

Abstract
CTCF, a nuclear transcriptional factor, is a multifunctional protein and involves regulation of growth factor- and cytokine-induced cell proliferation/differentiation. In the present study, we investigated the role of CTCF in protecting stress-induced apoptosis in various human cell types. We found that UV irradiation and hyper-osmotic stress induced human corneal epithelial (HCE) and hematopoietic myeloid cell apoptosis detected by significantly increased caspase 3 activity and decreased cell viability. The stress-induced apoptotic response in these cells requires down-regulation of CTCF at both mRNA and protein levels, suggesting that CTCF may play an important role in downstream events of stress-induced signaling pathways. Inhibition of NFκB activity prevented stress-induced down-regulation of CTCF and increased cell viability against stress-induced apoptosis. The anti-apoptotic effect of CTCF was further studied by manipulating CTCF activities in HCE and hematopoietic cells. Transient transfection of cDNAs encoding full-length human CTCF markedly suppressed stress-induced apoptosis in these cells. In contrast, knocking down of CTCF mRNA using siRNA specific to CTCF significantly promoted stress-induced apoptosis. Thus, our results reveal that CTCF is a down stream target of stress-induced signaling cascades and it plays a significant anti-apoptotic role in regulation of stress-induced cellular responses in HCE and hematopoietic myeloid cells.
Keywords: Gene expression, gene transfection, siRNA, programmed cell death, UV irradiation, hyper-osmotic pressure
Introduction
CTCF, a CCCTC binding factor, is a DNA binding protein which consists of 11 zinc finger domains [1–3]. The protein is a highly evolutional conserved transcriptional factor and is involved in multiple aspects of gene transcriptional regulations [1, 4]. CTCF was initially cloned as a repressive transcriptional factor to regulate c-Myc expression cross species of chicken, mouse and human [1]. CTCF regulates several genes important for regulations of development [5, 6]. Interestingly, the regulatory function of CTCF on its target genes appears mainly inhibitory or insulation effects, including repressing expressions of c-Myc, β-globin and chicken lysozyme genes [1–3]. With an exception, CTCF can up-regulate expression of amyloid protein precursor gene [7]. In the past years, remarkable finding reveals that CTCF plays an important role in controlling mono-allelic expression of imprinted genes, such as Igf-2 and H19 genes through a DNA methylation-sensitive mechanism [8, 9]. In addition, CTCF is also a candidate for the trans-acting factor determining X-inactivation choices [10]. Our previous studies demonstrate that CTCF activity is necessary for eye development in mice [5, 6]. CTCF interacts with binding motifs upstream from the P0 promoter of Pax6 gene and down-regulates Pax6 expression to control eye development [11]. As evidenced by our previous studies, CTCF is up-regulated by EGF-stimulated activation of the Erk signaling pathway resulting in suppression of Pax6 expression during EGF-induced corneal epithelial cell proliferation [12]. Moreover, CTCF is a phosphoprotein with different phosphorylated forms. The phosphorylation of CTCF is associated with cell proliferation/differentiation [13, 14]. However, the role of CTCF in regulating cell growth and apoptosis is still not clearly understood. In lymphocyte B cells, increased expression of CTCF is associated with down-regulation of c-Myc, resulting in cell growth arrest and cell death [15]. Accumulation of CTCF in the nucleoli is related to growth arrest and apoptosis during differentiation in human K562 myeloid cells and in human breast carcinoma cells, respectively [16, 17]. These results suggest that CTCF may be a determinant factor to death signaling pathways in these cells. However, other studies demonstrate contrary to the pro-apoptotic role of CTCF that knockdown and over-expression of CTCF in breast cancer cells result in triggering apoptosis and protecting ectopic expression of Bax-induced apoptosis, respectively [18]. In addition, CTCF expression levels are elevated in corneal epithelial cells in growth factor-induced proliferation [12]. In order to understand how CTCF regulates the cell growth and survival, it is necessary to further study the role of CTCF in stress-induced signaling and death pathways in various cell and tissue types.
Ultraviolet (UV) and hyper-osmotic stresses can induce activations of different signaling pathways, such as JNK and p38 signaling pathways, resulting in programmed cell death (apoptosis) [19]. Stress-induced activation of specific signaling pathways are also often resulted from stimulation of the cell membrane Kv channels and cytokine receptors, including epidermal growth factor (EGF), tumor necrosis factor (TNF) and interleukin-1 (IL-1) receptors [20–23]. UV and hyper-osmotic stress-induced corneal epithelial cell death is associated with activation of c-Jun NH2-terminal kinase/stress-activated protein kinase (JNK/SAPK) and p38 cascades that elicit cellular apoptotic responses [19, 22, 24]. On the other hand, stress-induced stimulation of signaling pathways that are upstream of MAP kinases includes activation of Ras, a GTP binding protein and Src, a nonreceptor tyrosine kinase [25–28]. In addition, the membrane-associated protein tyrosine phosphatases can be inhibited by UV irradiation through targeting an essential -SH group in the tyrosine phosphatase, resulting in inhibition of dephosphorylation and enhancement of autophosphorylation of cytokine receptors [29]. Further downstream, the stress response is characterized by increase of caspase 3 activities and triggering apoptosis. However, the regulatory mechanism regarding altered CTCF activities in response to cytokines and environmental stress is still largely unknown. In the present study, we demonstrate that UV irradiation and hyper-osmotic stresses induced down-regulation of CTCF activity. Down-regulation of CTCF activity is required for the effect of stress-induced NFκB activation on apoptosis in HCE and hematopoietic myeloid cells. These results reveal a new and functional role of CTCF which acts as an anti-apoptotic factor in mediating stress-related cell survival and death.
Materials and Methods
Cell culture and apoptosis induction
Human corneal epithelial (HCE) cells were cultured in a humidified incubator gassed with 5% CO2 at 37 °C and fed with DMEM/F12 medium contained 10% FBS and 5 ng/ml insulin (Sigma). HCE cells were serum-starved in the serum-free culture medium for 24 h to synchronize cell at the G1 phase of the cell cycle. HCE cells were detached by treatment with 0.05% trypsin-EDTA and passed at a seeding density of 105/ml. Several hematopoietic myeloid cell lines were used in our study including ML-1 [30], K562 (ATCC), U937 (ATCC) and Himeg-1 [31]. Hematopoietic myeloid cells were cultured in RPMI 1640 supplemented with 7.5% heat-inactivated fetal bovine serum (FBS; Invitrogen™ Life Technologies, Grand Island, NY, USA) in a humidified incubator constantly filled with 5% CO2 at 37°C. Hematopoietic myeloid cells were passed at 3×105 seeding density and synchronized by serum starvation that was achieved by maintaining cells in a medium containing 0.3% FBS for 36 h. For UV irradiation experiments, cells were placed in a tissue culture hood at a distance of 60 cm from the UV-C light source and exposed at an intensity of 42 μJ/cm2. For hyper-osmotic pressure experiments, cells were exposed to osmotic pressures from 300 to 600 mOsm created by increasing concentrations of sorbital in the medium.
Northern blot experiments
Total RNAs from HCE and hematopoietic myeloid cells were extracted by using a guanidine thiocyanate procedure [32]. Briefly, 1×107 cells were collected and rinsed with ice-cold phosphate buffered saline (PBS). Cells were lysed in 1 ml of guanidium solution (5 M guanidine hydrochloride, 50 mM Tris-HCl, pH 8, 0.5% N-lauroylsarcosine, 100 mM β-mercaptoethanol). Lysates were extracted with 50/50 phenol/chloroform for three times. Finally, RNAs were precipitated by centrifugation at 12,000 rpm for 15 min after pre-incubated with ethanol at −80 °C. RNA (20 μg) for each sample was loaded in 1% agarose gel denatured with 2.2 M formaldehyde. The fractionated RNA was transferred onto nylon membrane. The membrane was subsequently hybridized with [α-32P] labeled corresponding DNA probe using a Random Primer Labeling Kit (NE Biolabs, Beverly, MA). Signals in the membrane were visualized by exposure of the membrane to X-ray film overnight at −80 °C.
Western blot experiments
Western analysis was performed using a previously described protocol [22]. In brief, HCE and hematopoietic myeloid cells (5×106) were rinsed twice with PBS and harvested in 0.5 ml lysis buffer (20 mM Tris, pH 7.5, 137 mM NaCl, 1.5 mM MgCl2, 2 mM EDTA, 10 mM sodium pyrophosphate, 25 mM β-glycerophosphate, 10% glycerol, 1% Triton X-100, 1 mM Na-orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 mg/mL leupeptin). After centrifugation at 13,000 g for 15 min, cell lysates were denatured by an equal volume of 2× Laemmli buffer, and then by boiling for 5 min. Each sample containing 20 μg protein was electrophoresed in a 10% SDS-polyacrylamide gel, and then transferred to a polyvinylidene difluoride membrane. The membrane was incubated with respective antibodies against CTCF, cleaved caspase 3 and PARP fragments (Upstate, Chicago, IL). Secondary anti-rabbit immunoglobulin G conjugated with HRP was used to visualize the positive signals in the membrane (Santa Cruz Biotech, Santa Cruz, CA). These membranes were re-hybridized with mouse anti-β-actin and goat anti-mouse IgG antibodies after being stripped following a standard tripping protocol.
Over-expression and knockdown mRNA of CTCF
Full-length cDNA encoding human CTCF were cloned into pcDNA4-to-A vector (Gibco/Invitrogen, Carlsbad, CA), named as pcDNA4-CTCF. Both pcDNA4-CTCF and pcDNA4-To-A vector (served as control) were transfected into HCE cells by a lipofection protocol as follows: 1) subconfluent cells (60%) in 60 mm culture dishes were rinsed twice with PBS and placed in 1.2 ml of serum free DMEM/F12 medium; 2) DNA mixture samples containing 5 μg DNA each, 10 Plus Reagent and 10 μl lipfectamine (Invitrogen) in 200 μl serum free medium were added into the dishes. After 6 h incubation in transfection conditions, the cells were replaced to the normal culture condition for 2 days. For RNA interference (RNAi) experiments, a human CTCF specific double strand short RNA was synthesized by using a Silencer™ siRNA Construction kit (Ambion, Austin, TX). A pair of primers was used with a sense strand sequence of “aaggaaugucuucuuuacacc”, and an antisense strand of “aagguguaaagaagacauucc”. In addition, a control double strand siRNA (sense: “aacauucgguagauuccucgc” and antisense: “aagcgaggaaucuaccgaaug”) was also synthesized using the same method. The sequence homologies of siRNA primers were examined by using a NIH Blast program. The siRNAs were transfected into HCE and hematopoietic cells by using a siPORT Lipid system (Ambion, Austin, TX) following a standard protocol. Briefly, subconfluent cells in 6 well culture plates or in 35 mm culture dishes were washed twice with PBS and then 0.6 ml serum free DMEM/F12 were added to each well or dish. The transfection mixtures containing siRNA (25 nM) and siPORT lipid (10 μl) in 200 μl serum free medium were applied into each well or dish of cells. The cells were incubated at 37 °C for 6 h before replaced with DMEM/F12 medium containing 10% FBS for two days. Transfected were synchronized by serum starvation for 24 h before experiments.
Determination of cell apoptosis
Cell death was analyzed by detections of caspase 3 activation and cell survival index using MTT assays. Caspase 3 activity in cells was determined by Western analysis using specific antibodies against cleaved caspase 3 and PARP fragments (see Western blot experiments above), and by using a colorimetric CaspACE™ Assay system (Promega, Madison, WI). HCE and ML-1 cells (107) were washed twice with cold PBS and lysed in 100 μl lysis buffer by 3 cycles of freeze thawing. Following centrifugation at 15,000 g for 15 min at 4°C, 50 μl lysates were added into a 96 well plate containing reaction mix to measure caspase 3 activity. After incubation for 4 h at 37 °C, OD405nm of each sample was measured with a microplate reader (Molecular Devices Corporation, Sunnyvale, CA). Based on a standard curve obtained with pNA standards, pNA produced in the reaction was calculated as specific caspase activity. Their activities are expressed as pico-mole/106cell. MTT assay was performed following an established protocol [12]. Briefly, a colorimetric assay system was used to measure the reduction of a tetrazolium component (MTT) into an insoluble formazan product by the mitochondria in viable cells. The culture medium was replaced with 1 ml of serum free medium. MTT solution (100 μl, 5 mg/ml in PBS) was added into each wells and incubated in a CO2 incubator for 1 h. The medium was replaced by 0.4 ml acidic isopropanol (0.04 M HCl in absolute isopropanol) to solubilize the colored crystals. Samples were read using an ELISA plate reader (Labsystems Multiskan MCC/340, Fisher Scientific) at a wavelength of 570 nm with the background subtraction at 650 nm. The amount of color produced normalized with the background was directly proportional to the number of viable cells.
Results
Stress-induced alterations of CTCF expression
In previous studies, we found that CTCF activity in human corneal epithelial (HCE) cells was altered in cytokine-stimulated proliferation. Expression of CTCF was up-regulated by EGF-induced activation of MAP kinase cascades, suggesting that CTCF may be also involved in cellular responses stimulated by stresses. To pursue studying the role of CTCF in stress-induced cellular response, HCE and hematopoietic myeloid cells were exposed to UV irradiation in the normal culture medium and to hyper-osmotic pressure containing high concentrations of sorbital. CTCF mRNA and protein expression levels in UV irradiation-induced HCE cells were detected following a time course by Northern and Western blots, respectively (Fig. 1A). Expression of CTCF mRNA in HCE cells was largely diminished in 2 h and completely disappeared in 4 h after UV irradiation. In parallel to altered CTCF mRNA expression, expression of CTCF protein in these cells was also markedly decreased within 2–4 h after UV exposure. Four types of human hematopoietic myeloid cells were used in our experiments. Upon exposure of these cells to UV irradiation, expressions of CTCF mRNA and protein were markedly suppressed down to very low levels in 6 h (Fig. 1B). Stimulation of cells by hyper-osmotic pressures was carried out by increased sorbital concentrations from 300 to 600 mOsm in the culture. Expressions of CTCF mRNA and protein were strongly diminished following increased osmotic pressures and following a time course (Fig. 1C). The results indicate that cellular CTCF activities were sensitive to UV and hyper-osmotic stresses, suggesting that CTCF is very likely to play a functional role in stress-induced cellular response.
Figure 1.
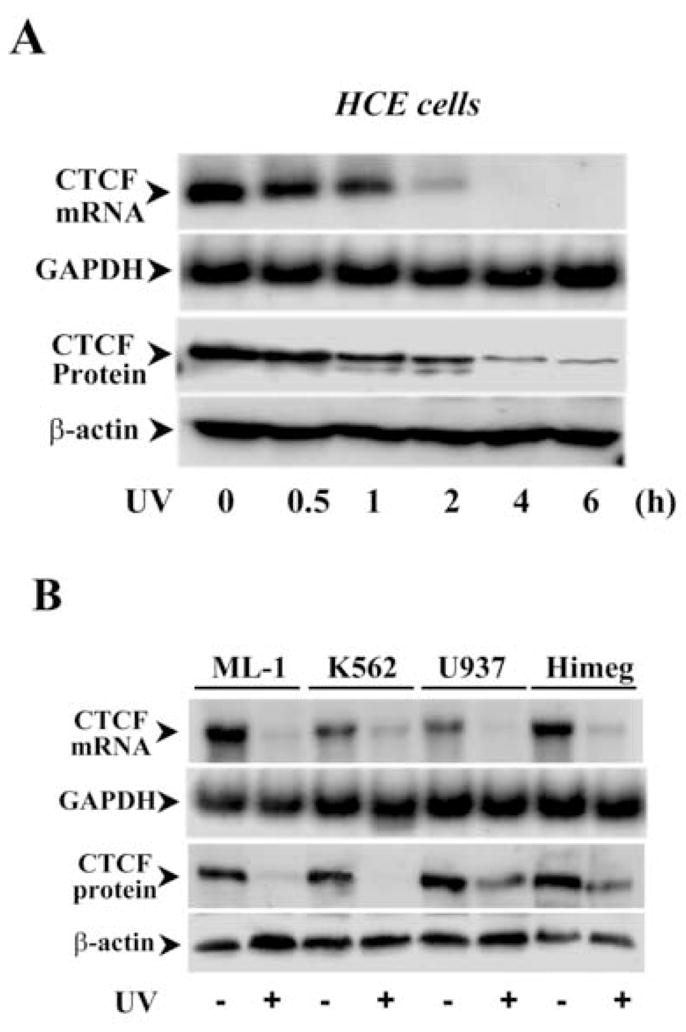
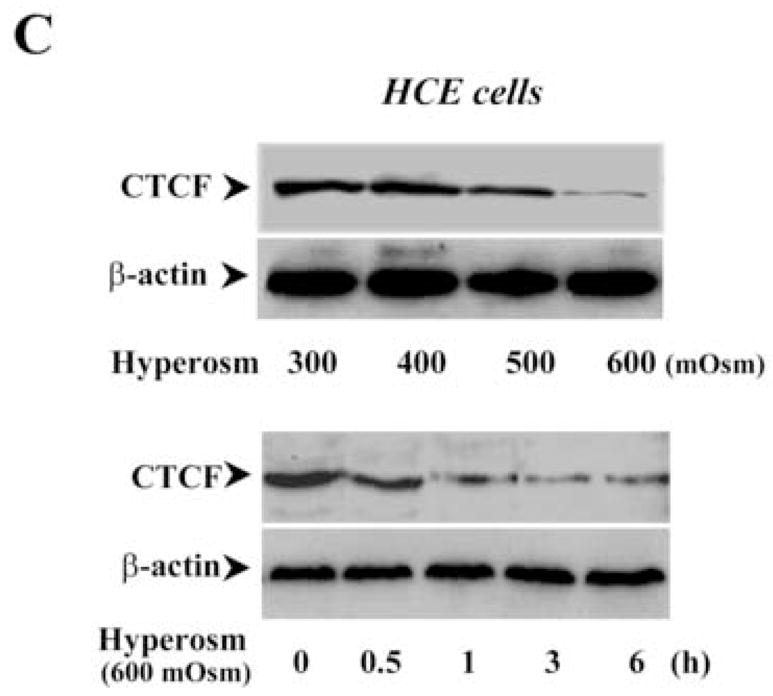
Effect of stress stimulation on CTCF expression. (A) Time course of UV irradiation-induced down-regulation of CTCF in HCE cells. (B) UV irradiation-induced down-regulation of CTCF in various hematopoietic myeloid cell types. Expression of CTCF levels was detected 1 h after the cells were exposed to UV irradiation. (C) Time course and dose-dependent response of hyper-osmotic stress-induced down-regulation of CTCF in HCE cells. CTCF mRNA and protein expression in HCE and hematopoietic myeloid cells were determined by Northern and Western analysis, respectively. GAPDH and β-actin levels were measured as the loading controls.
Effect of over-expressing CTCF on UV-induced cell death
UV irradiation-induced corneal epithelial apoptosis has been examined by using different methods in our lab including measurement of cell survival index, detection of DNA fragmentation, TUNEL stain, nuclear condensation stain and determination of caspase 3 activities [19, 23, 33]. In the present study, UV irradiation-induced HCE cell death was determined by analyzing caspase 3 activities using Western blots to measure contents of cleaved caspase 3 and PARP (a substrate for caspase 3) fragments. Apoptotic cells were determined by cell survival index via MTT assay. An expression vector containing cDNA encoding full-length CTCF gene (termed as pcDNA4-CTCF) was transfected into HCE cells to enhance cellular CTCF expression levels by using lipofection, while cells in the control group were transfected with pcDNA4 vector only. Exposure of HCE cells to UV irradiation induced decreased CTCF expression and increased caspase 3 activity in control cells. However, over-expression of CTCF in transfected HCE cells resulted in a counter effect against UV irradiation-induced caspase 3 activation and decreased CTCF expression (Fig. 2A). In addition, cell viability was significantly reduced by UV irradiation in control cells. Over-expression of CTCF in transfected cells significantly protected the cells from UV irradiation-induced death following a time course (Fig. 2B&2C). In consistence with results obtained from HCE cells, CTCF mRNA and protein expression levels were markedly enhanced by CTCF over-expression in hematopoietic myeloid ML-1 cells (Fig. 2D). UV irradiation-induced increases in caspase 3 activity were significantly suppressed in CTCF over-expressed ML-1 cells following a time course (Fig. 2E). The results suggest that UV irradiation-induced decrease in cellular CTCF levels is functionally correlated with the effectiveness of protecting cell death.
Figure 2.
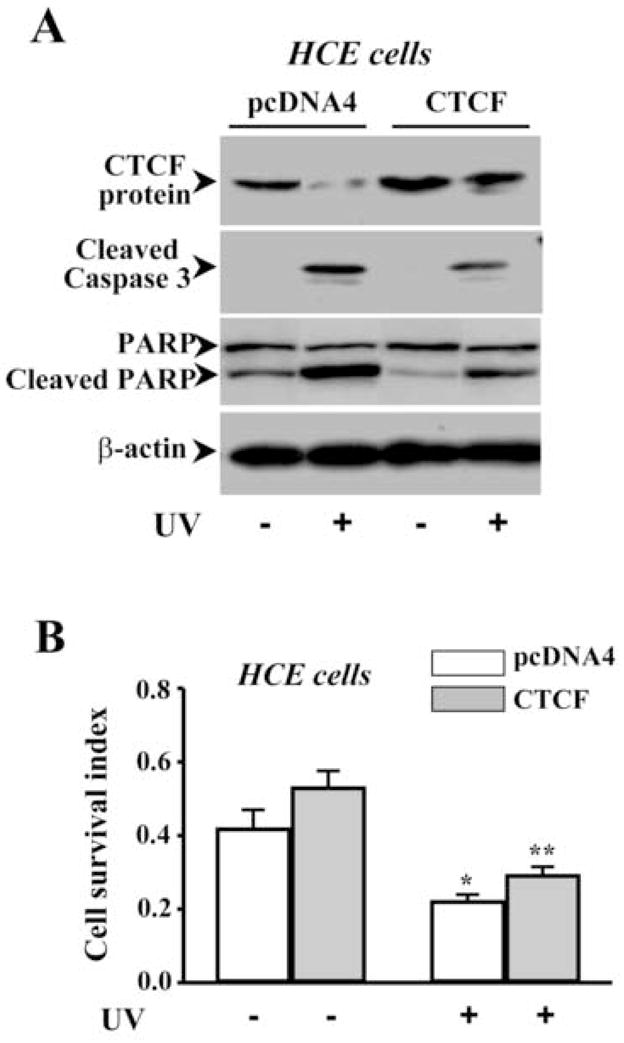
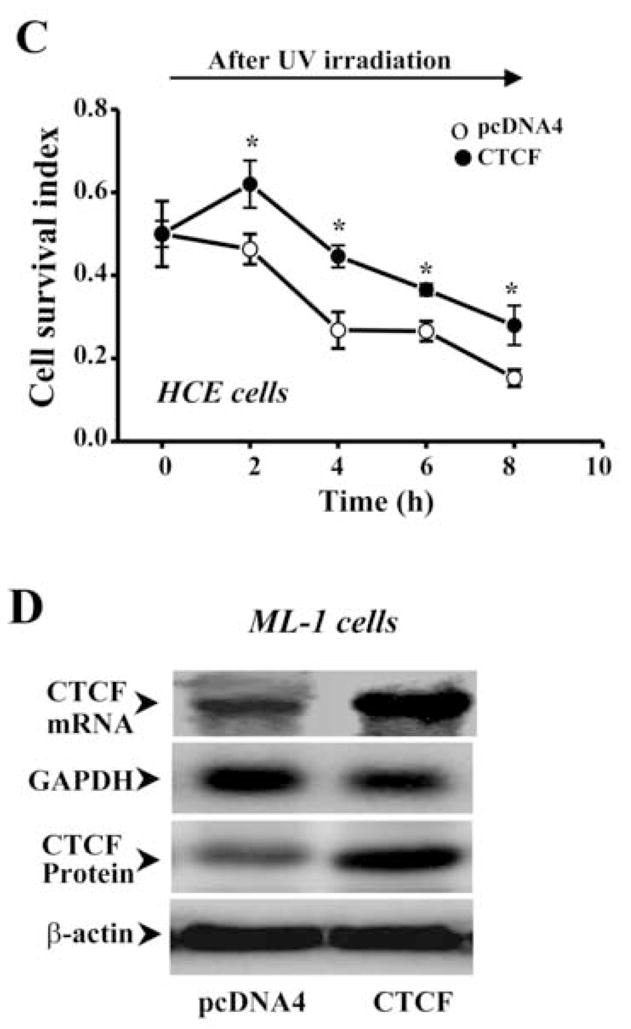
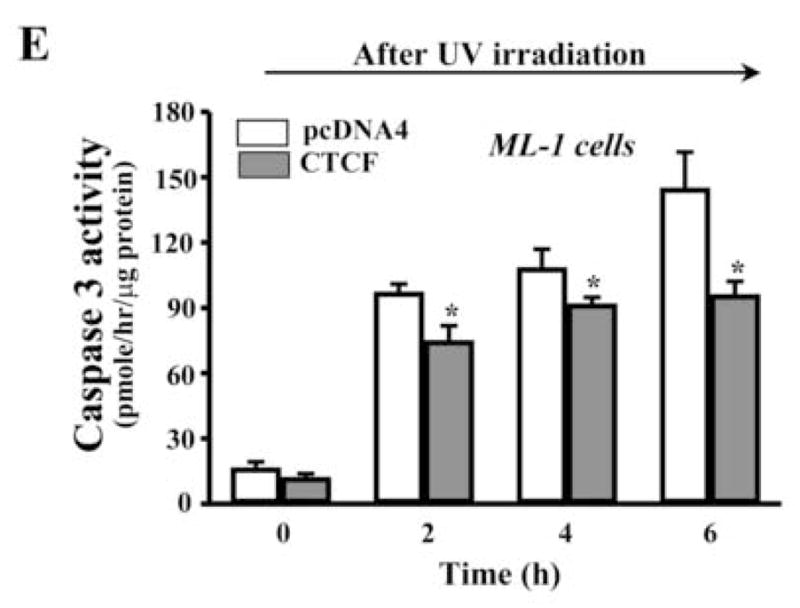
Effect of CTCF over-expression on UV irradiation-induced cell death. (A) Effect of over-expressing CTCF on UV irradiation-induced caspase 3 activation in HCE cells. (B) Effect of over-expressing CTCF on viability of HCE cells. (C) Time-dependent effect of UV irradiation on viabilities of CTCF over-expressed and vector-transfected HCE cells. (D) Over-expression of CTCF in transfected ML-1 cells. (E) Effect of over-expressing CTCF on UV irradiation-induced caspase 3 activation in ML-1 cells. HCE and ML-1 cells were transfected with pcDNA 4-CTCF cDNA, or transfected with pcDNA4 vector for control experiments. Cells were transfected for 3 days before exposure of the cells to UV irradiation. Cell survival index was determined by MTT assays. Symbols “*’ represent the statistical significance (n=6, p<0.05) between control and CTCF cDNA transfected cells in the presence and absence of UV irradiation, respectively.
Effect of Knocking down CTCF mRNA on UV-induced cell death
To further determining the effect of altered CTCF activity on UV irradiation-induced cell death, CTCF mRNA was knocked down by siRNA techniques. In previous studies, CTCF has been successfully knocked down to last at least for 5 days in HCE cells by using selected CTCF sequence-specific RNA primers [12]. In the present study, HCE cells were transfected with siRNA nucleotides specific to CTCF for 3 days and the cellular level of CTCF protein was markedly reduced compared to control cells transfected with nonspecific siRNA primers (Fig. 3A). As expected, knocking down CTCF mRNA in HCE cells by transfection of CTCF sequence-specific siRNA increased UV irradiation-induced caspase 3 activity and promoted UV irradiation-induced HCE cell death (Fig. 3B&3C). Similar approaches were also applied to ML-1 cells. Knocking down CTCF mRNA suppressed CTCF expression in both mRNA and protein levels in ML-1 cells (Fig. 3D). In addition, knocking down CTCF mRNA in ML-1 cells resulted in enhanced effects of UV irradiation on increased caspase 3 activity and decreased cell viability following a time course, respectively (Fig. 3E&3F). Results obtained from both types of CTCF knocking down cells were fairly consistent. The data provide further supporting evidence that CTCF activity is involved in UV irradiation-induced cell death.
Figure 3.
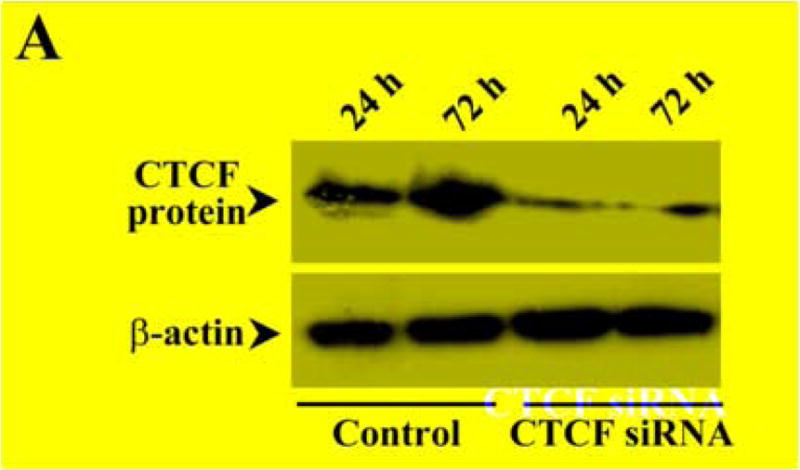
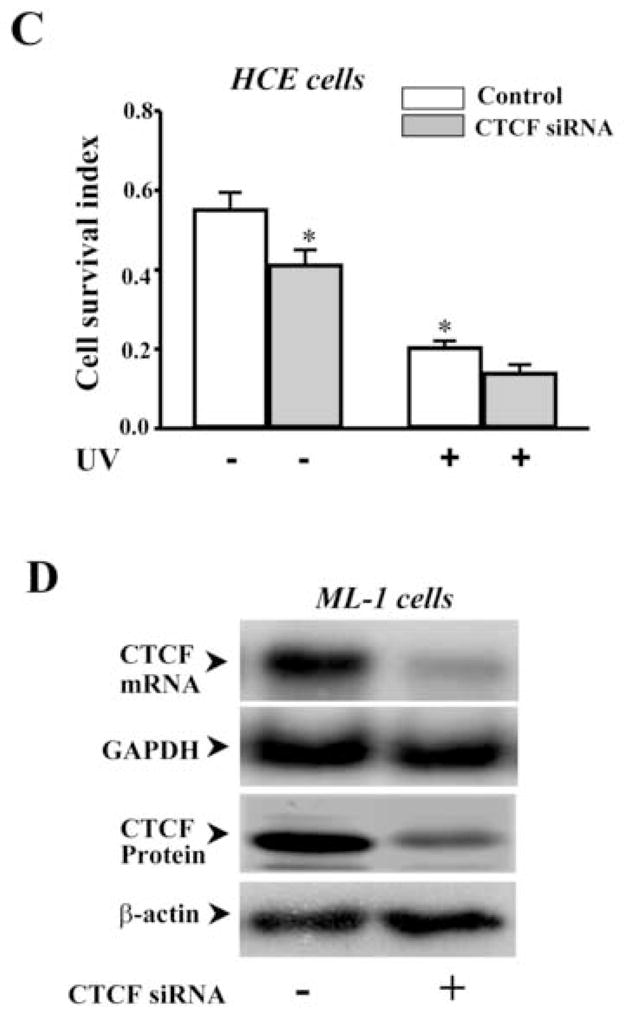
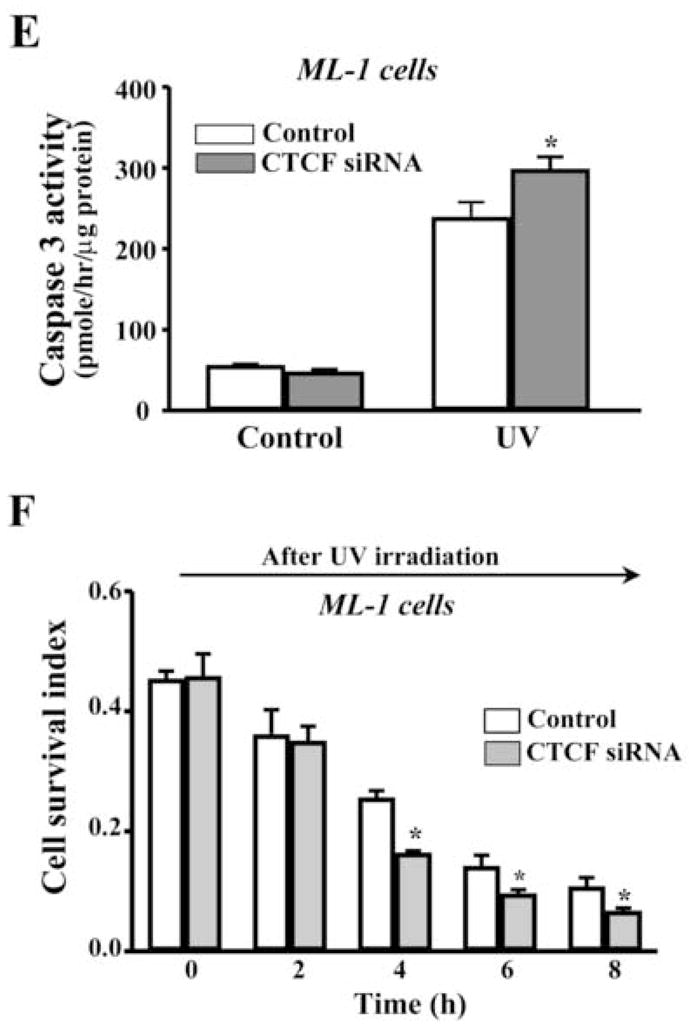
Effect of knocking down CTCF mRNA on UV irradiation-induced cell death. (A) Effect of knocking down CTCF mRNA on CTCF expression in HCE cells. (B) Effect of knocking down CTCF mRNA on UV irradiation-induced caspase 3 activation in HCE cells. (C) Effect of knocking down CTCF mRNA on viability of UV irradiation-induced HCE cells. (D) Effect of knocking down CTCF mRNA on CTCF mRNA and protein expression in ML-1 cells. (E) Effect of knocking down CTCF on UV irradiation-induced caspase 3 activation in ML-1 cells. (F) Effect of knocking down CTCF mRNA on viability of UV irradiation-induced ML-1 cells. HCE and ML-1 cells were transfected with non-specific siRNA (for the controls) or CTCF-specific siRNA for 3 days before performing experiments. Caspase 3 activity and cell viability were determined by using Western analysis and MTT assay, respectively. Symbols “*’ represent the statistical significance (n=6, p<0.05) between control and siRNA transfected cells in the presence and absence of UV irradiation, respectively.
Effect of over-expressing and knocking down CTCF on hyper-osmotic stress-induced cell death
It has reported that hyper-osmotic stress-induced decreased volume (cell shrinkage) can induce apoptotic responses specifically through activation of the p38 limb in the MAP kinase family [22, 24, 34]. The effect of hyper-osmotic stress on expressions of CTCF mRNA and protein was examined by exposure of HCE cells to high concentrations of sorbital ranging from 300 to 600 mOsm as shown in Fig. 1B. To further study the possible involvement of CTCF in hyper-osmotic stress-induced apoptosis, the effect of hyper-osmotic stress on caspase 3 activities was examined in CTCF over-expressed HCE cells. Over-expression of CTCF effectively prevented hyper-osmotic stress-induced increases in caspase 3 activities and significantly enhanced HCE cell viability in response to hyper-osmotic stress stimulation, respectively (Fig. 4A&4B). In contrast, Knockdown of CTCF mRNA in HCE cells markedly enhanced hyper-osmotic stress-stimulated increases in caspase 3 activity and significantly promoted hyper-osmotic stress-induced HCE cell apoptosis, respectively (Fig. 5A&5B). The effects of altered CTCF expression levels on hyper-osmotic stress-induced increases in caspase 3 activity and decreases in cell viability are consistent with those results observed in UV irradiation-induced cells. The data in this section provide additional evidence to support the notion that CTCF is a multifunctional protein and plays a functional role in stress-induced cell fate.
Figure 4. Effect of over-expressing CTCF on hyper-osmotic stress-induced cell death.
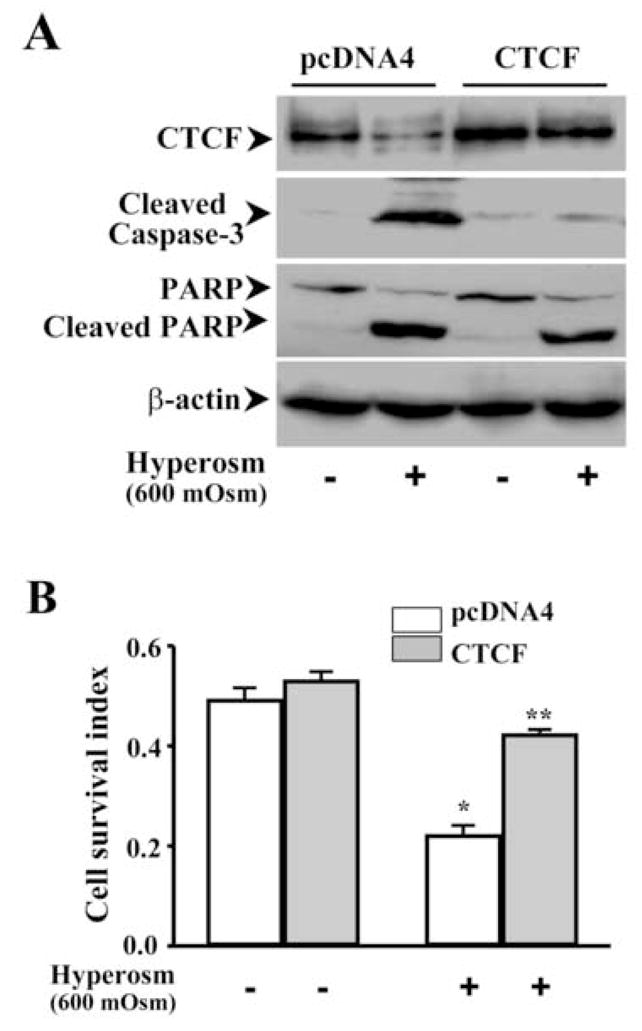
(A) Effect of over-expressing CTCF on hyper-osmotic stress-induced caspase 3 activation in HCE cells. (B) Effect of over-expressing CTCF on viability of hyper-osmotic stress-induced HCE cells. HCE cells were transfected with pcDNA 4-CTCF cDNA and transfected with pcDNA4 vector for the controls. Three days after transfection, HCE cells were exposed to 600 mOsm sorbital for 3 h. Caspase 3 activity was determined by measuring cleaved caspase 3 and PARP fragments. Cell survival index was determined by MTT assays. Symbols “*’ and “**” represent the statistical significance (n=6, p<0.05) between control and CTCF cNDA transfected cells in the presence and absence of hyper-osmotic stress, respectively.
Figure 5. Effect of knocking down CTCF on hyper-osmotic stress-induced cell death.
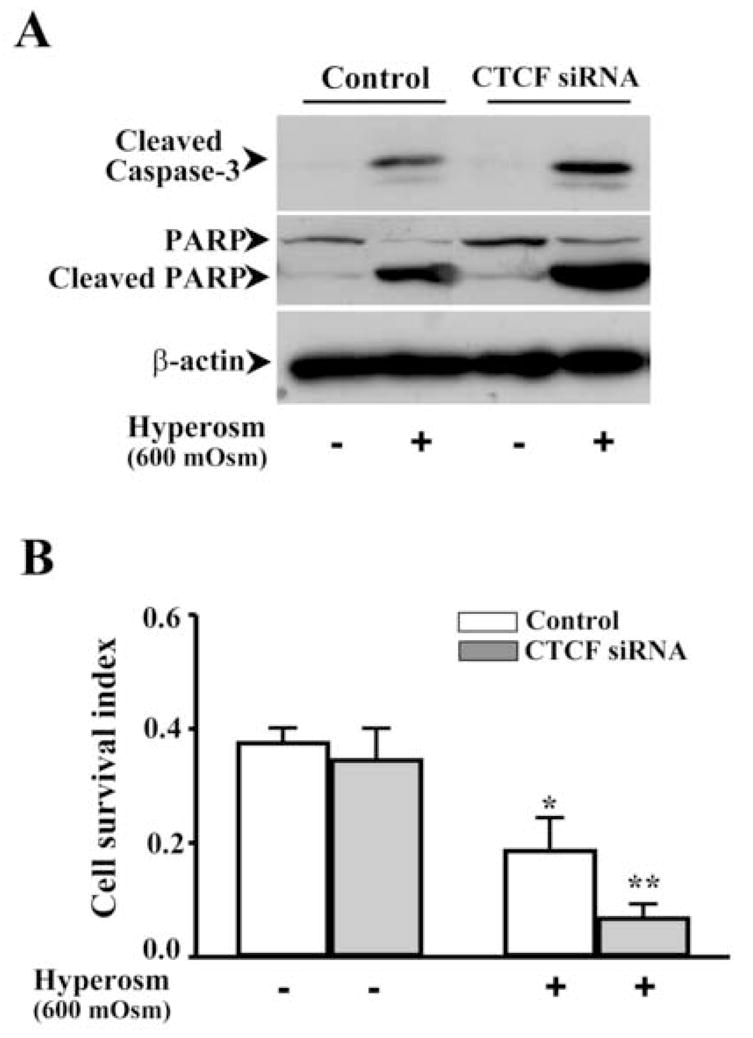
(A) Effect of knocking down CTCF mRNA on hyper-osmotic stress-induced caspase 3 activation in HCE cells. (B) Effect of knocking down CTCF mRNA on viability of hyper-osmotic stress-induced HCE cells. HCE cells were collected 3 days after transfection with CTCF-specific siRNA and non-specific siRNA for the controls. Caspase 3 activity and cell viability were determined by using Western analysis and MTT assay, respectively. Symbols “*’ represent the statistical significance (n=6, p<0.05) between control and siRNA transfected cells in the presence and absence of hyper-osmotic stress stimulation.
Effect of inhibiting UV-induced activation of NFκB on CTCF expression and apoptosis
Previous studies demonstrate that NFκB plays important roles in stress-induced death pathways. In HCE cells, UV irradiation induced activation of NFκB within 30 min detected by EMSA and the specificity of NFκB activation was verified by competitive binding of NFκB in EMSA (Fig. 6A). Nuclear protein binding of NFκB in UV irradiation-induced cells was only competitively inhibited by cold NFκB probes, but not by those of non-specific oligonucleotides. It is very likely that CTCF is involved in the stress-induced NFκB pathway. The effect of UV irradiation-activated NFκB on CTCF expression was examined by inhibition of NFκB activity with PDTC, a NFκB specific inhibitor. Inhibition of UV irradiation-induced NFκB activity with PDTC effectively reversed the effect of UV irradiation on suppression of CTCF expression in HCE and ML-1 cells (Fig. 6B). Furthermore, inhibition of NFκB with PDTC also minimized the effect of UV irradiation on HCE and ML-1 cell apoptosis (Fig. 6C). This result is consistent to the data showing the protective effect of CTCF on UV irradiation-induced cell apoptosis, which suggests that CTCF may play a role downstream in the stress-induced NFκB pathway.
Figure 6. Effect of inhibiting NFκB on UV-induced CTCF expression and apoptosis.
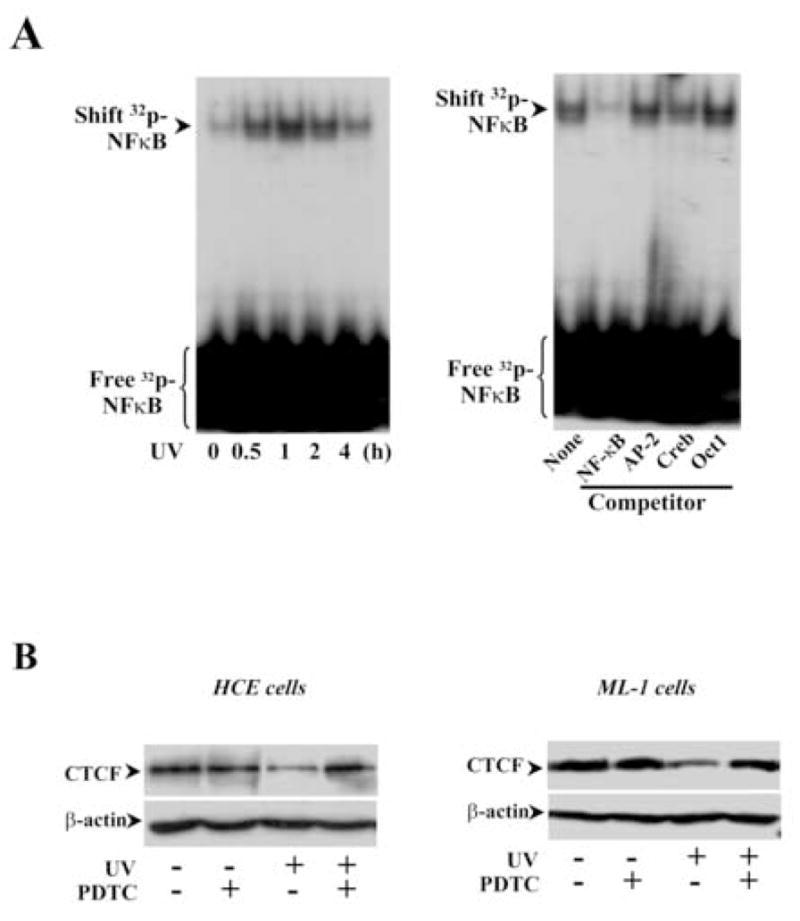
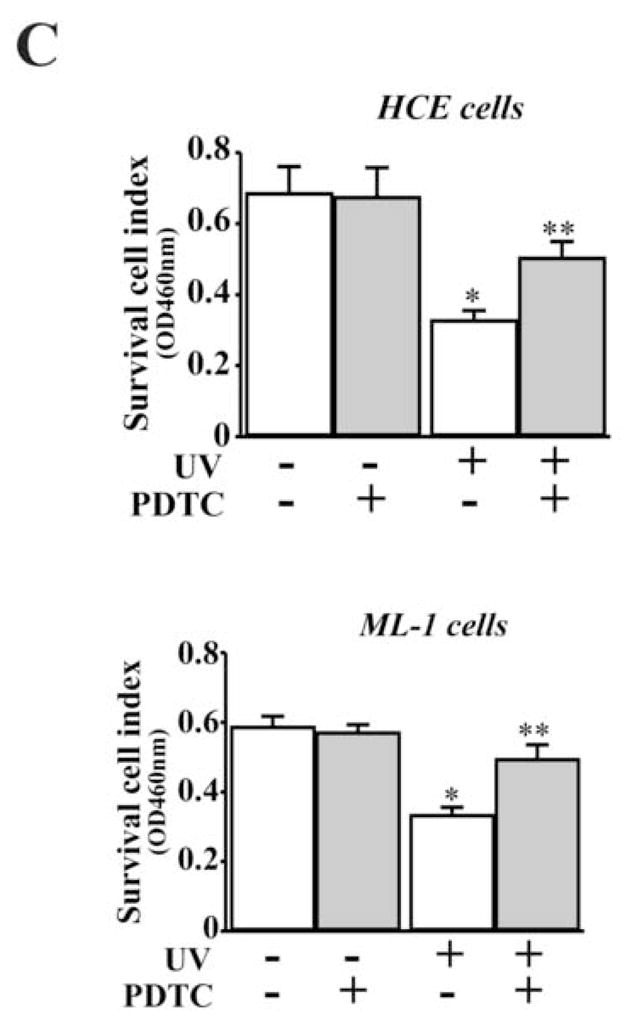
(A) Detection of UV irradiation-induced NFκB activation. The nuclear extracts from HCE cells were used in EMSA and unlabeled nucleotides of NFκB and other control probes were used in competitive experiments. (B) Effect of inhibiting NFκB on UV irradiation-induced decline of CTCF expression in HCE and ML-1 cells. (C) Effect of inhibiting NFκB on viabilities of UV irradiation-induced HCE and ML-1 cells. Cell viability was detected by MTT assays. Symbols “*’ and “**” represent the statistical significance (n=6, p<0.05) between control and UV irradiation-induced HCE and ML-1 cells in the absence and presence of PDTC, respectively.
Discussion
Environmental stress constitutes a major insult to all exposed tissues of the body, including those comprising the cornea and other cell types. The most common environmental stress includes exposure to UV irradiation, hyper-osmotic pressure and other biohazards. Apoptosis induced by UV irradiation and hyper-osmotic stress in various cells is a complex response at early times resulting from activation of cell signaling pathways that are distinct from those activated in the nucleus at later times [33]. In the present study, we found that expression of CTCF was significantly diminished following a time course in response to UV and hyper-osmotic stress stimulation (Fig. 1). CTCF is a transcription factor and multifunctional protein in controls of DNA imprinting and in regulations of gene expression. In previous studies, we report that CTCF is a downstream component in the Erk signaling pathway and plays important roles in growth factor-induced cell proliferation. The result that down-regulation of CTCF in events of stress-induced apoptotic responses is consistent with the previous studies that show up-regulation of CTCF downstream from the EGF receptor-linked Erk signaling pathway to promote HCE cell proliferation [12]. In addition, it has been shown that increased Erk activity can effectively against apoptosis in various cell types. In fact, increased cellular CTCF activity resulted from EGF-induced Erk activation plays functional roles in inhibition of Pax6 since suppression of Pax6 activity is required for EGF to stimulate corneal epithelial proliferation [12]. We believe that stress-induced down-regulation of CTCF activity is essential for HCE and hematopoietic cells to commit to UV irradiation and hyper-osmotic stress-stimulated apoptosis. In addition, activation of JNK and p38 cascades can result in apoptotic responses in corneal epithelial and hematopoietic myeloid cells [19, 22, 24]. In fact, the time course for stress-induced suppression of CTCF activity from our results is very similar to the time courses of stress-induced JNK and p38 activation in these cells [19, 22, 24]. It suggests that UV irradiation and hyper-osmotic stress-activated intracellular signaling pathways include the JNK and p38 pathways.
The effect of UV irradiation and hyper-osmotic stress stimulation on down-regulation of CTCF suggests that CTCF is a downstream component in stress-induced signaling pathways. Up to date, there is a little knowledge about the upstream signaling components that are involved in regulation of CTCF. Consistent with data from previous studies, we found that UV irradiation induced increased NF-κB DNA binding activity (Fig. 6). It is known that that increased NF-κB DNA binding activity can result in corresponding downstream effects. To test whether CTCF is one of these downstream events, UV irradiation-induced activation of NFκB was suppressed by application of NFκB-specific inhibitor. Suppression of NFκB activity effectively reversed the inhibitory effect of UV irradiation on CTCF expression and protected HCE and ML-1 cells from UV irradiation-induced apoptosis, suggesting CTCF may be a target gene of NFκB. The new finding of the regulatory relationship between NFκB and CTCF may explain the contrary effects of CTCF in pro- and anti-apoptosis in various cells in response to different stimuli because a specific set of NF-κB target genes is selectively activated or repressed based on the nature of stimuli [35]. However, the regulatory relationship between NFκB and CTCF and the role of CTCF in downstream events of stress-induced signaling pathways require further investigations in future studies.
CTCF is an important multifunctional factor that regulates cell function by controlling gene expression and by interacting with other cell signaling components. To verify the notion that stress-induced down-regulation of CTCF is essential for eliciting apoptotic response, cellular CTCF activity was manipulated to increase and to decrease by over-expression of CTCF and by knockdown of CTCF mRNA, respectively. Over-expression of full-length cDNA encoding full length CTCF effectively suppressed the effect of UV and hyper-osmotic stress-induced apoptosis. In contrast, knockdown of CTCF mRNA using CTCF specific siRNA significantly enhanced UV and hyper-osmotic stress-induced apoptosis. The results confirmed the effect of UV and hyper-osmotic stress stimulation on diminishing CTCF expression, indicating that down-regulation of CTCF in stress-induced cells is required for these cells to commit apoptosis. Taken together, CTCF activity in HCE and hematopoietic myeloid cells is regulated by stress stimulation. It is very likely that CTCF involves tress-induced cellular response by regulating stress-induced alterations of immediate early gene expression. The present study provides evidence for a new functional role of CTCF that involves regulating UV irradiation and hyper-osmotic stress-induced apoptosis in these cells. In addition, the anti-apoptotic function of CTCF in mediating stress-induced cellular response is consistent with previous findings that CTCF is regulated by growth factors and cytokines, resulting in controlling cell fate.
Acknowledgments
This study was supported by NIH grant EY15282 to L.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Filippova GN, Fagerlie S, Klenova EM, Myers C, Dehner Y, Goodwin G, Neiman PE, Collins SJ, Lobanenkovs VV. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol. 1996;16:2802–13. doi: 10.1128/mcb.16.6.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Awad TA, Bigler J, Ulmer JE, Hu YJ, Moore JM, Lutz M, Neiman PE, Collins SJ, Renkawitz R, Lobanenkov VV, Filippovas GN. Negative transcriptional regulation mediated by thyroid hormone response element 144 requires binding of the multivalent factor CTCF to a novel target DNA sequence. J Biol Chem. 1999;274:27092–8. doi: 10.1074/jbc.274.38.27092. [DOI] [PubMed] [Google Scholar]
- 3.Burcin M, Arnold R, Lutz M, Kaiser B, Runge D, Lottspeich F, Filippova GN, Lobanenkov VV, Renkawitzs R. Negative protein 1, which is required for function of the chicken lysozyme gene silencer in conjunction with hormone receptors, is identical to the multivalent zinc finger repressor CTCF. Mol Cell Biol. 1997;17:1281–8. doi: 10.1128/mcb.17.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Filippova GN, Lindblom A, Meincke LJ, Klenova EM, Neiman PE, Collins SJ, Doggett NA, Lobanenkovs VV. A widely expressed transcription factor with multiple DNA sequence specificity, CTCF, is localized at chromosome segment 16q22.1 within one of the smallest regions of overlap for common deletions in breast and prostate cancers. Genes Chromosomes Cancer. 1998;22:26–36. [PubMed] [Google Scholar]
- 5.Li T, Lu Z, Lus L. Regulation of eye development by transcription control of CCCTC binding factor (CTCF) J Biol Chem. 2004;279:27575–83. doi: 10.1074/jbc.M313942200. [DOI] [PubMed] [Google Scholar]
- 6.Burke LJ, Hollemann T, Pieler T, Renkawitzs R. Molecular cloning and expression of the chromatin insulator protein CTCF in Xenopus laevis. Mech Dev. 2002;113:95–8. doi: 10.1016/s0925-4773(02)00005-9. [DOI] [PubMed] [Google Scholar]
- 7.Vostrov AA, Quitschkes WW. The zinc finger protein CTCF binds to the APBbeta domain of the amyloid beta-protein precursor promoter. Evidence for a role in transcriptional activation. J Biol Chem. 1997;272:33353–9. doi: 10.1074/jbc.272.52.33353. [DOI] [PubMed] [Google Scholar]
- 8.Bell AC, Felsenfelds G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–5. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- 9.Nakagawa H, Chadwick RB, Peltomaki P, Plass C, Nakamura Y, de La Chapelles A. Loss of imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. Proc Natl Acad Sci U S A. 2001;98:591–6. doi: 10.1073/pnas.011528698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Percec I, Bartolomeis MS. Genetics. Do X chromosomes set boundaries? Science. 2002;295:287–8. doi: 10.1126/science.1068663. [DOI] [PubMed] [Google Scholar]
- 11.Wu D, Li T, Lu Z, Dai W, Xu M, Lus L. Effect of CTCF-binding motif on regulation of PAX6 transcription. Invest Ophthalmol Vis Sci. 2006;47:2422–9. doi: 10.1167/iovs.05-0536. [DOI] [PubMed] [Google Scholar]
- 12.Li T, Lus L. Epidermal growth factor-induced proliferation requires down-regulation of Pax6 in corneal epithelial cells. J Biol Chem. 2005;280:12988–91295. doi: 10.1074/jbc.M412458200. [DOI] [PubMed] [Google Scholar]
- 13.Delgado MD, Chernukhin IV, Bigas A, Klenova EM, Leons J. Differential expression and phosphorylation of CTCF, a c-myc transcriptional regulator, during differentiation of human myeloid cells. FEBS Lett. 1999;444:5–10. doi: 10.1016/s0014-5793(99)00013-7. [DOI] [PubMed] [Google Scholar]
- 14.El-Kady A, Klenovas E. Regulation of the transcription factor, CTCF, by phosphorylation with protein kinase CK2. FEBS Lett. 2005;579:1424–34. doi: 10.1016/j.febslet.2005.01.044. [DOI] [PubMed] [Google Scholar]
- 15.Qi CF, Martensson A, Mattioli M, Dalla-Favera R, Lobanenkov VV, Morse HC., 3rds CTCF functions as a critical regulator of cell-cycle arrest and death after ligation of the B cell receptor on immature B cells. Proc Natl Acad Sci U S A. 2003;100:633–8. doi: 10.1073/pnas.0237127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torrano V, Chernukhin I, Docquier F, D’Arcy V, Leon J, Klenova E, Delgados MD. CTCF regulates growth and erythroid differentiation of human myeloid leukemia cells. J Biol Chem. 2005;280:28152–61. doi: 10.1074/jbc.M501481200. [DOI] [PubMed] [Google Scholar]
- 17.Torrano V, Navascues J, Docquier F, Zhang R, Burke LJ, Chernukhin I, Farrar D, Leon J, Berciano MT, Renkawitz R, Klenova E, Lafarga M, Delgados MD. Targeting of CTCF to the nucleolus inhibits nucleolar transcription through a poly(ADP-ribosyl)ation-dependent mechanism. J Cell Sci. 2006;119:1746–59. doi: 10.1242/jcs.02890. [DOI] [PubMed] [Google Scholar]
- 18.Docquier F, Farrar D, D’Arcy V, Chernukhin I, Robinson AF, Loukinov D, Vatolin S, Pack S, Mackay A, Harris RA, Dorricott H, O’Hare MJ, Lobanenkov V, Klenovas E. Heightened expression of CTCF in breast cancer cells is associated with resistance to apoptosis. Cancer Res. 2005;65:5112–22. doi: 10.1158/0008-5472.CAN-03-3498. [DOI] [PubMed] [Google Scholar]
- 19.Lu L, Wang L, Shells B. UV-induced signaling pathways associated with corneal epithelial cell apoptosis. Invest Ophthalmol Vis Sci. 2003;44:5102–9. doi: 10.1167/iovs.03-0591. [DOI] [PubMed] [Google Scholar]
- 20.Kleiman NJ, Wang RR, Spectors A. Ultraviolet light induced DNA damage and repair in bovine lens epithelial cells. Curr Eye Res. 1990;9:1185–93. doi: 10.3109/02713689009003475. [DOI] [PubMed] [Google Scholar]
- 21.Rosette C, Karins M. Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science. 1996;274:1194–7. doi: 10.1126/science.274.5290.1194. [DOI] [PubMed] [Google Scholar]
- 22.Wang L, Xu D, Dai W, Lus L. An ultraviolet-activated K+ channel mediates apoptosis of myeloblastic leukemia cells. J Biol Chem. 1999;274:3678–85. doi: 10.1074/jbc.274.6.3678. [DOI] [PubMed] [Google Scholar]
- 23.Wang L, Li T, Lus L. UV-induced corneal epithelial cell death by activation of potassium channels. Invest Ophthalmol Vis Sci. 2003;44:5095–101. doi: 10.1167/iovs.03-0590. [DOI] [PubMed] [Google Scholar]
- 24.Xu D, Wang L, Olson JE, Lus L. Asymmetrical response of p38 kinase activation to volume changes in primary rat astrocytes. Exp Biol Med (Maywood) 2001;226:927–33. doi: 10.1177/153537020122601008. [DOI] [PubMed] [Google Scholar]
- 25.Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Daviss RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–37. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 26.Galcheva-Gargova Z, Derijard B, Wu IH, Daviss RJ. An osmosensing signal transduction pathway in mammalian cells. Science. 1994;265:806–8. doi: 10.1126/science.8047888. [DOI] [PubMed] [Google Scholar]
- 27.Devary Y, Gottlieb RA, Smeal T, Karins M. The mammalian ultraviolet response is triggered by activation of Src tyrosine kinases. Cell. 1992;71:1081–91. doi: 10.1016/s0092-8674(05)80058-3. [DOI] [PubMed] [Google Scholar]
- 28.Engelberg D, Klein C, Martinetto H, Struhl K, Karins M. The UV response involving the Ras signaling pathway and AP-1 transcription factors is conserved between yeast and mammals. Cell. 1994;77:381–90. doi: 10.1016/0092-8674(94)90153-8. [DOI] [PubMed] [Google Scholar]
- 29.Knebel A, Rahmsdorf HJ, Ullrich A, Herrlichs P. Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO Journal. 1996;15:5314–25. [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, Xu D, Dai W, Lus L. An ultraviolet-activated K+ channel mediates apoptosis of myeloblastic leukemia cells. J Biol Chem. 1999;274:3678–85. doi: 10.1074/jbc.274.6.3678. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Franco RS, Wang Y, Pan HQ, Eaton D, Cheng T, Kaushansky K, Dais W. Megakaryocytic differentiation of HIMeg-1 cells induced by interferon gamma and tumour necrosis factor alpha but not by thrombopoietin. Cytokine. 1998;10:880–9. doi: 10.1006/cyto.1998.0345. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Gong B, Dai W, Lus L. Identification of immediate early genes during TPA-induced human myeloblastic leukemia ML-1 cell differentiation. Gene. 1998;216:293–302. doi: 10.1016/s0378-1119(98)00345-x. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Dai W, Lus L. Ultraviolet irradiation-induced K(+) channel activity involving p53 activation in corneal epithelial cells. Oncogene. 2005;24:3020–7. doi: 10.1038/sj.onc.1208547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friis MB, Friborg CR, Schneider L, Nielsen MB, Lambert IH, Christensen ST, Hoffmanns EK. Cell shrinkage as a signal to apoptosis in NIH 3T3 fibroblasts. J Physiol. 2005;567:427–43. doi: 10.1113/jphysiol.2005.087130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyamotos S. RelA life and death decisions. Mol Cell. 2004;13:763–4. doi: 10.1016/s1097-2765(04)00158-3. [DOI] [PubMed] [Google Scholar]