

Abstract
Mutational activation of BRAF is the earliest and most common genetic alteration in human melanoma. Hence, to build a model of human melanoma, we generated mice with conditional melanocyte-specific expression of BRafV600E. Upon induction of BRafV600E expression, mice developed benign melanocytic hyperplasias that failed to progress to melanoma over 15-20 months. By contrast, expression of BRafV600E combined with Pten tumor suppressor gene silencing elicited development of melanoma with 100% penetrance, short latency and with metastases observed in lymph nodes and lungs. Melanoma was prevented by inhibitors of mTorc1 (Rapamycin) or MEK1/2 (PD325901) but, upon cessation of drug administration, mice developed melanoma indicating the presence of long-lived melanoma-initiating cells in this system. Importantly, combined treatment with Rapamycin and PD325901 led to shrinkage of established melanomas. These mice, engineered with a common genetic profile to human melanoma, provide an excellent system to study melanoma’s cardinal feature of metastasis and for pre-clinical evaluation of agents designed to prevent or treat metastatic disease.
Malignant melanoma is noted for its aggressive clinical behavior, a propensity for lethal metastasis and striking therapeutic resistance. This dismal clinical picture, coupled with an alarming increase in incidence, has motivated efforts to understand the genetic underpinnings of melanoma initiation and progression and translate such insights into effective preventive and therapeutic strategies for this dreaded disease 1,2.
Several key genetic lesions governing melanoma initiation and progression have been identified, the earliest and most common being a point mutation (T1799A) in the BRAF proto-oncogene, which is detected in ∼65% of patients 1,3-7. BRAFT1799A encodes BRAFV600E, a constitutively active protein serine kinase that elicits sustained activation of the BRAF→MEK1/2→ERK1/2 MAP kinase pathway. While contributing to the aberrant pathophysiological characteristics of the melanoma cell 8,9, oncogenic activation of BRAF in melanocytes is insufficient for full malignant conversion owing to activation of senescence 10,11. Hence, progression to malignant melanoma is invariably accompanied by silencing of one or more tumor suppressor genes, most commonly PTEN, INK4A and/or ARF 5,12-14. Of relevance to this study, the combination of mutated BRAF and silencing of PTEN expression is common in human melanoma (∼20%), although genetic evidence for BRAF/PTEN cooperation in this disease has not been obtained to date 14-16.
To understand and validate the role of such genetic events and their interactions in melanoma initiation, progression and response to therapy, we have generated a conditional mouse model of BRafV600E-induced/Pten deficient metastatic melanoma. This genetic model recapitulates key pathophysiological aspects of the human disease and offers exquisite control over the timing and location of melanoma initiation. Moreover, we demonstrate the utility of this model in the evaluation of pharmacologic agents that target key signaling pathways linked to these signature events in the melanoma cell, demonstrating the utility of this disease model in pre-clinical experimental therapeutics.
RESULTS
BRafV600E promotes benign melanocytic hyperplasia in vivo
BRafCA mice harbor a germ-line conditional BRafV600E allele, the expression of which is initiated at physiological levels under the control of the gene’s chromosomal promoter by the action of Cre recombinase (Fig. 1a) 17. When the BRafCA allele was combined with a constitutively active Cre transgene under the control of the Tyrosinase promoter (Tyr::Cre, Tec1) no viable offspring were obtained (Supplementary table 1) 18. Similarly, compound BRafCA and Dopachrome tautomerase promoter-directed Cre (Dct::Cre) transgenic mice succumbed within 3 months of age 19 (Matzen et al., Submitted). To circumvent lethality associated with embryonic expression of BRafV600E we utilized a Tyr::CreER transgene, which encodes conditionally active CreERT2 specifically in melanocytes (Fig. 1a) 20. In the absence of 4-hydroxytamoxifen (4-HT), Tyr::CreER; BRafCA/+ mice displayed no discernable phenotype over 18 months of observation (n≥50, Fig. 1b; i). However, topical administration of 4-HT to either fur bearing or glabrous skin or systemic 4-HT administration led to the appearance of highly pigmented lesions that were detected by 21-28 days after 4-HT administration (Fig. 1c; i). Such lesions were not detected in 4-HT treated Tyr::CreER mice indicating that they require the conversion of the BRafCA allele to BRafV600E (Supplementary Fig. 1a) 20.
Figure 1. Benign hyperplasias induced by melanocyte specific expression of BRafV600E.

(a) Mice carrying various conditional alleles of BRaf (BRafCA) and/or Pten (Ptenlox4-5 or Ptenlox5) were crossed to Tyr::CreER mice with melanocyte specific expression of a hormone dependent form of Cre recombinase (CreERT2) 17,20. 4-HT dependent activation of CreER leads to melanocyte specific conversion of BRafCA→BRafV600E and the conversion of the Ptenlox alleles to null alleles.
BRafCA/+ (b), Tyr::CreER; BRafCA/+ (c) and Tyr::CreER; BRafCA/CA (d) mice were treated topically with 4-HT and monitored for 75 weeks for signs of melanocytic proliferation (i). Mice were euthanized and skin from the ear (ii, iii) and flank (iv) was stained with hematoxylin and eosin and inspected for the presence of pigmented cells. (e) A Tyr::CreER; BRafCA/+ mouse developed a papular pigmented lesion ∼60 weeks after topical administration of 4-HT (i). This mouse was euthanized and skin sections encompassing the lesion were stained with hematoxylin and eosin and inspected for the presence of pigmented cells (ii & iii).
BRafV600E-induced pigmented lesions were frequently located at the dermal/epidermal junction with extension into the dermis, as is seen in human compound melanocytic nevi (Fig. 1c; iii). Other melanocytic proliferations formed in the dermis with no apparent junctional component. BRafV600E-induced pigmented lesions appeared only in regions of the skin to which 4-HT was applied and were not induced by topical administration of solvent as control (Supplementary Fig. 1). Occasionally, when mice were topically administered with high concentrations of 4-HT, we observed periocular lesions on the face and on the glabrous skin of the paws, most likely due to grooming behavior (Supplementary Fig. 1b & c). However, by reducing the amount of 4-HT administered, we could elicit spatially restricted melanocytic lesions with no evidence of inappropriate spreading to untreated sites.
To determine if BRafV600E-induced pigmented lesions would progress spontaneously to malignant melanoma, we monitored 35 4-HT treated Tyr::CreER; BRafCA mice over 15-20 months. Although 2 mice developed small papular pigmented lesions, these lesions were not considered malignant in nature based on their lack of significant invasion or metastasis, the small/self-limited tumor growth, the banal cytologic features of the cells and the absence of aberrant mitotic figures within the lesions (Fig. 1e; i-iii). These data are consistent with the hypothesis that BRAFV600E expression in human melanocytes promotes an initial phase of proliferation that is subsequently restrained from malignant progression by engagement of a cell cycle arrest program with features of senescence, reflecting the clinical observation of growth arrest of pigmented nevi in humans 10,21.
In addition, we generated a small number of Tyr::CreER mice that were homozygous for BRafCA to assess the impact of two copies of the BRafV600E allele (n=6). Topical 4-HT administration to the ear, tail or flank of these mice led to the appearance of benign melanocytic hyperplasias that were significantly larger and more highly pigmented than in mice heterozygous for BRafCA expressing only one copy of BRafV600E (Fig. 1d; i-iv respectively). Indeed, this phenotypic presentation was so reproducible that mice could be genotyped (BRafCA/+ vs. BRafCA/CA) based on the size and pigmentation of the melanocytic lesions that developed. These data indicate that BRafV600E gene dosage exerts a physiologically relevant impact on melanocyte proliferation, an experimental observation that may rationalize the observation of increased copy number of oncogenic BRAF in human melanoma and other tumors 22,23.
BRafV600E cooperates with Pten silencing to promote invasive, metastatic melanoma
Human melanomas expressing BRAFV600E often display activation of the PI3′-kinase→PDK→AKT pathway through silenced expression of the PTEN tumor suppressor gene 14,15,22. To model this genetic profile, we generated Tyr::CreER; BRafCA mice that were also homozygous for either one of two different conditional Pten alleles. One allele (PtenloxP-neo) conditionally deletes Pten exons 4 and 5 24. The second allele (not previously described) permits Cre-mediated deletion of Pten exon 5. (For purposes of clarity we refer to these alleles as Ptenlox4-5 and Ptenlox5 respectively). Importantly, the action of Cre recombinase on both alleles leads to deletion of exons that encode amino acids essential for the Pten’s PI3′-lipid phosphatase activity 24.
In striking contrast to BRafV600E-induced melanocytic neoplasia (Fig 1), 4-HT treatment of Tyr::CreER; Ptenlox4-5/lox4-5 or Tyr::CreER; Ptenlox5/lox5 mice failed to elicit any melanocytic phenotype over ∼18 months (n=20). These data are consistent with observations that Dct::Cre; Ptenlox/lox mice do not develop melanoma unless treated with carcinogen 25. However, 7-10 days following 4-HT administration, all Tyr::CreER; BRafCA; Ptenlox/lox mice displayed a dramatic expansion of highly pigmented cells regardless of where the 4-HT was administered to the skin and regardless of what Pten allele was used (Figs. 2a-c). Indeed, so dramatic was the cooperation between BRafV600E and Pten silencing that confluent melanocytic proliferation/hyperplasia was observed in 4-HT treated regions of the skin of Tyr::CreER; BRafCA; Ptenlox/lox mice. By contrast, treatment with DMSO solvent control was without effect (Figs. 2g-i and Supplementary Fig. 3b). Abrogation of Pten expression was confirmed by Western blotting analysis of extracts of primary lesions derived from 4-HT treated Tyr::CreER; BRafCA; Ptenlox5/lox5 mice (Supplementary Fig. 4). Pigmented melanoma cells were found throughout the dermis, subcutis and displayed pagetoid spread into the epidermis (Figs. 2h, i & k). Moreover, there was rapid emergence of highly pigmented malignant lesions elicited by 4-HT in the face, flank or tail skin, the location of which could be highly restricted by topical administration of low dose 4-HT (Figs. 2d-f and Supplementary Figs. 3a & c). These lesions progressed rapidly to advanced malignancy such that all Tyr::CreER; BRafCA; Ptenlox/lox mice required euthanasia 25-50 days following 4-HT administration (Fig. 2m).
Figure 2. BRafV600E cooperates with Pten loss in the induction of malignant melanoma.

(a-c) Tyr::CreER; BRafCA/+; Ptenlox5/lox5 mice were treated topically on the paw with 4-HT to elicit BRafV600E and to silence Pten expression. The presence of pigmented lesions was assessed 6 (a), 8 (b) and 10 (c) days following 4-HT administration.
(d) Tyr::CreER; BRafCA/+; Ptenlox4-5/lox4-5 (i) were treated topically with 4-HT on the right ear. Mice were monitored for 7 weeks.
Tyr::CreER; BRafCA/+; Pten+/+ (e) and Tyr::CreER; BRafCA/+; Ptenlox4-5/lox4-5 (f) were treated topically with 4-HT on the right flank. Mice were euthanized at ∼7 weeks with the presence of malignant melanoma lesions assessed by visual inspection of the underside of the ventral/lateral skin.
Tyr::CreER; BRafCA/+; Ptenlox5/lox5 mice were treated topically on the ear, flank and tail with 4-HT. 25 days later mice were euthanized, skin sections were prepared, stained with hematoxylin and eosin and examined for the presence of pigmented cells. Almost confluent proliferation of densely pigmented cells was observed in the ear and skin (g) that penetrated deep into the dermis and the subcutis (h & i) and showed signs of pagetoid spread into the epidermis (h & k). Cells in these lesions stained positive with α-PEP1 antisera that detects expression of Tyrp1 (j & k). Pigmented cells in the lesions displayed histological characteristics of malignant melanoma, including marked cytological atypia, prominent nucleoli and aberrant mitotic figures (l)
(j) Kaplan-Meier survival analysis of 4-HT treated Tyr::CreER; BRafCA/+; Pten+/+ (n=22), Tyr::CreER; BRaf+/+; Ptenlox4-5/lox4-5 (n=5) and Tyr::CreER; BRafCA/+; Ptenlox4-5/lox4-5 (n=22) mice. Log rank tests of survival plots of the data indicated a statistically significant difference between the following survival curves: Tyr::CreER; BRafCA/+; Pten+/+ versus Tyr::CreER; BRafCA/+; Ptenlox4-5/lox4-5 (p<0.0001) and; Tyr::CreER; BRaf+/+; Ptenlox4-5/lox4-5 versus Tyr::CreER; BRafCA/+; Ptenlox4-5/lox4-5 (p<0.0002)
Histological analysis of primary skin lesions revealed highly pigmented cells with variably shaped enlarged nuclei containing clumped chromatin, prominent nucleoli and atypical mitoses (Fig. 2l). In addition, the melanocytic origin of cells in the lesions was confirmed by the presence of abundant melanin and by immunostaining with α-PEP1 to detect expression of Tyrosinase related protein 1 (Figs. 2j & k) 26.
To further document the rate-limiting role of Pten in constraining malignant progression, we followed a small number of 4-HT treated Tyr::CreER; BRafCA mice that were heterozygous for Ptenlox4/5 allele. These mice initially developed benign melanocytic lesions similar to those observed in Tyr::CreER; BRafCA mice. However, they eventually developed focal malignant melanomas that we believe are most likely due to stochastic silencing of the wild-type Pten allele, as has been observed with other tumor suppressor genes (Supplementary Fig. 2).
BRafV600E; Pten-/- melanoma cells are invasive and metastatic
At euthanasia Tyr::CreER; BRafCA; Ptenlox/lox mice were subjected to complete histopathologic survey for evidence of melanoma invasion and metastasis. As described above, all mice analyzed had evidence of melanoma cells throughout the dermis with evidence of invasion into the subcutis (Fig. 2). In addition, spread of melanoma was observed into the regional draining lymph nodes in 100% of mice with expansile growth noted in most mice (Fig. 3a-c). This was also noted in mice with localized tumors in the distal region of the tail that had clear evidence of pigmented cells in the iliac lymph nodes (Fig. 3d and Supplementary Fig. 3c) and lungs (data not shown). Examination of the lungs revealed visible evidence of at least one and, in many cases, multiple nests of metastatic melanoma cells (Figs. 3e-g). These data indicate that activated BRafV600E and Pten deficiency cooperate to produce primary cutaneous malignant melanoma with robust capacity to invade into the skin and to metastasize to draining lymph nodes and distal organs that are commonly targeted in the human disease.
Figure 3. BRafV600E cooperates with Pten loss in the induction of invasive and metastatic melanoma.

(a) Tyr::CreER; BRafCA/+; Ptenlox4-5/lox4-5 mice were treated topically on the ear, flank and tail with 4-HT. 6 weeks later mice were euthanized and inspected for evidence of pigmented cells in the mammary gland lymph node. Tyr::CreER; BRafCA/+; Ptenlox5/lox5 mice were treated similarly to those described in (a), euthanized and then inspected by microscopy at low (b) and high (c) power for the presence of pigmented cells in the lymph nodes.
(d) A Tyr::CreER; BRafCA/+; Ptenlox5/lox5 mouse was treated topically with 5mM 4-HT in 100%(v/v) ethanol on the distal tail. The mouse had a visible tumor within 4 weeks but did not require euthanasia until 24 weeks later after 4-HT administration. The presence of pigmented cells in the Iliac, lumbar, inguinal, axillary and submandibular lymph nodes was assessed by stereomicroscopy. Tumor cells were restricted to the iliac nodes (pictured).
(e-f) Tyr::CreER; BRafCA/+; Ptenlox4-5/lox4-5 mice were treated topically on the ear, flank and tail with 4-HT. Mice were euthanized 6-7 weeks after 4-HT treatment at which times the lungs were excised, cleared of blood and visually inspected for the presence of pigmented lesions (e) and (f). Lung sections were prepared, stained with hematoxylin and eosin and examined for the presence of pigmented cells in the lung parenchyma (g).
Prevention and therapy of BRafV600E-induced melanomas by pharmacological inhibition of MEK1/2 and mTorc1
Melanoma is noted for its striking resistance to chemotherapy, although there are anecdotal reports of dramatic responses to therapy targeted against mutated c-KIT in humans 13,27. Consequently, we wished to determine if targeted inhibition of downstream effectors of BRafV600E or PI3′-kinase signaling could prevent the development of melanoma in this model system. To do so, we utilized specific and selective inhibitors of MEK1/2 (PD325901) or mTorc1 (Rapamycin), which are downstream of BRafV600E or PI3′-kinase respectively 28,29. Melanoma was initiated in adult Tyr::CreER; BRafCA; Ptenlox/lox mice by topical administration with 4-HT. Either 1 day (Rapamycin, Fig. 4a) or 1 week (PD325901, Fig. 5a) later the mice were treated systemically either with the relevant solvent control or with Rapamycin (7.5mg/kg) or PD325901 (15mg/kg) for 3 or 6 weeks respectively. Solvent treated mice rapidly developed melanoma that required euthanasia within 3-6 weeks (Figs. 4b & 5b, panel i). By contrast, treatment with either Rapamycin or PD325901 dramatically inhibited melanoma formation, such that by the time all of the solvent treated mice were euthanized, drug treated mice were alive and showed no signs of melanoma formation (Figs. 4b & 5b, panel ii).
Figure 4. Prevention of BRafV600E-induced melanomas by Rapamycin.

(a) Adult Tyr::CreER; BRafCA/+; Ptenlox5/lox5 mice were treated topically on the ear, flank and tail with 4-HT. The next day, mice were randomly assigned to be administered Rapamycin (7.5mg/kg, n=5) or solvent control (n=4) for 21 days. Mice were monitored daily for the presence of cutaneous malignant melanoma and euthanized according to a standard body conditioning score. After 3 weeks of Rapamycin treatment, drug administration was ceased and mice were monitored for the presence of cutaneous malignant melanoma as described above. Mouse survival was plotted using a Kaplan-Meier survival curve (c).
(b) Representative images of a vehicle control (i) or a Rapamycin treated mouse (ii) immediately after the end of the 21 day drug administration are presented.
(c) Kaplan-Meier survival curve of vehicle (n=4) or Rapamycin (n=5) treated cohorts of mice analyzed in this experiment. All mice were treated for the indicated 21 days as indicated in (a).
Figure 5. Prevention of BRafV600E-induced melanomas by PD325901.

(a) Adult Tyr::CreER; BRafCA/+; Ptenlox4-5/lox4-5 mice were treated topically on the ear with 4-HT. One week later, mice were randomly assigned to be administered PD325901 (12.5mg/kg, n=12) or the solvent control (n=12) for 6 weeks. Mice were analyzed daily for the presence of cutaneous malignant melanoma and euthanized according to a standard body conditioning score. After 6 weeks of PD325901 treatment 7 drug treated and 6 control mice were euthanized for analysis of skin sections. A further 5 PD325901 treated and 6 control mice were monitored over the course of a further 11 weeks without any further drug administration. These mice were analyzed for the presence of cutaneous malignant melanoma as described above. Mouse survival was plotted using a Kaplan-Meier survival curve (c).
(b) Representative anatomical and histological images of skin from 4-HT treated Tyr::CreER; BRafCA/+; Ptenlox4-5/lox4-5 mice analyzed after 6 weeks of treatment with control solvent (n=6, i) or PD325910 (n=7, ii) or from PD325901 treated mice left without further drug administration for an additional 9-11 weeks (iii).
(c) Kaplan-Meier survival curve of vehicle (n=6) or PD325901 (n=5) treated cohorts of mice analyzed in this experiment. All mice were treated for the indicated 6 weeks as in (b). Log rank tests of survival plots of the data demonstrate a statistically significant difference between vehicle and PD325901 treated animals (p=0.0024).
(d) 3 week old Tyr::CreER; BRafCA/+; Ptenlox4-5/lox4-5 mice (n=10) were treated topically on the right ear with 4-HT. 10 days later all mice were administered PD325901 (12.5mg/kg) for 6 weeks. Drug administration was then ceased for 8 weeks. Mice were then randomized into two groups: Group A (blue) was administered vehicle control and Group B (red) was administered PD325901 for a further 6 weeks. At the end of this period mice were monitored prospectively for disease progression as described above. Mouse survival was plotted using a Kaplan-Meier survival curve that demonstrated statistically significant difference (p=0.0018) between the groups by log rank tests (e).
To determine if treatment with either Rapamycin or PD325901 had completely eliminated melanoma cells initiated by 4-HT administration, drug treatment was ceased in both groups of animals with the animals monitored for an additional 3-11 weeks (Figs. 4c & 5c). Regardless of the drug employed, all of the animals subsequently developed malignant melanoma requiring their euthanasia within 5-11 weeks following cessation of drug administration (Figs. 4c, 5b, panel iii & 5c). These data indicate that the prevention of melanoma in the drug treated mice was not due to a technical flaw in the activation and function of CreER following 4-HT administration. Moreover, these data are consistent with the presence of latent melanoma-initiating cells that arise shortly after the CreER activation and that can survive extended exposure to agents that target signaling pathways essential for the melanoma cell division cycle and survival.
To determine if melanomas that recur following cessation of MEK1/2 inhibitor treatment are resistant to re-treatment of mice with this agent, melanoma was initiated in a cohort of 10 three week old Tyr::Cre; BRafCA; Ptenlox4-5/lox4-5 mice. 10 days later all mice were treated systemically with PD325901 for a period of 6 weeks. At this time, consistent with results described above, none of the mice displayed any signs of melanoma. Upon cessation of PD325901 administration the mice were left untreated for a period of 8 weeks. At this time mice were randomized into two groups of 5, one of which was administered solvent control (Group A) and the other PD325901 (Group B) for a further 6 weeks. As described above, Group A mice treated only once with PD325901 succumbed to disease 8-12 weeks following cessation of drug administration (Fig. 5d). By contrast, Group B mice that received 2 cycles of PD325901 remained alive at a time when all of the Group A mice had been euthanized. However, all Group B mice displayed signs of melanoma re-growth which, while not requiring euthanasia, strongly suggested that these mice would eventually succumb to disease. Regardless, these data indicate that mice treated once with PD325901 to prevent melanomagenesis remain sensitive to the anti-melanoma action of a second round of PD325901 administration.
Results described above document the potent melanoma prevention activity of PD325901 and Rapamycin. However, to determine if these agents can elicit regression of pre-existing tumors in mice, melanoma was initiated in a group of 20 Tyr::Cre; BRafCA; Ptenlox5/lox5 mice by local administration of 4-HT to back skin (Supplementary Fig. 3a). 3 weeks later, when the animals had readily measurable melanoma lesions, mice were randomly divided into 4 groups that were administered either solvent control (Vehicle), PD325901 (PD, 12.5mg/kg), Rapamycin (Rapa, 7.5mg/kg) or the combination of both Rapamycin and PD325901 (PD+Rapa) at the same dose used in the single agent arms (Fig. 6a). Mice were treated with these agents for 3 weeks with tumor size measured every day. Melanoma lesions in the vehicle treated animals progressed steadily over the time course analyzed and had expanded in size in all animals by ∼40% by the end of the experiment (Fig. 6b). By contrast, mice treated with either PD325901 or Rapamycin as single agents led to striking inhibition of tumor growth but with little or no evidence of tumor regression. By contrast, mice treated with a combination of Rapamycin and PD325901 displayed a ∼20% reduction in tumor size that was statistically significant. These data suggest that the combination of MEK1/2 and mTorc1 inhibition can lead to significant tumor shrinkage in the BRafV600E; Ptennull melanoma model.
Figure 6. Melanoma regression in response to combination treatment with PD325901 and Rapamycin.

(a) Melanoma was initiated in a group of 20 Tyr::Cre; BRafCA; Ptenlox/lox mice by local administration of 1-2μl of 5mM 4-HT to back skin. 3 weeks later, when the animals had readily measurable melanoma lesions, mice were randomly divided into 4 groups that were administered either: 1. Solvent control (Vehicle, blue); 2. PD325901 (PD, 12.5mg/kg, red); 3. Rapamycin (Rapa, 7.5mg/kg, green) or; 4. The combination of both Rapamycin and PD325901 (PD+Rapa, orange) for 3 weeks as indicated.
(b) Tumor size in each of the treatment groups described in (a) was measured daily and plotted as percentage change in tumor size compared to the starting size. Two-way ANOVA analysis demonstrates statistical significance: 1. Vehicle vs Rapa or PD or PD+Rapa (p<0.0001); 2. PD vs PD+Rapa (p=0.0024) and; 3. Rapa vs PD+Rapa (p=0.0120).
(c) 2 hours following the final administration of the various agents, mice were euthanized and melanoma specimens were prepared for staining with H&E or antisera against Ki67, pERK1/2 or p4EBP1 as indicated.
Two hours after the final administration of the various agents in the experiment described above, tumor specimens were prepared and stained with: 1. Hematoxylin and Eosin; 2. Antisera against Ki67 to assess cell proliferation; 3. Antisera against phospho-ERK1/2 (pERK1/2) and; 4. Antisera against phospho-4EBP1 (p4EBP1) as indicated (Fig. 6c). The last two were to assess the effects of PD325901 or Rapamycin on the relevant signaling pathways. Histological analysis of H&E stained sections revealed the presence of viable tumor cells in all treatment groups but with an increase in the number of cells with pyknotic features of apoptosis in mice treated with PD325901 or PD325901 in combination with Rapamycin. Administration of PD325901 or Rapamycin, either alone or in combination led to a reduction in cell proliferation as measured by Ki67 staining. As expected, treatment with PD325901 led to decreased pERK1/2 staining but had little or no effect on p4EBP1. Conversely, treatment with Rapamycin led to decreased p4EBP1 staining but had little or no effect on pERK1/2. As expected, the combination of PD325901 and Rapamycin led to decreased staining of both pERK1/2 and p4EBP1. These data suggest that, as single agents, PD325901 and Rapamycin have predominantly a cytostatic effect on melanoma initiated by expression of BRafV600E and extinction of Pten. By contrast, evidence of melanoma regression in mice treated with the combination of PD325901 and Rapamycin suggests that these agents may combine to elicit a cytotoxic effect on pre-existing lesions, supported by the decrease in tumor size accompanied by an increase in pyknotic tumor cells. However, it remains possible that both tumor cell autonomous and non-autonomous effects of these agents may contribute to melanoma regression in this experimental system.
Response of BRafV600E-induced melanoma cell lines to pharmacological inhibition of MEK1/2 and mTorc1
To characterize potential melanoma cell autonomous effects of Rapamycin and/or PD325901, a small number of melanoma derived cell lines were established. One such line that grew well in culture (2697T) was derived from a primary cutaneous lesion arising in a 4-HT treated Tyr::CreER; BRafCA; Ptenlox5/lox5 white mouse and was confirmed to have recombined the BRafCA and the Ptenlox alleles by PCR (Supplementary Fig. 5). These cells displayed evidence of phosphorylated MEK1/2, ERK1/2, p70S6K and 4EBP1 consistent with the genetic alterations in BRaf and Pten that promote melanoma initiation and progression in this model (Figs. 7c & d). Treatment of melanoma cells with either Rapamycin (25nM) for 24 or 48 hours led to 50% inhibition of cell proliferation after 72 hours compared to the control (Fig. 7a). The anti-proliferative effects of Rapamycin were accompanied by robust inhibition of phospho-p70S6K and 4EBP1, even at the lowest dose tested (2.5nM, Fig. 7d). Rapamycin had no effect on the phosphorylation of upstream Akt (T308 or S473) nor on the phosphorylation of MEK1/2. Treatment of 2697T cells with PD325901 (2μM) led to complete abrogation of cell proliferation (Fig. 7a), a dramatic reduction in S phase cells (Fig. 7b) and a modest reduction in cell number at the latest time point. Consistent with this, MEK1/2 inhibition elicited a striking reduction of phospho-ERK1/2 with no effect on phospho-Akt (Fig. 7c). Moreover, MEK inhibition was accompanied by induction of the pro-apoptotic Bcl-2 family protein Bim-EL, cleavage of Caspase 3 (Fig. 7c) and an increase in cells with a sub-G1 DNA content (Fig. 7b). These data are similar to those obtained in bona fide human melanoma cell lines where regulation of BIM-EL expression downstream of oncogenic NRAS or BRAF contributes to suppression of melanoma cell apoptosis 30,31.
Figure 7. Inhibition of mouse melanoma cell proliferation by Rapamycin or PD325901 in vitro.
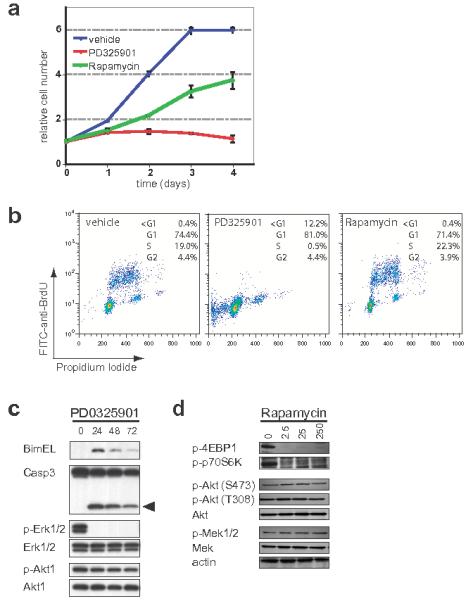
A melanoma cell line (2697T) was derived from a 4-HT induced lesion that developed on the skin of a white Tyr::CreER; BRafCA/+; Ptenlox5/lox5 mouse. 2697T cells growing asynchronously in full media were treated with PD325901 (2μM) or Rapamycin (50nM) as indicated. (a) Cell growth was measured using a standard Crystal Violet staining assay. (b) DNA synthesis and content was assessed at 48 hours using standard anti-BrdU/Propidium Iodide FACS analysis following a 2 hour BrdU pulse. The percentage of cells in each portion of the cell cycle is indicated.
(c) 2697T cells, growing asynchronously in full media, were treated with 2μM PD325901 for 24-72 hours at which time cell extracts were prepared. Expression of Bim-EL, ERK1/2, Akt1, Caspase 3 and its active cleavage product (arrowhead) as well as the phosphorylation of ERK1/2 and Akt1 was assessed by immunoblotting.
(d) Serum deprived 2697T cells were pre-treated with different concentrations of Rapamycin (2.5-250nM) for 30 minutes prior to stimulation with 10%(v/v) fetal calf serum for a further 20 minutes. Expression of Akt1, MEK1/2 and Actin as well as the phosphorylation of 4EBP1, p70S6K, and Akt1 (S473 and T308) was assessed by immunoblotting.
DISCUSSION
Mouse models have provided key insights into cancer initiation, progression and therapy, and the model described here offers additional distinct and important advantages in the study of melanoma and its treatment 32-34. Here, we provide evidence of cooperative interactions of two signature mutations found in human melanoma, enabling the generation of a mouse that recapitulates hallmark features of the disease, including metastases to relevant organ sites. Moreover, the use of BRafCA mice allows accurate modeling of the earliest effects of physiological levels of expression of BRafV600E, the purported initiating mutation that promotes the conversion of normal human melanocytes to benign nevus cells 4,7. Since expression of BRafV600E promotes the development of benign melanocytic hyperplasias that fail to progress to malignant melanoma, this system may be used to explore mechanisms constraining progression in vivo, to discover and validate tumor suppressor genes governing such progression checkpoints and to define the mechanisms driving invasion and metastasis 10,35,36. Next, use of the Tyr::CreERT2 transgene affords the experimental advantage of exquisite spatial and temporal control over the genetic alterations that initiate melanoma formation in mice. Finally, and most notably, we provide evidence that this model is useful to study the signaling axis of these key genetic lesions and to assess the impact of agents targeting these pathways that may facilitate the development of new strategies to target malignant melanoma using conventional and/or targeted therapies 37.
Data presented here are consistent with effects of transgenic over-expression of BRAFV600E in zebrafish, which leads to the development of “fish nevi” 38. Moreover, when combined with loss of Trp53 expression, BRAFV600E elicited malignant fish melanoma. However, the relevance of this combination of genetic events to human melanoma is unclear given the relatively low frequency of TP53 mutations in the human disease 1,13. However, these data stand in contrast to the reported effects of BRAFV600E over-expression in human melanocytes engineered to express the catalytic subunit of telomerase (hTERT), dominant-negative TP53R248W and CDK4R24C, the last being resistant to the inhibitory effects of the melanoma suppressor, p16INK4A 39. In this context, the effects of ectopic BRAFV600E expression were benign inducing only mild junctional melanocyte nesting. By contrast, ectopic expression of oncogenic variants of RAS (NRASG12V or HRASG12V) or the catalytic subunit of PI3′-kinase-α (p110-CAAX) led to striking malignant transformation of such “initiated” human melanocytes. It is unclear why the effect of BRAFV600E expression is so benign in this context, especially given the dramatic effects of BRAFV600E in zebrafish and mice.
Although genetic analysis of human melanomas suggest that mutated BRAF cooperates with loss of PTEN expression in melanomagenesis, it has yet to be unequivocally demonstrated that such is the case 14,15. However, data presented here demonstrates the ability of BRafV600E expression to cooperate with Pten silencing in the genesis of metastatic melanoma. It remains to be seen whether PI3′-lipid phosphatase activity is key to Pten’s tumor suppressor activity, especially in view of recent demonstrations of effects of Pten on other signaling pathways that are independent of its lipid phosphatase activity 40.
A key feature of this model is the ability to control melanoma initiation and progression in response to expression of oncogenic BRafV600E in combination with alterations in other genes implicated in melanoma initiation, progression or response to therapy such as Ink4a/Arf, β-catenin, Mitf or various alleles of Mc1r 5,41,42. Given the high frequency of invasion and metastasis observed in this model, it may be possible to initiate a primary malignant melanoma in a readily accessible site, resect the primary lesion and then monitor the mice prospectively for local or distant recurrence. This aspect of the model may afford a detailed analysis of mechanisms that promote melanoma cell migration, local invasion and metastasis in a manner that more closely recapitulates the clinical course of melanoma recurrence in humans.
Metastatic melanoma presents one of the most difficult diseases to treat in medical oncology. Indeed, there has not been a major breakthrough in the treatment of this disease in over 25 years. Despite the current grim outlook for metastatic melanoma patients, considerable excitement has been generated by the identification of genetic and epigenetic alterations that contribute to melanomagenesis. All the more so given the accelerated pace of development of pharmacological agents that target key signaling pathways in the melanoma cell. However, it remains unclear how best to use such agents in the clinic. Although studies with the broad spectrum Raf inhibitor Sorafenib, either as a single agent or in combination chemotherapy, have proven disappointing in treating metastatic melanoma this may be due to incomplete inhibition of BRAFV600E activity 43,44. Moreover, individual case reports indicate that even highly aggressive melanomas expressing mutationally activated c-KIT can show a dramatic response to the potent c-KIT inhibitor, Gleevec 27,45. Such observations raise hope that pharmacological targeting of relevant molecular target(s) may lead to dramatic clinical responses even in late stage melanoma patients. Hence, given the prevalence of BRAF and PTEN alterations in melanoma, it may inform the clinical development of various new and more selective BRAF, MEK, PI3′-kinase, AKT and mTor inhibitors by testing such agents in a validated pre-clinical mouse model 46-48. Experiments presented here suggest such agents may have value in melanoma prevention and therapy, but the relapse of mice following cessation of drug administration indicates that single agent therapy with MEK1/2 or mTor inhibitors is unlikely to cure patients with this disease. However, this model system is readily tractable to test combinations of targeted agents or targeted therapy combined with conventional cytotoxics or other agents such as biochemotherapy or immune based approaches 49. Such studies might provide useful guides to the best dose and scheduling of anti-melanoma therapeutics in clinical trials in humans.
METHODS
Mouse breeding, activation of Tyr::CreER transgene and treatment of mice with targeted therapeutics
BRafCA, Tyr::CreER and Ptenlox4-5 mice were genotyped as previously described 17,20,24. Cre-mediated conversion of BRafCA to BRafVE and the deletion of exons 4 and 5 of Pten were assessed by PCR as previously described. Topical administration of 4-hydroxytamoxifen (4-HT) was performed by preparing a 25-50mg/ml (65-130mM) solution of 4-HT (70% Z-isomer, Sigma) in DMSO and applying enough solution to wet the right ear, right flank and tail with a small paint brush on post-natal days 2, 3, and 4. For localized melanoma induction on the back skin, adult (6-8 weeks of age) mice were treated topically with 1-2 μl of 1.9mg/ml (5mM) 4-HT at 6-8 weeks of age using a similar protocol. Generalized induction in adult mice was performed by intra-peritoneal injection of 1mg of tamoxifen/40g mouse on 3 consecutive days. In this case tamoxifen was prepared as a 10mg/ml suspension in peanut oil. PD352901 was dissolved in 0.5%(w/v) Hydroxy-propyl-methylcellulose, 0.2%(v/v) Tween 80 (Sigma) and administered to mice daily by oral gavage at a dose of 12.5mg/kg. Rapamycin (LC Laboratories, Woburn, MA) was suspended in 0.5%(w/v) methylcellulose and administered to mice daily by oral gavage at a dose of 7.5mg/kg. Control animals in the melanoma prevention studies were administered with the relevant solvent. Tissues were prepared for analysis as previously described 17,20
Generation of mice carrying a Cre-inactivated allele of Pten
A targeting vector was generated in pKO SelectDT-915 using a 12kb fragment of the mouse Pten gene by standard techniques. A cassette carrying Pten exon 5 flanked by loxP sites and PGK::Neo flanked by FRT sites was then subcloned between the genomic DNA arms. This construct was linearized, transfected into TC1 ES cells and genomic DNA from G418 and Diphtheria toxin resistant ES cells were screened by Southern blot and PCR for homologous recombination events. Blastocyst injections were carried out with three different targeted clones, and transmitting chimaeric mice were bred from CAGG—Flpe transgenic mice to generate the Ptenlox5 allele 50. Chimeric mice were mated with FVB/N mice and transmission verified by Southern blotting, and PCR with the following primers: 5′-CTTCGGAGCATGTCTGGCAATGC-3′; 5′-CTGCACGAGACTAGTGAGACGTGC-3′ and; 5′-AAGGAAGAGGGTGGGGATAC-3′. All mice were maintained on a mixed C57Bl/6 and FVB/N background by random interbreeding.
Melanoma cell line derivation and culture
Melanoma cell lines were derived by physical resection of primary cutaneous lesions followed by trituration through a 20-gauge syringe needle and filtration through a 100μm nylon cell strainer (BD/Falcon, Bedford, MA before being placed into media. Cell suspensions were plated in media (a 1:1 mix of DMEM and Hams F12) supplemented with fetal calf serum (5%v/v) and penicillin, streptomycin and glutamine. Serum starved cells were treated with different doses of Rapamycin (2.5-250nM as indicated) for 30 minutes prior to 10%(v/v) serum stimulation for an additional 20 minutes. Asynchronously growing cells in full media were treated with PD325901 (2μM) for different periods of time (24-96 hours as indicated) with cell proliferation quantified by staining of fixed cells with Crystal Violet. Data presented in Figure 6a are results from two separate experiments in which each time point was measured in triplicate. Subconfluent cells grown in full melanocyte media treated with PD325901 (2uM) or Rapamycin (50nM) were cultured for 46 hours when BrdU was added to 20uM for 2 hours. Single cell suspensions fixed in ethanol were stained with FITC-labelled BrdU (BD Pharmingen 556028), incubated with propidium iodide (10ug/ml), and analyzed with a BD—FACS caliber. Cell extracts were prepared by standard techniques, resolved using SDS-polyacryamide gel electrophoresis, Western blots prepared by standard techniques, which were probed with commercially available antisera against various proteins as indicated 31.
Immunochemistry and immunoblotting
The expression of Tyrosinase (Tyr, Albino), Tyrosinase related protein 1 (Tyrp1, Brown) or Tyrosinase related protein 2 (Tyrp2, Slaty) was detected by staining with , or antisera respectively (gift of Dr. Vincent Hearing) 26. Slides were developed using and di-amino-benzidine-peroxidase substrate kit (Vector Labs, Burlingame, CA), according to the manufacturer’s instructions. The expression and phosphorylation of proteins was detected by immunoblotting using appropriate commercially available antisera: phospho-4EBP1, phospho-p70S6K, phospho-MEK1/2, total MEK1/2, phospho-ERK1/2; total ERK1/2; phospho S473-AKT; phospho-T308-AKT; total AKT; Caspase 3 (all from Cell Signaling Technology), Bim-EL (Calbiochem mAb 14A8); and Actin (Sigma).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the members of the McMahon and Bosenberg labs as well as Boris Bastian, Lynda Chin, Elena Filenova, Byron Hann, Meenhard Herlyn, Leisa Johnson, Pier Paolo Pandolfi, Vincent Hearing, Matthew Held, Graeme Kay and Dana Matzen for the provision of mouse strains, reagents, advice and support. M.M. thanks Alejandro Ricart and Judy Sebolt-Leopold (Pfizer Inc.) for provision of PD325901 and acknowledges the support of the UCSF/Helen Diller Family Comprehensive Cancer Center Mouse Pathology and Pre-Clinical Therapeutics cores. RAD is an American Cancer Society Research Professor. This work was supported by grants from the Melanoma Research Foundation, U.C. Discovery Award and from the N.I.H. (CA 108972) to M.M., (CA 84313) to R.A.D. and (CA 89124 & CA 112054) to M.B. respectively.
LITERATURE CITED
- 1.Chin L, Merlino G, DePinho RA. Malignant melanoma: modern black plague and genetic black box. Genes Dev. 1998;12:3467–81. doi: 10.1101/gad.12.22.3467. [DOI] [PubMed] [Google Scholar]
- 2.Gray-Schopfer VC, da Rocha Dias S, Marais R. The role of B-RAF in melanoma. Cancer Metastasis Rev. 2005;24:165–83. doi: 10.1007/s10555-005-5865-1. [DOI] [PubMed] [Google Scholar]
- 3.Chin L. The genetics of malignant melanoma: lessons from mouse and man. Nat Rev Cancer. 2003;3:559–70. doi: 10.1038/nrc1145. [DOI] [PubMed] [Google Scholar]
- 4.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 5.Garraway LA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–22. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 6.Hayward NK. Genetics of melanoma predisposition. Oncogene. 2003;22:3053–62. doi: 10.1038/sj.onc.1206445. [DOI] [PubMed] [Google Scholar]
- 7.Pollock PM, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 8.Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–85. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 9.Mercer KE, Pritchard CA. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim Biophys Acta. 2003;1653:25–40. doi: 10.1016/s0304-419x(03)00016-7. [DOI] [PubMed] [Google Scholar]
- 10.Michaloglou C, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 11.Sviderskaya EV, et al. p16(Ink4a) in melanocyte senescence and differentiation. J Natl Cancer Inst. 2002;94:446–54. doi: 10.1093/jnci/94.6.446. [DOI] [PubMed] [Google Scholar]
- 12.Wellbrock C, et al. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004;64:2338–42. doi: 10.1158/0008-5472.can-03-3433. [DOI] [PubMed] [Google Scholar]
- 13.Chin L, Garraway LA, Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006;20:2149–82. doi: 10.1101/gad.1437206. [DOI] [PubMed] [Google Scholar]
- 14.Lin WM, et al. Modeling genomic diversity and tumor dependency in malignant melanoma. Cancer Res. 2008;68:664–73. doi: 10.1158/0008-5472.CAN-07-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol. 2004;122:337–41. doi: 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsao H, Zhang X, Fowlkes K, Haluska FG. Relative reciprocity of NRAS and PTEN/MMAC1 alterations in cutaneous melanoma cell lines. Cancer Res. 2000;60:1800–4. [PubMed] [Google Scholar]
- 17.Dankort D, et al. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21:379–84. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tonks ID, et al. Tyrosinase-Cre mice for tissue-specific gene ablation in neural crest and neuroepithelial-derived tissues. Genesis. 2003;37:131–8. doi: 10.1002/gene.10242. [DOI] [PubMed] [Google Scholar]
- 19.Guyonneau L, Murisier F, Rossier A, Moulin A, Beermann F. Melanocytes and pigmentation are affected in dopachrome tautomerase knockout mice. Mol Cell Biol. 2004;24:3396–403. doi: 10.1128/MCB.24.8.3396-3403.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bosenberg M, et al. Characterization of melanocyte-specific inducible Cre recombinase transgenic mice. Genesis. 2006;44:262–7. doi: 10.1002/dvg.20205. [DOI] [PubMed] [Google Scholar]
- 21.Bennett DC. Human melanocyte senescence and melanoma susceptibility genes. Oncogene. 2003;22:3063–9. doi: 10.1038/sj.onc.1206446. [DOI] [PubMed] [Google Scholar]
- 22.Jonsson G, et al. Genomic profiling of malignant melanoma using tiling-resolution arrayCGH. Oncogene. 2007;26:4738–48. doi: 10.1038/sj.onc.1210252. [DOI] [PubMed] [Google Scholar]
- 23.Willmore-Payne C, Holden JA, Hirschowitz S, Layfield LJ. BRAF and c-kit gene copy number in mutation-positive malignant melanoma. Hum Pathol. 2006;37:520–7. doi: 10.1016/j.humpath.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 24.Trotman LC, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inoue-Narita T, et al. Pten deficiency in melanocytes results in resistance to hair graying and susceptibility to carcinogen-induced melanomagenesis. Cancer Res. 2008;68:5760–8. doi: 10.1158/0008-5472.CAN-08-0889. [DOI] [PubMed] [Google Scholar]
- 26.Jimenez M, Tsukamoto K, Hearing VJ. Tyrosinases from two different loci are expressed by normal and by transformed melanocytes. J Biol Chem. 1991;266:1147–56. [PubMed] [Google Scholar]
- 27.Hodi FS, et al. Major response to imatinib mesylate in KIT-mutated melanoma. J Clin Oncol. 2008;26:2046–51. doi: 10.1200/JCO.2007.14.0707. [DOI] [PubMed] [Google Scholar]
- 28.Ohren JF, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11:1192–7. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- 29.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 30.Sheridan C, Brumatti G, Martin SJ. Oncogenic B-RafV600E Inhibits Apoptosis and Promotes ERK-dependent Inactivation of Bad and Bim. J Biol Chem. 2008;283:22128–35. doi: 10.1074/jbc.M800271200. [DOI] [PubMed] [Google Scholar]
- 31.Cartlidge RA, et al. Oncogenic BRAF(V600E) inhibits BIM expression to promote melanoma cell survival. Pigment Cell Melanoma Res. 2008;21:534–44. doi: 10.1111/j.1755-148X.2008.00491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walker GJ, Hayward NK. Pathways to melanoma development: lessons from the mouse. J Invest Dermatol. 2002;119:783–92. doi: 10.1046/j.1523-1747.2002.00217.x. [DOI] [PubMed] [Google Scholar]
- 33.Bardeesy N, Wong KK, DePinho RA, Chin L. Animal models of melanoma: recent advances and future prospects. Adv Cancer Res. 2000;79:123–56. doi: 10.1016/s0065-230x(00)79004-x. [DOI] [PubMed] [Google Scholar]
- 34.Tietze MK, Chin L. Murine models of malignant melanoma. Mol Med Today. 2000;6:408–10. doi: 10.1016/s1357-4310(00)01781-0. [DOI] [PubMed] [Google Scholar]
- 35.Woods D, et al. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol. 1997;17:5598–611. doi: 10.1128/mcb.17.9.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu J, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998;12:2997–3007. doi: 10.1101/gad.12.19.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olive KP, Tuveson DA. The use of targeted mouse models for preclinical testing of novel cancer therapeutics. Clin Cancer Res. 2006;12:5277–87. doi: 10.1158/1078-0432.CCR-06-0436. [DOI] [PubMed] [Google Scholar]
- 38.Patton EE, et al. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr Biol. 2005;15:249–54. doi: 10.1016/j.cub.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 39.Chudnovsky Y, Adams AE, Robbins PB, Lin Q, Khavari PA. Use of human tissue to assess the oncogenic activity of melanoma-associated mutations. Nat Genet. 2005;37:745–9. doi: 10.1038/ng1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freeman DJ, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3:117–30. doi: 10.1016/s1535-6108(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 41.Larue L, Delmas V. The WNT/Beta?catenin pathway in melanoma. Front Biosci. 2006;11:733–42. doi: 10.2741/1831. [DOI] [PubMed] [Google Scholar]
- 42.Landi MT, et al. MC1R germline variants confer risk for BRAF-mutant melanoma. Science. 2006;313:521–2. doi: 10.1126/science.1127515. [DOI] [PubMed] [Google Scholar]
- 43.Eisen T, et al. Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis. Br J Cancer. 2006;95:581–6. doi: 10.1038/sj.bjc.6603291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flaherty KT. Chemotherapy and targeted therapy combinations in advanced melanoma. Clin Cancer Res. 2006;12:2366s–2370s. doi: 10.1158/1078-0432.CCR-05-2505. [DOI] [PubMed] [Google Scholar]
- 45.Lutzky J, Bauer J, Bastian BC. Dose-dependent, complete response to imatinib of a metastatic mucosal melanoma with a K642E KIT mutation. Pigment Cell Melanoma Res. 2008;21:492–3. doi: 10.1111/j.1755-148X.2008.00475.x. [DOI] [PubMed] [Google Scholar]
- 46.Maira SM, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–63. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 47.Tsai J, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105:3041–6. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yeh TC, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–83. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 49.Sosman JA, Puzanov I. Molecular targets in melanoma from angiogenesis to apoptosis. Clin Cancer Res. 2006;12:2376s–2383s. doi: 10.1158/1078-0432.CCR-05-2558. [DOI] [PubMed] [Google Scholar]
- 50.Rodriguez CI, et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25:139–40. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.