

Abstract
Autotaxin (ATX) catalyzes the hydrolysis of lysophosphatidylcholine (LPC) to form the bioactive lipid lysophosphatidic acid (LPA). LPA stimulates cell proliferation, cell survival, and cell migration and is involved in obesity, rheumatoid arthritis, neuropathic pain, atherosclerosis and various cancers, suggesting that ATX inhibitors have broad therapeutic potential. Product feedback inhibition of ATX by LPA has stimulated structure activity studies focused on LPA analogs. However, LPA displays mixed mode inhibition, indicating it can bind to both the enzyme and the enzyme-substrate complex. This suggests that LPA may not interact solely with the catalytic site. In this report we have prepared LPC analogs to help map out substrate structure activity relationships. The structural variances include length and unsaturation of the fatty tail, choline and polar linker presence, acyl versus ether linkage of the hydrocarbon chain, and methylene and nitrogen replacement of the choline oxygen. All LPC analogs were assayed in competition with the synthetic substrate, FS-3, to show the preference ATX has for each alteration. Choline presence and methylene replacement of the choline oxygen were detrimental to ATX recognition. These findings provide insights into the structure of the enzyme in the vicinity of the catalytic site as well as suggesting that ATX produces rate enhancement, at least in part, by substrate destabilization.
1. Introduction
Autotaxin (ATX) has recently become an attractive target for therapeutic development efforts. ATX is a 125 kDa glycoprotein originally isolated from the human melanoma cell line A20581 and is upregulated in many tumor cell lines.2 ATX, a lysophospholipase D enzyme, hydrolyzes lysophosphatidylcholine (LPC) to form the bioactive lipid lysophosphatidic acid (LPA).3,4 ATX elicits its biological activity through its product LPA.3,4 LPA induces many biological events by activating specific G protein-coupled receptors, LPA1-8,5-10 and a nuclear hormone receptor, PPARγ.10,11 The effects of LPA include stimulation of cell proliferation, cell migration and cell survival.2 LPA-induced cell motility is mediated through the LPA1 receptor.12 These are detrimental cellular responses as it pertains to cancer cell biology. LPA is also implicated in obesity,13 rheumatoid arthritis,14 neuropathic pain,15 and atherosclerosis,16 a precursor to cardiovascular disease.
The ATX protein has yet to be crystallized; therefore little is directly known about the three-dimensional structure of the enzyme. In contrast, indirect insights have been obtained from homology models17,18 constructed using alkaline phosphatase19 and later a bacterial nucleotide pyrophosphatase/phosphodiesterase (NPP) homolog.20 Additional indirect structural insights can be obtained from substrates, substrate analogs and inhibitors. Until Parrill et al., Moulharat et al. and Saunders et al. recently described non-lipid ATX inhibitors,21-23 LPA analogs and metal chelators24 were the only known ATX inhibitors. LPA analogs showing ATX inhibition include sphingosine 1-phosphate,25 phosphonates,26-28 thiophosphates,27 cyclic glycerothiophosphates,29 carba cyclic phosphatidic acids,30 alpha-substituted phosphonic acids,31 and FTY720-phosphate.32 Since all published phospholipid ATX inhibitors are analogs of LPA, which does not exhibit simple competitive inhibition but rather shows mixed mode inhibition,25 they may not interact solely with the active site. LPC analogs, (i.e analogs of the natural substrate) can provide structure activity relationships that more directly reflect the ATX active site.
In this study, multiple LPC analogs have been examined to help define ATX substrate recognition. The structural features of LPC that were varied are length and unsaturation of the fatty tail, choline and polar linker presence, acyl versus ether linkage of the hydrocarbon chain, and methylene and nitrogen replacement of the choline oxygen (Figure 1). All compounds were tested for their ability to inhibit hydrolysis of a fluorescent synthetic ATX substrate, FS-3,33 to show the preference ATX has for each modification.
Figure 1.
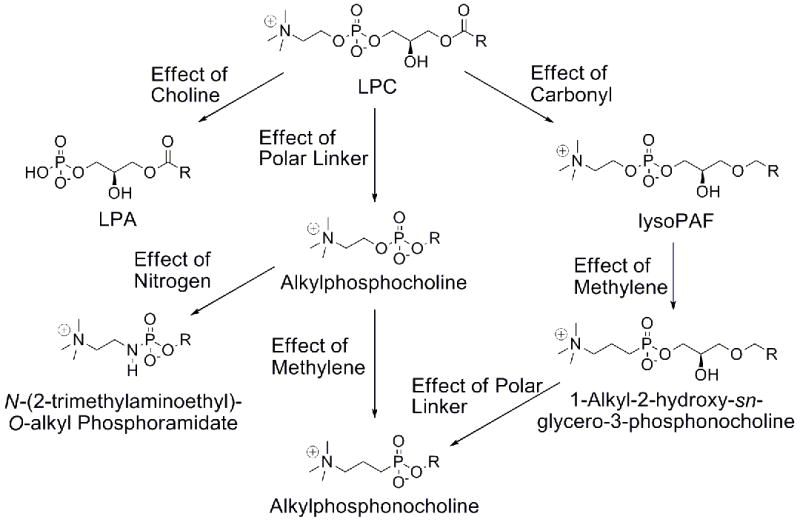
LPC, LPA, and LPC analogs examined.
2. Results
2.1. Determination of acyl chain length and unsaturation effects on ATX recognition
Initially, multiple LPC compounds were assayed to investigate ATX preferences for hydrophobic tail length and degree of unsaturation. The assayed LPC compounds had 6:0, 8:0, 10:0, 12:0, 14:0, 16:0, 18:0 and 18:1 acyl chains. All compounds were tested at 0.1, 1 and 10 μM concentrations using concentrated conditioned media collected from MD-MBA 435 breast cancer cell cultures as the source of ATX and 1 μM FS-3 as the ATX substrate. Since LPC is converted to LPA , a potent ATX inhibitor, the time courses of ATX activity were carefully examined to verify that no changes in rate occurred prior to the time point used to calculate inhibition. A decrease in rate during the course of the assay would suggest that LPA was generated in sufficient amounts to mask LPC effects. Figure 2 shows that both LPC and LPA inhibition of ATX were linear over the time frame examined. Thus, LPC activities are due to LPC and not due to metabolism to LPA. The 6:0, 8:0 and 10:0 LPC compounds failed to inhibit ATX-catalyzed hydrolysis of FS-3 (Table 1). Beginning with LPC 12:0 (10 μM), suppression of ATX-mediated FS-3 hydrolysis (Table 1) was noted. LPC 12:0, 14:0, 16:0, 18:0 and 18:1 (10 μM) inhibited FS-3 hydrolysis by 34 ± 4.3%, 64 ± 3.9%, 64 ± 16.9%, 41 ± 17.8%, and 83 ± 8.4%, respectively (Table 1). There is no statistical difference in ATX inhibition by LPC 12:0 and 18:0 (p = 0.7154) or by LPC 16:0 and 18:1 (p = 0.0907). LPC 18:1 inhibited ATX-mediated FS-3 hydrolysis the greatest at both 10 and 1 μM, indicating a preference by ATX for unsaturation in the hydrophobic tail. With respect to the saturated chains, both LPC 14:0 and 16:0 inhibited ATX-mediated hydrolysis of FS-3 similarly at 64 ± 3.9% and 64 ± 16.9%, respectively, whereas LPC 12:0 and 18:0 were less efficacious (Table 1). The optimal unbranched chain lengths for LPC as ATX inhibitors are 14:0, 16:0, and 18:1.
Figure 2.
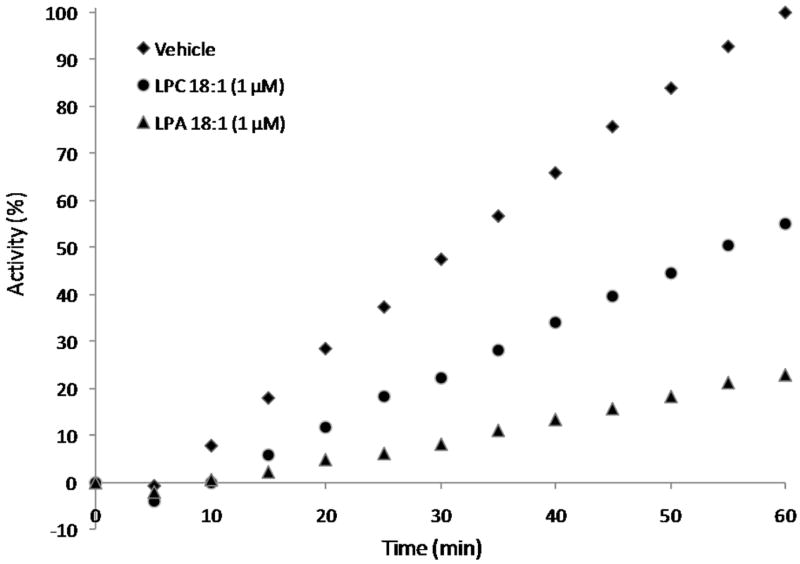
LPC and LPA time course. FS-3 inhibition remained linear during the course of the assay, indicating that LPA accumulation due to LPC hydrolysis was low and did not affect ATX activity.
Table 1.
ATX percent activity.
| Percent Activity ± S.D | |||
|---|---|---|---|
| LPC | 0.1 μM | 1 μM | 10 μM |
| 6:0 Acyl | 104 ± 4.2 | 100 ± 4.8 | 97 ± 4.3 |
| 8:0 Acyl | 94 ± 5.3 | 81 ± 5.6 | 101 ± 4.1 |
| 10:0 Acyl | 101 ± 4.1 | 101 ± 3.7 | 85 ± 5.4 |
| 12:0 Acyl | 102 ± 3.8 | 97 ± 4.5 | 66 ± 4.3 |
| 14:0 Acyl | 72 ± 4.8 | 80 ± 4.5 | 36 ± 3.9 |
| 16:0 Acyl | 109 ± 17.2 | 89 ± 16.8 | 36 ± 16.9 |
| 18:0 Acyl | 111 ± 17.0 | 99 ± 17.2 | 59 ± 17.8 |
| 18:1 Acyl | 92 ± 8.4 | 55 ± 7.9 | 17 ± 8.4 |
| LPA | 0.1 μM | 1 μM | 10 μM |
| 16:0 Acyl | 74 ± 16.9 | 49 ± 8.6 | 5 ± 8.7 |
| 18:0 Acyl | 89 ± 15.6 | 74 ± 8.1 | 36 ± 8.6 |
| 18:1 Acyl | 90 ± 17.1 | 23 ± 17.4 | -2 ± 16.9 |
| lysoPAF | 0.1 μM | 1 μM | 10 μM |
| 16:0 Alkyl | 100 ± 5.3 | 89 ± 4.8 | 49 ± 4.2 |
| 18:0 Alkyl | 92 ± 18.7 | 86 ± 17.2 | 44 ± 18.1 |
| Alkylphosphocholine | 0.1 μM | 1 μM | 10 μM |
| 2a (14:0 Alkyl) | 110 ± 11.2 | 112 ± 10.2 | 91 ± 10.4 |
| 2b (16:0 Alkyl) | 114 ± 10.1 | 113 ± 10.9 | 92 ± 13.2 |
| 2c (18:0 Alkyl) | 110 ± 10.0 | 96 ± 9.7 | 44 ± 10.0 |
| 2d (18:1 Alkyl) | 105 ± 11.1 | 59 ± 10.5 | 8 ± 9.9 |
| Alkylglycero-phosphonocholine | 0.1 μM | 1 μM | 10 μM |
| 6a (R 10:0 Alkyl) | 101 ± 4.7 | 95 ± 9.8 | 103 ± 4.2 |
| 6b (R 12:0 Alkyl) | 99 ± 10.5 | 96 ± 10.0 | 89 ± 6.6 |
| 6c (R 14:0 Alkyl) | 98 ± 6.7 | 85 ± 9.4 | 76 ± 6.0 |
| 6d (R 16:0 Alkyl) | 83 ± 4.5 | 82 ± 4.1 | 72 ± 4.7 |
| 6e (R 18:0 Alkyl) | 81 ± 5.1 | 89 ± 5.5 | 76 ± 4.2 |
| 6f (S 10:0 Alkyl) | 97 ± 6.4 | 96 ± 6.1 | 89 ± 6.6 |
| 6g (S 12:0 Alkyl) | 98 ± 6.4 | 95 ± 6.6 | 87 ± 6.2 |
| 6h (S 14:0 Alkyl) | 97 ± 5.7 | 95 ± 5.5 | 80 ± 6.2 |
| 6i (S 16:0 Alkyl) | 82 ± 6.1 | 86 ± 4.7 | 76 ± 4.7 |
| 6j (S 18:0 Alkyl) | 78 ± 5.6 | 77 ± 6.7 | 68 ± 6.6 |
| Alkylphosphonocholine | 0.1 μM | 1 μM | 10 μM |
| 8a (14:0 Alkyl) | 94 ± 17.7 | 90 ± 14.3 | 71 ± 14.9 |
| 8b (16:0 Alkyl) | 97 ± 18.0 | 94 ± 17.6 | 91 ± 22.3 |
| 8c (18:0 Alkyl) | 114 ± 16.9 | 116 ± 20.7 | 102 ± 24.4 |
| 8d (18:1 Alkyl) | 101 ± 18.0 | 100 ± 16.3 | 79 ± 16.3 |
| Phosphonamidate | 0.1 μM | 1 μM | 10 μM |
| 11a (14:0 Alkyl) | 89 ± 8.8 | 90 ± 9.1 | 75 ± 8.7 |
| 11b (16:0 Alkyl) | 81 ± 12.9 | 81 ± 9.9 | 77 ± 9.6 |
| 11c (18:0 Alkyl) | 86 ± 8.9 | 74 ± 7.9 | 29 ± 7.2 |
| 11d (18:1 Alkyl) | 93 ± 9.4 | 89 ± 11.2 | 74 ± 9.7 |
2.2. ATX dependence on choline and ester carbonyl functional groups
After establishing the optimal LPC chain lengths, the effect of the choline group was examined (Figure 1). To do this, commercially available LPA compounds with the same chain lengths as commercially available LPC were tested. LPA 16:0, 18:0 and 18:1 (10 μM) inhibited ATX by 95 ± 8.7%, 64 ± 8.6% and 102 ± 16.9% respectively, compared with inhibition by LPC 16:0, 18:0 and 18:1 of 64 ± 16.9%, 41 ± 17.8% and 83 ± 8.4%, respectively (Table 1). In all three cases, LPA inhibited ATX-mediated FS-3 hydrolysis, as well as, or better than the corresponding LPC, indicating that the choline functional group is detrimental to ATX recognition (Table 1).
To examine the effect of an ester versus an ether linkage to the hydrocarbon chain we tested two commercially available lysoPAF compounds, 16:0 and 18:0 (Figure 1). There was no statistical difference in ATX inhibition for 16:0 lysoPAF and LPC (p = 0.1150) and 18:0 lysoPAF and LPC (p = 0.1058) (Table 1). This indicates that the carbonyl group is not required for ATX recognition.
2.3. ATX recognition of the polar linker
Alkylphosphocholines were synthesized to show the importance of substrate polar linker to ATX recognition (Figure 1). Scheme 1 shows the synthesis for these alkylphosphocholines. Phosphorus oxychloride was reacted first with a long chain alcohol (tetradecanol, hexadecanol, octadecanol, or oleyl alcohol), second with 2-bromoethanol and third with water all in the presence of triethylamine. Finally, intermediates 1a – d were reacted with trimethylamine to produce final products 2a – d.
Scheme 1.
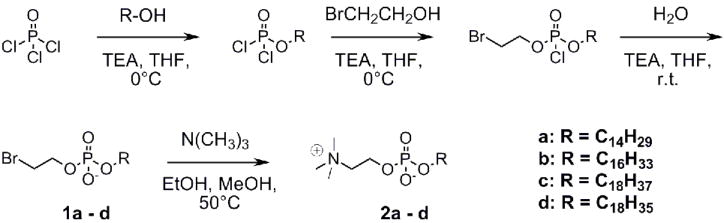
Alkylphosphocholine synthesis.
To compare an LPC, which is a glycerophospholipid, to an alkylphosphocholine, the alkylphosphocholine must contain four extra carbons in the hydrocarbon chain to compensate for the four additional heavy atoms within the polar linker of LPC. For example, LPC 14:0 has the same number of atoms extending from the phosphate as alkylphosphocholine 18:0. When assayed in the presence of FS-3, the 10 μM concentrations of saturated alkylphosphocholines showed similar ATX inhibition compared to the corresponding LPC. LPC 10:0, 12:0 and 14:0 inhibited ATX-mediated hydrolysis of FS-3 by 15 ± 5.4%, 34 ± 4.3% and 64 ± 3.9%, respectively (Table 1). Compounds 2a, 2b and 2c, with 14:0, 16:0 and 18:0 alkyl chains, inhibited ATX-mediated hydrolysis by 9 ± 10.4%, 8 ± 13.2% and 56 ± 10.0%, respectively (Table 1). Only LPC 12:0 showed significantly greater ATX inhibition by an extra 26% compared to alkylphosphocholine 16:0 (P = 0.0097) (Table 1). The polar linker is therefore not detrimental to ATX recognition, but also is not universally beneficial.
Alkylphosphocholine 18:1 (2d) inhibited ATX activity by 92 ± 9.9% at 10 μM (Table 1). This confirms that ATX has a preference for unsaturation in a hydrophobic tail regardless of overall chain length. LPC 18:1 inhibits ATX by 83 ± 8.4% at 10 μM (Table 1). Comparison of LPC 18:1 to alkylphosphocholine 18:1 suggests that the position of unsaturation is not critical.
2.4. The effect of methylene replacement for the choline oxygen on ATX recognition
The necessity of the choline oxygen in the presence and absence of the polar linker was examined via the synthesis and evaluation of phosphonocholines and alkylglycerophosphonocholines, respectively (Figure 1). Such compounds would act as nonhydrolyzable ATX inhibitors if the choline oxygen proved unnecessary. We have shown that LPC 12:0 inhibits ATX-mediated FS-3 hydrolysis more effectively than alkylphosphocholine 16:0 suggesting that the polar linker can be beneficial, therefore compounds with and without the polar linker were evaluated.
Intermediate alkylglycerols were stereospecifically synthesized, according to Erukulla and colleagues, by the treatment of a long chain alcohol with DIBAL-H and subsequent addition of glycidol.34 To synthesize the alkyl phosphonocholines and alkylglycerophosphonocholines, 3-chloropropylphosphonic acid was synthesized using the procedure of Kley and colleagues.35 Attachment of a long chain alcohol or alkylglycerol (3a – j) to 3-chloropropylphosphonic acid (4) was accomplished using DCC/DMAP (Scheme 2).35 Finally, treatment of coupled products (5a – j and 7a – d) with trimethylamine produced final products (6a – j and 8a – d)35. The alkylglycerophosphonocholines were isolated as a mixture of sn2 and sn3 coupled isomers.
Scheme 2.
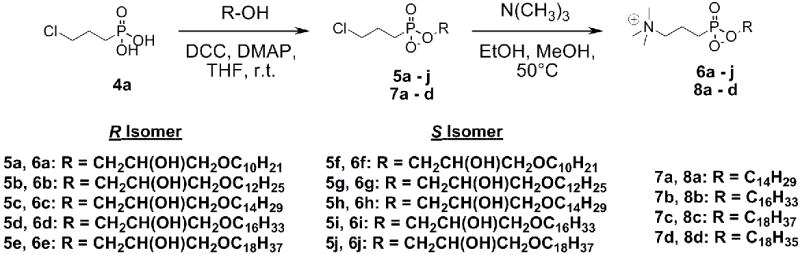
Alkylglycerophosphonocholine and alkylphosphonocholine synthesis.
The alkylphosphonocholines displayed little to no ATX inhibition (Table 1). Alkylphosphonocholines 8a and 8d inhibited ATX by 29 ± 14.9% and 21 ± 16.3%, respectively (Table 1). The corresponding alkylphosphocholines (2a and 2d) inhibited ATX-mediated FS-3 hydrolysis by 9 ± 10.4% and 92 ± 9.9%, respectively (Table 1). The apparently better ATX inhibition of 8a compared to 2a was not statistically significant (p = 0.0778). The 18:0 and 18:1 alkylphosphonocholine/alkylphosphocholine (8c / 2c and 8d / 2d) pairs did show a statistically significant difference in inhibition (p = 0.0003 for 8d and 2d, p = 0.0044 for 8c and 2c). Alkylphosphonocholines (8b and 8c) failed to inhibit ATX. These alkyl phosphonocholines (8a – d), when compared to the alkyl phosphocholines (2a – d), show that regardless of chain length and degree of unsaturation, methylene substitution for the choline oxygen is detrimental to ATX recognition.
The alkylglycerophosphonocholines were also compared to the alkylphosphonocholines (Figure 1). None of the compounds, 6a-c/f-h, significantly inhibited ATX-mediated FS-3 hydrolysis (Table 1). Comparing compounds 8a-c to 6a-f, it is evident that the glycerol backbone is not beneficial to a phosphonate head group for ATX recognition.
When compared to lysoPAF, the enantiomeric alkylglycerophosphonocholine mixtures indicate the importance of the substrate leaving group oxygen atom and the chiral center orientation. LysoPAF and LPC naturally occur as the R isomer. The 16:0 chain length enantiomers, 6d and 6i, inhibited ATX by 28 ± 4.7% and 24 ± 4.7%, respectively at 10 μM (Table 1). The 18:0 chain length enantiomers, 6e and 6j, inhibited ATX by 24 ± 4.2% and 32 ± 6.6%, respectively at 10 μM (Table 1). None of the enantiomeric pairs showed statistically different inhibition (p = 0.2964 for the 16:0 pair and p = 0.0868 for the 18:0 pair), suggesting that ATX is not stereoselective in its recognition of glycerol-based phospholipids. However these compounds failed to show statistically significant dose-dependence, so future stereoselectivity investigations with LPC or PAF are warranted. LysoPAF 16:0 and 18:0 inhibited ATX by 51 ± 4.2% and 56 ± 16.8% respectively at 10 μM. Compared to the naturally occurring lysoPAF compounds, the alkylglycerophosphonates were not as effective (Table 1), indicating that the substrate leaving group oxygen plays a role in recognition. The alkylglycerophosphonates were isolated as a mixture of two constitutional isomers. After testing the mixture we decided not to pursue further purification due to their failure to substantially inhibit ATX-mediated FS-3 hydrolysis. These results confirm that ATX has a preference for the choline oxygen over the methylene group. Inclusion of a glycerol backbone, regardless of stereochemistry, did not improve ATX recognition for the phosphonate head group.
2.5. The effect of nitrogen substitution for the choline oxygen on ATX recognition
A phosphonamidate is electrostatically more similar to a phosphate than is a phosphonate (Figure 1). Scheme 3 shows a modified synthesis from Garrido-Hernandez and colleagues for the phosphonamidate series.36 Phosphorus oxychloride, in the presence of triethylamine, was successively reacted with a long chain alcohol (tetradecanol, hexadecanol, octadecanol and oleyl alcohol), phenol, and the hydrogen bromide salt of 2-bromoethylamine. Finally, treatment of 10a – d with trimethylamine35 afforded final products 11a – d.
Scheme 3.
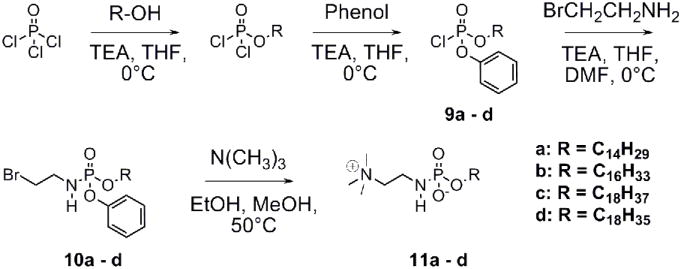
Alkylphosphoramidate synthesis.
Evaluation of the alkyl phosphonamide series gave varying results. Compounds 11a, 11b, 11c and 11d inhibited ATX by 25 ± 8.7%, 23 ± 9.6%, 71 ± 7.2% and 26 ± 9.7%, respectively (Table 1). For the alkyl phosphonamide series, only the 18:0 chain length at 10 μM was effective at inhibiting ATX. Comparing the 18:0 and 18:1 chain lengths for both series, the alkyl phosphonamide 18:0 (11c) and 18:1 (11d) inhibited ATX by 71 ± 7.2% and 26 ± 9.7%, respectively, and alkyl phosphocholine 18:0 (2c) and 18:1 (2d) by 56 ± 10.0% and 92 ± 9.9%, respectively (Table 1). Alkyl phosphonamide 18:0 (11c) inhibited ATX more effectively than the corresponding alkyl phosphocholine 18:0 (2c) by an extra 15% (p = 0.0476). On the other hand, alkyl phosphocholine 18:1 (2d) inhibited ATX more effectively than the corresponding alkyl phosphonamide 18:1 (11d) by an additonal 66% (p = 0.0001). This shows that nitrogen recognition is optimal with long saturated hydrocarbon chains among the alkyl phosphonamides.
3. Discussion
Multiple LPC compounds were tested to evaluate the effect of chain length and monounsaturation on ATX recognition. Tokumura and colleagues report that LPC 12:0 and 14:0 are optimal chains lengths for LPC as an ATX substrate.4 We report optimal chain lengths of 14:0 and 16:0 for the saturated chains when LPC is evaluated as an inhibitor, but ultimately the 18:1 chain length was optimal. The difference we see here is due to the bioassay utilized. When LPC is assayed in competition with FS-3, as in the present study, LPC 14:0 and 16:0 most effectively reduce FS-3 hydrolysis. When formation of choline directly from added LPC is monitored, as in the work reported by the Tokumura group, LPC 12:0 and 14:0 were optimal. This suggests that LPC 16:0 binds to ATX more effectively than LPC 12:0, but is not as well positioned for enzymatic hydrolysis. Furthermore, the catalytic product of LPC hydrolysis is LPA, a known inhibitor of ATX. We considered that LPA may have masked direct LPC effects by inhibiting ATX, since its concentration would increase during the assay. However, the accumulation of FS-3 hydrolytic products remained linear at the one-hour time point utilized to compute inhibition (Figure 2), indicating that the concentration of LPA did not amass sufficiently to affect the reported results.
There are two ways to lower the activation energy in an enzyme catalyzed reaction. The activation energy barrier can be reduced either by transition state stabilization or by substrate destabilization. LPA lacks the choline group of the substrate analogs examined and inhibited ATX most effectively, indicating that the choline group is detrimental to ATX recognition and may serve to destabilize the substrate complex in order to reduce the activation energy barrier of the enzymatic phosphate hydrolysis reaction.
Oleyl phosphocholine (2d) showed the most ATX inhibition of the choline-containing analogs. This molecule has a phosphodiester linkage and may be hydrolyzed by ATX. The resulting product, oleyl phosphate, may be inhibiting ATX. Durgam and colleagues reported the synthesis of a series of fatty alcohol phosphates, their effect on the LPA receptors, and on ATX activity for selected compounds.27 Oleyl phosphate had been reported to have no effect on the LPA1-3 receptors, although it was not evaluated as an ATX inhibitor.27 The related oleyl thiophosphate, however, was an effective ATX inhibitor.27 The catalytic product of oleyl phosphocholine (oleyl phosphate), might inhibit ATX similarly to oleyl thiophosphate. As was justified for LPA production from LPC above, oleyl phosphate did not amass sufficiently to affect the reported results.
Phosphonate isosteric replacement for phosphate has long been used to probe inhibitory effects for phosphatases. Therefore, we used phosphonate LPC analogs to test their inhibitory effect on the phosphodiesterase activity of ATX. All LPC phosphonate analogs tested possessed modest activity. Similar results have been shown by Chen and colleagues for phosphonomethyl phenylalanine as a protein-tyrosine phosphatase inhibitor.37 When the phosphonate methylene was converted to a difluoromethylene group, the inhibitory effect was greatly enhanced.37 The fluorine atoms likely mimic the partial negative charge of the phosphate oxygen and therefore more tightly interact with the active site. Choline is the leaving group when ATX hydrolyzes LPC and therefore leaves with a negative charge. A difluoromethylene group would more closely imitate the negative electrostatic potential of an oxygen atom, and might therefore be recognized more readily by ATX.
Various LPA phosphonate analogs have been tested by multiple groups as ATX inhibitors. These phosphonates have the methylene group on the opposite side of the phosphorus atom than do our LPC phosphonate analogs. Our best LPC phosphonate analogs were (S)-alkylglycerophosphonocholine 18:0 (6j) , alkylphosphonocholine 14:0 (8a), and (R)-alkylglycerophosphonocholine 16:0 (6d) inhibiting ATX at 10 μM by 32 ± 6.6%, 29 ± 14.9%, and 28 ± 4.7%, respectively. Cui and colleagues identified two LPA phosphonate analogs that inhibit ATX by 87% and 79% at 10 μM26. Durgam and colleagues also identified an LPA phosphonate analog that inhibited ATX by approximately 72% at 10 μM.27 These results suggest that methylene replacement of the choline oxygen is detrimental to ATX inhibition, but methylene replacement on the opposite side of the phosphodiester bond is better tolerated.
4. Conclusions
Multiple LPC analogs were evaluated to determine ATX substrate structure activity relationships. The alkyl phosphocholines, alkyl phosphonamides, alkylglycerophosphonates and alkyl phosphonates were synthetic LPC analogs. Choline has been proven detrimental to ATX recognition (Figure 3). Methylene replacement of the choline oxygen proved ineffective for ATX inhibition, with or without a polar linker, which is not required for ATX recognition/inhibition (Figure 3). These results indicate that substrate recognition involves direct interaction between the leaving group oxygen atom and the enzyme. The stereochemistry of the glycerol backbone has been shown to be irrelevant to ATX recognition (Figure 3). Nitrogen substitution for the choline oxygen proved to be context dependent, displaying improved ATX recognition coupled with some chain lengths and impaired ATX recognition in other contexts (Figure 3).
Figure 3.
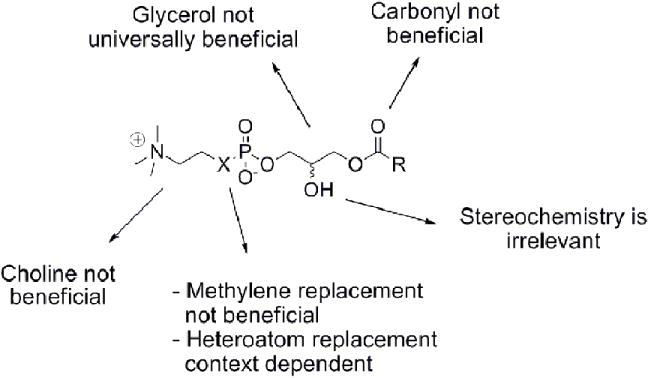
Functional group relevance to ATX recognition.
5. Experimental
5.1. Materials and Instrumentation
All reactions were carried out in clean glassware that was dried in a 175°C oven for at least eight hours. All solvents and chemicals were purchased from either Aldrich (St. Louis, Missouri) or Fisher Scientific (Pittsburgh, Pennsylvania) except for the lysophosphatidylcholines, lysophosphatidic acids, and lyso platelet activation factors, which were purchased from Avanti Polar Lipids, Inc (Alabaster, Alabama). All long chain alcohols, 3-chloropropylphosphonic acid, and DMAP were dried using magnesium sulfate or over phosphorus pentoxide in a vacuum desicator for at least eight hours prior to use. Mass spectra were acquired with a Thermoelectron LTQ XL LC-MS equipped with an electrospray ionization (ESI) source and a linear ion trap mass analyzer. All 1H NMR spectra were obtained on a JEOL 270 MHz spectrometer and all 13C NMR and 31P NMR were obtained on a Varian 500 MHz spectrometer. All NMR spectra were reported in parts per million (ppm) relative to solvent peak. Elemental analysis was performed by Columbia Analytical Services, Inc. (Tucson, AZ). Assay data was produced using a BioTek Synergy-2 plate reader. TLC plates and silica gel columns (24 g and 40 g SupraFlash Cartridges) were purchase from Sorbent Technologies (Atlanta, Georgia). The TLC plates were sprayed with 20% sulfuric acid and heated at 175°C to visualize spots. The removal of solvents was performed by evaporation under reduced pressure.
The student’s t test was used for all statistical analyses comparing two sets of data, where p < 0.05 was considered significant. GraphPad software was used to calculate statistical significance.
5.2. Compound Syntheses
5.2.1. General Procedure for Alkylphosphocholine Synthesis
A mixture of 2-bromoethanol (460 μL, 6.5 mmol), triethylamine (2.27 mL, 16.3 mmol), and THF (6 mL) was added dropwise over 15 minutes to a solution of phosphorus oxychloride (1.031 g, 6.5 mmol) and THF (5 mL) on an ice bath. A mixture of fatty alcohol (6.5 mmol), triethylamine (2.27 mL, 16.3 mmol), and THF (6 mL) was added dropwise to the reaction mixture. After stirring for one hour, a mixture of water (362 μL, 20.1 mmol) and triethylamine (2.27 mL, 16.3 mmol) was added, the ice bath was removed, and the mixture was stirred for one hour. The white precipitate was removed by gravity filtration and the solvent was evaporated. The residue was dissolved in a mixture of chloroform (10 mL) and methanol (24 mL) and extracted with water (20 mL). The aqueous phase was extracted twice with a mixture of chloroform (10 mL) and methanol (2 mL). The solvents were removed from the combined organic layers. The residue was loaded onto a 24 g SupraFlash column and eluted with ethyl acetate:hexane (2:3) then chloroform:methanol:ammonium hydroxide (60:40:7) collecting 20 mL fractions to remove unreacted alcohol. After evaporation of solvents, the residue was dissolved in 4.2 M trimethylamine (77 eq.) in ethanol and diluted with twice the volume with methanol and stirred at 50°C for 72 hours. The solvents were evaporated and the final product was purified by flash column chromatography starting with chloroform:methanol:ammonium hydroxide (60:40:7) then switching to chloroform:methanol:ammonium hydroxide (60:40:12).
5.2.2. Tetradecylphosphocholine (2a)
Yield: 70 mg of white solid (2.8%). Rf = 0.20 (chloroform:methanol:ammonium hydroxide 60:40:7). 1H NMR (chloroform-d:methanol-d4 3:2): δ = 0.64 (t, 3H), 1.03 (br, 22H), 1.38 (m, 2H), 2.98 (s, 9H), 3.35 (m, 2H), 3.61 (dt, 2H), 3.97 (m, 2H). 31P NMR: δ = 4.83. MS (ESI): m/z = 380.36 [M]+. Elemental Analysis: calcd = 56.70% C, 10.52% H, 3.48% N, 7.70% P; found = 56.92% C, 10.81% H, 3.68% N, 7.80% P.
5.2.3. Hexadecylphosphocholine (2b)
Yield: 136 mg of white solid (5.1%). Rf = 0.23 (chloroform:methanol:ammonium hydroxide 60:40:7). 1H NMR (chloroform-d:methanol-d4 3:2): δ = 0.64 (t, 3H), 1.07 (br, 26H), 1.39 (m, 2H), 2.98 (s, 9H), 3.37 (m, 2H), 3.61 (dt, 2H), 3.99 (m, 2H). 31P NMR: δ = 4.82. MS (ESI): m/z = 408.41 [M]+. Elemental analysis: calcd = 58.58% C, 10.77% H, 3.25% N, 7.19% P; found = 59.09% C, 11.36% H, 3.37% N, 7.50% P.
5.2.4. Octadecylphosphocholine (2c)
Yield: 21 mg of white solid (0.74%). Rf = 0.19 (chloroform:methanol:ammonium hydroxide 60:40:7). 1H NMR (chloroform-d:methanol-d4 3:2): δ = 0.65 (t, 3H), 1.03 (br, 30H), 1.39 (m, 2H), 2.98 (s, 9H), 3.38 (m, 2H), 3.61 (dt, 2H), 3.99 (m, 2H). 31P NMR: δ = 4.93. MS (ESI): m/z = 436.43 [M]+. Elemental analysis: calcd = 60.24% C, 10.99% H, 3.05% N, 6.75% P; found = 59.82% C, 11.08% H, 3.52% N, 7.10% P.
5.2.5. Oleylphosphocholine (2d)
Yield: 47 mg of off-white waxy solid (1.7%). Rf = 0.31 (chloroform:methanol:ammonium hydroxide 60:40:7). 1H NMR (chloroform-d:methanol-d4 3:2): δ = 0.64 (t, 3H), 1.03 (br, 22H), 1.39 (m, 2H), 1.72 (m, 4H), 2.98 (s, 9H), 3.37 (m, 2H), 3.61 (dt, 2H), 3.99 (m, 2H), 5.09 (m, 2H). 31P NMR: δ = 5.02. MS (ESI): m/z = 434.43 [M]+. Elemental analysis: calcd = 60.50% C, 10.60% H, 3.07% N, 6.78% P; found = 50.40% C, 9.20% H, 2.60% N, 5.00% P. Insufficient sample remained for repeat CHN and P analysis.
5.2.6. General Procedure for Alkylglycerol Synthesis
The long chain alcohol (17.8 mmol) was dissolved in methylene chloride (6.5 mL) and cooled on an ice bath. DIBAL-H (1 M in hexane, 7.7 mL) was added and the reaction mixture was removed from the ice bath and stirred for one hour. The appropriate glycidol (500 μL, 7.7 mmol) was added to the reaction mixture and was stirred for 72 hours at room temperature. Sodium potassium tartrate (1.64 g, 7.8 mmol) was dissolved in water (3 mL) and added to the reaction mixture. The reaction mixture was stirred for 30 minutes and the solvents were removed. The residue was dissolved in ethyl acetate (100 mL) and extracted with water (40 mL). The organic phase was dried over magnesium sulfate and the solvent was then removed. The crude products were purified by flash column chromatography starting with ethyl acetate:hexane (2:3) then switching to 100% ethyl acetate as the eluents to afford a white solid after evaporation of solvents.
5.2.7. (R)-Decylglycerol (3a)
Yield: 209 mg (11.7%). Rf = 0.23 (ethyl acetate:hexane 2:3). 1H NMR (chloroform-d): δ = 0.87 (t, 3H), 1.24 (br, 14H), 1.57 (m, 2H), 2.16 (dd, 1H), 2.59 (d, 1H), 3.48 (overlapping multiplets, 4H), 3.67 (dq, 2H), 3.84 (m, 1H).
5.2.8. (S)-Decylglycerol (3b)
Yield: 228 mg (12.7%). Rf = 0.22 (ethyl acetate:hexane 2:3). 1H NMR (chloroform-d): δ = 0.86 (t, 3H), 1.24 (br, 14H), 1.56 (m, 2H), 2.17 (dd, 1H), 2.59 (d, 1H), 3.47 (overlapping mulitiplets, 4H), 3.67 (dq, 2H), 3.85 (m, 1H).
5.2.9. (R)-Dodecylglycerol (3c)
Yield: 638 mg (31.8%). Rf = 0.24 (ethyl acetate:hexane 2:3). 1H NMR (chloroform-d): δ = 0.86 (t, 3H), 1.25 (br, 18H), 1.57 (m, 2H), 2.18 (dd, 1H), 2.61 (d, 1H), 3.49 (overlapping multiplets, 4H), 3.68 (dq, 2H), 3.84 (m, 1H).
5.2.10. (S)-Dodecylglycerol (3d)
Yield: 176 mg (8.8%). Rf = 0.25 (ethyl acetate:hexane 2:3). 1H NMR (chloroform-d): δ = 0.87 (t, 3H), 1.25 (br, 18H), 1.56 (m, 2H), 2.14 (dd, 1H), 2.57 (d, 1H), 3.48 (overlapping multiplets, 4H), 3.68 (dq, 2H), 3.84 (m, 1H).
5.2.11. (R)-Tetradecylglycerol (3e)
Yield: 483 mg (21.7%). Rf = 0.27 (ethyl acetate:hexane 2:3). 1H NMR (chloroform-d): δ = 0.87 (t, 3H), 1.25 (br, 22H), 1.57 (m, 2H), 2.15 (dd, 1H), 2.58 (d, 1H), 3.48 (overlapping multiplets, 4H), 3.67 (dq, 2H), 3.84 (m, 1H).
5.2.12. (S)-Tetradecylglycerol (3f)
Yield: 458 mg (20.6%). Rf = 0.28 (ethyl acetate:hexane 2:3). 1H NMR (chloroform-d): δ = 0.87 (t, 3H), 1.25 (br, 22H), 1.57 (m, 2H), 2.15 (dd, 1H), 2.58 (d, 1H), 3.48 (overlapping multiplets, 4H), 3.67 (dq, 2H), 3.84 (m, 1H).
5.2.13. (R)-Hexadecylglycerol (3g)
Yield: 513 mg (21%). Rf = 0.28 (ethyl acetate:hexane 2:3). 1H NMR (chloroform-d): δ = 0.86 (t, 3H), 1.24 (br, 26H), 1.56 (m, 2H), 3.48 (overlapping multiplets, 4H), 3.67 (dq, 2H), 3.84 (m, 1H).
5.2.14. (S)-Hexadecylglycerol (3h)
Yield: 913 mg (35%). Rf = 0.27 (ethyl acetate:hexane 2:3). 1H NMR (chloroform-d): δ = 0.86 (t, 3H), 1.24 (br, 26H), 1.56 (m, 2H), 3.48 (overlapping multiplets, 4H), 3.68 (dq, 2H), 3.85 (m, 1H).
5.2.15. (R)-Octadecylglycerol (3i)
Yield: 785 mg (30%). Rf = 0.26 (ethyl acetate:hexane 2:3). 1H NMR (chloroform-d): δ = 0.87 (t, 3H), 1.24 (br, 30H), 1.56 (m, 2H), 3.48 (overlapping multiplets, 4H), 3.67 (dq, 2H), 3.85 (p, 1H).
5.2.16. (S)-Octadecylglycerol (3j)
Yield: 298 mg (11%). Rf = 0.26 (ethyl acetate:hexane 2:3). 1H NMR (chloroform-d): δ = 0.87 (t, 3H), 1.24 (br, 30H), 1.56 (m, 2H), 3.48 (m, 4H), 3.67 (dq, 2H), 3.84 (m, 1H).
5.2.17. 3-Chloropropylphosphonic acid (4a)
Dimethylphosphite (4.5 mL, 49.0 mmol) was added dropwise to a solution of potassium t-butoxide (5.150 g, 45.9 mmol) and THF (18 mL) within 15 minutes. The resulting thick grey gel was added dropwise to a solution of 1-bromo-3-chloropropane (5.7 mL, 57.6 mmol) and THF (9 mL). The reaction mixture was heated under reflux for 15 minutes. The white precipitate was filtered using gravity filtration. The precipitate was washed twice with two portions of diisopropyl ether (18 mL). The solvents were evaporated to produce a clear liquid residue. The resulting residue was dissolved in concentrated HCl (69 mL) and heated under reflux for 9 hours. The solvent was evaporated under reduced pressure to produce 4.908 g (67% yield) of waxy amber-colored crude final product. 1H NMR (D2O): δ = 1.70 – 2.10 (overlapping multiplets, 4H), 3.64 (m, 2H).
5.2.18. General Procedure for 1-Alkyl-2-hydroxy-sn-glycero-3-phosphonocholine Synthesis
All reactions were carried out with at least 2.0 mmol of alkylglycerol. Alkylglycerol (1 eq.), 3-chloropropylphosphonic acid (1.1 eq.), DCC (2.2 eq.), and DMAP (0.1 eq.) were dissolved in THF (9.7 mL/mmol alkylglycerol) and stirred for 48 hours at room temperature. Water (84 eq.) was added to the reaction mixture and it was allowed to stir for an additional 24 hours. The precipitate was removed by gravity filtration and the solvents were evaporated. The residue was dissolved in a mixture of chloroform (3.8 mL/mmol alkylglycerol) and methanol (4.5 mL/mmol alkylglycerol) and extracted with water (3.8 mL/mmol alkylglycerol). The aqueous phase was extracted twice with mixtures of chloroform (3.8 mL/mmol alkylglycerol) and methanol (1 mL/mmol alkylglycerol). The solvents were removed from the combined organic layers. The residue was loaded onto a 24 g SupraFlash column and eluted with ethyl acetate then methanol collecting 20 mL fractions to remove unreacted alkylglycerol. After evaporation of solvents, the residue was dissolved in 4.2 M trimethylamine (77 eq.) in ethanol and diluted with twice the volume with methanol and stirred at 50°C for 72 hours. The solvents were evaporated and the final product was purified by flash column chromatography using methylene chloride:methanol:water (65:35:6). Products were isolated as oils after evaporation of solvent. Yields were generally low, in the range of 1-9%.
5.2.19. (R)-1-Decyl-2-hydroxy-sn-glycero-3-phosphonocholine (6a)
Rf = 0.09 (methylene chloride:methanol:water 65:35:6). 1H NMR (chloroform-d:methanol-d4 1:1): δ = 0.50 (t, 3H), 0.94 (br, 14H), 1.23 (overlapping multiplets, 4H), 1.63 (m, 2H), 2.75 (s, 9H), 2.97 - 3.40 (overlapping multiplets, 8H), 3.51 (late fraction, m, 1H), 3.94 (early fraction, m, 1H). 31P NMR: δ = 28.61 (early fraction), 28.84 (late fraction). MS (ESI): m/z = 396.33 [M]+. Elemental analysis: calcd = 54.53% C, 10.12% H, 3.35% N; found = 55.15% C, 10.81% H, 3.08% N (Insufficient sample for P analysis).
5.2.20. (R)-1-Dodecyl-2-hydroxy-sn-glycero-3-phosphonocholine (6b)
Rf = 0.09 (methylene chloride:methanol:water 65:35:6). 1H NMR (chloroform-d:methanol-d4 1:1): δ = 0.50 (t, 3H), 0.90 (br, 18H), 1.21 (overlapping multiplets, 4H), 1.65 (m, 2H), 2.76 (s, 9H), 2.98 - 3.42 (overlapping multiplets, 8H), 3.52 (late fraction, m, 1H), 3.96 (early fraction, m, 1H). 31P NMR: δ = 28.26 (early fraction), 28.52 (late fraction). MS (ESI): m/z = 424.38 [M]+. Elemental analysis: calcd = 56.48% C, 10.38% H, 3.14% N, 6.94% P; found = 52.65% C, 10.24% H, 2.81% N, 6.90% P.
5.2.21. (R)-1-Tetradecyl-2-hydroxy-sn-glycero-3-phosphonocholine (6c)
Rf = 0.09 (methylene chloride:methanol:water 65:35:6). 1H NMR (chloroform-d:methanol-d4 1:1): δ = 0.51 (t, 3H), 0.88 (br, 22H), 1.23 (overlapping multiplets, 4H), 1.62 (m, 2H), 2.74 (s, 9H), 2.95 - 3.39 (overlapping multiplets, 8H), 3.49 (late fraction, m, 1H), 3.92 (early fraction, m, 1H). 31P NMR: δ = 28.27 (early fraction), 28.55 (late fraction). MS (ESI): m/z = 452.42 [M]+. Elemental analysis: calcd = 58.21% C, 10.62% H, 2.95% N, 6.53% P; found = 59.59% C, 10.96% H, 3.13% N, 6.20% P.
5.2.22. (R)-1-Hexadecyl-2-hydroxy-sn-glycero-3-phosphonocholine (6d)
Rf = 0.09 (methylene chloride:methanol:water 65:35:6). 1H NMR (chloroform-d:methanol-d4 1:1): δ = 0.53 (t, 3H), 0.99 (br, 26H), 1.21-1.52 (overlapping multiplets, 4H), 1.65 (m, 2H), 2.75 (s, 9H), 3.11-3.40 (overlapping multiplets, 8H), 3.55 (m, 1H). 31P NMR: δ = 29.38. MS (ESI): m/z = 480.45 [M]+. Elemental analysis: calcd = 59.74% C, 10.83% H, 2.79% N, 6.16% P; found = 59.90% C, 9.35% H, 2.17% N, 5.10% P.
5.2.23. (R)-1-Octadecyl-2-hydroxy-sn-glycero-3-phosphonocholine (6e)
Rf = 0.09 (methylene chloride:methanol:water 65:35:6). 1H NMR (chloroform-d:methanol-d4 1:1): δ = 0.50 (t, 3H), 0.90 (br, 30H), 1.21 (overlapping multiplets, 4H), 1.64 (m, 2H), 2.75 (s, 9H), 2.97-3.40 (overlapping multiplets, 8H), 3.51 (late fraction, m, 1H), 3.95 (early fraction, m, 1H). 31P NMR: δ = 28.83. MS (ESI): m/z = 508.50 [M]+. Elemental analysis: calcd = 61.10% C, 11.02% H, 2.64% N, 5.84% P; found = 58.58% C, 10.83% H, 3.37% N, 5.70% P.
5.2.24. (S)-1-Decyl-2-hydroxy-sn-glycero-3-phosphonocholine (6f)
Rf = 0.09 (methylene chloride:methanol:water 65:35:6). 1H NMR (chloroform-d:methanol-d4 1:1): δ = 0.52 (t, 3H), 0.92 (br, 14H), 1.22 (overlapping multiplets, 4H), 1.65 (m, 2H), 2.76 (s, 9H), 3.00 - 3.44 (overlapping multiplets, 8H), 3.54 (late fraction, m, 1H), 3.96 (early fraction, m, 1H). 31P NMR: δ = 33.63 (early fraction), 34.08 (late fraction). MS (ESI): m/z = 396.36 [M]+. Elemental analysis: calcd = 54.53% C, 10.12% H, 3.35% N, 7.40% P; found = 50.42% C, 10.20% H, 3.02% N, 7.00% P.
5.2.25. (S)-1-Dodecyl-2-hydroxy-sn-glycero-3-phosphonocholine (6g)
Rf = 0.09 (methylene chloride:methanol:water 65:35:6). 1H NMR (chloroform-d:methanol-d4 1:1): δ = 0.50 (t, 3H), 0.88 (br, 18H), 1.20 (overlapping multiplets, 4H), 1.62 (m, 2H), 2.72 (s, 9H), 2.92 - 3.38 (overlapping multiplets, 8H), 3.58 (late fraction, m, 1H), 3.91 (early fraction, m, 1H). 31P NMR: δ = 28.25 (early fraction), 28.53 (late fraction). MS (ESI): m/z = 424.38 [M]+. Elemental analysis: calcd = 56.48% C, 10.38% H, 3.14% N, 6.94% P; found = 56.97% C, 11.11% H, 3.33% N, 7.20% P.
5.2.26. (S)-1-Tetradecyl-2-hydroxy-sn-glycero-3-phosphonocholine (6h)
Rf = 0.09 (methylene chloride:methanol:water 65:35:6). 1H NMR (chloroform-d:methanol-d4 1:1): δ = 0.50 (t, 3H), 0.90 (br, 22H), 1.22 (overlapping multiplets, 4H), 1.65 (m, 2H), 2.76 (s, 9H), 2.98 - 3.42 (overlapping multiplets, 8H), 3.54 (late fraction, m, 1H), 3.94 (early fraction, m, 1H). 31P NMR: δ = 28.22 (early fraction), 28.61 (late fraction). MS (ESI): m/z = 452.40 [M]+. Elemental analysis: calcd = 58.21% C, 10.62% H, 2.95% N, 6.53% P; found = 54.62% C, 11.40% H, 2.72% N, 6.10% P.
5.2.27. (S)-1-Hexadecyl-2-hydroxy-sn-glycero-3-phosphonocholine (6i)
Rf = 0.09 (methylene chloride:methanol:water 65:35:6). 1H NMR (chloroform-d:methanol-d4 1:1): δ = 0.58 (t, 3H), 0.98 (br, 26H), 1.16-1.43 (overlapping multiplets, 4H), 1.71 (m, 2H), 2.83 (s, 9H), 3.07-3.49 (overlapping multiplets, 8H), 3.61 (late fraction, m, 1H), 4.04 (early fraction, m, 1H). 31P NMR: δ = 28.57 (early fraction), 28.88 (late fraction). MS (ESI): m/z = 480.45 [M]+. Elemental analysis: calcd = 59.74% C, 10.83% H, 2.79% N, 6.16% P; found = 50.31% C, 10.72% H, 5.11% N, 6.10% P.
5.2.28. (S)-1-Octadecyl-2-hydroxy-sn-glycero-3-phosphonocholine (6j)
Rf = 0.09 (methylene chloride:methanol:water 65:35:6). 1H NMR (chloroform-d:methanol-d4 1:1): δ = 0.51 (t, 3H), 0.89 (br, 30H), 1.12-1.34 (overlapping multiplets, 4H), 1.65 (m, 2H), 2.76 (s, 9H), 2.98-3.42 (overlapping multiplets, 8H), 3.53 (late fraction, m, 1H), 3.96 (early fraction, m, 1H). 31P NMR: δ = 28.69 (early fraction), 28.95 (late fraction). MS (ESI): m/z = 508.50 [M]+. Elemental analysis: calcd = 61.10% C, 11.02% H, 2.64% N, 5.84% P; found = 52.00% C, 10.32% H, 5.01% N, 5.40% P.
5.2.29. General Procedure for Alkyphosphonocholine Synthesis
All reactions were carried out with at least 8.8 mmol of long chain alcohol. Alcohol (1 eq.), 3-chloropropylphosphonic acid (1.1 eq.), DCC (2.2 eq.), and DMAP (0.1 eq.) were dissolved in THF (9.3 mL/mmol alcohol) and stirred for 48 hours at room temperature. Water (84 eq.) was added to the reaction mixture and it was allowed to stir for an additional 24 hours. The precipitate was removed by gravity filtration and the solvents were evaporated. The residue was dissolved in a mixture of chloroform (41 mL) and methanol (49 mL) and extracted with water (40 mL). The aqueous phase was extracted twice with mixtures of chloroform (40 mL) and methanol (10 mL). The solvents were removed from the combined organic layers. The organic phase was evaporated and the residue was loaded onto a 24 g SupraFlash column and eluted with ethyl acetate/hexane (2:3) then chloroform:methanol:ammonium hydroxide (60:40:7), collecting 20 mL fractions to remove unreacted alcohol. After evaporation of solvents, the residue was dissolved in 4.2 M trimethylamine (77 eq.) in ethanol and diluted with twice the volume of methanol and stirred at 50°C for 72 hours. The solvents were evaporated and the final product was purified by flash column chromatography starting with chloroform:methanol:ammonium hydroxide (60:40:7) then switching to chloroform:methanol:ammonium hydroxide (60:40:12).
5.2.30. Tetradecylphosphonocholine (8a)
Yield: 47 mg (1.2% yield) of white solid. Rf = 0.15 (chloroform:methanol:ammonium hydroxide 60:40:7). 1H NMR (chloroform-d:methanol-d4 2:1): δ = 0.65 (t, 3H), 1.03 (br, 22 H), 1.26-1.44 (overlapping multiplets, 4H), 1.75 (m, 2H), 2.88 (s, 9H), 3.19 (m, 2H), 3.62 (m, 2H). 31P NMR: δ = 27.60. MS (ESI): m/z = 378.36 [M]+. Elemental analysis: calcd = 59.97% C, 11.07% H, 3.50% N, 7.73% P; found = 54.68% C, 18.81% H, 4.02% N, 5.00% P (Insufficient sample remained for repeat CHN and P analysis).
5.2.31. Hexadecylphosphonocholine (8b)
Yield: 15 mg (0.4% yield) of white solid. Rf = 0.21 (chloroform:methanol:ammonium hydroxide 60:40:7). 1H NMR (chloroform-d:methanol-d4 2:1): δ = 0.65 (t, 3H), 1.03 (br, 26 H), 1.22-1.42 (overlapping multiplets, 4H), 1.71 (m, 2H), 2.88 (s, 9H), 3.20 (m, 2H), 3.60 (m, 2H). 31P NMR: δ = 27.51. MS (ESI): m/z = 406.38 [M]+. Elemental analysis: calcd = 7.23% P; found = 6.10% P (Insufficient sample available for initial CHN analysis).
5.2.32. Octadecylphosphonocholine (8c)
Yield: 7 mg (0.2% yield) of white solid. Rf = 0.13 (chloroform:methanol:ammonium hydroxide 60:40:7). 1H NMR (chloroform-d:methanol-d4 2:1): δ = 0.65 (t, 3H), 1.05 (br, 30 H), 1.22-1.42 (overlapping multiplets, 4H), 1.71 (m, 2H), 2.90 (s, 9H), 3.22 (m, 2H), 3.62 (m, 2H). 31P NMR: δ = 27.19. MS (ESI): m/z = 434.43 [M]+. Elemental analysis: calcd = 63.13% C, 11.48% H, 3.07% N, 6.78% P; found = 55.40% C, 10.60% H, 2.70% N, 5.30% P (Limited sample availability resulted in larger reporting limits and analytical error).
5.2.33. Oleylphosphonocholine (8d)
Yield: 145 mg (6.6% yield) of white solid. Rf = 0.15 (chloroform:methanol:ammonium hydroxide 60:40:7). 1H NMR (chloroform-d:methanol-d4 2:1): δ = 0.56 (t, 3H), 0.95 (br, 22 H), 1.25-1.45 (overlapping multiplets, 4H), 1.65-1.85 (overlapping multiplets, 6H), 2.88 (s, 9H), 3.10 (m, 2H), 3.50 (m, 2H), 5.00 (t, 2H). 31P NMR: δ = 27.66. MS (ESI): m/z = 432.42 [M]+. Elemental analysis: calcd = 63.41% C, 11.09% H, 3.08% N, 6.81% P; found = 38.60% C, 7.00% H, 10.10% N, 5.00% P (Insufficient sample remained for repeat CHN and P analysis).
5.2.34. General Procedure for Phenyl-O-alkylphosphoromonochloridate Synthesis
Triethylamine (841 μL, 5.9 mmol) in methylene chloride (2.3 mL) was added dropwise over 15 minutes to a freshly prepared solution of phosphorus oxychloride (500 μL, 5.5 mmol), alcohol (5.5 mmol), and methylene chloride (13.6 mL) on an ice bath. Once all of the triethylamine solution was added, the reaction mixture was stirred for 30 minutes. Phenol (5.5 mmol) was added, followed by dropwise addition of triethylamine (841 μL, 5.9 mmol) in methylene chloride (2.3 mL). The reaction mixture was stirred for one hour at 0° C, then quenched by addition of 20 mL of saturated ammonium chloride. The mixture was extracted with three portions of methylene chloride (11 mL ea.). The combined organic layers were washed with 20 mL of brine and dried over magnesium sulfate. The solvents were removed to afford crude monochloro products. Partial purification of monochloro products was achieved using flash column chromatography and ethyl acetate:hexane (9:1) as the eluent.
5.2.35. Phenyl-O-tetradecylphosphoromonochloridate (9a)
Yield: 1.222 g (58% yield). Rf = 0.59 (ethyl acetate:hexane 9:1). 1H NMR (chloroform-d): δ = 0.84 (t, 3H), 1.30 (br, 22H), 1.72 (m, 2H), 4.31 (tq, 2H), 7.26 (m, 3H), 7.36 (m, 2H).
5.2.36. Phenyl-O-hexadecylphosphoromonochloridate (9b)
Yield: 607 mg (27% yield). Rf = 0.45 (ethyl acetate:hexane 9:1). 1H NMR (chloroform-d): δ = 0.87 (t, 3H), 1.24 (br, 26H), 1.77 (m, 2H), 4.30 (tq, 2H), 7.24 (m, 3H), 7.35 (m, 2H).
5.2.37. Phenyl-O-octadecylphosphoromonochloridate (9c)
Yield: 671 mg (28% yield). Rf = 0.48 (ethyl acetate:hexane 9:1). 1H NMR (chloroform-d): δ = 0.87 (t, 3H), 1.24 (br, 30H), 1.77 (m, 2H), 4.30 (tq, 2H), 7.24 (m, 3H), 7.34 (m, 2H).
5.2.38. Phenyl-oleylphosphoromonochloridate (9d)
Yield: 829 mg (35% yield). Rf = 0.51 (ethyl acetate:hexane 9:1). 1H NMR (chloroform-d): δ = 0.87 (t, 3H), 1.25 (br, 22H), 1.79 (m, 2H), 2.00 (m, 4H), 4.29 (tq, 2H), 5.34 (m, 2H), 7.24 (m, 3H), 7.36 (m, 2H).
5.2.39. General Procedure for N-(2-trimethylaminoethyl)-O-alkyl Phosphoramidate Synthesis
All reactions were carried out with at least 1.46 mmol of phenyl-alkylphosphoromonochloridate. 2-Bromoethylamine hydrogen bromide (1 eq.) was dissolved in a minimal amount of DMF and diluted with THF (0.355 mL/mmol phenyl-alkylphosphoromonochloridate). Phenyl-alkylphosphoromonochloridate (1 eq.) was added to the DMF/THF mixture. Triethylamine (2.3 eq.) was added dropwise to the reaction mixture at room temperature. Once all of the triethylamine had been added, the reaction mixture was stirred for 80 minutes. The precipitate was removed using gravity filtration and the solvents were evaporated. The residue was dissolved in chloroform (20 mL) and extracted with water (10 mL). The organic phase was evaporated and the residue was loaded onto a 24 g SupraFlash column and eluted with chloroform/ethyl acetate (10:1) collecting 20 mL fractions to remove unreacted phenyl-alkylphosphoromonochloridate. After evaporation of solvents, the residue was dissolved in 4.2 M trimethylamine (77 eq.) in ethanol and diluted with twice the volume with methanol and stirred at 50°C for 72 hours. The solvents were evaporated and the final product was purified by flash column chromatography starting with chloroform:methanol:ammonium hydroxide (60:40:7) then changing to chloroform:methanol:ammonium hydroxide (60:40:12). Products were isolated as white solids after evaporation of solvents. Yields were generally low, in the range of 2-8%.
5.2.40. N-(2-trimethylaminoethyl)-O-tetradecyl Phosphoramidate (11a)
Rf = 0.16 (chloroform:methanol:ammonium hydroxide 60:40:7). 1H NMR (chloroform-d:methanol-d4 2:1), δ = 0.65 (t, 3H), 1.03 (br, 22 H), 1.34 (m, 2H), 2.96 (s, 9H), 2.98-3.23 (overlapping multiplets, 4H), 3.53 (q, 2H). 31P NMR: δ = 11.61. MS (ESI): m/z = 379.38 [M]+. Elemental analysis: calcd = 56.84% C, 10.79% H, 6.98% N, 7.71% P; found = 54.70% C, 10.10% H, 6.90% N, 7.50% P.
5.2.41. N-(2-trimethylaminoethyl)-O-hexadecyl Phosphoramidate (11b)
Rf = 0.16 (chloroform:methanol:ammonium hydroxide 60:40:7). 1H NMR (chloroform-d:methanol-d4 2:1), δ = 0.64 (t, 3H), 1.02 (br, 26 H), 1.37 (m, 2H), 2.95 (s, 9H), 2.98-3.24 (overlapping multiplets, 4H), 3.52 (q, 2H), 3.87 (s, 1H). 31P NMR: δ = 11.61. MS (ESI): m/z = 407.40 [M]+. Elemental analysis: calcd = 58.72% C, 11.03% H, 6.52% N, 7.21% P; found = 55.67% C, 10.90% H, 6.47% N, 6.40% P.
5.2.42. N-(2-trimethylaminoethyl)-O-octadecyl Phosphoramidate (11c)
Rf = 0.15 (chloroform:methanol:ammonium hydroxide 60:40:7). 1H NMR (chloroform-d:methanol-d4 2:1), δ = 0.74 (t, 3H), 1.13 (br, 30 H), 1.47 (m, 2H), 3.06 (s, 9H), 3.14-3.38 (overlapping multiplets, 4H), 3.63 (q, 2H). 31P NMR: δ = 11.60. MS (ESI): m/z = 435.45 [M]+. Elemental analysis: calcd = 60.37% C, 11.23% H, 6.12% N, 6.77% P; found = 58.38% C, 11.16% H, 6.12% N, 6.20% P.
5.2.43. N-(2-trimethylaminoethyl)-O-oleyl Phosphoramidate (11d)
Rf = 0.16 (chloroform:methanol:ammonium hydroxide 60:40:7). 1H NMR (chloroform-d:methanol-d4 2:1), δ = 0.66 (t, 3H), 1.05 (br, 22 H), 1.37 (m, 2H), 1.77 (overlapping multiplets, 4H), 2.97 (s, 9H), 3.04-3.28 (overlapping multiplets, 4H), 3.54 (q, 2H), 5.10 (t, 2H). 31P NMR: δ = 11.60. MS (ESI): m/z = 433.43 [M]+. Elemental analysis: calcd = 60.63% C, 10.84% H, 6.15% N, 6.80% P; found = 56.10% C, 10.64% H, 5.68% N, 6.60% P.
5.3. Cell Culture
MDA-MB-435 cells were cultured at 37°C and 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM) (MediaTech, Herndon, VA) containing 5% fetal bovine serum (Hyclone, Logan, UT), 100 U/ml penicillin-streptomycin (Hyclone, Logan, UT) and 292 μg/ml L-glutamine (Hyclone, Logan, UT). Cells were grown to ~80% confluence at which time the cells were washed twice with sterile phosphate buffered saline prior to the addition of serum free DMEM containing L-glutamine. Conditioned medium was collected after 24-30 hours, supplemented with 20% ethylene glycol and was clarified by centrifugation at 3000×g and 4°C for 10 min. The media was concentrated ~10 fold and buffer exchanged into Tris (50 mM, pH 7.4) containing 20% ethylene glycol using an Amicon 8050 cell fitted with a PM30 filter (Millipore, Billerica, MA). Aliquots of concentrated conditioned media were stored at 4°C until needed.
5.4. ATX Inhibition Assay
ATX inhibition was determined utilizing the synthetic substrate FS-3 (Echelon Biosciences, Inc., Salt Lake City, Utah, USA), concentrated conditioned media, and LPC analog, each comprising one third of the total volume. Final concentrations of FS-3 and charcoal-stripped fatty acid free bovine serum albumin were 1 μM and 30 μM, respectively, in assay buffer (1mM MgCl2, 1 mM CaCl2, 5 mM KCl, 140 mM NaCl, 50 mM Tris, pH 8). LPC analogs had final concentrations of 0.1, 1, and 10 μM in assay buffer. All assays were carried out in 96 well plates in a BioTek Synergy-2 plate reader (BioTek, Winooski, VT, USA) with excitation and emission wavelengths of 485 and 538 nm, respectively. Fluorescent emission detection occurred every five minutes and data was shown as percent ATX inhibition, with respect to vehicle control after subtraction of fluorescence with no CCM, at the one hour time point, where fluorescence detection with respect to time is linear. All data were reported as mean ± standard deviation with at least three wells.
Acknowledgments
The authors acknowledge the National Institutes of Health (NIH R01 HL 084007) for their financial support and the National Science Foundation (NSF CHE 0443627, NSF CHE 0619682) for their financial aid in the acquisition of a Varian 500 MHz NMR and a Thermoelectron LTQ-XL LC-MS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stracke M, Krutzch H, Unsworth E, Arestad A, Cioce V, Schiffmann E, Liotta L. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J Biol Chem. 1992;267:2524–2529. [PubMed] [Google Scholar]
- 2.van Meeteren LA, Moolenaar WH. Regulation and biological activities of the autotaxin-LPA axis. Prog Lipid Res. 2007;46:145–160. doi: 10.1016/j.plipres.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, Yamori T, Mills G, Inoue K, Aoki J, Arai H. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J Cell Biol. 2002;158:227–233. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, Fukuzawa K. Identification of Human Plasma Lysophospholipase D, a Lysophosphatidic acid-producing Enzyme, as Autotaxin, a Multifunctional Phosphodiesterase. J Biol Chem. 2002;277:39436–39442. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- 5.Tabata K-i, Baba K, Shiraishi A, Ito M, Fujita N. The orphan GPCR GPR87 was deorphanized and shown to be a lysophosphatidic acid receptor. Biochem Biophys Res Commun. 2007;363:861–866. doi: 10.1016/j.bbrc.2007.09.063. [DOI] [PubMed] [Google Scholar]
- 6.Mills Ga, M W. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 7.Lee C-W, Rivera R, Gardell S, Dubin AE, Chun J. GPR92 as a New G12/13 - and Gq - coupled Lysophosphatidic Acid Receptor That Increases cAMP, LPA5. J Biol Chem. 2006;281:23589–23597. doi: 10.1074/jbc.M603670200. [DOI] [PubMed] [Google Scholar]
- 8.Pasternack SM, von Kugelgen I, Aboud KA, Lee Y-A, Ruschendorf F, Voss K, Hillmer AM, Molderings GJ, Franz T, Ramirez A, Nurnberg P, Nothen MM, Betz RC. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat Genet. 2008;40:329–334. doi: 10.1038/ng.84. [DOI] [PubMed] [Google Scholar]
- 9.Murakami M, Shiraishi A, Tabata K, Fujita N. Identification of the orphan GPCR, P2Y10 receptor as the sphingosine-1-phosphate and lysophosphatidic acid receptor. Biochem Biophys Res Commun. 2008 doi: 10.1016/j.bbrc.2008.04.145. [DOI] [PubMed] [Google Scholar]
- 10.Parrill AL. Lysophospholipid interactions with protein targets. Biochim Biophys Acta. 2008;1781:540–546. doi: 10.1016/j.bbalip.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McIntyre TM, Pontsler AV, Silva AR, Hilaire AS, Xu Y, Hinshaw JC, Zimmerman GA, Hama K, Aoki J, Arai H, Prestwich GD. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc Natl Acad Sci U S A. 2003;100:131–136. doi: 10.1073/pnas.0135855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hama K, Aoki J, Fukaya M, Kishi Y, Sakai T, Suzuki R, Ohta H, Yamuri T, Watanabe M, Chun J, Arai H. Lysophosphatidic acid and autotaxin stimulate cell motility of neoplastic and non-neoplastic cells through LPA1. J Biol Chem. 2004;279:17634–17639. doi: 10.1074/jbc.M313927200. [DOI] [PubMed] [Google Scholar]
- 13.Boucher J, Quilliot D, Pradere J-P, Simon M-F, Gres S, Guigne C, Prevot D, Ferry G, Boutin JA, Carpene C, Valet P, Saulnier-Blache JS. Potential involvement of adipocyte insulin resistance in obesity-associated up-regulation of adipocyte lysophospholipase D/autotaxin expression. Diabetologia. 2005;48:569–577. doi: 10.1007/s00125-004-1660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao C, Fernandes MJ, Prestwich GD, Turgeon M, Battista JD, Clair T, Poubelle PE, Bourgoin SG. Regulation of Lysophosphatidic Acid Receptor Expression and Function in Human Synoviocytes: Implications for Rheumatiod Arthritis? Mol Pharmacol. 2008;73:587–600. doi: 10.1124/mol.107.038216. [DOI] [PubMed] [Google Scholar]
- 15.Inoue M, Ma L, Aoki J, Chun J, Ueda H. Autotaxin, a synthetic enzyme of lysophosphatidic acid (LPA), mediates the induction of nerve-injured neuropathic pain. Molecular Pain. 2008;4 doi: 10.1186/1744-8069-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rother E, Brandl R, Baker DL, Goyal P, Gebhard H, Tigyi G, Siess W. Subtype-Selective Antagonists of Lysophosphatidic Acid Receptors Inhibit Platelet Activation Triggered by the Lipid Core of Atherosclerotic Plaques. Circulation. 2003;108:741–747. doi: 10.1161/01.CIR.0000083715.37658.C4. [DOI] [PubMed] [Google Scholar]
- 17.Gijsbers R, Ceulemans H, Stalmans W, Bollen M. Structural and catalytic similarities between nucleotide pyrophosphatases/phosphodiesterases and alkaline phosphatases. J Biol Chem. 2001;276:1361–1368. doi: 10.1074/jbc.M007552200. [DOI] [PubMed] [Google Scholar]
- 18.Jansen S, Callewaert N, Dewerte I, Andries M, Ceulemans H, Bollen M. An Essential Oligomannosidic Glycan Chain in the Catalytic Domain of Autotaxin, a Secreted Lysophospholipase-D. J Biol Chem. 2007;282:11084–11091. doi: 10.1074/jbc.M611503200. [DOI] [PubMed] [Google Scholar]
- 19.Galperin MY, Jedrzejas MJ. Conserved Core Structure and Active Site Residues in Alkaline Phosphatase Superfamily Enzymes. Proteins: Structure, Function, and Genetics. 2001;45:318–324. doi: 10.1002/prot.1152. [DOI] [PubMed] [Google Scholar]
- 20.Zalatan JG, Fenn TD, Brunger AT, Herschlag D. Structural and Functional Comparisons of Nucleotide Pyrophosphatase/Phosphodiesterase and Alkaline Phosphatase: Implications for Mechanism and Evolution. Biochemistry. 2006;45:9788–9803. doi: 10.1021/bi060847t. [DOI] [PubMed] [Google Scholar]
- 21.Saunders LP, Ouellette A, Bandle R, Chang WC, Zhou H, Misra RN, Cruz EMDL, Braddock DT. Identification of small-molecule inhibitors of autotaxin that inhibit melanoma cell migration and invasion. Mol Cancer Ther. 2008;7:3352–3362. doi: 10.1158/1535-7163.MCT-08-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parrill AL, Echols U, Nguyen T, Pham T-CT, Hoeglund A, Baker DL. Virtual screening approaches for the identification of non-lipid autotaxin inhibitors. Bioorg Med Chem. 2008;16:1784–1795. doi: 10.1016/j.bmc.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 23.Moulharat N, Fould B, Giganti A, Boutin JA, Ferry G. Molecular pharmacology of adipocyte-secreted autotaxin. Chem-Biol Interact. 2008;172:115–124. doi: 10.1016/j.cbi.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 24.Clair T, Koh E, Ptaszynska M, Bandle R, Liotta L, Schiffmann E, Stracke M. L-histidine inhibits production of lysophosphatidic acid by the tumor-associated cytokine, autotaxin. Lipids Health Dis. 2005;4 doi: 10.1186/1476-511X-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Meeteren L, Ruurs P, Christodoulou E, Goding J, Takakusa H, Kikuchi K, Perrakis A, Nagano T, Moolenaar W. Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1-phosphate. J Biol Chem. 2005;280:21155–21161. doi: 10.1074/jbc.M413183200. [DOI] [PubMed] [Google Scholar]
- 26.Cui P, Tomsig JL, McCalmont WF, Lee S, Becker CJ, Lynch KR, Macdonald TL. Synthesis and biological evaluation of phosphonate derivatives as autotaxin (ATX) inhibitors. Bioorg Med Chem Lett. 2007;17:1634–1640. doi: 10.1016/j.bmcl.2006.12.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durgam G, Virag T, Walker M, Tsukahara R, Yasuda S, Liliom K, van Meeteren L, Moolenaar W, Wilke N, Siess W, Tigyi G, Miller D. Synthesis, structure-activity relationships, and biological evaluation of fatty alcohol phosphates as lysophosphatidic acid receptor ligands, activators of PPAR gamma, and inhibitors of autotaxin. J Med Chem. 2005;48:4919–4930. doi: 10.1021/jm049609r. [DOI] [PubMed] [Google Scholar]
- 28.Ferry G, Moulharat N, Pradere J-P, Desos P, Try A, Genton A, Giganti A, Beucher-Gaudin M, Lonchampt M, Bertrand M, Saulnier-Blache J-S, Tucker GC, Cordi A, Boutin JA. S32826: A Nanomolar Inhibitor of Autotaxin. Discovery, Synthesis and Applications as a Pharmacological Tool. J Pharmacol Exp Ther. 2008 doi: 10.1124/jpet.108.141911. Published Ahead of Print. [DOI] [PubMed] [Google Scholar]
- 29.Gududuru V, Zeng K, Tsukahara R, Makarova N, Fujiwara Y, Pigg KR, Baker DL, Tigyi G, Miller DD. Identification of Darmstoff analogs as selective agonists and antagonists of lysophosphatidic acid receptors. Bioorg Med Chem Lett. 2006;16:451–456. doi: 10.1016/j.bmcl.2005.08.096. [DOI] [PubMed] [Google Scholar]
- 30.Baker DL, Fujiwara Y, Pigg KR, Tsukahara R, Kobayashi S, Murofushi H, Uchiyama A, Murakami-Murofushi K, Koh E, Bandle RW, Byun H-S, Bittman R, Fan D, Murph M, Mills GB, Tigyi G. Carba Analogs of Cyclic Phosphatidic Acid Are Selective Inhibitors of Autotaxin and Cancer Cell Invasion and Metastasis. J Biol Chem. 2006;281:22786–22793. doi: 10.1074/jbc.M512486200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang G, Xu Y, Fujiwara Y, Tsukahara T, Tsukahara R, Gajewiak J, Tigyi G, Prestwich GD. alpha-Substituted Phosphonate Analoques of Lysophosphatidic Acid (LPA) Selectively Inhibit Production and Action of LPA. ChemMedChem. 2007;2:679–690. doi: 10.1002/cmdc.200600280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Meeteren LA, Brinkmann V, Saulnier-Blache J-S, Lynch KR, Moolenaar WH. Anticancer activity of FTY720: Phosphorylated FTY720 inhibits autotaxin, a metastasis-enhancing and angiogenic lysophospholipase D. Cancer Lett. 2008;266:203–208. doi: 10.1016/j.canlet.2008.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferguson CG, Bigman CS, Richardson RD, van Meeteran LA, Moolenaar WH, Prestwich GD. Fluorogenic Phospholipid Substrate to Detect Lysophospholipase D/Autotaxin Activity. Org Lett. 2006;8:2023–2026. doi: 10.1021/ol060414i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erukulla RK, Byun H-S, Locke DC, Bittman R. Stereospecific and regiospecific ring opening of glycidol with primary and secondary alcohols mediated by diisobutylaluminium hydride. J Chem Soc Perkin Trans 1. 1995;18:2199–2200. [Google Scholar]
- 35.Kley J, Unger C, Massing U. Synthesis of Isosteric Phosphono Analogs of Biologically Active Alkylphosphocholines. Monatshefte fur Chemie. 1998;129:173–185. [Google Scholar]
- 36.Garrido-Hernandez H, Moon KD, Geahlen RL, Borch RF. Design and Synthesis of Phosphotyrosine Peptidomimetic Prodrugs. J Med Chem. 2006;49:3368–3376. doi: 10.1021/jm060142p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen L, Wu L, Otaka A, Smyth MS, Roller PP, Burke TR, Jr, Hertog Jd, Zhang Z-Y. Why Is Phosphonodifluoromethyl Phenylalanine a More Potent Inhibitory Moiety Than Phosphonomethyl Phenylalanine toward Protein-Tyrosine Phosphatases? Biochem Biophys Res Commun. 1995;216:976–984. doi: 10.1006/bbrc.1995.2716. [DOI] [PubMed] [Google Scholar]