis and Hemorrhage. Philadelphia: Lippincott, Williams & Wilkins; 2002. pp. 1–21. [Google Scholar]
- 12.Colman RW, Marder VJ, Salzman EW, Hirsh J. Overview of Hemostasis. In: Colman RW, Marder VJ, Salzman EW, Hirsh J, editors. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Third ed. Philadelphia: J.B Lippincott Company; 1994. pp. 3–18. [Google Scholar]
- 13.Nemerson Y. The Tissue Factor Pathway of Blood Coagulation. In: Colman RW, Marder VJ, Salzman EW, Hirsh J, editors. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Third ed. Philadelphia: J.B Lippincott Company; 1994. pp. 81–93. [Google Scholar]
- 14.Colman RW, Scott CF, Schmaier AH, Wachtfogel TT, Pixley RA, Edmunds LH. Initiation of Blood Coagulation at Artificial Surfaces. Annals New York Academy of Sciences. 1987;516:253–267. doi: 10.1111/j.1749-6632.1987.tb33046.x. [DOI] [PubMed] [Google Scholar]
- 15.Malone PC, Agutter PS. The Aetiology of Deep Venous Thrombosis. Dordrecht: Springer; 2008. The Coagulation Cascade and the Consensus Model of DVT; pp. 11–29. [Google Scholar]
- 16.Roberts HR. Oscar Ratnoff: His Contributions to the Golden Era of Coagulation Research. British Journal of Haematology. 2003;122:180–192. doi: 10.1046/j.1365-2141.2003.04459.x. [DOI] [PubMed] [Google Scholar]
- 17.Schmaier AH. The Elusive Physiologic Role of Factor XII. J Clin Invest. 2008;118(9):3006–3009. doi: 10.1172/JCI36617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sainz IM, Pixley RA, Colman RW. Fifty Years of Research on the Plasma Kallikrein-Kinin System: from Protein Structure and Function to Cell Biology and in-vivo Pathophysiology. Thromb Haemost. 2007;98(1):77–83. [PubMed] [Google Scholar]
- 19.Ratnoff OD. Hemostasis and Blood Coagulation. In: Berne RM, Levy MN, editors. Physiology. 3. St. Louis: Mosby Year Book; 1993. pp. 327–357. [Google Scholar]
- 20.Colman RW, Schmaier AH. Contact System: A Vascular Biology Modulator With Anticoagulant, Profibrinolytic, Antiadhesive, and Proinflammatory Attributes. Blood. 1997;90(10):3819–3843. [PubMed] [Google Scholar]
- 21.Fuhrer GGM, Heller W, Hoffmeister HE. FXII. Blut. 1990;61(5):258–266. doi: 10.1007/BF01732874. [DOI] [PubMed] [Google Scholar]
- 22.Cadena RAD, Wachtfogel YT, Coleman RW. Contact Activation Pathway: Inflammation and Coagulation. In: Colman RW, Hirsh J, Mardner BJ, Salzman EW, editors. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 3. Philadelphia: J B Lippincott; 1994. [Google Scholar]
- 23.Pokhilko AV, Ataullakhanov FI. Contact Activation of Blood Coagulation: Trigger Properties and Hysteresis: Kinetic Recognition of Foreign Surfaces upon Contact Activation of Blood Coagulation: A Hypothesis. J Theor Biol. 1998;191:213–219. doi: 10.1006/jtbi.1997.0584. [DOI] [PubMed] [Google Scholar]
- 24.Colman RW. Contact Activation Pathway: Inflammatory Fibrinolytic, Anticoagulant, Antiadhesive, and Antiangiogenic Activities. In: Colman RW, Marder VJ, Hirsh J, Clowes AW, editors. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 4. Philadelphia: J.B Lippincott Company; 2000. pp. 103–121. [Google Scholar]
- 25.Samuel M, Pixley RA, Villanueva MA, Colman RW, Villanueva GB. Human factor XII (Hageman factor) Autoactivation by Dextran Sulfate. Circular Dichroism, Fluorescence, and Ultraviolet Difference Spectroscopic Studies. J Biol Chem. 1992;267:19691–19697. [PubMed] [Google Scholar]
- 26.Martorana F, Moro A. On the Kinetics of Enzyme Amplifier Systems with Negative Feedback. Mathematical Biosciences. 1974;21:77–84. [Google Scholar]
- 27.Martorana F. Some considerations on the Enzyme Amplifier System: the Blood Clotting. J Nuclear Med and Allied Sci. 1978;22(4):181–183. [PubMed] [Google Scholar]
- 28.Levine SN. Enzyme Amplifier Kinetics. Science. 1966;152:651–653. doi: 10.1126/science.152.3722.651. [DOI] [PubMed] [Google Scholar]
- 29.Kirchhofer D, Tschopp TB, Baumgartner HR. Active Site-Blocked Factors VIIa and IXa Differentially Inhibit Fibrin Formation in a Human Ex Vivo Thrombosis Model. Arteriosclerosis, Thrombosis, and Vascular Biology. 1995;15:1098–1106. doi: 10.1161/01.atv.15.8.1098. [DOI] [PubMed] [Google Scholar]
- 30.Rand MD, Lock JB. Blood Clotting in Minimally Altered Whole Blood. Annual meeting of the American Society of Hematology; 1995; San Diego, California. 1995. [Google Scholar]
- 31.Peyrou V, Lormeau JC, Herault JP, Gaich C, Pfliegger AM, Herbert JM. Contribution of Erythrocytes to Thrombin Generation. Thrombosis Haemostasis. 1999;81:400–406. [PubMed] [Google Scholar]
- 32.Xu CQ, Zeng YJ, Gregersen H. Dynamic Model of the Role of Platelets in the Blood Coagulation System. Medical engineering & Physics. 2002;24:587–593. doi: 10.1016/s1350-4533(02)00047-4. [DOI] [PubMed] [Google Scholar]
- 33.Butenas S, Veer Cvt, Mann KG. Evaluation of the Initiation Phase of Blood Coagulation Using Ultrasensitive Assays for Serine Proteases. The Journal of Biological Chemistry. 1997;272(34):21527–21533. doi: 10.1074/jbc.272.34.21527. [DOI] [PubMed] [Google Scholar]
- 34.Engelmann B, Luther T, Muller I. Intravascular Tissue Factor Pathway - A Model for Rapid Initiation of Coagulation within the Blood Vessel. Thrombosis Haemostasis. 2003;89:3–8. [PubMed] [Google Scholar]
- 35.Jesty J, Beltrami E, Willems G. Mathematical analysis of a proteolytic positive-feedback loop: Dependence of lag time and enzyme yields on the initial conditions and kinetic parameters. Biochemistry. 1993;32:6266–6274. doi: 10.1021/bi00075a021. [DOI] [PubMed] [Google Scholar]
- 36.Beltrami E, Jesty J. Mathematical analysis of activation thresholds in enzyme-catalyzed positive feedbacks: Application of the feedbacks of blood coagulation. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:8744–8748. doi: 10.1073/pnas.92.19.8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khanin MA, Semenov VV. A Mathematical Model of the Kinetics of Blood Coagulation. J Theor Biol. 1989;136:127–134. doi: 10.1016/s0022-5193(89)80220-6. [DOI] [PubMed] [Google Scholar]
- 38.Hockin MF, Jones KC, Everse SJ, Mann KG. A Model for the Stoichiometric Regulation of Blood Coagulation. The Journal of Biological Chemistry. 2002;277(21):18322–18333. doi: 10.1074/jbc.M201173200. [DOI] [PubMed] [Google Scholar]
- 39.Jones KC, Mann KG. A Model for the Tissue Factor Pathway to Thrombin. The Journal of Biological Chemistry. 1994;269(37):23367–23373. [PubMed] [Google Scholar]
- 40.Lawson JH, Kalafatis M, Stram S, Mann KG. A Model for the tissue factor pathway to thrombin I. An Empirical Study. The Journal of Biological Chemistry. 1994;269(37):23357–23366. [PubMed] [Google Scholar]
- 41.Pohl B, Beringer C, Bomhard M, Keller F. The quick machine - a mathematical model for the extrinsic activation of coagulation. Haemostasis. 1994;24(6):325–337. doi: 10.1159/000217122. [DOI] [PubMed] [Google Scholar]
- 42.Kuharsky AL, Fogelson AL. Surface-Mediated Control of Blood Coagulation: The Role of Binding Site Densities and Platelet Deposition. Biophysical journal. 2001;80:1050–1074. doi: 10.1016/S0006-3495(01)76085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ataullakhanov FI, Molchanova DA, Pokhilko AV. A simulated mathematical model of the blood coagulation system intrinsic pathway. Biofizika. 1995;40(2):434–442. [PubMed] [Google Scholar]
- 44.Gregory G, Basmadjian D. An Analysis of the Contact Phase of Blood Coagulation: Effects of Shear Rate and Surface Are Intertwined. Annals of Biomedical Engineering. 1994;22:184–193. doi: 10.1007/BF02390376. [DOI] [PubMed] [Google Scholar]
- 45.Vogler EA, Graper JC, Harper GR, Lander LM, Brittain WJ. Contact Activation of the Plasma Coagulation Cascade.1. Procoagulant Surface Energy and Chemistry. J Biomed Mat Res. 1995;29:1005–1016. doi: 10.1002/jbm.820290813. [DOI] [PubMed] [Google Scholar]
- 46.Vogler EA, Graper JC, Sugg HW, Lander LM, Brittain WJ. Contact Activation of the Plasma Coagulation Cascade.2. Protein Adsorption on Procoagulant Surfaces. J Biomed Mat Res. 1995;29:1017–1028. doi: 10.1002/jbm.820290814. [DOI] [PubMed] [Google Scholar]
- 47.Zhuo R, Miller R, Bussard KM, Siedlecki CA, Vogler EA. Procoagulant Stimulus Processing by the Intrinsic Pathway of Blood Plasma Coagulation. Biomaterials. 2005;26:2965–2973. doi: 10.1016/j.biomaterials.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 48.Guo Z, Bussard K, Vogler EA, Siedlecki CA. Mathematical Modeling of Material-Induced Blood Plasma Coagulation. Biomaterials. 2006;27:796–806. doi: 10.1016/j.biomaterials.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 49.Colman RW, Esterl EW. Subcommittee on Contact Activation. Nomenclature of Kininogens. Thromb Haemost. 1988;60(2):340–341. [PubMed] [Google Scholar]
- 50.Saito H, Kojima T. Factor XII, Prekallikrein, and High-Molecular-Weight Kininogen. In: High KA, Roberts HR, editors. Molecular Basis of Thrombosis and Hemostasis. New York: Marcel Dekker; 1995. pp. 269–285. [Google Scholar]
- 51.Griep MA, Fujikawa K, Nelsestuen GL. Possible Basis for the Apparent Surface Selectivity of the Contact Activation of Human Blood Coagulation Factor XII. Biochemistry. 1986;25:6688–6694. doi: 10.1021/bi00369a054. [DOI] [PubMed] [Google Scholar]
- 52.Mitropoulos KA. High Affinity Binding of Factor FXIIa to an Electronegative Surface Controls the Rates of Factor XII and Prekallikrein Activation in vitro. Thrombosis Research. 1999;94:117–129. doi: 10.1016/s0049-3848(98)00207-2. [DOI] [PubMed] [Google Scholar]
- 53.Vogler EA, Nadeau JG, Graper JC. Contact Activation of the Plasma Coagulation Cascade. 3. Biophysical Aspects of Thrombin Binding Anticoagulants. J Biomed Mat Res. 1997;40(1):92–103. doi: 10.1002/(sici)1097-4636(199804)40:1<92::aid-jbm11>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 54.Zhuo R, Colombo P, Pantano C, Vogler EA. Silicon Oxycarbide Glasses for Blood-Contact Applications. Acta Biomaterialia. 2005;1:583–589. doi: 10.1016/j.actbio.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 55.Miller R, Guo Z, Vogler EA, Siedlecki CA. Plasma Coagulation Response to Surfaces with Nanoscale Chemical Heterogeneity. Biomaterials. 2006;27:208–215. doi: 10.1016/j.biomaterials.2005.05.087. [DOI] [PubMed] [Google Scholar]
- 56.Conley CL, Hartmann RC, Morse WI. The Clotting Behavior of Human “Platelet-Free” Plasma: Evidence for the Existence of a “Plasma Thromboplastin”. J Clin Inv. 1949;28:340–352. doi: 10.1172/JCI102077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vogler EA. How Water Wets Biomaterials. In: Morra M, editor. Water in Biomaterials Surface Science. New York: John Wiley and Sons; 2001. pp. 269–290. [Google Scholar]
- 58.Vogler EA. Water and the Acute Biological Response to Surfaces. J Biomat Sci Polym Edn. 1999;10(10):1015–1045. doi: 10.1163/156856299x00667. [DOI] [PubMed] [Google Scholar]
- 59.Vogler EA. Structure and Reactivity of Water at Biomaterial Surfaces. Adv Colloid and Interface Sci. 1998;74(13):69–117. doi: 10.1016/s0001-8686(97)00040-7. [DOI] [PubMed] [Google Scholar]
- 60.Putnam FW. Alpha, Beta, Gamma, Omega - The Roster of the Plasma Proteins. In: Putnam FW, editor. The Plasma Proteins: Structure, Function, and Genetic Control. New York: Academic Press; 1975. pp. 58–131. [Google Scholar]
- 61.Anderson NL, Anderson NG. The Human Plasma Proteome: History, Character, and Diagnostic Prospects. Molecular and Cellular Proteomics. 2002;1(11):845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 62.Revak SD, C C, Johnston AR, Hugli TE. Structural changes accompanying enzymatic activation of human Hageman factor. Journal of Clinical Investigation. 1974;54(3):619–27. doi: 10.1172/JCI107799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cochrane CG, Revak SD, Wuepper KD. Activation of Hageman Factor in Solid and Fluid Phases. J Exp Med. 1973;138:1564–1583. doi: 10.1084/jem.138.6.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vogler EA. Interfacial Chemistry in Biomaterials Science. In: Berg J, editor. Wettability. New York: Marcel Dekker; 1993. pp. 184–250. [Google Scholar]
- 65.Aveyard R, Haydon DA. An Introduction to the Principles of Surface Chemistry. London: Cambridge University Press; 1973. [Google Scholar]
- 66.Rosen MJ. Surfactants and Interfacial Phenomena. New York: Wiley; 1978. [Google Scholar]
- 67.Birdi KS, editor. Handbook of Surface and Colloid Chemistry. Boca Raton, FL: CRC Press; 1997. [Google Scholar]
- 68.Vogler EA. On the Origins of Water Wetting Terminology. In: Morra M, editor. Water in Biomaterials Surface Science. New York: John Wiley and Sons; 2001. pp. 150–182. [Google Scholar]
- 69.Vroman L. What Factors Determine Thrombogenicity? Bull N Y Acad Med. 1972;48:302–310. [PMC free article] [PubMed] [Google Scholar]
- 70.Vogler EA. Practical Use of Concentration-Dependent Contact Angles as a Measure of Solid-Liquid Adsorption II: Experimental Aspects. Langmuir. 1992;8:2013–2020. [Google Scholar]
- 71.Vogler EA, Martin DA, Montgomery DB, Graper JC, Sugg HW. A Graphical Method for Predicting Protein and Surfactant Adsorption Properties. Langmuir. 1993;9:497–507. [Google Scholar]
- 72.Noh H, Vogler EA. Volumetric Interpretation of Protein Adsorption: Mass and Energy Balance for Albumin Adsorption to Particulate Adsorbents with Incrementally-Increasing Hydrophilicity. Biomaterials. 2006;27:5801–5812. doi: 10.1016/j.biomaterials.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 73.Gao L, McCarthy TJ. Teflon is Hydrophilic. Comments on Definitions of Hydrophobic, Shear versus Tensile Hydrophobicity, and Wettability Characterization. Langmuir. 2008;24(17):9183–9188. doi: 10.1021/la8014578. [DOI] [PubMed] [Google Scholar]
- 74.Noh H, Vogler EA. Volumetric Interpretation of Protein Adsorption: Partition Coefficients, Interphase Volumes, and Free Energies of Adsorption to Hydrophobic Surfaces. Biomaterials. 2006;27:5780–5793. doi: 10.1016/j.biomaterials.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 75.Yaminsky VV, Thuresson K, Ninham BW. Marangoni “Precursor” in Dynamic Dewetting Transition: Adsorption Pressure Effect. 1999 [Google Scholar]
- 76.Yaminsky V, Ninham B, Karaman M. Dewetting of Mica Induced by Simple Organic Ions. Kinetic and Thermodynamic Study. Langmuir. 1997;13:5979–5990. [Google Scholar]
- 77.Frank B, Garoff S. Temporal and Spatial Development of Surfactant Self Assemblies Controlling Spreading of Surfactant Solutions. Langmuir. 1995;11:4333–4340. [Google Scholar]
- 78.Cha P, Krishnan A, Fiore VF, Vogler EA. Interfacial Energetics of Protein Adsorption from Aqueous Buffer to Surfaces with Varying Hydrophilicity. Langmuir. 2008;24:2553–2563. doi: 10.1021/la703310k. [DOI] [PubMed] [Google Scholar]
- 79.Vogler EA. Practical Use of Concentration-Dependent Contact Angles as a Measure of Solid-Liquid Adsorption I: Theoretical Aspects. Langmuir. 1992;8:2005–2012. [Google Scholar]
- 80.Zhuo R, Siedlecki CA, Vogler EA. Autoactivation of Blood Factor XII at Hydrophilic and Hydrophobic Surfaces. Biomaterials. 2006;27:4325–4332. doi: 10.1016/j.biomaterials.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 81.Mitropoulos KA. The Levels of FXIIa Generated in Hyman Plasma on an Electronegative Surface are Insensitive to Wide Variation in the Concentration of FXII, Prekallikrein, High Molecular Weight Kininogen or FXI. Thromb Haemost. 1999;82:1033–1040. [PubMed] [Google Scholar]
- 82.Noh H, Vogler EA. Volumetric Interpretation of Protein Adsorption: Competition from Mixtures and the Vroman Effect. Biomaterials. 2007;28:405–422. doi: 10.1016/j.biomaterials.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barnthip N, Noh H, Leibner E, Vogler EA. Volumetric Interpretation of Protein Adsorption: Kinetic Consequences of a Slowly-Concentrating Interphase. Biomaterials. 2008;29:3062–3074. doi: 10.1016/j.biomaterials.2008.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen X, Wang J, Pszti Z, Wang F, Schrauben JN, Tarabara VV, et al. Ordered adsorption of coagulation factor XII on negatively charged polymer surfaces probed by sum frequency generation vibrational spectroscopy. Analytical and Bioanalytical Chemistry. 2007;388(1):65–72. doi: 10.1007/s00216-006-0999-8. [DOI] [PubMed] [Google Scholar]
- 85.Collins KD. Sticky Ions in Biological Systems. Proc Natl Acad Sci USA. 1995;92:5553–5557. doi: 10.1073/pnas.92.12.5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Krishnan A, Sturgeon J, Siedlecki CA, Vogler EA. Scaled Interfacial Activity of Proteins at the Liquid-Vapor Interface. J Biomed Mat Res. 2004;68A:544–557. doi: 10.1002/jbm.a.20104. [DOI] [PubMed] [Google Scholar]
- 87.Krishnan A, Siedlecki C, Vogler EA. Traube-Rule Interpretation of Protein Adsorption to the Liquid-Vapor Interface. Langmuir. 2003;19:10342–10352. [Google Scholar]
- 88.Krishnan A, Siedlecki CA, Vogler EA. Mixology of Protein Solutions and the Vroman Effect. Langmuir. 2004;20(12):5071–5078. doi: 10.1021/la036218r. [DOI] [PubMed] [Google Scholar]
- 89.Krishnan A, Wilson A, Sturgeon J, Siedlecki CA, Vogler EA. Liquid-Vapor Interfacial Tension of Blood Plasma, Serum and Purified Protein Constituents Thereof. Biomaterials. 2005;26:3445–3453. doi: 10.1016/j.biomaterials.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 90.Krishnan A, Liu Y-H, Cha P, Allara DL, Vogler EA. Interfacial Energetics of Globular-Blood Protein Adsorption to a Hydrophobic Surface from Aqueous-Buffer Solution. Journal of the Royal Society Interface. 2006;3:283–301. doi: 10.1098/rsif.2005.0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krishnan A, Liu Y-H, Cha P, Allara DL, Vogler EA. Scaled Interfacial Activity of Proteins at a Hydrophobic Solid/Aqueous-Buffer Interface. J Biomed Mater Res. 2005;75A(2):445–457. doi: 10.1002/jbm.a.30444. [DOI] [PubMed] [Google Scholar]
- 92.Krishnan A, Liu Y-H, Cha P, Allara DL, Vogler EA. Interfacial Energetics of Blood Plasma and Serum Adsorption to a Hydrophobic Self-Assembled Monolayer Surface. Biomaterials. 2006;27:3187–3194. doi: 10.1016/j.biomaterials.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 93.Ariola F, Krishnan A, Vogler EA. Interfacial Rheology of Blood Proteins Adsorbed to the Aqueous-Buffer/Air Interface. Biomaterials. 2006;27:3404–3412. doi: 10.1016/j.biomaterials.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 94.Zhuo R, Vogler EA. Practical Application of a Chromogenic FXIIa Assay. Biomaterials. 2006;27:4840–4845. doi: 10.1016/j.biomaterials.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 95.Sheiko SS, Sun FC, Randall A, Shirvanyants D, Rubinstein M, Lee H-i, et al. Adsorption-induced scission of carbon–carbon bonds. Nature. 2006;440(7081):191–194. doi: 10.1038/nature04576. [DOI] [PubMed] [Google Scholar]
- 96.Ishitoya H, Kawamura M, Linneweber J, Motomura T, Ichikawa S, Nishimura I, et al. Titania Gel Reduces Thrombin Generation. Artif Organs. 2002;26(11):959–963. doi: 10.1046/j.1525-1594.2002.07115.x. [DOI] [PubMed] [Google Scholar]
- 97.Chatterjee K, Guo Z, Vogler EA, Siedlecki CA. Contributions of Contact Activation Pathways of Coagulation Factor XII in Plasma. J Biomed, Mat Res Part A. 2008 doi: 10.1002/jbm.a.32076:NA. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Clark AJ, Kotlicki A, Haynes CA, Whitehead LA. A New Model of Protein Adsorption Kinetics Derived from Simultaneous Measurement of Mass Loading and Changes in Surface Energy. Langmuir. 2007;23(10):5591–5600. doi: 10.1021/la0635350. [DOI] [PubMed] [Google Scholar]
- 99.Brash JL, Lyman DJ. Adsorption of Plasma Proteins in Solution to Uncharged, Hydrophobic Polymer Surfaces. J Biomed Mat Res. 1969;3:175–189. doi: 10.1002/jbm.820030114. [DOI] [PubMed] [Google Scholar]
- 100.Revak SD, Cochrane CG, Griffin JH. The binding and cleavage characteristics of human Hageman factor during contact activation: A comparison of normal plasma with plasmas deficient in Factor XI, Prekallikrein, or High Molecular Weight Kininogen. The Journal of Clinical Investigation. 1977;59:1167–1175. doi: 10.1172/JCI108741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pixley RA, Stumpo LG, Birkmeyer K, Silver LD, Colman RW. A Monoclonal Antibody Recognizing an Icosapeptide Sequence in the Heavy Chain of Human Factor XII Inhibits Surface-Catalyzed Activation. Journal of Biological Chemistry. 1987;262(21):10140–10145. [PubMed] [Google Scholar]
- 102.Fair BD, Saito H, Ratnoff OD, Rippon WB. Detection by fluorescence of structural changes accompanying the activation of Hageman factor (factor XII) Proceedings of the Society for Experimental Biology and Medicine. 1977;155:199–202. doi: 10.3181/00379727-155-39773. [DOI] [PubMed] [Google Scholar]
- 103.Vroman L. Effects of Hydrophobic Surfaces Upon Blood Coagulation. Thrombosis et Diathesis Haemorrhagica. 1964;10:455–493. [PubMed] [Google Scholar]
- 104.Silverberg M, Kaplan AP. Enzymatic activity of activated and zymogen forms of human Hageman factor (factor XII) Blood. 1982;60(1):64–70. [PubMed] [Google Scholar]
- 105.Griffin JH. Role of Surface in Surface-Dependent Activation of Hageman Factor (Blood Coagulation Factor XIII) Proc Natl Acad Sci USA. 1978;75(4):1998–2002. doi: 10.1073/pnas.75.4.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Silverberg M, Dunn JT, Garen L, Kaplan AP. Autoactivation of Human Hageman Factor. Demonstration Utilizing a Synthetic substrate. J Biol Chem. 1980;255(15):7281–7286. [PubMed] [Google Scholar]
- 107.Ratnoff OD, Saito H. Interactions Among Hageman factor, Plasma Prekallikrein, High Molecular Weight Kininogen, and Plasma Thromboplastin Antecedent. Proceedings of the National Academy of Sciences USA. 1979;76(2):958–961. doi: 10.1073/pnas.76.2.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bouma BN, Griffin JH. Human Blood Coagulation Factor XI. Purification, Properties, and Mechanism of Activation by Activated Factor XII. The Journal of Biological Chemistry. 1977;252(18):6432–6437. [PubMed] [Google Scholar]
- 109.Griffin JH. Role of surface in surface-dependent activation of Hageman factor (blood coagulation factor XII) Proc Natl Acad Sci U S A. 1978;75:1998–2002. doi: 10.1073/pnas.75.4.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brown B. Hematology: Principles and Procedures. 3. Philadelphia: Lea and Febiger; 1980. [Google Scholar]