

Abstract
Mutations in TRPC6, a member of the transient repeptor potential (TRP) superfamily of non-selective cation channels, have been identified as causing a familial form of focal segmental glomerulosclerosis, a disease characterized by proteinuria and progressive renal failure. Here we review the effect of disease-associated mutations on TRPC6 function and place TRPC6 within the context of other proteins central to glomerular and podocyte function. Finally, the known roles of TRPC6 in the kidney and other organ systems are used as a framework to discuss possible signaling pathways that TRPC6 may modulate during normal glomerular function and in disease states.
Keywords: focal segmental glomerulosclerosis, transient receptor potential, podocyte, calcium
Review of the glomerulus
The glomerulus is the individual blood filtration unit within the kidney and is the first component involved in regulating the composition of urine. An intricate structure, it consists of multiple capillary loops suspended within a fluid filled capsule, the urinary space (see Figure 1A). The capillary wall’s three major components - a fenestrated endothelium, a specialized basement membrane, and an outer layer of glomerular epithelial cells, also known as podocytes- together constitute a highly selective filtration barrier (Figure 1B), representing a low resistance for the passage of water and small molecules into the urinary space, while being highly resistant to the passage of serum proteins, such as albumin.
Figure 1.
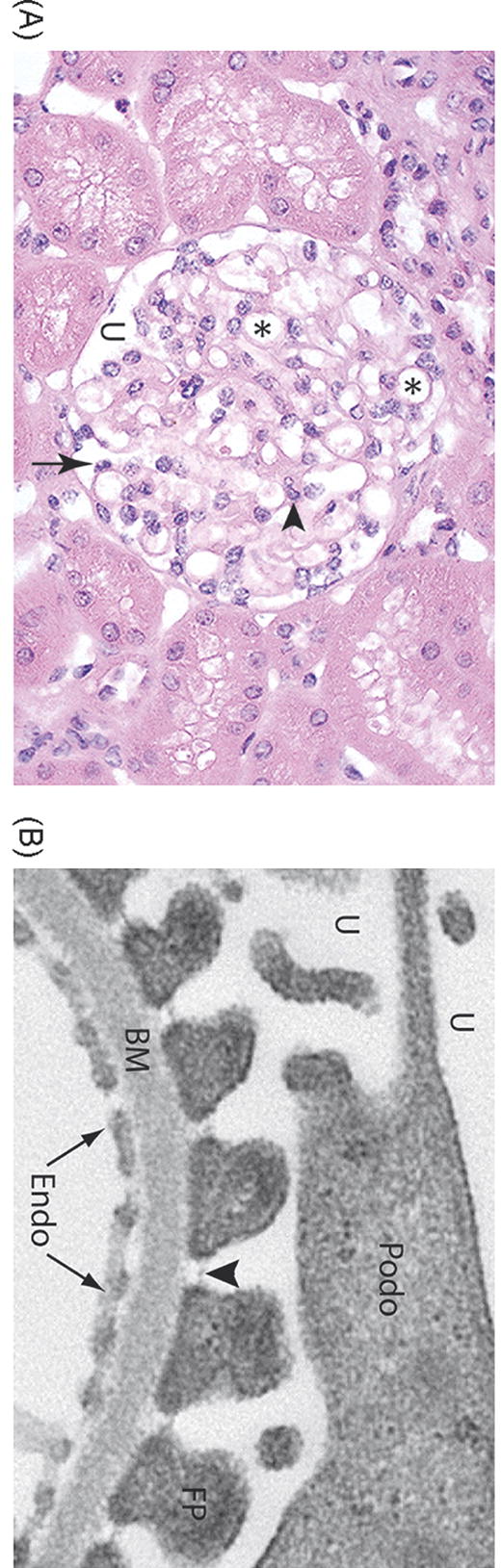
Structure of the glomerulus. A. Light micrograph of a normal glomerulus. The glomerulus consists of a tuft of capillary loops (*), which are situated within Bowman’s capsule. The capillaries are covered on the urinary space (U) by specialized glomerular epithelial cells, also known as podocytes (arrow). Mesangial cells and their associated matrix (arrowhead) provide further structural support. B. Electron microscopy of the glomerular filtration barrier of the kidney. The capillary lumen is lined by fenestrated endothelial cells (Endo), which sit atop a specialized basement membrane (BM). Podocytes (Podo) project interdigitating foot processes (FP) which attach to the basement membrane. The intercellular adhesion complex formed between adjacent foot processes is termed the slit diaphragm (arrowhead).
Disruption of the glomerular filtration barrier is a common outcome of many kidney diseases, including diabetic nephropathy, IgA nephritis, lupus nephritis and focal segmental glomerulosclerosis (FSGS). Proteinuria (the presence of protein in the urine) is a hallmark of loss of the glomerulus’ permselectivity. In many cases, persistent dysfunction of the glomerular filtration barrier leads to progressive renal failure[1]. Together, glomerular diseases represent a major cause of renal failure and need for dialysis or kidney transplantation.
Both because of its clinical importance and because of its rather unique biological properties, understanding the glomerular filtration barrier has been a longstanding interest of the nephrology community. However, the complex architecture of the structure, requiring the interplay of several cell types and their specialized matrix, has limited in vitro approaches to understanding the filtration barrier.
Over the last decade, studies of rare familial forms of glomerular diseases have helped identify key genes involved in establishing and maintaining the glomerular filtration barrier[2]. Interestingly, several of the proteins affected in these diseases are expressed by podocytes. Podocytes are characterized by several elongated cytoplasmic elements termed major processes, from which multiple actin-based projections, termed minor processes or foot processes, sprout. Together, these foot processes create an intricate branched structure that completely encircles the capillary loops. In addition to binding to the basement membrane, they form a network of lateral connections with other foot processes through specialized cell-cell contacts termed the slit diaphragm (Figure 1B). The slit diaphragm appears to be a specialized cell-cell contact with a critical role in the filtration barrier’s size and charge selectivity[3]. Coupled with the long-standing observation that foot process effacement (the loss of the podocytes’ intricate architecture and slit diaphragm) occurrs in many glomerular diseases characterized by proteinuria, the genetic studies have thrust the podocyte into the spotlight of glomerular research[4–6].
TRPC6 mutations as an etiology of familial focal segmental glomerulosclerosis
Winn et al [7] reported the identification of a mutation in TRPC6 in a large family with an autosomal dominant pattern of adult onset FSGS, a clinicopathologic pattern of kidney injury characterized by proteinuria and a signature glomerular lesion under microscopic examination[8]. A total of six different families have now been identified with distinct mutations in the TRPC6 gene [7, 9], with several potential additional mutations being evaluated (our unpublished data). All show a dominant mode of inheritance with adult onset of disease and variable penetrance.
TRPC6 is a member of the large transient receptor potential superfamily of non-selective cation channels (reviewed in [10, 11]). This superfamily consists of a group of six transmembrane domain-containing ion channels and has been subdivided into six subfamilies, including the classical TRP or TRPC proteins. TRPC proteins contain several amino-terminal ankrin repeats prior to the first transmembrane domain, a short sequence termed the TRP box, of unknown function, in the carboxy-terminal cytoplasmic domain, and potential coiled-coil structures in both the amino- and carboxy-sequences (see Figure 2). Within the TRPC subfamily, TRPC3, 6 and 7 have been grouped together based on sequence similarity, the ability to heteromerize with each other, and their responsiveness to diacylglycerol (DAG)[12]. TRPC6 appears to be a receptor-operated channel, leading to the influx of calcium (possibly directly or indirectly[13, 14]) in response to phospholipase C (PLC)-mediated signals. Although all TRPC members appear to be PLC-dependent, TRPC 3, 6 and 7 can also be directly activated by DAG[15, 16].
Figure 2.
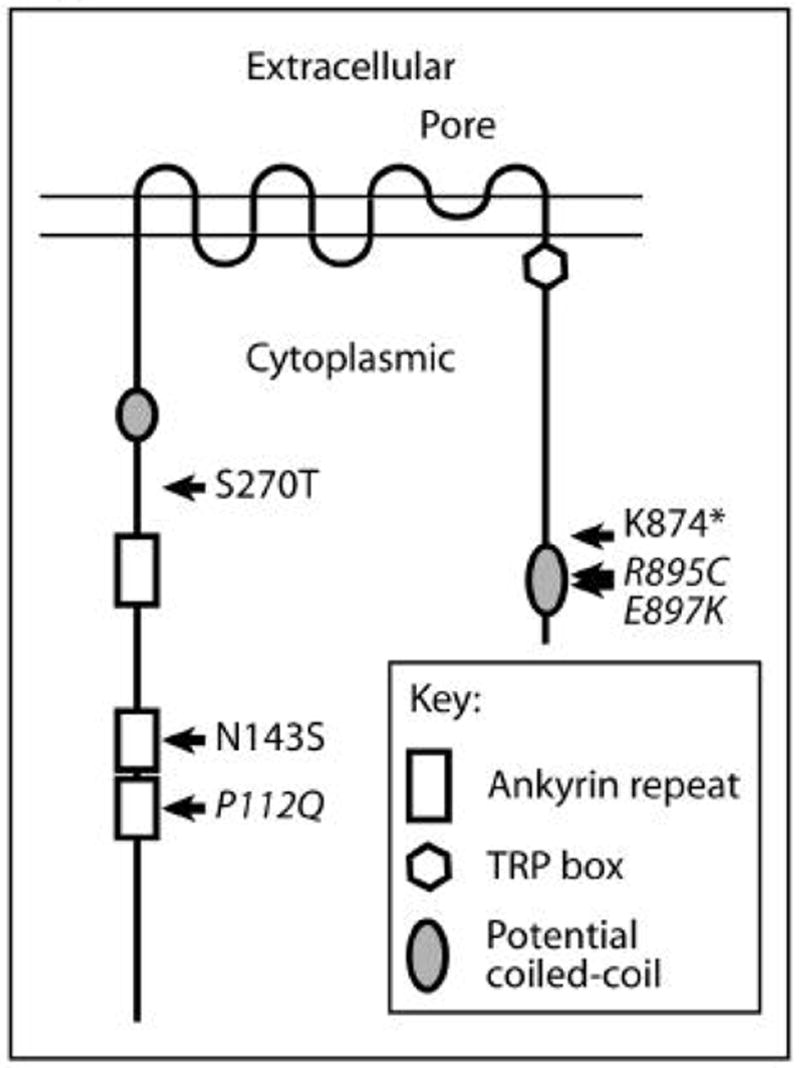
Schematic representation of TRPC6. The amino-terminal cytoplasmic sequence contains several ankyrin repeats and a potential coiled-coil region. TRP channels have six transmembrane domains, with a putative pore structure between the fifth and sixth transmembrane domains. The carboxy-terminal cytoplasmic domain contains a conserved sequence found in TRPC, TRPV and TRPM proteins, termed the TRP box, and a potential coiled-coil sequence. The function of the TRP box remains to be elucidated. The location and identity of mutations associated with familial forms of FSGS are indicated. Mutations which have been reported to alter channel activity are presented in italics.
To date, the FSGS-associated TRPC6 mutations are located within either the amino- or carboxy-terminal cytoplasmic domains of the protein (see Figure 2). With the exception of one mutation leading to truncation of the last 58 amino acids, all are point mutations. Two of the amino-terminal mutations lie within the ankrin repeats. The carboxy terminal 58 amino acids, which are affected by the known carboxy-terminal mutations, may include a coiled-coil region.
The effects of FSGS-associated mutations on TRPC6 activity are still poorly understood. None of the mutations appear to disrupt channel activity[7, 9]. Interestingly, the P112Q mutation leads to both increased amplitude and duration of calcium influx when stimulated in a heterologous overexpression system[7], while two of the carboxy-terminal mutations, R895C and E897K, also demonstrated increased current amplitude upon activation[9]. The remaining three mutations show current amplitudes comparable to that of the wild-type channel. Basal, unstimulated channel activity does not appear to be substantially altered by any of the mutations. It would appear that the FSGS-associated mutations represent gain-of-function alleles. Although this will require further study, two additional findings are consistent with this conclusion: (i) TRPC6-deficient mice have not been reported to have any obvious glomerular phenotype[17], and (ii) transient overexpression of wild-type TRPC6 in murine glomeruli leads to proteinuria (J. Reiser, C. Moeller, personal communication).
How might FSGS-associated mutations lead to increased TRPC6 channel activity? The gating of TRPC channels is still poorly understood. Several members of the TRPC family, including TRPC6, appear to be translocated from a subcellular compartment to the cell surface upon stimulation of Gq protein coupled receptors or receptor tyrosine kinases, and this likely plays some role in regulating their activity[12, 18–22]. In addition to being translocated to the cell surface, TRPC6 is also rapidly internalized upon inhibition of the muscarinic receptor by atropine[19]. Winn et al demonstrated that expression of the P112Q TRPC6 mutant at the cell surface was increased compared to wild-type TRPC6[7]. Although surface expression after cell stimulation was not assessed, it is intriguing to speculate that altered subcellular trafficking might explain the mutant’s enhanced channel activity. The mechanism whereby the mutation could alter surface expression also remains unclear, and one could postulate either a loss of effective intracellular sequestration or an inability to effectively recycle protein from the cell surface.
TRPC6 expression in the kidney
TRPC6 is expressed widely in the kidney, having been reported in the microcirculation, all three cell types within the glomerulus (capillary endothelial cells, mesangial cells, and podocytes), and the tubulointerstitial compartment[7, 9, 23–25]. In most of these cells, it is expressed in conjunction with several other TRPC members, including one of its potential binding partners, TRPC3.
To begin to assess the potential role of TRPC6 in non-hereditary glomerular disease, Reiser and colleagues have investigated the expression of TRPC6 in various glomerular diseases (J. Reiser, C. Moeller; personal communication). TRPC6 expression within the glomerulus, and podocytes specifically, is increased in several diseases, including minimal change disease, FSGS and membranous nephropathy. Increases in expression also occur in animal models of membranous nephropathy and in vitro in podocytes exposed to activated complement. In the cell culture system, the increased TRPC6 expression was associated with increased calcium influx in response to a DAG-analog, a known activator of TRPC6. Finally, as noted above, transient overexpression of TRPC6 in glomeruli leads to an increase in proteinuria in mice, arguing for more than just a correlation between proteinuric disease and TRPC6 expression (J. Reiser, C. Moeller; personal communication).
Although TRPC6 has a broad expression pattern, an initial bias within the field has been to focus on TRPC6 within the podocyte. Previous studies of both familial forms of FSGS and murine models of this glomerular disease have largely implicated podocyte dysfunction as a common event in the disease pathology[4–6]. Podocyte dysfunction appears to be central to the disease pathogenesis both when mutations affect proteins whose expression is largely restricted to podocytes (e.g. nephrin, podocin) and when they affect proteins with a broad expression pattern (e.g. α-actinin-4). Within podocytes, TRPC6 appears to localize within both major processes and foot processes by immunogold labeling, and at least some TRPC6 localizes to the slit diaphragm[9]. Two additional pieces of data suggest TRPC6 may function at the slit diaphragm: (i) when overexpressed in cultured podocytes, TRPC6 colocalizes with nephrin, podocin, and CD2AP, all proteins previous reported to localize to the slit diaphragm; and (ii) nephrin and podocin can be co-immunoprecipitated with TRPC6 from podocytes grown in culture[9].
Taken together, there is some circumstantial evidence to suggest that it is the abnormal function of TRPC6 within the podocyte that ultimately leads to disease in families with FSGS-associated TRPC6 mutations. Furthermore, abnormal TRPC6 function may also play a role in non-hereditary forms of glomerular disease. Ultimately, cell-type specific TRPC6 mutant “knock-in” or transgenic animals will need to be generated to answer these questions definitively.
TRPC6: functions beyond the kidney
TRPC6 has been of interest to investigators studying smooth muscle (reviewed in [26]) as well as axonal guidance[27] before its implication in glomerular function. TRPC6 is expressed in multiple organs, with the highest levels of transcript detected in lungs[28]. It is expressed in both pulmonary and vascular smooth muscle cells. Evidence suggests that TRPC6 is activated downstream of the α1-adrenergic receptor[29] and the vasopressin receptor[30], and it is believed to be important in mediating vascular and airway tone[26]. TRPC6 has been found to be upregulated in pulmonary artery smooth muscle cells in primary pulmonary hypertension and in response to hypoxia[31, 32], a response that appears to be mediated by HIF-1[33].
Available analysis of TRPC6-deficient mice has focused on its role in regulating smooth muscle tone. Interestingly, the animals were found to have hypertension and airway hyperreactivity, contrary to what had been hypothesized [17]. This appears to be due to a compensatory upregulation of TRPC3 expression, leading to higher basal smooth muscle tone. At least partially due to differences in their glycosylation patterns, TRPC3 has greater constitutive channel activity than TRPC6[34], thus explaining how loss of TRPC6 might paradoxically lead to enhanced baseline vascular tone.
Several of the TRPC channels have been shown to play a role in nerve growth cone guidance and neurite extension, including XTRPC1[22, 35], TRPC3[27], and TRPC5[36]. By comparison, the importance of TRPC6 is less clear: overexpression of a TRPC6 dominant negative construct inhibits brain-derived neurotrophic factor mediated growth cone turning, but may do so by interfering with TRPC3 function[27]. Growth cone guidance in neurons from TRPC6-deficient mice has not yet been addressed.
Potential regulators of TRPC6 in the glomerulus
The genetic evidence implicating TRPC6 in certain familial forms of FSGS is robust, and circumstantial evidence suggests that TRPC6 plays an important role in the podocyte. On a molecular level, though, the signaling pathways and cellular functions altered by the FSGS-associated TRPC6 mutations remain unknown. Possibilities abound, and insights into this aspect of TRPC6 function will likely be forthcoming in the near future. For the time being, we are left to speculate.
The slit diaphragm, the specialized cell-cell contact between podocytes, is a critical component of the glomerular filtration barrier[3]. Over a half dozen proteins have been shown to specifically localize to the slit diaphragm[2]. In many cases, loss of one of these slit diaphragm proteins leads to proteinuria and glomerular dysfunction. For instance, disruption of either nephrin[37] or podocin[38] has been shown to lead to early onset, or congenital, nephrotic syndrome. Besides providing a specialized cell-cell contact, and presumably contributing to the permselectivity of the glomerular filration barrier, the slit diaphragm is also believed to act as a signaling platform[39, 40]. Nephrin is felt to be a key component of this platform[41]. Clustering of nephrin leads to the activation of multiple downstream signaling pathways, including activation of Src family kinase Fyn[42–45], and the PI3K/Akt pathway[46]. As mentioned above, nephrin associates with TRPC6 in podocytes[9]. Furthermore, Fyn has been reported to bind to TRPC6[47], and Src mediated phosphorylation of TRPC3 is critical for channel activation[48]. Finally, TRPC6 has been shown to bind to phosphatidylinositol 3,4,5-trisphosphate (PIP(3)), a lipid product of PI3K, and PIP(3)-induced calcium influx can be modulated by altering the expression level of TRPC6[49]. It is thus tempting to speculate that TRPC6 is a component and downstream target of the nephrin signaling platform involved in monitoring the integrity of the slit diaphragm in podocytes.
Alternatively, numerous G protein-coupled receptors, potential activators of TRPC channels, have been implicated in regulating glomerular perfusion and filtration (reviewed in [6, 50]). The angiotensin receptors, in particular, are felt to play a central role in regulating glomerular filtration pressures, and in so modulating proteinuria[51]. Rats modestly overexpressing the angiotensin II type 1 receptor (AT1) on podocytes develop proteinuria and pathologic lesions characteristic of FSGS[52]. The P112Q mutation has been shown to induce higher and more sustained calcium influx than wild-type TRPC6 upon stimulation of AT1 when co-expressed in HEK cells[7]. It is not unreasonable, therefore, to postulate that it is enhanced calcium influx downstream of AT1 that leads to FSGS in individuals carrying TRPC6 mutations. Along these lines, it would be interesting to discover if TRPC6-deficient mice are protected from renal damage caused by an overactive renin-angiotensin system.
VEGF is another potential activator of TRPC channels. VEGF has recently been shown to play a critical role in maintaining the structure and function of the glomerulus[53]. Loss of even one of the VEGF-A alleles in podocytes leads to glomerular capillary endothelial cell dysfunction and proteinuria, while overexpression of the VEGF-164 isoform by podocytes leads to collapsing glomerulopathy and renal failure[54]. In vitro, VEGF-A and -C appear to enhance podocyte survival[55, 56]. TRPC channels have been implicated in mediating the calcium influx into endothelial cells, and changes in vascular permeability, seen in response to VEGF[57–59]. The ion channels stimulated by VEGF in microvascular endothelial cells share characteristics with TRPC family members[57]. TRPC6 and TRPC3 are capable of inducing calcium influx downstream of VEGFR2 when coexpressed in tissue culture[57, 59], while other studies have suggested TRPC1 as an important mediator of VEGF-induced calcium influx[58]. It is therefore reasonable to speculate that if glomerular function is disrupted by even modest changes in the level of VEGF, alterations in signaling downstream of VEGF receptors might similarly lead to glomerular disease.
Based on their location within the glomerulus, podocytes are subject to significant hydraulic forces[60]. There is substantial transmural pressure forcing fluid from the capillary lumen into the urinary space, with a resulting force tending to stretch and lift the podocyte off of the basement membrane. It has been postulated that podocytes are able to alter the filtration characteristics of the glomerulus by adjusting the total surface area between its processes through which the filtrate must pass[61, 62]. In addition, fluid flow within the urinary space of the glomerulus toward the proximal tubule will subject podocytes to shear stress. Podocytes have been found to respond to both stretch[63, 64] and shear stress[65]. While the mechanosensor(s) in podocytes remain unknown, multiple TRP family members have been implicated in this process in other systems (reviewed in [66]). Perhaps TRPC6 plays a similar role in podocytes? Could flow or stretch be sensed through the slit diaphragm by TRPC6, in conjunction with podocin, whose homologue, mec-2, is known to function in mechanosensation in C. elegans[67–69]? Alternatively, might TRPC6 function in sensing flow through the urinary space in a manner analagous to TRPP2 sensing flow in renal tubules[70]? The possibility of TRPC6 being involved in sensing, and thereby perhaps regulating, glomerular filtration is particularly appealing as glomerular hyperfiltration over time is thought to predispose to glomerular dysfunction and lesions of secondary FSGS[8].
What lies downstream of TRPC6?
Identifying the physiologic signals which regulate TRPC6 activity in glomeruli is a clear priority. Discovering the consequences of TRPC6 activation on glomerular function and how abnormal TRPC6 activity leads to the pathological lesions of FSGS are of equal importance.
What is known about the effects of activating TRPC channels in non-renal systems? As discussed above, activation of TRPC channels have been shown to be involved in smooth muscle contraction, neurite extension and growth cone guidance, and changes in vascular permeability. In the case of vascular smooth muscle contraction, TRPC activation leads to depolarization, activation of voltage-gated L-type Ca2+ channels, a rise in intracellular calcium and subsequent activation of the actin-myosin contractile apparatus (reviewed in [71]). In endothelial cells, increased intracellular calcium leads to the activation of PKCα and the Ca2+/calmodulin(CaM)-dependent myosin light chain kinase. These in turn enhance endothelial cell contraction and mediate disassembly of VE-cadherin cell-cell junctions, together allowing for increased fluid flow between cells[72]. Loss of TRPC4 limits thrombin-induced endothelial cell retraction and stress fiber formation, and prevents thrombin-mediated increases in microvascular permeability[73]. The situation in growth cone guidance is more complex (reviewed in [74]): (i) Channels in addition to TRPCs are likely also involved in modulating intracellular calcium levels. (ii) Calcium influx can lead to either growth cone repulsion or attraction, possibly based on the amplitude of calcium influx and the relationship of local and global intracellular calcium concentrations. (iii) Multiple distinct proteins have been shown to modulate events downstream of calcium influx, some of which appear to be antagonistic. Among these are regulators of actin bundling (α-actinin, fodrin), Ca2+/CaM-dependent protein kinases (CaMK), calcineurin (a Ca2+/CaM-dependent phosphatase), protein kinase C, Rho-family GTPases, and the calcium-dependent protease, calpain[74]. While it is not known which of these are modulated specifically in response to TRPC-activation, it should be noted that TRPC6 has been reported to bind α-actinin-1 and -4, α-fodrin[75], PKC, and calcineurin[76]. Based on results from these disparate systems, a common role for TRPC6 may be regulation of the actin-based cytoskeleton.
The possibilities that TRPC6 functions as a modulator of the actin cytoskeleton is particularly intriguing, as a central role for the actin cytoskeleton has emerged in maintaining podocyte structure and function (reviewed in [64, 77]). Foot processes contain a central actin bundle as well as cortical actin[78]. In addition to providing the structural framework for the foot processes, the actin cytoskeleton is also critical for effective integrin- and dystroglycan-mediated contact to the basement membrane and for stabilizing the components of the slit diaphragm (reviewed in [77]). Mutations is α-actinin-4 which alter the protein’s actin binding affinity and disrupt normal actin architecture and dynamics[79, 80] are a cause of adult-onset FSGS[81, 82], a phenotype that is mimicked in mice bearing a similar mutation[80], lacking α-actinin-4[83], or overexpressing the mutant protein specifically in podocytes[84]. In addition, integrin-mediated cell-matrix adhesion is compromised in α-actinin-4 deficient podocytes (our unpublished data). Nck, which binds to phosphorylated nephrin and is required for proper podocyte function in vivo, appears to be involved in mediating actin polymerization and reorganization[44, 85]. Synaptopodin, another actin-bundling protein important for regulating Rho-mediated actin reorganization[86], is critical for both dendritic spine formation[87] and for proper podocyte function[88, 89]. Rho, in turn, is believed to be critical for proper podocyte function as mice deficient in the Rho-inactivator Rho GDIα develop proteinuria and renal failure[90]. Finally, integrin linked kinase (ILK), deficiency of which in podocytes leads to progressive proteinuria and renal failure[91, 92], can complex with α-actinin-4[91] and modulate the podocyte cytoskeleton in vitro[93]. Taken as a whole, there is a wealth of evidence suggesting that persistent regulation of the podocyte actin cytoskeleton is critical for maintenance of the glomerular filtration barrier.
Preliminary evidence suggests that there is a reciprocal relationship between the actin cytoskeleton and TRPC6 in podocytes (C. Moeller and J. Reiser; personal communication). When the actin cytoskeleton is depolymerized in podocytes by treatment with cytochalasin D, TRPC6 was found to relocate to areas of actin aggregates. In contrast, TRPC3 is internalized in HEK cells treated with calyculin-A, but not in cells treated with cytochalasin[94]. Overexpression of TRPC6 in differentiated podocytes in vitro disrupts the formation of stress fibers normally present in these cells (C. Moeller, J. Reiser; personal communication). Further investigation is clearly needed to build upon these intriguing observations, including assessment of the ability of TRPC6 to alter the actin-myosin based contractile machinery in podocytes.
Proteinuria is often induced by alterations in the slit diaphragm[2]. The slit diaphragm has been compared to a modified adherens junction, containing P-cadherin, alpha- and beta-catenins, ZO-1[95] and the cadherin superfamily members Fat1[96] and cadherin-5[97]. It is tempting to suggest, then, that activation of TRPC6 may lead to calcium-dependent phosphorylation and disassembly of the slit diaphragm with resulting development of proteinuria, much as calcium-dependent disassembly of VE-cadherin based endothelial cell-cell junctions is thought to increased vascular permeability (reviewed in [72]). Experiments shedding light onto this possibility are eagerly awaited.
Conclusion
The list of TRPs involved in various renal diseases has expanded over the years, with TRPC6 now joining the company of TRPP2 (polycystin 2), TRPV5, and TRPM6 (reviewed in [98–101]). Members of this superfamily are now implicated in the maintenance and function of the glomerular filtration barrier, tubular architecture and proliferation, and tubular transport. As is so often the case, identifying a genetic lesion involved in a hereditary disease is only the first, though critical, step in furthering our understanding of pathophysiology. Exciting times are surely ahead as we delve into the details of TRPC6’s role in maintaining the glomerular filtration barrier, and when we look back at this review in time, we will surely be struck by what Thomas Huxley termed “the great tragedy of science - the slaying of a beautiful hypothesis by an ugly fact.”
Acknowledgments
M.P. is supported by grants from the NIH and the American Heart Association. J.S. has been supported by NIH training grants T32 DK07527 and 1F32DK074308-01. We thank Jochen Reiser and Clemens Moeller for sharing unpublished data, and members of the Pollak lab for many stimulating discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tryggvason K, Pettersson E. Causes and consequences of proteinuria: the kidney filtration barrier and progressive renal failure. J Intern Med. 2003;254:216–224. doi: 10.1046/j.1365-2796.2003.01207.x. [DOI] [PubMed] [Google Scholar]
- 2.Tryggvason K, Patrakka J, Wartiovaara J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med. 2006;354:1387–1401. doi: 10.1056/NEJMra052131. [DOI] [PubMed] [Google Scholar]
- 3.Tryggvason K, Wartiovaara J. How does the kidney filter plasma? Physiology (Bethesda) 2005;20:96–101. doi: 10.1152/physiol.00045.2004. [DOI] [PubMed] [Google Scholar]
- 4.Barisoni L, Mundel P. Podocyte biology and the emerging understanding of podocyte diseases. Am J Nephrol. 2003;23:353–360. doi: 10.1159/000072917. [DOI] [PubMed] [Google Scholar]
- 5.Ly J, Alexander M, Quaggin SE. A podocentric view of nephrology. Curr Opin Nephrol Hypertens. 2004;13:299–305. doi: 10.1097/00041552-200405000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 7.Winn MP, Conlon PJ, Lynn KL, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 8.Meyrier A. Mechanisms of disease: focal segmental glomerulosclerosis. Nat Clin Pract Nephrol. 2005;1:44–54. doi: 10.1038/ncpneph0025. [DOI] [PubMed] [Google Scholar]
- 9.Reiser J, Polu KR, Moller CC, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37:739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montell C. The TRP superfamily of cation channels. Sci STKE. 2005;re3:2005. doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- 11.Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- 12.Dietrich A, Kalwa H, Rost BR, et al. The diacylgylcerol-sensitive TRPC3/6/7 subfamily of cation channels: functional characterization and physiological relevance. Pflugers Arch. 2005;451:72–80. doi: 10.1007/s00424-005-1460-0. [DOI] [PubMed] [Google Scholar]
- 13.Eder P, Poteser M, Romanin C, et al. Na(+) entry and modulation of Na(+)/Ca(2+) exchange as a key mechanism of TRPC signaling. Pflugers Arch. 2005;451:99–104. doi: 10.1007/s00424-005-1434-2. [DOI] [PubMed] [Google Scholar]
- 14.Estacion M, Sinkins WG, Jones SW, et al. Human TRPC6 expressed in HEK 293 cells forms non-selective cation channels with limited Ca2+ permeability. J Physiol. 2006;572:359–377. doi: 10.1113/jphysiol.2005.103143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hofmann T, Obukhov AG, Schaefer M, et al. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 16.Okada T, Inoue R, Yamazaki K, et al. Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca(2+)-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J Biol Chem. 1999;274:27359–27370. doi: 10.1074/jbc.274.39.27359. [DOI] [PubMed] [Google Scholar]
- 17.Dietrich A, Mederos YSM, Gollasch M, et al. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol Cell Biol. 2005;25:6980–6989. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bezzerides VJ, Ramsey IS, Kotecha S, et al. Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol. 2004;6:709–720. doi: 10.1038/ncb1150. [DOI] [PubMed] [Google Scholar]
- 19.Cayouette S, Lussier MP, Mathieu EL, et al. Exocytotic insertion of TRPC6 channel into the plasma membrane upon Gq protein-coupled receptor activation. J Biol Chem. 2004;279:7241–7246. doi: 10.1074/jbc.M312042200. [DOI] [PubMed] [Google Scholar]
- 20.Smyth JT, Lemonnier L, Vazquez G, et al. Dissociation of regulated trafficking of TRPC3 channels to the plasma membrane from their activation by phospholipase C. J Biol Chem. 2006;281:11712–11720. doi: 10.1074/jbc.M510541200. [DOI] [PubMed] [Google Scholar]
- 21.van Rossum DB, Patterson RL, Sharma S, et al. Phospholipase Cgamma1 controls surface expression of TRPC3 through an intermolecular PH domain. Nature. 2005;434:99–104. doi: 10.1038/nature03340. [DOI] [PubMed] [Google Scholar]
- 22.Wang GX, Poo MM. Requirement of TRPC channels in netrin-1-induced chemotropic turning of nerve growth cones. Nature. 2005;434:898–904. doi: 10.1038/nature03478. [DOI] [PubMed] [Google Scholar]
- 23.Facemire CS, Mohler PJ, Arendshorst WJ. Expression and relative abundance of short transient receptor potential channels in the rat renal microcirculation. Am J Physiol Renal Physiol. 2004;286:F546–551. doi: 10.1152/ajprenal.00338.2003. [DOI] [PubMed] [Google Scholar]
- 24.Goel M, Sinkins WG, Zuo CD, et al. Identification and localization of TRPC channels in the rat kidney. Am J Physiol Renal Physiol. 2006;290:F1241–1252. doi: 10.1152/ajprenal.00376.2005. [DOI] [PubMed] [Google Scholar]
- 25.Sours S, Du J, Chu S, et al. Expression of canonical transient receptor potential (TRPC) proteins in human glomerular mesangial cells. Am J Physiol Renal Physiol. 2006;290:F1507–1515. doi: 10.1152/ajprenal.00268.2005. [DOI] [PubMed] [Google Scholar]
- 26.Dietrich A, Chubanov V, Kalwa H, et al. Cation channels of the transient receptor potential superfamily: Their role in physiological and pathophysiological processes of smooth muscle cells. Pharmacol Ther. 2006 doi: 10.1016/j.pharmthera.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Jia YC, Cui K, et al. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature. 2005;434:894–898. doi: 10.1038/nature03477. [DOI] [PubMed] [Google Scholar]
- 28.Boulay G, Zhu X, Peyton M, et al. Cloning and expression of a novel mammalian homolog of Drosophila transient receptor potential (Trp) involved in calcium entry secondary to activation of receptors coupled by the Gq class of G protein. J Biol Chem. 1997;272:29672–29680. doi: 10.1074/jbc.272.47.29672. [DOI] [PubMed] [Google Scholar]
- 29.Inoue R, Okada T, Onoue H, et al. The transient receptor potential protein homologue TRP6 is the essential component of vascular alpha(1)-adrenoceptor-activated Ca(2+)-permeable cation channel. Circ Res. 2001;88:325–332. doi: 10.1161/01.res.88.3.325. [DOI] [PubMed] [Google Scholar]
- 30.Jung S, Strotmann R, Schultz G, et al. TRPC6 is a candidate channel involved in receptor-stimulated cation currents in A7r5 smooth muscle cells. Am J Physiol Cell Physiol. 2002;282:C347–359. doi: 10.1152/ajpcell.00283.2001. [DOI] [PubMed] [Google Scholar]
- 31.Lin MJ, Leung GP, Zhang WM, et al. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res. 2004;95:496–505. doi: 10.1161/01.RES.0000138952.16382.ad. [DOI] [PubMed] [Google Scholar]
- 32.Yu Y, Fantozzi I, Remillard CV, et al. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci U S A. 2004;101:13861–13866. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Weigand L, Lu W, et al. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res. 2006;98:1528–1537. doi: 10.1161/01.RES.0000227551.68124.98. [DOI] [PubMed] [Google Scholar]
- 34.Dietrich A, Mederos y Schnitzler M, Emmel J, et al. N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J Biol Chem. 2003;278:47842–47852. doi: 10.1074/jbc.M302983200. [DOI] [PubMed] [Google Scholar]
- 35.Shim S, Goh EL, Ge S, et al. XTRPC1-dependent chemotropic guidance of neuronal growth cones. Nat Neurosci. 2005;8:730–735. doi: 10.1038/nn1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greka A, Navarro B, Oancea E, et al. TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat Neurosci. 2003;6:837–845. doi: 10.1038/nn1092. [DOI] [PubMed] [Google Scholar]
- 37.Kestila M, Lenkkeri U, Mannikko M, et al. Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- 38.Boute N, Gribouval O, Roselli S, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24:349–354. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 39.Benzing T. Signaling at the slit diaphragm. J Am Soc Nephrol. 2004;15:1382–1391. doi: 10.1097/01.asn.0000130167.30769.55. [DOI] [PubMed] [Google Scholar]
- 40.Huber TB, Benzing T. The slit diaphragm: a signaling platform to regulate podocyte function. Curr Opin Nephrol Hypertens. 2005;14:211–216. doi: 10.1097/01.mnh.0000165885.85803.a8. [DOI] [PubMed] [Google Scholar]
- 41.Mathieson PW. Nephrin sends us signals. Kidney Int. 2003;64:756–757. doi: 10.1046/j.1523-1755.2003.00141.x. [DOI] [PubMed] [Google Scholar]
- 42.Lahdenpera J, Kilpelainen P, Liu XL, et al. Clustering-induced tyrosine phosphorylation of nephrin by Src family kinases. Kidney Int. 2003;64:404–413. doi: 10.1046/j.1523-1755.2003.00097.x. [DOI] [PubMed] [Google Scholar]
- 43.Li H, Lemay S, Aoudjit L, et al. SRC-family kinase Fyn phosphorylates the cytoplasmic domain of nephrin and modulates its interaction with podocin. J Am Soc Nephrol. 2004;15:3006–3015. doi: 10.1097/01.ASN.0000146689.88078.80. [DOI] [PubMed] [Google Scholar]
- 44.Verma R, Kovari I, Soofi A, et al. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J Clin Invest. 2006;116:1346–1359. doi: 10.1172/JCI27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verma R, Wharram B, Kovari I, et al. Fyn binds to and phosphorylates the kidney slit diaphragm component Nephrin. J Biol Chem. 2003;278:20716–20723. doi: 10.1074/jbc.M301689200. [DOI] [PubMed] [Google Scholar]
- 46.Huber TB, Hartleben B, Kim J, et al. Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol Cell Biol. 2003;23:4917–4928. doi: 10.1128/MCB.23.14.4917-4928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hisatsune C, Kuroda Y, Nakamura K, et al. Regulation of TRPC6 channel activity by tyrosine phosphorylation. J Biol Chem. 2004;279:18887–18894. doi: 10.1074/jbc.M311274200. [DOI] [PubMed] [Google Scholar]
- 48.Kawasaki BT, Liao Y, Birnbaumer L. Role of Src in C3 transient receptor potential channel function and evidence for a heterogeneous makeup of receptor- and store-operated Ca2+ entry channels. Proc Natl Acad Sci U S A. 2006;103:335–340. doi: 10.1073/pnas.0508030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tseng PH, Lin HP, Hu H, et al. The canonical transient receptor potential 6 channel as a putative phosphatidylinositol 3,4,5-trisphosphate-sensitive calcium entry system. Biochemistry. 2004;43:11701–11708. doi: 10.1021/bi049349f. [DOI] [PubMed] [Google Scholar]
- 50.Winn MP, Daskalakis N, Spurney RF, et al. Unexpected role of TRPC6 channel in familial nephrotic syndrome: does it have clinical implications? J Am Soc Nephrol. 2006;17:378–387. doi: 10.1681/ASN.2005090962. [DOI] [PubMed] [Google Scholar]
- 51.Pavenstadt H. The charge for going by foot: modifying the surface of podocytes. Exp Nephrol. 1998;6:98–103. doi: 10.1159/000020511. [DOI] [PubMed] [Google Scholar]
- 52.Hoffmann S, Podlich D, Hahnel B, et al. Angiotensin II type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats. J Am Soc Nephrol. 2004;15:1475–1487. doi: 10.1097/01.asn.0000127988.42710.a7. [DOI] [PubMed] [Google Scholar]
- 53.Eremina V, Quaggin SE. The role of VEGF-A in glomerular development and function. Curr Opin Nephrol Hypertens. 2004;13:9–15. doi: 10.1097/00041552-200401000-00002. [DOI] [PubMed] [Google Scholar]
- 54.Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Foster RR, Satchell SC, Seckley J, et al. VEGF-C promotes survival in podocytes. Am J Physiol Renal Physiol. 2006;291:F196–207. doi: 10.1152/ajprenal.00431.2005. [DOI] [PubMed] [Google Scholar]
- 56.Guan F, Villegas G, Teichman J, et al. Autocrine VEGF-A system in podocytes regulates podocin and its interaction with CD2AP. Am J Physiol Renal Physiol. 2006;291:F422–428. doi: 10.1152/ajprenal.00448.2005. [DOI] [PubMed] [Google Scholar]
- 57.Cheng HW, James AF, Foster RR, et al. VEGF activates receptor-operated cation channels in human microvascular endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:1768–1776. doi: 10.1161/01.ATV.0000231518.86795.0f. [DOI] [PubMed] [Google Scholar]
- 58.Jho D, Mehta D, Ahmmed G, et al. Angiopoietin-1 opposes VEGF-induced increase in endothelial permeability by inhibiting TRPC1-dependent Ca2 influx. Circ Res. 2005;96:1282–1290. doi: 10.1161/01.RES.0000171894.03801.03. [DOI] [PubMed] [Google Scholar]
- 59.Pocock TM, Foster RR, Bates DO. Evidence of a role for TRPC channels in VEGF-mediated increased vascular permeability in vivo. Am J Physiol Heart Circ Physiol. 2004;286:H1015–1026. doi: 10.1152/ajpheart.00826.2003. [DOI] [PubMed] [Google Scholar]
- 60.Kriz W, Endlich K. Hypertrophy of podocytes: a mechanism to cope with increased glomerular capillary pressures? Kidney Int. 2005;67:373–374. doi: 10.1111/j.1523-1755.2005.00141.x. [DOI] [PubMed] [Google Scholar]
- 61.Andrews PM, Coffey AK. Cytoplasmic contractile elements in glomerular cells. Fed Proc. 1983;42:3046–3052. [PubMed] [Google Scholar]
- 62.Neal CR, Crook H, Bell E, et al. Three-dimensional reconstruction of glomeruli by electron microscopy reveals a distinct restrictive urinary subpodocyte space. J Am Soc Nephrol. 2005;16:1223–1235. doi: 10.1681/ASN.2004100822. [DOI] [PubMed] [Google Scholar]
- 63.Endlich N, Kress KR, Reiser J, et al. Podocytes respond to mechanical stress in vitro. J Am Soc Nephrol. 2001;12:413–422. doi: 10.1681/ASN.V123413. [DOI] [PubMed] [Google Scholar]
- 64.Endlich N, Endlich K. Stretch, tension and adhesion - adaptive mechanisms of the actin cytoskeleton in podocytes. Eur J Cell Biol. 2006;85:229–234. doi: 10.1016/j.ejcb.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 65.Friedrich C, Endlich N, Kriz W, et al. Podocytes are sensitive to fluid shear stress in vitro. Am J Physiol Renal Physiol. 2006;291:F856–865. doi: 10.1152/ajprenal.00196.2005. [DOI] [PubMed] [Google Scholar]
- 66.Lin SY, Corey DP. TRP channels in mechanosensation. Curr Opin Neurobiol. 2005;15:350–357. doi: 10.1016/j.conb.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 67.Goodman MB, Ernstrom GG, Chelur DS, et al. MEC-2 regulates C. elegans DEG/ENaC channels needed for mechanosensation. Nature. 2002;415:1039–1042. doi: 10.1038/4151039a. [DOI] [PubMed] [Google Scholar]
- 68.Huang M, Gu G, Ferguson EL, et al. A stomatin-like protein necessary for mechanosensation in C. elegans. Nature. 1995;378:292–295. doi: 10.1038/378292a0. [DOI] [PubMed] [Google Scholar]
- 69.O'Hagan R, Chalfie M, Goodman MB. The MEC-4 DEG/ENaC channel of Caenorhabditis elegans touch receptor neurons transduces mechanical signals. Nat Neurosci. 2005;8:43–50. doi: 10.1038/nn1362. [DOI] [PubMed] [Google Scholar]
- 70.Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 71.Gudermann T, Mederos y Schnitzler M, Dietrich A. Receptor-operated cation entry--more than esoteric terminology? Sci STKE. 2004;2004:pe35. doi: 10.1126/stke.2432004pe35. [DOI] [PubMed] [Google Scholar]
- 72.Tiruppathi C, Minshall RD, Paria BC, et al. Role of Ca2+ signaling in the regulation of endothelial permeability. Vascul Pharmacol. 2002;39:173–185. doi: 10.1016/s1537-1891(03)00007-7. [DOI] [PubMed] [Google Scholar]
- 73.Tiruppathi C, Freichel M, Vogel SM, et al. Impairment of store-operated Ca2+ entry in TRPC4(−/−) mice interferes with increase in lung microvascular permeability. Circ Res. 2002;91:70–76. doi: 10.1161/01.res.0000023391.40106.a8. [DOI] [PubMed] [Google Scholar]
- 74.Gomez TM, Zheng JQ. The molecular basis for calcium-dependent axon pathfinding. Nat Rev Neurosci. 2006;7:115–125. doi: 10.1038/nrn1844. [DOI] [PubMed] [Google Scholar]
- 75.Goel M, Sinkins W, Keightley A, et al. Proteomic analysis of TRPC5- and TRPC6-binding partners reveals interaction with the plasmalemmal Na(+)/K(+)-ATPase. Pflugers Arch. 2005;451:87–98. doi: 10.1007/s00424-005-1454-y. [DOI] [PubMed] [Google Scholar]
- 76.Kim JY, Saffen D. Activation of M1 muscarinic acetylcholine receptors stimulates the formation of a multiprotein complex centered on TRPC6 channels. J Biol Chem. 2005;280:32035–32047. doi: 10.1074/jbc.M500429200. [DOI] [PubMed] [Google Scholar]
- 77.Oh J, Reiser J, Mundel P. Dynamic (re)organization of the podocyte actin cytoskeleton in the nephrotic syndrome. Pediatr Nephrol. 2004;19:130–137. doi: 10.1007/s00467-003-1367-y. [DOI] [PubMed] [Google Scholar]
- 78.Ichimura K, Kurihara H, Sakai T. Actin filament organization of foot processes in rat podocytes. J Histochem Cytochem. 2003;51:1589–1600. doi: 10.1177/002215540305101203. [DOI] [PubMed] [Google Scholar]
- 79.Michaud JL, Chaisson KM, Parks RJ, et al. FSGS-associated alpha-actinin-4 (K256E) impairs cytoskeletal dynamics in podocytes. Kidney Int. 2006;70:1054–1061. doi: 10.1038/sj.ki.5001665. [DOI] [PubMed] [Google Scholar]
- 80.Yao J, Le TC, Kos CH, et al. Alpha-actinin-4-mediated FSGS: an inherited kidney disease caused by an aggregated and rapidly degraded cytoskeletal protein. PLoS Biol. 2004;2:e167. doi: 10.1371/journal.pbio.0020167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kaplan JM, Kim SH, North KN, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24:251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 82.Weins A, Kenlan P, Herbert S, et al. Mutational and Biological Analysis of alpha-actinin-4 in focal segmental glomerulosclerosis. J Am Soc Nephrol. 2005;16:3694–3701. doi: 10.1681/ASN.2005070706. [DOI] [PubMed] [Google Scholar]
- 83.Kos CH, Le TC, Sinha S, et al. Mice deficient in alpha-actinin-4 have severe glomerular disease. J Clin Invest. 2003;111:1683–1690. doi: 10.1172/JCI17988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Michaud JL, Lemieux LI, Dube M, et al. Focal and segmental glomerulosclerosis in mice with podocyte-specific expression of mutant alpha-actinin-4. J Am Soc Nephrol. 2003;14:1200–1211. doi: 10.1097/01.asn.0000059864.88610.5e. [DOI] [PubMed] [Google Scholar]
- 85.Jones N, Blasutig IM, Eremina V, et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature. 2006;440:818–823. doi: 10.1038/nature04662. [DOI] [PubMed] [Google Scholar]
- 86.Asanuma K, Yanagida-Asanuma E, Faul C, et al. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. 2006;8:485–491. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- 87.Deller T, Korte M, Chabanis S, et al. Synaptopodin-deficient mice lack a spine apparatus and show deficits in synaptic plasticity. Proc Natl Acad Sci U S A. 2003;100:10494–10499. doi: 10.1073/pnas.1832384100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Asanuma K, Kim K, Oh J, et al. Synaptopodin regulates the actin-bundling activity of alpha-actinin in an isoform-specific manner. J Clin Invest. 2005;115:1188–1198. doi: 10.1172/JCI23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huber TB, Kwoh C, Wu H, et al. Bigenic mouse models of focal segmental glomerulosclerosis involving pairwise interaction of CD2AP, Fyn, and synaptopodin. J Clin Invest. 2006;116:1337–1345. doi: 10.1172/JCI27400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Togawa A, Miyoshi J, Ishizaki H, et al. Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIalpha. Oncogene. 1999;18:5373–5380. doi: 10.1038/sj.onc.1202921. [DOI] [PubMed] [Google Scholar]
- 91.Dai C, Stolz DB, Bastacky SI, et al. Essential role of integrin-linked kinase in podocyte biology: Bridging the integrin and slit diaphragm signaling. J Am Soc Nephrol. 2006;17:2164–2175. doi: 10.1681/ASN.2006010033. [DOI] [PubMed] [Google Scholar]
- 92.El-Aouni C, Herbach N, Blattner SM, et al. Podocyte-specific deletion of integrin-linked kinase results in severe glomerular basement membrane alterations and progressive glomerulosclerosis. J Am Soc Nephrol. 2006;17:1334–1344. doi: 10.1681/ASN.2005090921. [DOI] [PubMed] [Google Scholar]
- 93.Yang Y, Guo L, Blattner SM, et al. Formation and phosphorylation of the PINCH-1-integrin linked kinase-alpha-parvin complex are important for regulation of renal glomerular podocyte adhesion, architecture, and survival. J Am Soc Nephrol. 2005;16:1966–1976. doi: 10.1681/ASN.2004121112. [DOI] [PubMed] [Google Scholar]
- 94.Lockwich T, Singh BB, Liu X, et al. Stabilization of cortical actin induces internalization of transient receptor potential 3 (Trp3)-associated caveolar Ca2+ signaling complex and loss of Ca2+ influx without disruption of Trp3-inositol trisphosphate receptor association. J Biol Chem. 2001;276:42401–42408. doi: 10.1074/jbc.M106956200. [DOI] [PubMed] [Google Scholar]
- 95.Reiser J, Kriz W, Kretzler M, et al. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol. 2000;11:1–8. doi: 10.1681/ASN.V1111. [DOI] [PubMed] [Google Scholar]
- 96.Yaoita E, Kurihara H, Yoshida Y, et al. Role of Fat1 in cell-cell contact formation of podocytes in puromycin aminonucleoside nephrosis and neonatal kidney. Kidney Int. 2005;68:542–551. doi: 10.1111/j.1523-1755.2005.00432.x. [DOI] [PubMed] [Google Scholar]
- 97.Cohen CD, Klingenhoff A, Boucherot A, et al. Comparative promoter analysis allows de novo identification of specialized cell junction-associated proteins. Proc Natl Acad Sci U S A. 2006;103:5682–5687. doi: 10.1073/pnas.0511257103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nauli SM, Zhou J. Polycystins and mechanosensation in renal and nodal cilia. Bioessays. 2004;26:844–856. doi: 10.1002/bies.20069. [DOI] [PubMed] [Google Scholar]
- 99.Giamarchi A, Padilla F, Coste B, et al. The versatile nature of the calcium-permeable cation channel TRPP2. EMBO Rep. 2006;7:787–793. doi: 10.1038/sj.embor.7400745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mene P. Transient receptor potential channels in the kidney: calcium signaling, transport and beyond. J Nephrol. 2006;19:21–29. [PubMed] [Google Scholar]
- 101.Thebault S, Hoenderop JG, Bindels RJ. Epithelial Ca2+ and Mg2+ channels in kidney disease. Adv Chronic Kidney Dis. 2006;13:110–117. doi: 10.1053/j.ackd.2006.01.002. [DOI] [PubMed] [Google Scholar]