

Abstract
Intranasal application of vesicular stomatitis virus (VSV) causes acute infection of the central nervous system (CNS). However, VSV encephalitis is not invariably fatal, suggesting that the CNS may contain a professional antigen-presenting cell (APC) capable of inducing or propagating a protective antiviral immune response. To examine this possibility, we first characterized the cellular elements that infiltrate the brain as well as the activation status of resident microglia in the brains of normal and transgenic mice acutely ablated of peripheral dendritic cells (DCs) in vivo. VSV encephalitis was characterized by a pronounced infiltrate of myeloid cells (CD45highCD11b+) and CD8+ T cells containing a subset that was specific for the immunodominant VSV nuclear protein epitope. This T cell response correlated temporally with a rapid and sustained upregulation of MHC class I expression on microglia, whereas class II expression was markedly delayed. Ablation of peripheral DCs profoundly inhibited the inflammatory response as well as infiltration of virus-specific CD8+ T cells. Unexpectedly, the VSV-induced interferon-gamma (IFN-γ) response in the CNS remained intact in DC-deficient mice. Thus, both the inflammatory and certain components of the adaptive primary antiviral immune response in the CNS are dependent on peripheral DCs in vivo.
Keywords: microglia, dendritic cells, viral encephalitis, vesicular stomatitis virus
INTRODUCTION
The central nervous system (CNS) has long been held as an immunologically privileged site (Galea, Bechmann, and Perry, 2007). Understanding neurological infections is critical to the treatment of several human diseases including HIV, several herpes viruses, measles, rabies, and possibly multiple sclerosis (Nair, Hunzeker, and Bonneau, 2007; Nelson, Soma, and Lavi, 2002; Ponomarev et al., 2005a). Most current research in mouse models focuses on experimental autoimmune encephalomyelitis (EAE) and persistent infections such as Theiler’s murine encephalomyelitis virus (TMEV), primarily with regard to development of treatments for multiple sclerosis. Although these studies provide insights into the general inflammatory and adaptive immune responses in the CNS, they do not accurately reflect the events during acute viral infection.
Several studies suggest that during CNS inflammation, activated DCs migrate to the cervical lymph nodes (Bailey et al., 2007; Dimier-Poisson et al., 2006; Hatterer et al., 2006; Plakhov et al., 1995; Schwob et al., 2001; Velge-Roussel et al., 2000), where they activate naïve lymphocytes, which then emigrate to the site of inflammation. The origin of these cells, whether peripherally-derived or brain-resident, is still contentious. Most research fails to convincingly demonstrate the presence of DC in naïve brain parenchyma (Lauterbach et al., 2006; Matyszak and Perry, 1996; Perry, 1998; Serafini et al., 2000), although they are readily detected in perivascular spaces, cerebrospinal fluid, and areas unprotected by the blood-brain barrier (BBB) (Bailey et al., 2007; Fischer and Reichmann, 2001; Karman et al., 2006; Lauterbach et al., 2006; Matyszak and Perry, 1996; McMenamin, 1999; Miller et al., 2007; Newman et al., 2005; Perry, 1998; Serafini et al., 2000; Serot et al., 2000; Serot et al., 1997; Serot et al., 1998). Only a handful of recent studies, such as those by Fabry et al. and Bulloch et al. (Bulloch et al., 2008; Karman et al., 2006), have demonstrated DCs in the naïve CNS parenchyma. These studies further indicate that either resident or infiltrating DCs provide APC function essential for propagation of innate and adaptive immunity in the CNS. It should be noted that identification of DCs in the CNS relies on a phenotypic rather than a functional definition for DCs; many of these studies also note that another population of CNS-resident cells may fulfil the role of APC.
Microglia are widely regarded as the most critical resident CNS cells with immunological capacity and represent approximately 10-20% of the brain parenchyma (Havenith, Askew, and Walker, 1998; Lawson et al., 1990; Rock et al., 2004; Santambrogio et al., 2001; Town, Nikolic, and Tan, 2005). Several studies imply that microglia are capable of acquiring APC capacity and may be able to initiate and/or propagate the adaptive immune response in the CNS (Fischer and Reichmann, 2001; Juedes and Ruddle, 2001; Mack et al., 2003; Persidsky et al., 1999; Ponomarev et al., 2005a; Ponomarev et al., 2005b; Shortman and Liu, 2002). Identification of the true APC in CNS infections is therefore a controversial area given the conflicting data for DCs and/or microglia as APCs.
In a previous study, we demonstrated that approximately one-third of mice acutely depleted of DC in vivo and infected peripherally with vesicular stomatitis virus (VSV) developed persistent brain infections (Ciavarra et al., 2006). These data imply a crucial role for peripheral DCs in mediating CNS immunity. To more directly evaluate this possibility, we induced viral encephalitis in all mice by a single intranasal application of VSV (Barna et al., 1996; Bi et al., 1995a; Huneycutt et al., 1994; Plakhov et al., 1995). The innate and adaptive antiviral immune response in the CNS was then characterized in normal mice and transgenic mice rendered deficient of peripheral DCs.
RESULTS
VSV encephalitis is characterized by a prominent mixed cellular infiltrate
We first phenotyped the cells recruited into the brain of mice following intranasal application of VSV. CB6F1 mice were infected with VSV and monitored for signs of illness. Mice became ill approximately 8 days post-infection, and brains were harvested for flow cytometric analysis at this time. Microglia were gated as CD45low/intCD11b+ cells (Figure 1, box in panels a-b) which distinguished them from resident or infiltrating CD45highCD11b+ macrophages (mΦ) and CD45highCD11b- lymphocytes. Microglia accounted for about 20% of cells recovered from normal, uninfected (mock-infected) mice and comprised approximately 90% of CD11b+ cells. In contrast, brains from mice infected with VSV contained a prominent population of CD45highCD11b+ mΦ and a smaller population of lymphocytes (CD45highCD11b-). Microglia isolated from virus-infected, but not mock-infected brains, expressed MHC class II molecules suggesting an activated state (panels c-d). Mock-infected mice contained only trace numbers of conventional (CD11c+PDCA-1-) and pDCs (CD11c+PDCA-1+), CD4+ and CD8+ T cells, whereas VSV induced infiltration of conventional CD11c+ DCs (panels e-f), CD4+ and CD8+ T cells (panels g-h) but few NK cells, B cells (panels k-l) and pDCs (panels e-f). A population of B cells (CD45R+) were noted in naïve and infected mice (panels e-f), but these cells did not expand with VSV infection. Staining with tetramers revealed a trace tetramer+-population in the mock infected brain that expanded following infection with VSV (panels i, j). Although this is not the optimal time for a CD8+ T cell response (see Figure 3), there was still an impressive difference in the number of CD8+VSV-N T cells present in the mock versus virus infected animals (140 versus 11,000 cells/brain, respectively, data not shown).
Figure 1. Intranasal application of VSV induces a vigorous mixed cellular infiltrate in the brain.
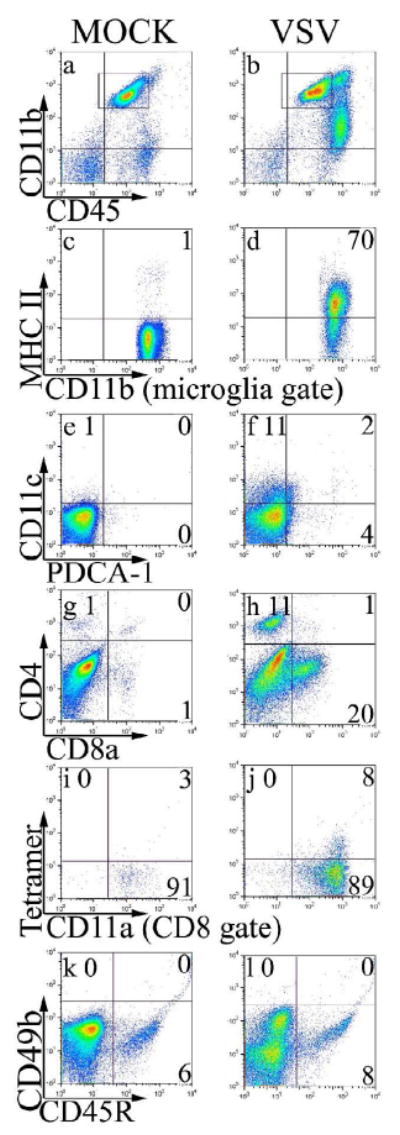
Mice were given either PBS (Mock) or intranasal VSV at 2×105 PFU (VSV). Eight days post-infection, leukocytes were isolated from the brain and the infiltrate characterized by flow cytometry. Microglia and infiltrating leukocytes were first identified by forward and side scatter profiles. Within this gate microglia were defined as CD11b+ and CD45low/int (box in panels a-b) and expression of MHC class II was evaluated on microglia-gated cells (panels c-d). To characterize other infiltrating cell types, we assessed gated leukocytes (forward and side scatter gate) for DCs (panels e-f) and T cell subsets (panels g-h). To identify VSV-N T cells, co-expression of CD11a and tetramers were assessed on gated CD8+ cells (panels i-j). NK cells and B cells were identified in the leukocyte gate as CD45highCD49b+ and CD45high CD45R+, respectively (panels k-l). This data is derived from the pooled brains of 4 mice per group.
Figure 3. Kinetics of T cell subset infiltration in the encephalitic brain.
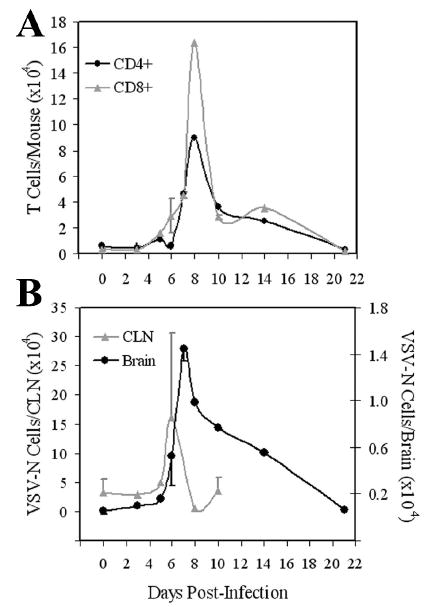
Mice were infected with VSV and at the indicated times post infection, brains were excised, pooled and leukocytes isolated by Percol gradient centrifugation. Single cell suspensions of pooled cervical lymph nodes (CLNs) were also prepared from the same animals. Cell populations were then phenotyped by flow cytometry. (A) Leukocytes infiltrating the brain were stained with mAbs to either CD8 or CD4 and the number of each T cell subset per brain calculated based on cell recoveries and percentage of each subset. (B) Cells were incubated with tetramers, washed and then stained with mAb to CD8. The absolute number of CD8+ VSV-N T cells present in the brain and CLN was then calculated based on the cell recoveries in each organ and percentage of CD8+tetramer+ cells. These values represent the means ± SEM of 2-8 experiments with 3-5 mice per time point.
Kinetics of the Inflammatory Response in the CNS
The above study demonstrates that VSV recruits a variety of blood cells into the virus-infected brain. However, this study did not provide any insights into either the status of resident microglia or the kinetics of this inflammatory response. To address these questions, mice were inoculated with VSV for various periods of time and the number of microglia and the identity of infiltrating blood cells in the CNS determined by flow cytometry. It is apparent from Figure 2A (panels a-c) that VSV induced an initial decrease in the number of microglia before a transient microgliosis became evident. Although the infiltrate population in naïve mice (panel a) appears to be smaller than that seen in Figures 1 and 5, this is reflective of a smaller number of events acquired by flow cytometry. This infiltrate population is quantified later (Figure 5) and is quite small in terms of both percentage and absolute number despite an apparently large population visible in the flow cytometry density plots. Similar kinetics were observed for CD45high blood cells (Figure 2B, panel a). We also detected a gradual and sustained increase in the number of conventional CD11c+ DCs, although their numbers were small relative to other myeloid and lymphoid elements in the brain (Figure 2B, panel b). VSV did not induce a significant infiltrate of pDCs, NK and NKT cells at any of the time points tested (Figure 2B, panel b and data not shown). It should be noted, however, that we have not formally excluded the possibility that the expansion of CD45high cells may have included proliferation of resident haematopoietic cells such as perivascular macrophages.
Figure 2. Kinetics of the inflammatory response following infection of the CNS.
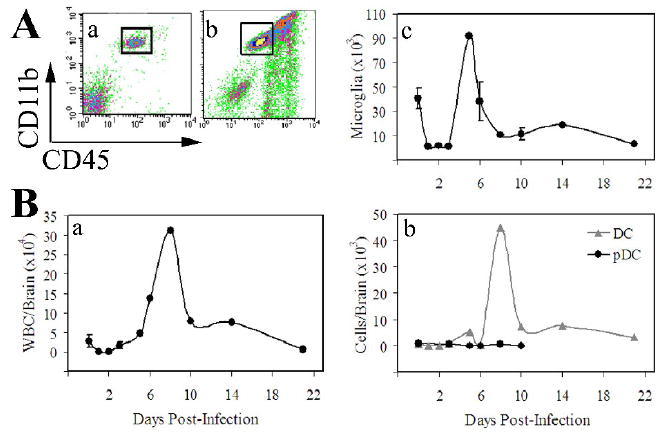
Mice were administered a single intranasal dose of VSV at the indicated times prior to euthanasia. Brains were excised, homogenized and the leukocyte fraction enriched by discontinuous Percoll gradient centrifugation. Cells were stained with the indicated mAbs and phenotyped by multiparameter flow cytometry. (A) Microglia isolated from mock (a) and VSV infected (b) brains were gated as CD11b+CD45low/int (box) and the absolute number of microglia calculated at each of the indicated time points (c). (B) A similar calculation to determine absolute numbers/brain was performed for infiltrating blood-derived leukocytes identified as CD45high cells (a). The absolute numbers of conventional (CD45highCD11c+PDCA-1-) and pDCs (CD45highCD11c+PDCA-1+) per brain were similarly determined (b). The values presented represent the mean ± S.E.M. cell yields from the pooled brains of 3-5 mice and 2-9 experiments per time point. Absolute numbers were calculated based on cell recoveries in each organ and the percentage of microglia at each of the indicated time points. Note that the scales in Figure 2B (panels a and b) are different.
Figure 5. Ablation of peripheral dendritic cells in vivo markedly suppresses the CNS innate and adaptive antiviral immune response.
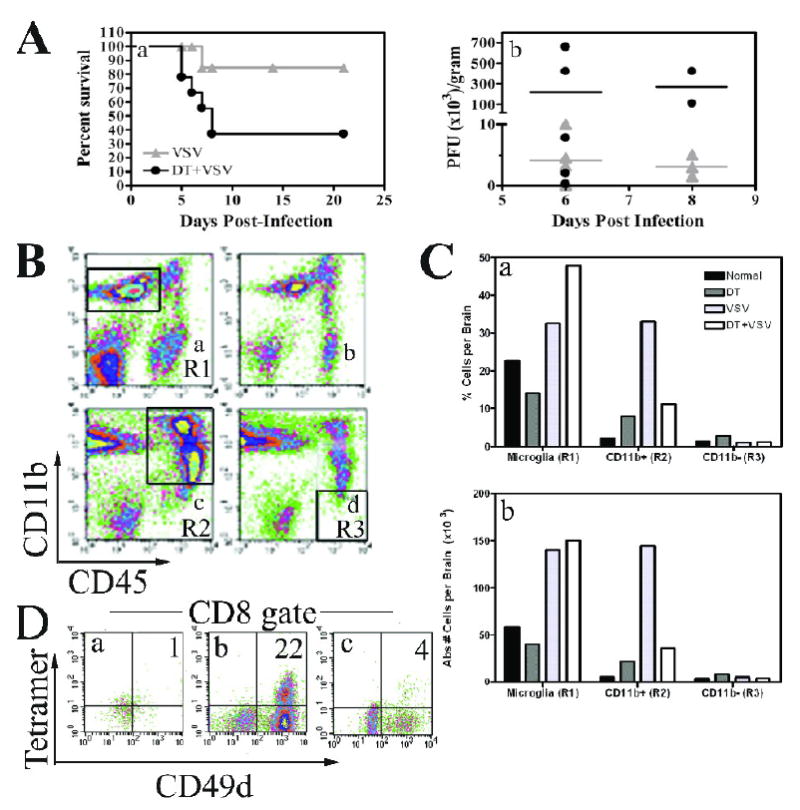
(A) DTRTg mice were given either PBS or DT one day before and after intranasal instillation of VSV (2×105 PFU). Mice were then monitored for morbidity (panel a). Mice were euthanized when moribund and brains and peripheral organs evaluated for VSV titres by plaque assay (panel b). This data is derived from 17 VSV-infected mice and 18 DT-treated, VSV-infected mice. (B) Mice were treated with either PBS (panels a, c) or DT (panels b, d). Cohorts either remained uninfected (panels a, b) or were given an intranasal inoculation of VSV at 2×105 PFU/mouse (panels c, d). Six days post-infection, brains were homogenized and then subjected to Percoll gradient centrifugation to enrich for leukocytes. Cells were then phenotyped by flow cytometry and a microglia gate defined as CD11b+CD45low/int cells (panel a, R1 gate). A second gate was established for peripheral mΦ/monocytes defined as CD11b+CD45high (panel c, R2). A final CD11b-CD45high gate was used to evaluate lymphocytes (panel d, R3). (C) The percent positive and absolute number of cells was then calculated within each of these gates and is summarized in the bar graphs. (D) To identify CD8+VSV-specific T cells, cells were first incubated with H-2Kb/VSV-N52-59 tetramers and then stained with mAbs to CD45, CD8, and the activation antigen CD49d. CD8+ cells were gated and the percentage of VSV-specific T cells within this gate determined by tetramer staining and co-expression of CD49d. Brains from 3-5 mice were pooled within each group. This experiment has been repeated two additional times and yielded similar results.
We next defined the kinetics of T cell subset infiltration into the CNS following infection with VSV. In addition, we assessed the specificity of infiltrating CD8+ cells using class I tetramers specific for the immunodominant epitope (VSV-N52-59). Figure 3 indicates that the brain contained a small basal population of T cells that did not expand for several days after virus infection (panel A). CD8+ T cells began to infiltrate the brain on day 6, peaked on day 8 and gradually returned to basal levels on day 21-post infection. Similar kinetics were observed for CD4+ cells. Virus-specific CD8+ T cells (VSV-N) were detectable in the draining CLN around day 5, reached maximal clonal expansion two days later and then their numbers rapidly diminished to achieve basal levels (panel B). The accumulation of VSV-N T cells in the brain followed similar kinetics; however, the response in terms of absolute numbers was much smaller relative to the draining CLN (panel B, note different scales). Thus, VSV induces expansion of both CD8+ and CD4+ T cell populations including CD8+ T cells specific for the nuclear protein of this virus. These kinetics, defined by flow cytometric analysis, are consistent with immunohistochemical studies reported by Reiss and her colleagues (Bi et al., 1995b; Forger et al., 1991). Based on these kinetics, we performed subsequent studies on day 6 post infection because leukocyte infiltration including tetramer+ cells were readily detectable in the CNS and morbidity/mortality were reduced relative to the peak of infection at days 7-8.
Phenotypic characterization of microglia isolated from encephalitic brains
The presence of an activated and expanded population of VSV-N T cells in the brains of VSV-infected mice suggests that primary antiviral immune responses may be either initiated and/or propagated in the CNS. If this is true, it implies that the brain possesses a professional APC capable of driving clonal expansion and differentiation of naïve CD8+ T cells. To examine the extent to which microglia may function as APCs, we evaluated microglial expression of several molecules essential for activation of naïve T cells during the early stages of the virus infection. Naïve microglia of mice express low to intermediate levels of CD45 (Ford et al., 1995; Ponomarev et al., 2005a; Ponomarev et al., 2005b) and CD11b (Ponomarev et al., 2005b), are CD11c-, and have a characteristic ramified morphology when resting (Ponomarev et al., 2005b; Santambrogio et al., 2001; Town, Nikolic, and Tan, 2005), although they are slightly smaller than peripheral CD45high leukocytes (Ford et al., 1995). As previously reported, the vast majority of microglia from naïve mice express undetectable to low levels of MHC class I antigens. However, virtually all microglia (96%) became class I+ by day 3 with significant (~50%) class I expression being detected as early as 48 hours following infection (Figure 4, panel A). Although the percentage of class I+ microglia dramatically increased, reduced yields of microglia during these early time points prevented a corresponding increase in the absolute number of microglia in the VSV-infected brain (Figure 4B, panel a). Class I expression was sustained for two weeks but eventually waned to achieve mock-infected levels, consistent with CD8+ infiltrate (Figure 3A).
Figure 4. Vesicular stomatitis virus induces the rapid activation of microglia and a delayed microgliosis.
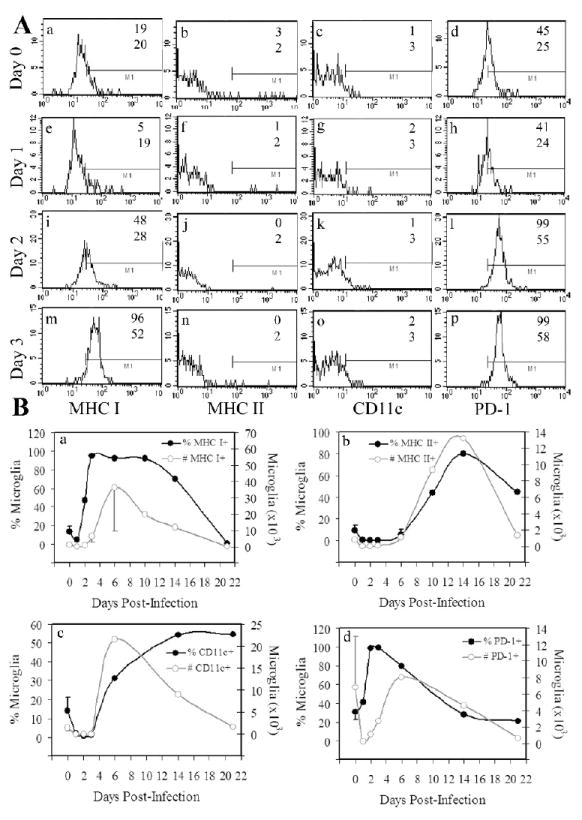
Mice were given a single intranasal instillation of VSV at the indicated times prior to euthanasia. Single cell suspensions of the brain were then prepared, subjected to Percoll gradient centrifugation and immunostained for flow cytometric analysis. (A) Microglia were defined as CD45low/intCD11b+ cells and expression of MHC I/II, CD11c, and PD-1 on gated microglia determined at early time points post-infection (4 mice per time point). The numbers in each panel refer to either % positive (upper) or MFI (bottom). Marker bars were set based on appropriate isotype controls (<4% positive). (B) Kinetics of VSV-induced upregulation of MHC class I (panel a), class II (panel b), CD11c (panel c), or PD-1 (panel d) molecules expressed either as a percentage of total leukocytes per brain or absolute number of microglia per brain (calculated from cell recoveries in each organ). The values in panel B represent the mean ± SEM of 2-7 experiments using the pooled brains of 3-5 mice at each time point.
Microglia slowly upregulated class II antigens (Figure 2B, panel b) and as a result significant co-expression of these molecules was not seen until day 10 (data not shown). Microglia also upregulated CD11c late during infection, corresponding with onset of morbidity and increased inflammation in the brain (Figure 4A and 4B, panel c). Interestingly, high constitutive levels of PD-1 were detected on microglia and virus infection induced further expression of this molecule so that essentially all microglia were PD-1+ two days post-infection (Figure 4A and B, panel d). The physiological significance of the negative regulator PD-1 during acute viral encephalitis is currently under investigation.
Impact of peripheral dendritic cell ablation on the inflammatory and primary antiviral immune response in the CNS
Our previous results demonstrated that microglia became activated in response to viral infection of the CNS and expressed surface molecules appropriate for antigen presentation. However, DCs also infiltrated the encephalitic CNS, complicating the contribution of each of these cell types to viral clearance and host survival. To more precisely define the role of microglia in viral clearance and survivability, DTRTg mice were treated with either PBS (mock) or DT to systemically deplete DCs and infected with VSV via the intranasal route. Mice were monitored for survival, euthanized when moribund, and virus titres determined on the brain and peripheral organs. It is apparent from Figure 5 (panel A) that the majority (63%) of mice depleted of peripheral DCs did not survive this dose of virus, whereas only 15% of control mice became moribund and had to be euthanized. Decreased survival was associated with delayed viral clearance in the brain in mice depleted of DCs (panel B). As previously reported, VSV was rapidly cleared from peripheral organs even in moribund mice depleted of DCs (Ciavarra et al., 2006). Thus, ablation of peripheral DCs specifically inhibits viral clearance from the CNS and as a result likely contributes to the observed increase in morbidity/mortality.
The inability of mice to efficiently clear VSV from the CNS suggests that the antiviral immune response was impaired in mice depleted of DCs. To assess this possibility, DTRTg mice were treated with either PBS or DT and then infected i.n. with VSV. Six days post-infection, the number of myeloid (CD11b+CD45high) and lymphoid (CD11b-CD45high) cells in the brain was examined by flow cytometry. As expected, microglia were readily detectable as a CD45low/int CD11b+ population (Region 1, R1) in mock-infected mice (Figure 5B, panel a). No significant changes in the number of microglia were observed in mice treated with DT alone, a result consistent with low endogenous CD11c expression on resting microglia (panel b). However, VSV infection was associated with a microgliosis that was not inhibited by prior DC depletion (compare R1, panels c and d). As expected, infection of the brain induced a potent inflammatory response revealed by the accumulation of a prominent population of CD45high CD11b+ cells (R2) in the brains of VSV infected mice. Surprisingly, prior DT treatment profoundly inhibited this infiltrate (panel d). This was evident whether data were expressed as a percentage or absolute number of infiltrating myeloid cells (panel C). Clonal expansion and/or infiltration of VSV-N T cells into the encephalitic brain were also profoundly suppressed by prior DC ablation (panel D). This response was also suppressed in the CLN of VSV infected mice because 9,940 and 1,902 CD8+ tetramer+ cells were detected in VSV and DT+VSV treated mice, respectively. Thus, DT treatment of transgenic mice ablates DCs but preserves resident microglia. In the absence of peripheral DCs, the inflammatory response as well as the accumulation of clonally expanded CD8+ VSV-specific T cells is markedly suppressed in the CNS.
As demonstrated in Figure 5 (panel D) a well-defined CD8+ tetramer+ population was present in the encephalitic brain despite the small infiltrate of CD45highCD11b- cells (panel C). This apparent contradiction reflects VSV-induced upregulation of CD11b on activated T cells at this time point (data not presented). Thus, most of the infiltrating CD8+ T cells are found in the CD45highCD11b+ gate. This finding is consistent with reports from other inflammation models (Andersson et al., 1994; Bullard et al., 2005; Christensen et al., 2001; Soilu-Hanninen et al., 1997).
Virus-induced cytokine response in the CNS is not dependent on peripheral dendritic cells
To determine the functional consequences of DC depletion in vivo, we evaluated the VSV-induced IFN-γ response in the CNS and CLNs in mice systemically depleted of DCs. IFN–γ is an important cytokine for host resistance to this virus because of its antiviral activity in the CNS. It should be noted that cells were cultured overnight in ELISPOT plates in the absence of exogenous virus or viral peptide to more accurately estimate the number of actual cytokine-producing cells in vivo. In control mice infected with VSV, few IL-2 or IL-4 -secreting cells were detected in the brain and draining CLNs (data not shown). IFN-γ-producing cells were also detected at very low frequencies in the brains of mock-infected mice. However, IFN-γ-producing cells were readily detected in the brains of mice infected with VSV (Figure 6). In striking contrast to the proliferative response of class I-restricted VSV-N T cells, mice depleted of DCs mounted a normal VSV-induced IFN-γ cytokine response in the CNS. This was consistently observed whether the data was expressed as frequency (upper panel) or total number of IFN-γ-producing cells per brain (lower panel). Indeed, in some experiments DC ablation actually enhanced this response (data not shown). Although an IFN-γ response could be detected in the CLNs, this response was modest at this time point relative to the CNS (data not presented). Thus, the VSV-induced IFN-γ cytokine response in the CNS is not inhibited by systemic depletion of conventional and pDCs and implicates that T cells are not essential for IFN-γ production; therefore, microglia or other resident CNS cells may be the source of this cytokine.
Figure 6. Systemic ablation of peripheral dendritic cells does not suppress the virus-induced IFN-γ response in the CNS.
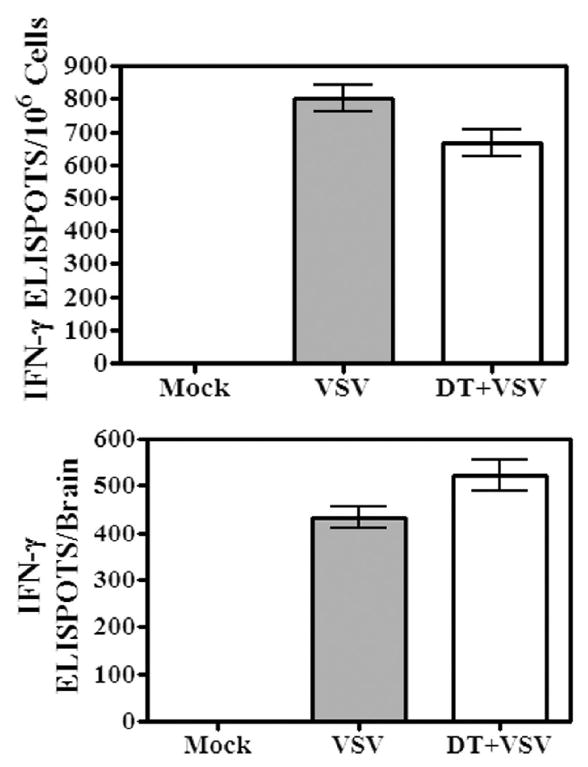
Mice were given a single intranasal instillation of VSV after being treated with either PBS (VSV) or DT (DT + VSV). Control mice were not infected (mock). Six days post-infection, brains were removed, pooled and leukocytes isolated by Percoll gradient centrifugation. Cells were seeded into ELISPOT plates in triplicate at up to 2×106 cells/well and incubated overnight. No exogenous virus or viral peptide was added to these cultures. The following day plates were developed and the number of ELISPOTs/input cell number determined under a dissecting microscope. The number of ELISPOTS/106 cells determined for each triplicate input cell number was averaged and expressed as the mean ± S.E.M. (top panel). The total number of IFN-γ-producing cells/brain was then calculated based on this value and cell recoveries (bottom panel) per organ. Brains from 4-5 mice were pooled within each group. This experiment is representative of two additional experiments.
DISCUSSION
The present understanding of the CNS as an immune privileged site is rapidly changing in response to closer scrutiny. It is no longer held that the BBB is impenetrable because several studies have demonstrated that some areas of the brain are unprotected by a BBB. These areas include the meninges, choroid plexus, circumventricular organs and ventricles (Farina, Aloisi, and Meinl, 2007; Galea, Bechmann, and Perry, 2007). Furthermore, the perivascular spaces in the CNS were initially termed lymphatic clefts by Goldman (Bechmann, Galea, and Perry, 2007). Current studies have clearly demonstrated localization of mΦ to these regions, which may produce a localized region architecturally similar to lymphoid tissue (also known as tertiary lymphoid regions, (Galea, Bechmann, and Perry, 2007)). The relevant cellular elements (DC, T cells, mΦ) that reside there may be sufficient to drive T cell activation and clonal expansion. This is in keeping with low numbers of activated T cells in the draining cervical lymph nodes despite their presence in the brain, and provides indirect evidence for more direct, site-specific activation of antigen-specific T cells.
The susceptibility to and kinetics of VSV infection in the CNS vary with mouse strain and gender (Barna et al., 1996). The work of Huneycutt et al. demonstrated that VSV antigen is detectable in the olfactory bulb as early as 12 hours post-infection and spreads caudally through the forebrain by 7 days post-infection, with only a few areas of the midbrain demonstrating antigen reactivity (Huneycutt et al., 1994). Previous studies by Reiss and colleagues demonstrated a high rate of morbidity/mortality in this model that correlated with high titres of VSV at 7 days post-infection and loss of the BBB function late in the infection. Surviving mice efficiently cleared VSV from the CNS, suggesting that the host can mount an efficient antiviral immune response in the CNS (Barna et al., 1996; Huneycutt et al., 1994; Plakhov et al., 1995). This view is further supported by immunohistochemical studies that demonstrated a VSV-induced CNS infiltrate composed primarily of mΦ and lymphocytes (Bi et al., 1995a). The kinetics we demonstrated in the CB6F1/DTRTg mouse are similar to those previously reported (Bi et al., 1995b). Starting as early as 3 days post-infection, we observed a mixed infiltrate of leukocytes in the CNS. Consistent with the findings of Bi et al. (Bi et al., 1995b), the infiltrate contained primarily mΦ, DCs, and T cells, but did not include B cells, NK or NKT cells. Macrophage and lymphocyte infiltration of the CNS increased sharply between days 6-8, corresponding with the peak of viral infection and onset of hind-limb paralysis, morbidity, and mortality. By 8 days post-infection, a significant number of both CD4+ and CD8+ T cells (both antigen-specific and nonspecific) had entered the brain. Our data demonstrate that CD8 infiltration coincides with CD4 entry into the brains of infected mice, consistent with previous studies (Ireland and Reiss, 2006).
Microglia become phenotypically similar to DC when activated (Ponomarev et al., 2005a; Ponomarev et al., 2005b; Shortman and Liu, 2002) and can upregulate several cell surface antigens, including MHC I and II, CD80, and CD40 (Ponomarev et al., 2005a; Ponomarev et al., 2005b), and assume a more spheroid shape. Activated microglia can present antigen to CD4+ T cells and secrete various chemokines (Persidsky et al., 1999) that help recruit activated lymphocytes. Additionally, exposure to GM-CSF has been reported to direct the phenotypic and morphologic maturation of naïve microglia into DC-like cells (Fischer and Reichmann, 2001). Juedes and Ruddle showed that CNS derived microglia can stimulate IFN-γ production in T-MOG (myelin oligodendrocyte glycoprotein)-specific lymphocytes (Juedes and Ruddle, 2001). Following these studies, Mack et al. demonstrated that microglia from the inflamed CNS in the presence of antigen can serve as antigen-presenting cells (APC) for myelin proteolipid protein (PLP139-151)-specific T cells, resulting in the production of IFN-γ and cellular proliferation (Mack et al., 2003). Our results demonstrated that microglia upregulated MHC I and II in response to infection, with MHC I appearing as early as 2 days post-infection and MHC II increasing much later during the course of infection (days 6-10). The prompt expression of class I antigens on microglia is consistent with their putative role as APCs in the CNS. Together, these data suggest that microglia express peptide/MHC class I molecules essential for antigen recognition by naïve CD8+ T cells. However, we have not detected expression of CD80 and CD86 on microglia isolated from VSV-infected brains although these are preliminary studies that have not examined multiple time points or specific brain regions to detect regional expression of costimulatory molecules on these cells. Nonetheless, even if microglia do not express costimulatory molecules, they can still function as APCs for T cells activated in the CLN or VSV memory cells to further propagate the immune response in the CNS. Thus, their role as functional APCs for a primary antiviral immune response in the CNS remains to be confirmed.
It is interesting to note that ≤ 25% of CD8+ T cells bound class I tetramers at the peak of the proliferative response. This suggests that most brain infiltrating CD8+ T cells are either not specific for VSV. However, it should be noted that while C57BL/6 mice recognise a single immunodominant epitope for VSV (H-2Db restricted), Balb/c mice can recognise two epitopes for VSV (H-2Ld and H-2Db restricted) (Forman et al., 1983). The CB6F1 mice used in these studies may therefore be able to recognise both VSV epitopes, whereas the tetramers used recognise only the H-2Db restricted antigen. A novel VSV cryptic determinant displayed in the CNS but not in the periphery may be another possible explanation for the lack of antigen specificity among infiltrating T cells. Recent studies demonstrated an antigen-specific pathway for CD8+ T cells across the BBB (Galea et al., 2007). It is perhaps not surprising that non-specific CD8+ T cells infiltrate the brain. VSV upregulates both early (CD25, CD69) and late (CD11a, CD49d) activation antigens on essentially all CD8+ and CD4+ T cells by a DC-independent mechanism (Figure 5, data not shown) and expression of some of these activation antigens (CD49d, VLA-4) may be required for penetration of the BBB. VSV also disrupts the BBB and this may also contribute to T cells penetration of the brain parenchyma (Bi et al., 1995b). Thus, both of these factors may contribute to the predominance of CD8+ T cells in the CNS that lack obvious specificity for the inducing virus. It is unclear why activated CD8+ T cells remain in the brain in the absence of cognate antigen. This study suggests that the paradigm that only T cells activated in the DLN infiltrate the CNS may not apply to VSV and other viruses with similar mitogenic properties. For these viruses, non-specific T cell activation and disruption of the BBB may allow CNS penetration of T cells with a variety of specificities. Activated microglia could then function as APCs to induce or propagate a primary antiviral T cell-mediated immune response within the CNS and not the CLN. Additional studies in this model are underway to test this hypothesis.
We previously reported that depletion of conventional and pDCs with DT treatment markedly inhibited clonal expansion of naïve CD8+ VSV-N T cells in non-neuronal sites (Ciavarra et al., 2006). However, recent studies by Probst and colleagues (Probst et al., 2005) questioned the specificity of this ablation model because they reported that DCs and mΦ were depleted by DT treatment of DTRTg mice. In our experience, the dose of DT used by these investigators was toxic and killed a significant percentage of mice prior to virus infection. Furthermore, we titrated the dose of DT administered to deplete DCs and found efficient and systemic depletion of DCs was achieved with as little as 0.5 ng/g DT (data not shown) without any detectable morbidity. We also observed that different commercial preparations of DT vary in toxicity and potency. The lowest dose (per preparation of DT) that efficiently and specifically depleted DCs in vivo was used in the studies presented in this report.
Our current studies expand the observed inhibition of CD8+ VSV-N T cells resulting from DT treatment to include the CNS. Thus, in both the periphery and the CNS, clonal expansion of naïve VSV-specific T cells is in DC-dependent, an observation consistent with studies demonstrating that CNS DCs are crucial for antigen presentation to CD4+ T cells (Bailey et al., 2007; Miller et al., 2007). Ablation of DCs also profoundly inhibited VSV encephalitis. These results were somewhat surprising, given that the traditional role for DCs is primarily as an activator of naïve T cells. The mechanistic basis for the failure of peripheral blood cells to infiltrate the brain in mice depleted of peripheral DCs remains to be clarified. Although DT can penetrate the blood-brain barrier and has been used to selectively kill oligodendrocytes in a similar DTR depletion model (Buch et al., 2005; Gropp et al., 2005), this required a high dose of DT (100ng/injection) and 3 injections/day for one week. Diphtheria toxin has a very short serum T1/2 life (90% cleared in 6 hours) with poor CNS penetrance (low blood/CNS transfer constant) (Wrobel et al., 1990)), and this may explain why multiple high dose injections were required to deplete oligodendrocytes. Thus, it is very unlikely that we depleted DCs in the brain because such treatment conditions were not employed in our studies (≤20ng/injection, two injections). This view is further supported by the observation that microgliosis was not diminished by DT treatment despite upregulation of CD11c during virus infection (data not shown). These studies imply that activated microglia are not sufficient for a normal inflammatory response to VSV indicating that peripheral DCs provide a unique and essential function in the CNS. This function could be early chemokine production by these cells or, alternatively, reflect a DC-glial cell interaction essential for chemokine production and blood cell infiltration into the CNS. Although the lack of T cell infiltration is in keeping with the paradigm of lymphocyte activation in the CLN, the infiltration of monocytic cells was also profoundly inhibited by depletion of DCs. Previous characterization of the DTRTg model and our titration of DT demonstrated that our dosage did not deplete macrophages (Ciavarra et al., 2006); therefore, peripheral DC apparently play a role above and beyond that of T cell activation in regulation of the CNS immune response.
Although DC ablation profoundly inhibited CNS inflammation and proliferation of VSV-N T cells, it reduced neither microgliosis nor the secretion of IFN-γ in response to viral infection. While IFN-γ is primarily considered a product of T cells, the levels observed in the brains of mice do not correspond with T cell infiltration and are not sensitive to DT-mediated loss of T cell infiltration. Thus, it is apparent that a cellular source of IFN-γ is present in the native CNS and studies in other models suggest that microglia may represent one source of non-lymphoid derived IFN-γ (Kawanokuchi et al., 2006; Suzuki et al., 2005; Wang and Suzuki, 2007). This view is further supported by the observation that IFN-γ production in response to EAE was not significantly reduced in CD11b-/- mice that also showed impaired T cell infiltrate (Bullard et al., 2005). Microglia can also produce significant amounts of IFN-γ in response to antigenic stimulation (Bi et al., 1995b; Fischer and Reichmann, 2001; Mack et al., 2003; Speth et al., 2007). Perhaps importantly, IFN-γ does not appear to have a significant protective effect in peripheral VSV infections (Muller et al., 1994);, however, it does appear to promote VSV clearance in the CNS.
In summary, VSV applied to the nasal mucosa causes reproducible encephalitis in mice characterized initially by microglia activation and microgliosis followed by a massive infiltrate of myeloid and lymphoid blood cells. CD8+ T cell infiltration into the brain correlates temporally with the rapid upregulation of MHC class I on microglia. Kinetic analysis of the development of VSV-N T cells support a model wherein VSV-N T cells become initially sensitized to VSV in the CLN, undergo clonal expansion and then emigrate from the CLN into the brain. This view is consistent with studies by Mendez-Fernandez and colleagues who demonstrated that sensitization of naïve CD8+ TMEV T cells requires the presence of peripheral lymph nodes (Mendez-Fernandez et al., 2005). It is possible that once in the CNS, sensitized VSV-N T cells undergo further clonal expansion and effector cell differentiation driven by peptide/MHC class I complexes displayed on activated microglia. Peripheral DCs, either conventional and/or pDCs, play an essential role promoting CNS inflammation and clonal expansion of virus-specific CD8+ T cells in both the CNS and CLN in vivo. However, VSV-induced production of IFN-γ is completely independent of conventional and pDCs suggesting that this response is driven by an APC resident in the CNS. The cellular interactions and underlying mechanism(s) that render both the innate and adaptive antiviral immune response dependent on peripheral DCs is currently being investigated.
MATERIALS AND METHODS
Mice
To assess DC function in vivo, we utilized a recently described transgenic mouse model that allows for the selective ablation of DCs in vivo. Diphtheria toxin (DT) receptor transgenic (DTRTg) mice (C.FVB-Tg(Itgax-DTR/EGFP)57Lan/J were purchased from Jackson Laboratories (Bar Harbor, ME) and subsequently bred to C57BL/6 mice (also purchased from Jackson Laboratories) to generate CB6F1 mice. The transgenic F1 mice are subsequently referred to DTRTg mice for simplicity. DTRTg mice possess a hybrid gene composed of the simian DTR and green fluorescent protein (GFP) under the control of the CD11c promoter. Mice injected with DT show rapid depletion of DCs from the spleen, lymph nodes, nasal mucosa, lungs, bladder, peritoneal fluid, thymus, and blood (Ciavarra et al., 2006; Engel et al., 2006; Jung et al., 2002; KleinJan et al., 2006). Expression of the simian diphtheria toxin receptor was confirmed by multiplex PCR as previously described (Ciavarra et al., 2006). Primer pairs were purchased from Integrated DNA Technologies, Coraville, IA. Mice lacking the transgene were used as non-DTRTg controls. Although flow cytometry for CD11c+ cells is often used to assess depletion of DCs, we have determined that the most sensitive indicator of DC depletion is the loss of T cell activation and clonal expansion (Ciavarra et al., 2006).
Virus and Cell Depletion
Wild-type VSV-Indiana strain, provided by Dr. Philip Marcus, University of Connecticut, was grown and assayed as previously described (Marvaldi et al., 1977). Virus was grown in confluent monolayers of Vero cells and virus titers determined by standard plaque assays (Sekellick and Marcus, 1979). Mice were infected with either 5×104 (male) or 2×105 (female) PFU VSV by i.n. inoculation of 5μL/nostril (Barna et al., 1996). For depletion of dendritic cells, mice were treated i.p. with 2ng/g DT (Sigma, St. Louis, MO) one day before and after virus infection. Mice were euthanized at various time points post-infection by CO2 asphyxiation. All mice were utilized at 6-10 weeks of age following protocols approved by the Institutional Animal Care and Use Committee according to federal guidelines.
Multicolour Flow Cytometry
Brains were excised and individually homogenized in a glass Tenbroek homogenizer with 2 mL PBS for 20 strokes. Cell suspensions were centrifuged for 8 min at 300xg. Supernatants were stored separately and the cell pellets were subjected to discontinuous Percoll centrifugation. Briefly, cells were resuspended in 70% Percoll overlayered with 35% Percoll and PBS, then centrifuged for 45 min at 20°C and 1200xg. The cells at the 35%-70% interface were collected, diluted in PBS, and centrifuged for 8 min at 300xg. Cervical lymph nodes were dissected and scrubbed through a 40μm nylon mesh cell strainer, then centrifuged at 300xg for 8 min. Cells were resuspended in flow cytometry wash buffer (1% goat serum, 0.1% sodium azide in PBS) for staining according to standard protocols. Monoclonal antibodies for leukocyte antigens were obtained from eBioscience (San Diego, CA): CD11b, clone M1/70; CD45, clone 30-F11; MHC II, clone M5/114.15.2; CD11c, clone N418; CD4, clone GK1.5; CD8α, clone 53-6.7; CD49b, clone DX5; CD45R, clone RA3-6B2; MHC I, clone 34-1-2S; PD-1, clone J43; and CD49d, clone R1-2. PDCA-1 (clone JF05-1C2.4.1) was purchased from Miltenyi Biotec, Auburn, CA. Tetramers directed against the immunodominant VSV nucleocapsid protein (VSV-N52-59, RGYVYQGL) were obtained from the NIH Tetramer Core Facility. Fluorophore conjugates varied based on staining profiles used. Acquisition of 20-200,000 events was performed using a Becton Dickinson (San Diego, CA) FACSCalibur in conjunction with CellQuest software (v3.3, Becton Dickinson). Nonspecific binding in the absence of additional Fc block was previously evaluated and did not affect staining patterns. To determine the absolute number of microglia and blood cells in the CNS, a gate was first defined for these cells based on forward and side scatter characteristics. The percentage of microglia (CD45low/int) or infiltrating blood cells (CD45high) within this gate was then used to calculate cell recoveries. All gates and quadrants were established with the use of appropriate isotype controls.
Cytokine ELISPOT Assay
Detection of IL-2, IL-4, and IFN-γ was performed with standard ELISPOT assays according to manufacturer’s protocols. Briefly, brain cells from a 70%-35% Percoll interface were incubated overnight in ELISPOT plates (Millipore) containing anti-cytokine capture mAbs (IL-2, clone JES6-5H4; IL-4, clone 11B11; IFN-γ, clone AN-18; all purchased from eBioscience). No exogenous virus was added during this incubation. The following day, ELISPOTs were detected with the appropriate biotin-conjugated detection mAb (IL-2, clone JES6-5H4; IL-4, clone BVD6-24G2; IFN-γ, clone R4-6A2; all purchased from eBioscience) and then revealed with horseradish peroxidase-conjugated avidin (Sigma-Aldrich) followed by AEC (3-amino-9-ethylcarbazole; Sigma-Aldrich) substrate.
Acknowledgments
This work was supported by a grant from the National Institutes of Health Grant (NIAID48700) awarded to R.P. C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Genotyping Protocol for Itgax-DTR/GFP. The Jackson Laboratory; 2007. [Google Scholar]
- Andersson EC, Christensen JP, Marker O, Thomsen AR. Changes in cell adhesion molecule expression on T cells associated with systemic virus infection. J Immunol. 1994;152(3):1237–1245. [PubMed] [Google Scholar]
- Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4(+) T(H)-17 cells in relapsing EAE. Nat Immunol. 2007;8(2):172–80. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- Barna M, Komatsu T, Bi Z, Reiss CS. Sex differences in susceptibility to viral infection of the central nervous system. J Neuroimmunol. 1996;67(1):31–9. doi: 10.1016/0165-5728(96)00022-7. [DOI] [PubMed] [Google Scholar]
- Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends in Immunology. 2007;28(1):5–11. doi: 10.1016/j.it.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Bi Z, Barna M, Komatsu T, Reiss C. Vesicular stomatitis virus infection of the central nervous system activates both innate and acquired immunity. J Virol. 1995a;69(10):6466–6472. doi: 10.1128/jvi.69.10.6466-6472.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi Z, Quandt P, Komatsu T, Barna M, Reiss CS. IL-12 promotes enhanced recovery from vesicular stomatitis virus infection of the central nervous system. J Immunol. 1995b;155(12):5684–9. [PubMed] [Google Scholar]
- Buch T, Heppner FL, Tertilt C, Heinen TJ, Kremer M, Wunderlich FT, Jung S, Waisman A. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat Methods. 2005;2(6):419–26. doi: 10.1038/nmeth762. [DOI] [PubMed] [Google Scholar]
- Bullard DC, Hu X, Schoeb TR, Axtell RC, Raman C, Barnum SR. Critical Requirement of CD11b (Mac-1) on T Cells and Accessory Cells for Development of Experimental Autoimmune Encephalomyelitis. J Immunol. 2005;175(10):6327–6333. doi: 10.4049/jimmunol.175.10.6327. [DOI] [PubMed] [Google Scholar]
- Bulloch K, Miller MM, Gal-Toth J, Milner TA, Gottfried-Blackmore A, Waters EM, Kaunzner UW, Liu K, Lindquist R, Nussenzweig MC, Steinman RM, McEwen BS. CD11c/EYFP transgene illuminates a discrete network of dendritic cells within the embryonic, neonatal, adult, and injured mouse brain. J Comp Neurol. 2008;508(5):687–710. doi: 10.1002/cne.21668. [DOI] [PubMed] [Google Scholar]
- Christensen JE, Andreasen SO, Christensen JP, Thomsen AR. CD11b expression as a marker to distinguish between recently activated effector CD8(+) T cells and memory cells. Int Immunol. 2001;13(4):593–600. doi: 10.1093/intimm/13.4.593. [DOI] [PubMed] [Google Scholar]
- Ciavarra RP, Stephens A, Nagy S, Sekellick M, Steel C. Evaluation of Immunological Paradigms in a Virus Model: Are Dendritic Cells Critical for Antiviral Immunity and Viral Clearance? J Immunol. 2006;177(1):492–500. doi: 10.4049/jimmunol.177.1.492. [DOI] [PubMed] [Google Scholar]
- Dimier-Poisson I, Aline F, Bout D, Mevelec MN. Induction of protective immunity against toxoplasmosis in mice by immunization with Toxoplasma gondii RNA. Vaccine. 2006;24(10):1705–9. doi: 10.1016/j.vaccine.2005.09.053. [DOI] [PubMed] [Google Scholar]
- Engel D, Dobrindt U, Tittel A, Peters P, Maurer J, Gutgemann I, Kaissling B, Kuziel W, Jung S, Kurts C. Tumor necrosis factor alpha- and inducible nitric oxide synthase-producing dendritic cells are rapidly recruited to the bladder in urinary tract infection but are dispensable for bacterial clearance. Infect Immun. 2006;74(11):6100–7. doi: 10.1128/IAI.00881-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends in Immunology. 2007;28(3):138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Fischer H-G, Reichmann G. Brain Dendritic Cells and Macrophages/Microglia in Central Nervous System Inflammation. J Immunol. 2001;166(4):2717–2726. doi: 10.4049/jimmunol.166.4.2717. [DOI] [PubMed] [Google Scholar]
- Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein- reactive CD4+ T cells compared. J Immunol. 1995;154(9):4309–4321. [PubMed] [Google Scholar]
- Forger JM, 3rd, Bronson RT, Huang AS, Reiss CS. Murine infection by vesicular stomatitis virus: initial characterization of the H-2d system. J Virol. 1991;65(9):4950–8. doi: 10.1128/jvi.65.9.4950-4958.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman J, Goodenow RS, Hood L, Ciavarra R. Use of DNA-mediated gene transfer to analyze the role of H-2Ld in controlling the specificity of anti-vesicular stomatitis virus cytotoxic T cells. J Exp Med. 1983;157(4):1261–72. doi: 10.1084/jem.157.4.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends in Immunology. 2007;28(1):12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, Perry VH. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med. 2007;204(9):2023–30. doi: 10.1084/jem.20070064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, Barsh GS, Horvath TL, Bruning JC. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci. 2005;8(10):1289–91. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- Hatterer E, Davoust N, Didier-Bazes M, Vuaillat C, Malcus C, Belin M-F, Nataf S. How to drain without lymphatics? Dendritic cells migrate from the cerebrospinal fluid to the B-cell follicles of cervical lymph nodes. Blood. 2006;107(2):806–812. doi: 10.1182/blood-2005-01-0154. [DOI] [PubMed] [Google Scholar]
- Havenith CE, Askew D, Walker WS. Mouse resident microglia: isolation and characterization of immunoregulatory properties with naive CD4+ and CD8+ T-cells. Glia. 1998;22(4):348–59. [PubMed] [Google Scholar]
- Huneycutt BS, Plakhov IV, Shusterman Z, Bartido SM, Huang A, Reiss CS, Aoki C. Distribution of vesicular stomatitis virus proteins in the brains of BALB/c mice following intranasal inoculation: an immunohistochemical analysis. Brain Res. 1994;635(12):81–95. doi: 10.1016/0006-8993(94)91426-5. [DOI] [PubMed] [Google Scholar]
- Ireland DD, Reiss CS. Gene expression contributing to recruitment of circulating cells in response to vesicular stomatitis virus infection of the CNS. Viral Immunol. 2006;19(3):536–45. doi: 10.1089/vim.2006.19.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juedes AE, Ruddle NH. Resident and Infiltrating Central Nervous System APCs Regulate the Emergence and Resolution of Experimental Autoimmune Encephalomyelitis. J Immunol. 2001;166(8):5168–5175. doi: 10.4049/jimmunol.166.8.5168. [DOI] [PubMed] [Google Scholar]
- Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17(2):211–20. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karman J, Chu HH, Co DO, Seroogy CM, Sandor M, Fabry Z. Dendritic cells amplify T cell-mediated immune responses in the central nervous system. J Immunol. 2006;177(11):7750–60. doi: 10.4049/jimmunol.177.11.7750. [DOI] [PubMed] [Google Scholar]
- Kawanokuchi J, Mizuno T, Takeuchi H, Kato H, Wang J, Mitsuma N, Suzumura A. Production of interferon-gamma by microglia. Mult Scler. 2006;12(5):558–64. doi: 10.1177/1352458506070763. [DOI] [PubMed] [Google Scholar]
- KleinJan A, Willart M, van Rijt LS, Braunstahl GJ, Leman K, Jung S, Hoogsteden HC, Lambrecht BN. An essential role for dendritic cells in human and experimental allergic rhinitis. J Allergy Clin Immunol. 2006;118(5):1117–25. doi: 10.1016/j.jaci.2006.05.030. [DOI] [PubMed] [Google Scholar]
- Lauterbach H, Zuniga EI, Truong P, Oldstone MB, McGavern DB. Adoptive immunotherapy induces CNS dendritic cell recruitment and antigen presentation during clearance of a persistent viral infection. J Exp Med. 2006;203(8):1963–75. doi: 10.1084/jem.20060039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–70. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Mack CL, Vanderlugt-Castaneda CL, Neville KL, Miller SD. Microglia are activated to become competent antigen presenting and effector cells in the inflammatory environment of the Theiler’s virus model of multiple sclerosis. J Neuroimmunol. 2003;144(12):68–79. doi: 10.1016/j.jneuroim.2003.08.032. [DOI] [PubMed] [Google Scholar]
- Marvaldi JL, Lucas-Lenard J, Sekellick MJ, Marcus PI. Cell killing by viruses. IV. Cell killing and protein synthesis inhibition by vesicular stomatitis virus require the same gene functions. Virology. 1977;79(2):267–80. doi: 10.1016/0042-6822(77)90354-3. [DOI] [PubMed] [Google Scholar]
- Matyszak MK, Perry VH. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience. 1996;74(2):599–608. doi: 10.1016/0306-4522(96)00160-1. [DOI] [PubMed] [Google Scholar]
- McMenamin PG. Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J Comp Neurol. 1999;405(4):553–62. [PubMed] [Google Scholar]
- Mendez-Fernandez YV, Hansen MJ, Rodriguez M, Pease LR. Anatomical and cellular requirements for the activation and migration of virus-specific CD8+ T cells to the brain during Theiler’s virus infection. J Virol. 2005;79(5):3063–70. doi: 10.1128/JVI.79.5.3063-3070.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SD, McMahon EJ, Schreiner B, Bailey SL. Antigen Presentation in the CNS by Myeloid Dendritic Cells Drives Progression of Relapsing Experimental Autoimmune Encephalomyelitis. Ann NY Acad Sci. 2007;1103(1):179–191. doi: 10.1196/annals.1394.023. [DOI] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264(5167):1918–21. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Nair A, Hunzeker J, Bonneau RH. Modulation of microglia and CD8(+) T cell activation during the development of stress-induced herpes simplex virus type-1 encephalitis. Brain Behav Immun. 2007;21(6):791–806. doi: 10.1016/j.bbi.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Soma LA, Lavi E. Microglia in diseases of the central nervous system. Ann Med. 2002;34(78):491–500. doi: 10.1080/078538902321117698. [DOI] [PubMed] [Google Scholar]
- Newman TA, Galea I, van Rooijen N, Perry VH. Blood-derived dendritic cells in an acute brain injury. J Neuroimmunol. 2005;166(12):167–72. doi: 10.1016/j.jneuroim.2005.04.026. [DOI] [PubMed] [Google Scholar]
- Perry VH. A revised view of the central nervous system microenvironment and major histocompatibility complex class II antigen presentation. J Neuroimmunol. 1998;90(2):113–21. doi: 10.1016/s0165-5728(98)00145-3. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Ghorpade A, Rasmussen J, Limoges J, Liu XJ, Stins M, Fiala M, Way D, Kim KS, Witte MH, Weinand M, Carhart L, Gendelman HE. Microglial and astrocyte chemokines regulate monocyte migration through the blood-brain barrier in human immunodeficiency virus-1 encephalitis. Am J Pathol. 1999;155(5):1599–611. doi: 10.1016/S0002-9440(10)65476-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plakhov IV, Arlund EE, Aoki C, Reiss CS. The earliest events in vesicular stomatitis virus infection of the murine olfactory neuroepithelium and entry of the central nervous system. Virology. 1995;209(1):257–62. doi: 10.1006/viro.1995.1252. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Novikova M, Maresz K, Shriver LP, Dittel BN. Development of a culture system that supports adult microglial cell proliferation and maintenance in the resting state. J Immunol Methods. 2005a;300(12):32–46. doi: 10.1016/j.jim.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res. 2005b;81(3):374–89. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- Probst HC, Tschannen K, Odermatt B, Schwendener R, Zinkernagel RM, Van Den Broek M. Histological analysis of CD11c-DTR/GFP mice after in vivo depletion of dendritic cells. Clin Exp Immunol. 2005;141(3):398–404. doi: 10.1111/j.1365-2249.2005.02868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. Role of microglia in central nervous system infections. Clin Microbiol Rev. 2004;17(4):942–64. doi: 10.1128/CMR.17.4.942-964.2004. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santambrogio L, Belyanskaya SL, Fischer FR, Cipriani B, Brosnan CF, Ricciardi-Castagnoli P, Stern LJ, Strominger JL, Riese R. Developmental plasticity of CNS microglia. Proc Natl Acad Sci U S A. 2001;98(11):6295–300. doi: 10.1073/pnas.111152498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwob JE, Saha S, Youngentob SL, Jubelt B. Intranasal inoculation with the olfactory bulb line variant of mouse hepatitis virus causes extensive destruction of the olfactory bulb and accelerated turnover of neurons in the olfactory epithelium of mice. Chem Senses. 2001;26(8):937–52. doi: 10.1093/chemse/26.8.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekellick MJ, Marcus PI. Persistent infection. II. Interferon-inducing temperature-sensitive mutants as mediators of cell sparing: possible role in persistent infection by vesicular stomatitis virus. Virology. 1979;95(1):36–47. doi: 10.1016/0042-6822(79)90399-4. [DOI] [PubMed] [Google Scholar]
- Serafini B, Columba-Cabezas S, Di Rosa F, Aloisi F. Intracerebral recruitment and maturation of dendritic cells in the onset and progression of experimental autoimmune encephalomyelitis. Am J Pathol. 2000;157(6):1991–2002. doi: 10.1016/S0002-9440(10)64838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serot JM, Bene MC, Foliguet B, Faure GC. Monocyte-derived IL-10-secreting dendritic cells in choroid plexus epithelium. J Neuroimmunol. 2000;105(2):115–9. doi: 10.1016/s0165-5728(99)00240-4. [DOI] [PubMed] [Google Scholar]
- Serot JM, Foliguet B, Bene MC, Faure GC. Ultrastructural and immunohistological evidence for dendritic-like cells within human choroid plexus epithelium. Neuroreport. 1997;8(8):1995–8. doi: 10.1097/00001756-199705260-00039. [DOI] [PubMed] [Google Scholar]
- Serot JM, Foliguet B, Bene MC, Faure GC. Intraepithelial and stromal dendritic cells in human choroid plexus. Hum Pathol. 1998;29(10):1174–5. doi: 10.1016/s0046-8177(98)90437-3. [DOI] [PubMed] [Google Scholar]
- Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2(3):151–61. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- Soilu-Hanninen M, Roytta M, Salmi AA, Salonen R. Semliki Forest virus infection leads to increased expression of adhesion molecules on splenic T-cells and on brain vascular endothelium. J Neurovirol. 1997;3(5):350–60. doi: 10.3109/13550289709030749. [DOI] [PubMed] [Google Scholar]
- Speth C, Rambach G, Hagleitner M, Konstanzer K, Hollmuller I, Dierich MP, Mohsenipour I, Maier H. Immune response to retroviral infections of the brain. Front Biosci. 2007;12:1508–19. doi: 10.2741/2164. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Claflin J, Wang X, Lengi A, Kikuchi T. Microglia and macrophages as innate producers of interferon-gamma in the brain following infection with Toxoplasma gondii. Int J Parasitol. 2005;35(1):83–90. doi: 10.1016/j.ijpara.2004.10.020. [DOI] [PubMed] [Google Scholar]
- Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velge-Roussel F, Marcelo P, Lepage AC, Buzoni-Gatel D, Bout DT. Intranasal immunization with Toxoplasma gondii SAG1 induces protective cells into both NALT and GALT compartments. Infect Immun. 2000;68(2):969–72. doi: 10.1128/iai.68.2.969-972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Suzuki Y. Microglia produce IFN-gamma independently from T cells during acute toxoplasmosis in the brain. J Interferon Cytokine Res. 2007;27(7):599–605. doi: 10.1089/jir.2006.0157. [DOI] [PubMed] [Google Scholar]
- Wrobel CJ, Wright DC, Dedrick RL, Youle RJ. Diphtheria toxin effects on brain-tumor xenografts. Implications for protein-based brain-tumor chemotherapy. J Neurosurg. 1990;72(6):946–50. doi: 10.3171/jns.1990.72.6.0946. [DOI] [PubMed] [Google Scholar]