

Summary
COMMD1 is the prototype of a new protein family that plays a role in several important cellular processes, including NF-κB signaling, sodium transport, and copper metabolism. The COMMD proteins interact with one another via a conserved C-terminal domain, whereas distinct functions are predicted to result from a variable N-terminal domain. The COMMD proteins have not been characterized biochemically or structurally. Here we present the solution structure of the N-terminal domain of COMMD1 (N-COMMD1, residues 1-108). This domain adopts an α-helical structure that bears little resemblance to other all helical proteins. The compact nature of N-COMMD1 suggests that full-length COMMD proteins are modular, consistent with specific functional properties for each domain. Interactions between N-COMMD1 and partner proteins may occur via complementary electrostatic surfaces. These data provide a new foundation for biochemical characterization of COMMD proteins and for probing COMMD1 protein-protein interactions at the molecular level.
Keywords: MURR1, COMMD1, COMMD6, NF-κB, Wilson disease protein
Introduction
The COMMD1 protein, also known as MURR1, is the founding member of the COMMD (copper metabolism gene MURR1 domain) family. COMMD1 was originally identified as the protein encoded by the defective gene in canine copper toxicosis,1 and human COMMD1 has been demonstrated to interact with the copper binding N-terminus of the Wilson disease protein.2 On the basis of this interaction, its subcellular localization pattern,3 and the canine copper toxicosis phenotype,4, 5 COMMD1 has been proposed to play a role in the copper-dependent intracellular trafficking of the Wilson disease protein.1, 3, 6, 7 COMMD1 also interacts with the X-linked inhibitor of apoptosis (XIAP), which regulates the degradation of COMMD1 by acting as an E3 ubiquitin ligase.8 Notably, copper binding by XIAP has been linked to a lowering of the apoptotic threshold.9
In addition to its apparent role in copper homeostasis, COMMD1 is involved in sodium transport and in NF-κB transcriptional regulation.6 Interactions of COMMD1 with the human epithelial sodium channel (ENaC) have been detected, and coexpression of COMMD1 and ENaC inhibits the sodium current.10 Similar to the Wilson protein, ENaC activity is regulated by relocalization,11 and it is possible that COMMD1 plays a role in this trafficking.6, 10 In the case of NF-κB, COMMD1 interacts with the NF-κB inhibitor IκBα as well as with the cullin 1 component of the E3 ligase complex, which is required for degradation of phospho-IκBα12 These data suggest that COMMD1 might prevent the degradation of IκBα and subsequent translocation of NF-κB to the nucleus. COMMD1 does not suppress nuclear translocation of NF-κB, however, and has recently been shown to affect the interaction between NF-κB and chromatin.13 Interestingly, the interaction of COMMD1 with NF-κB requires full-length COMMD1, suggesting that the N-terminus as well the C-terminal COMM domain is involved.13 The role of COMMD1 in this pathway has attracted much attention because by inhibiting NF-κB, COMMD1 restricts HIV-1 replication in resting CD4+ lymphocytes.12
A screen to identify additional COMMD1 binding partners led to the identification of nine proteins with homology to CMD1.13 These proteins, designated COMMD2-COMMD10, share a highly conserved 70-85 residue C-terminal domain, the COMM-domain. The N-terminal regions of the 10 COMMD proteins vary in sequence and length ranging from 18 residues for COMMD6 to 151 residues for COMMD5. All 10 COMMD proteins interact with each other, and the C-terminal COMM domain serves as an interface for these COMMD:COMMD interactions.13, 14 Genes encoding COMMD proteins are found in mammals and lower metazoans, but not in unicellular eukaryotes or bacteria.13 In human tissues, they are expressed widely with a different relative abundance profile for each COMMD protein.3, 13. Several COMMD proteins in addition to COMMD1 inhibit NF-κB activation, and different COMMD proteins interact preferentially with specific NF-κB subunits.13
Despite the variety of protein partners and the identification of 9 homologs, the exact function of the COMMD proteins remains unknown. One major obstacle is that the COMMD proteins are not homologous to any known proteins and contain no previously characterized functional domains or motifs. Furthermore, no COMMD proteins have been characterized biochemically. The extensive conservation of COMMD proteins, the different tissue preferences, and the differential binding to NF-κB subunits suggest that each COMMD protein has a distinct function. Since the N-terminal regions vary, these regions might confer specific functions on each COMMD protein. To gain insight into the cellular function of COMMD1, we have determined the solution structure of the N-terminal region of human COMMD1 (residues 1-108, N-COMMD1).
Results and Discussion
Preparation of N-COMMD1
The full-length COMMD1 protein with a predicted molecular mass of 21 kDa is prone to aggregation and proteolysis (data not shown). Gel filtration chromatography shows the presence of higher aggregates that elute with the void volume and a second major peak with an apparent molecular mass of 57 kDa (Supplemental Figure S1). This second peak may correspond to a dimer of non-spherical shape. Addition of glucose, sucrose, n-octyl-β-D-glucopyranoside, or imidazole did not reduce aggregation, and precipitation occurred upon storage at concentrations higher than 5 mg/ml. Since the C-terminal COMM-domain is involved in COMMD:COMMD protein interactions, this domain likely aggregates at the high protein concentrations required for structure determination. To obtain a soluble sample, three constructs for the N-terminal domain comprising residues 1-108, 1-115, and 1-118 were generated. The variant 1-118 excludes the predicted COMM-domain, whereas the construct ending at residue 108 omits an additional stretch including some hydrophobic residues (LMNQSRWNSG). The variant 1-108 (N-COMMD1) exhibited the best soluble expression profile and was folded with a high α-helical content by CD spectroscopy (Supplemental Figure S2). In addition, N-COMMD1 was a single, monomeric species by gel filtration (apparent molecular mass 19 kDa), and no aggregates were observed (Supplemental Figure S3).
NMR Structure Determination
The backbone amide resonances of N-COMMD1 were completely assigned except for residues Met1, Ala20, and the five proline residues (Pro11, Pro28, Pro41, Pro44, Pro45, and Pro50). More than 95% of all detectable sidechain 1H and 13C resonances were assigned. From the integration of the 3D 15N-and 13C-edited NOESY and 2D aromatic NOESY spectra, a total of 2436 distance constraints were obtained (Table 1). In addition, 47 torsion angle backbone constraints derived from TALOS were employed for the structure calculation. Figure 1 depicts the backbones of the 20 energy minimized conformers that represent the solution structure of N-COMMD1. The atomic root mean square (r.m.s.) deviation about the mean coordinates for the 20 conformers for all residues is 1.665 Å for the backbone nuclei and 2.106 Å for all heavy atoms. These values decrease to 0.643 Å and 1.155 Å, respectively, if the disordered regions (residues 1-8, 20-29, and 102-108) of the protein are excluded. An analysis of the 20 conformers with PROCHECK-NMR indicates that 98.8% of all residues are in allowed regions of the Ramachandran plot.
Table 1.
NMR Structure Determination Statistics
| Restraint Statistics | |
|---|---|
| Distance restraints | |
| Unambiguous NOE-based restraints | 1892 |
| Intraresidue | 957 |
| Sequential (|i-j|=1) | 370 |
| Medium-range (1<|i-j| ≤ 4) | 353 |
| Long-range (|i-j| > 4) | 212 |
| Ambiguous NOE-based restraints | 544 |
| Torsion angle restraints | |
| Protein backbone Φ and Ψ restraints | 47 |
| Ensemble statistics for structure quality | |
| Restraint satisfaction | |
| RMS differences for distance restraints (Å) | 0.0143 +/-0.0003 |
| RMS differences for torsion angle restraints (°) | 0.4592 +/-0.0145 |
| Deviations from ideal covalent geometry | |
| Bond length (Å) | 0.0019 +/-0.0001 |
| Bond angles (°) | 0.3910 +/-0.0020 |
| Impropers (°) | 0.2452 +/-0.0029 |
| Ramachandran plot statistics (%) | |
| Residues in most favored regions | 70.4 |
| Residues in allowed regions | 28.4 |
| Residues in disallowed regions | 1.2 |
| Average atomic rmsds from the average structure (Å) | |
| All atoms | 2.106 |
| All atoms in ordered regions* | 1.155 |
| Backbone atoms (N, Cα, C’) | |
| All atoms | 1.665 |
| Residues in ordered regions* | 0.643 |
Ordered regions include residues 9-19 and 30-101.
Figure 1.
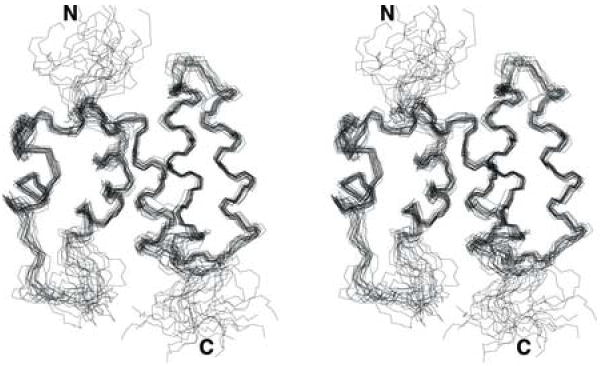
Stereoview of the 20 conformers representing the solution structure of N-COMMD1. The well-ordered regions comprise residues 9-19 and 30-101.
The 15N-HSQC spectrum of N-COMMD1 revealed the presence of 22 additional peaks of lower intensity indicating the presence of a second, minor conformation. Of these additional peaks, 17 were assigned to residues Ala3, Gly4, Glu5, Leu6, Glu7, Ala18, Gln21, Gly26, Gly29, Ile30, Thr31, Leu34, Ser37, Gln38, Tyr40, Val43, and Glu107 based on their corresponding signals in HNCACB, CBCA(CO)NH, C(CO)NH, HNCO and HC(CO)NH spectra. Since these residues show very weak traces in the 13C-NOESY and 15N-NOESY spectra and most of them are located in the disordered regions of the final structure, a calculation for this minor second conformation was not pursued.
Structure of N-COMMD1
N-COMMD1 adopts a compact, monomeric, and completely α-helical fold in the absence of the C-terminal COMM-domain (Figure 2). The dimensions of the protein are approximately 40 Å × 30 Å × 20 Å. The solvent-exposed surface area is 7366 Å2, which is greater than that predicted for a typical globular monomeric protein of this size (approximately 5950 Å2).15 Almost all the hydrophobic residues are sequestered within the protein interior. At the N-terminus, an irregular helix (α1) and a short helix (α2) are connected via a long disordered loop (residues 20-29). This loop as well as α2 can adopt a second minor conformation. Notably, this region shows some variability in the otherwise highly conserved COMMD1 sequence (Figure 3). Since this loop is adjacent to the C-terminus of N-COMMD1, it may contact the C-terminal COMM-domain in the full-length protein. This interaction could be mediated by a group of aromatic residues (Phe24, His25, Tyr27) located within the disordered loop.
Figure 2.
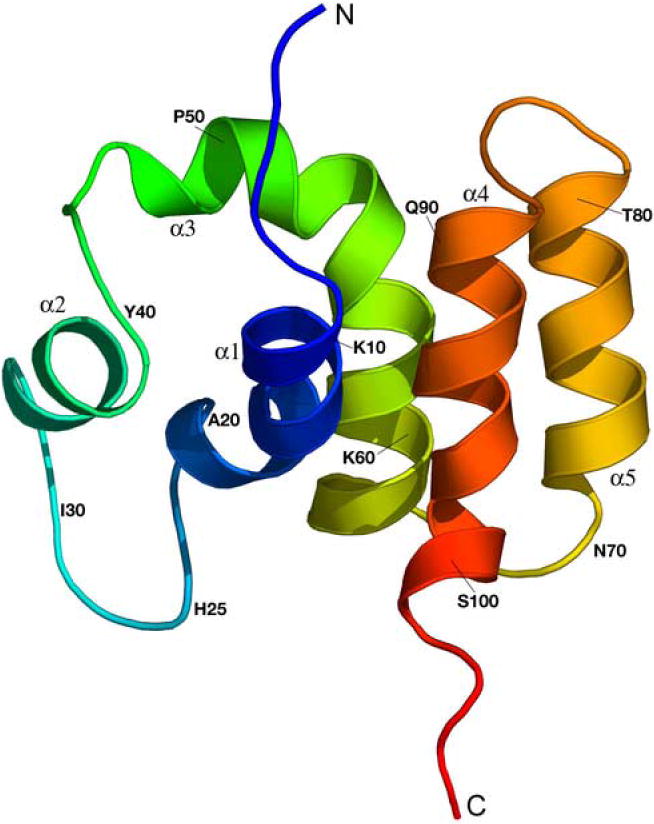
Ribbon diagram of the averaged and energy minimized structure of N-COMMD1 generated with PyMol. The N-terminus is colored in blue and the C-terminus in red.
Figure 3.
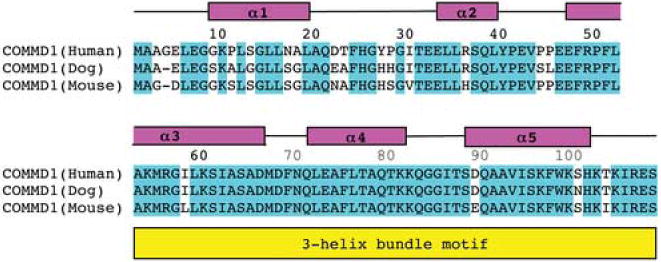
Sequence alignment of human N-COMMD1 with its homologs from dog and mouse showing the secondary structure elements and the pattern of conserved residues. The amino acid sequences were obtained form Swiss-Prot (entries Q8N668, Q8WMD0, and Q8K4M5) and the alignment was generated with ClustalW.40
The third helix, α3, is bent in the middle by almost 90°. The first of two intron boundaries in the gene encoding COMMD13 falls within this helix at residue 60. Although most intron boundaries are typically found in coils and at the ends of secondary structure elements, some do occur within α-helices and β-strands.16 The C-terminal half of helix α3 starting at residue 53 forms a three-helix bundle with helices α4 and α5. A comparison with other protein structures using DALI17 identified several proteins with a three-helix bundle segment similar to that in N-COMMD1, including the tyrosine phosphatase SptP from Salmonella enterica,18 the vesicular transport protein Sec17 from Saccharomyces cerevisiae,19 the Rab geranylgeranyltransferase from rat brain,20 and a de novo designed three-helix bundle protein.21 Three-helix bundles are ubiquitous and functionally diverse, occurring in DNA-binding proteins, enzymes, and structural proteins, often as subdomains of larger proteins.22 The other structural features of N-COMMD1 including the bend in helix α3 and the arrangement of the two N-terminal helices are unique, however, and the combination of these elements with a three-helix bundle has not been observed in any other structure.
Although COMMD1 has been implicated in copper homeostasis, its sequence does not contain any obvious copper binding motifs, and no potential metal binding sites are evident in the N-COMMD1 structure. There are no cysteine residues in N-COMMD1 (Figure 3) and the three methionine and two histidine residues are located in spatially distant positions. Thus, the role of COMMD1 in copper homeostasis is probably mediated indirectly through other proteins.
Protein-Protein Interactions
Calculation of the electrostatic potential surface of N-COMMD1 using PDB2PQR23 and APBS24 reveals two positively charged regions. Residues Arg49, Lys54, Arg56, and Lys60 from helices α3 and α4 and residue Lys82 comprise one positive patch (Figure 4, right). On the opposite face of the protein, Lys10 together with Lys96, Lys99, His101, and Lys104 from the C-terminus of α5 generate a second positively charged surface (Figure 4, left). These regions might serve as docking sites for partner proteins with complementary electrostatic surfaces.
Figure 4.
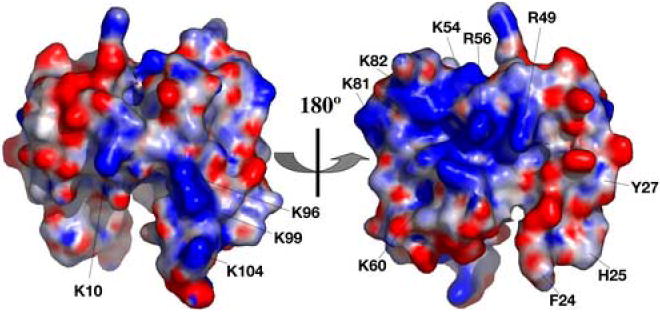
Electrostatic surface of N-COMMD1 calculated with the programs PDB2PQR23 and APBS24 and displayed with PyMOL. The surface on the left corresponds to the orientation shown in Figure 2. The surface is color-coded according to electrostatic potential: red, -10 kT; white, 0 kT; blue, +10 kT.
For several COMMD1 protein partners, the relative contributions of the COMMD1 N- or C-terminal domains have been investigated. First, the COMMD:COMMD interactions have been mapped to the C-terminal COMM-domain.13, 14 Second, the interaction between COMMD1 and the IκBα ankyrin repeats is mediated through COMMD1 residues 1-160, and an N-terminal construct comprising residues 1-80 is not sufficient for interaction.12 Analysis of the human IκBα crystal structures25, 26 reveals two negatively charged patches which could conceivably interact with the positively charged residues at the C-terminus of N-COMMD1 (vide supra). These patches, which are located at opposite ends of the ankyrin repeat domain, comprise residues Glu 85, Glu 86, Glu 125, and Glu 128 and residues Glu 282, Glu 284, Asp 285, Glu 286, and Glu 292. Finally, full length COMMD1 is required for interaction with the RelA subunit of NF-κB. The RelA surface is dominated by positive charge,27 however, suggesting that an electrostatic interaction with N-COMMD1 is unlikely. Furthermore, other COMMD proteins can also interact with the RelA subunit, indicating this interaction is probably mediated through the common C-terminal COMM-domain.
Several other interaction partners, for which no information regarding the relative involvement of the N-and C-terminal COMMD1 domains is available, also exhibit negatively charged surface patches. The cullin1 component of the E3 ligase complex has several areas of negative charge, but these are primarily located on a long stalk-like region that has been proposed to act as a spacer and not as a docking site.28 The BIR3 domain of XIAP has a small, elongated surface stretch of negative charge.29 Finally, homology models of the Wilson disease protein N-terminus predict patches of negative charges on metal binding domains 2, 4, and 5.30
In sum, the structure of N-COMMD1 reveals a novel α-helical fold with little resemblance to other all α-helical protein structures. The compact nature of N-COMMD1 suggests that full-length COMMD proteins are modular with specific functional properties for each domain. Several positively charged surface patches may provide interaction surfaces for partner proteins. Taken together, these observations provide a new starting point for probing COMMD1 protein-protein interactions at the molecular level.
Materials and Methods
Cloning, Expression, and Purification of COMMD1
A plasmid containing the cDNA for the entire coding region of COMMD1 was generously supplied by Dr. Jonathan Gitlin (Washington University School of Medicine). The constructs comprising full-length COMMD1 as well as different variations of the N-terminal domain (residues 1-108, 1-115, and 1-118) were generated by PCR using the forward primer (5’-GGTATTGAGGGTCGCATGGCGGCGGGCGAGCTTGAG-3’) for all constructs and the reverse primers (5’-AGAGGAGAGTTAGAGCCTCAGTTAGGCTGGCTGATCAGTG-3’) for full-length COMMD1, (5’-AGAGGAGAGTTAGAGCCCTAGCTCTCACGGATTTTTGTCTTGTG-3’) for residues 1-108, (5’-AGAGGAGAGTTAGAGCCCTACCAGCGGCTCTGGTTCATGAG-3’) for residues 1-115, and (5’-AGAGGAGAGTTAGAGCCCTACCCGCTATTCCAGCGGCTCTG-3’) for residues 1-118. The PCR products were gel purified and inserted into a pET32Xa/LIC vector following the ligation independent cloning protocol from Novagen. This vector includes an N-terminal thioredoxin tag followed by a (His)6 tag, a thrombin cleavage site, an S-tag, and a Factor Xa cleavage site. The plasmids were transformed into Rosetta(DE3)pLysS cells for expression. Cells containing plasmids for N-terminal COMMD1 constructs were grown at 30 °C and 250 rpm in Luria-Bertani (LB) medium supplemented with 34 μg/ml chloramphenicol and 100 μg/ml carbenicillin. Protein expression was induced with 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG) at an A600 of 0.7-0.9 in 1 l cultures. The cells were harvested after 3 hrs. The procedure for full-length COMMD1 was similar to that described for N-COMMD1 except for a reduction of the growth temperature to 16 °C before IPTG addition and a later harvest after 18-20 hrs.
The cells were resuspended in 50 ml 50 mM Tris-HCl (pH 8.0), 100 mM NaCl (buffer A), stored at -20 °C, and lysed by freeze-thawing. The lysate was supplemented with 1 mM phenylmethylsulfonylfluoride (PMSF) and clarified by ultracentrifugation at 125,000 × g for 1 hr at 4 °C. For full-length COMMD1, an additional protease inhibitor cocktail (P8849 from Sigma-Aldrich) was used. The proteins were bound to a slurry of Ni(II)-loaded Chelating Sepharose (Amersham Biosciences) in buffer A and washed several times with 50 mM imidazole, 50 mM Tris-HCl (pH 8.0), 100 mM NaCl to remove contaminating proteins. To cleave the affinity tags, the slurry was incubated with 200 units of Factor Xa (Novagen) in buffer A supplemented with 5 mM CaCl2 at room temperature for 16-18 hrs. At this point, 1 mM PMSF was added to stop the proteolytic cleavage. The affinity tags remained bound to the Ni(II)-loaded Chelating Sepharose, and the cleaved products were eluted with buffer A.
To determine the oligomerization state of full length COMMD1 and the N-terminal construct spanning residues 1-108, denoted N-COMMD1, Superdex 75 and Superdex 200 size exclusion columns were calibrated using the Amersham Biosciences gel filtration calibration kit. For full-length COMMD1, multiple buffer systems, including buffer A, buffer A supplemented with 10-20% glycerol, 10-20% sucrose, or 0.5-1% n-octyl-β-D-glucopyranoside, 100 mM imidazole (pH 8.0), 100 mM NaCl, 0.2 mM tris-(2-carboxyethyl)-phosphine (TCEP) and 300 mM imidazole (pH 8.0), 300 mM NaCl, 0.2 mM TCEP were tested. To check for secondary structure, CD spectra were recorded on a Jasco J-715 spectropolarimeter in a quartz cuvette of 5 mm path length at room temperature.
NMR Sample Preparation
Rosetta(DE3)pLys cells containing the N-COMMD1 plasmid were grown at 37 °C and 250 rpm in M9 minimal medium containing 1× BME vitamin solution (Sigma), 1.5 g/l 15N-ammonium sulfate (Spectra Stable Isotopes), and 4 g/l 13C6-D-glucose (Spectra Stable Isotopes). Chloramphenicol and carbenicillin were added to final concentrations of 34 μg/ml and 100 μg/ml, respectively. Protein expression was induced with 0.5 mM IPTG at an A600 of 0.7-0.9 and the cells were harvested 3 hrs thereafter.
N-COMMD1 was purified as described above with one additional step of reverse-phase high pressure liquid chromatography (HPLC) using a C18 column (Vydac) with a mobile phase gradient ranging from 0.1% trifluoroacetic acid (TFA) in 40% acetonitrile to 0.1% TFA in 80% acetonitrile. The main HPLC peak was collected and lyophilized resulting in a total protein yield of 10 mg. Sample 1 contained 0.6 mM protein in 25 mM sodium phosphate (pH 6.1), 50 mM NaCl, 0.2% NaN3 (buffer P) with 10% D2O (Sigma). Sample 2 contained 0.95 mM protein in buffer P with 100% D2O (Sigma). The protein concentration was determined by absorbance measurements at 280 nm using an extinction coefficient of 10.5 mM-1 cm-1 obtained from amino acid hydrolysis.
NMR Structure Determination
All NMR spectra were acquired at 25 °C on a Varian Inova 600 MHz spectrometer equipped with a triple resonance, xyz gradient probe. NMR data processing and analyses were performed with NMRPipe31 and SPARKY.31, 32 Backbone and side chain 1H, 15N, and 13C resonances were assigned from three-dimensional (3D) HNCACB, CBCA(CO)NH, C(CO)NH, HNCO, HC(CO)NH, HCCH-COSY, and HCCH-TOCSY spectra.33 Aromatic 1H and 13C resonances were assigned from 2D (HB)CB(CGCD)HD and (HB)CB(CGCDCE)HE spectra.34 NOE restraints were obtained from a 3D 15N-edited NOESY (in H2O) and a 13C-edited NOESY as well as a 2D aromatic NOESY (in D2O) with mixing times of 100ms for all spectra. Polypeptide backbone Φ and Ψ torsion angle restraints were derived from an analysis of Hα, Cα, Cβ, C’, and backbone 15N chemical shifts using TALOS.35 No additional restraints, such as hydrogen bonds, were included. Structures were calculated using ARIA (version 1.2).36 All NOEs were calibrated automatically and assigned iteratively by ARIA followed by a manual inspection for errors after every refinement cycle. Structures were calculated from extended backbone conformations as starting models. The default settings in the ARIA run.cns task file were used except for the following parameters. The number of steps in the simulated annealing protocol was doubled for improved convergence, and the final force constants for the distance and torsion angle restraints were set to 50 kcal mol-1 Å-2 and 200 kcal mol-1 rad-2, respectively. Eighty structures were calculated in the last iteration, of which 20 were chosen for further analysis. None of these structures have NOE or dihedral violations greater than 0.5 Å or 5°, respectively. The structures were analyzed with PROCHECK-NMR37 and figures were generated with MOLMOL38 or PyMOL.39
Accession Codes
The coordinates and constraints for N-COMMD1 have been deposited in the Protein Data Bank with accession code 2H2M. The chemical shift assignments have been deposited in the Biological Magnetic Resonance Data Bank (BMRB) with accession code 7300.
Supplementary Material
Acknowledgments
We thank Jonathan Gitlin for materials and valuable discussions. This work was supported by NIH grant GM58518.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Van De Sluis B, Rothuizen J, Pearson PL, Van Oost BA, Wijmenga C. Identification of a new copper metabolism gene by positional cloning in a purebred dog population. Hum Mol Genet. 2002;11:165–173. doi: 10.1093/hmg/11.2.165. [DOI] [PubMed] [Google Scholar]
- 2.Tao TY, Liu F, Klomp L, Wijmenga C, Gitlin JD. The copper toxicosis gene product Murr1 interacts directly with the Wilson disease protein. J Biol Chem. 2003;278:41593–41596. doi: 10.1074/jbc.C300391200. [DOI] [PubMed] [Google Scholar]
- 3.Klomp AEM, Van De Sluis B, Klomp LWJ, Wijmenga C. The ubiquitously expressed Murr1 protein is absent in canine copper toxicosis. J Hepatol. 2003;39:703–709. doi: 10.1016/s0168-8278(03)00380-5. [DOI] [PubMed] [Google Scholar]
- 4.Hyun C, Filippich LC. Inherited canine copper toxicosis in Australian Bedlington Terriers. J Vet Sci. 2004;5:19–28. [PubMed] [Google Scholar]
- 5.Fuentealba IC, Aburto EM. Animal models of copper-associated liver disease. Comp Hepatol. 2003;2:5–17. doi: 10.1186/1476-5926-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Bie P, Van De Sluis B, Klomp L, Wijmenga C. The many faces of the copper metabolism protein Murr1/COMMD1. J Heredity. 2005;96:803–811. doi: 10.1093/jhered/esi110. [DOI] [PubMed] [Google Scholar]
- 7.Wijmenga C, Klomp LWJ. Molecular regulation of copper excretion in the liver. Proc Nutr Soc. 2004;63:31–39. doi: 10.1079/pns2003316. [DOI] [PubMed] [Google Scholar]
- 8.Burstein E, Ganesh L, Dick RD, Van De Sluis B, Wilkinson JC, Klomp LWJ, et al. A novel role for XIAP in copper homeostasis through regulation of Murr1. EMBO J. 2004;23:244–254. doi: 10.1038/sj.emboj.7600031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mufti AR, Burstein E, Csomos RA, Graf PCF, Wilkinson JC, Dick RD, et al. XIAP is a copper binding protein deregulated in Wilson’s disease and other copper toxicosis disorders. Mol Cell. 2006;21:775–785. doi: 10.1016/j.molcel.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 10.Biasio W, Chang T, McIntosh CJ, McDonald FJ. Identification of Murr1 as a regulator of the human delta epithelial sodium channel. J Biol Chem. 2003;7:5429–5434. doi: 10.1074/jbc.M311155200. [DOI] [PubMed] [Google Scholar]
- 11.Hanwell D, Ishikawa T, Saleki R, Rotin D. Trafficking and cell surface stability of the epithelial Na+ channel expressed in epithelial Madin-Darby canine kidney cells. J Biol Chem. 2002;12:9772–9779. doi: 10.1074/jbc.M110904200. [DOI] [PubMed] [Google Scholar]
- 12.Ganesh L, Burstein E, Guha-Niyogi A, Louder MK, Mascola JR, Klomp LWJ, et al. The gene product Murr1 restricts HIV-1 replication in resting CD4+ lymphocytes. Nature. 2003;426:853–857. doi: 10.1038/nature02171. [DOI] [PubMed] [Google Scholar]
- 13.Burstein E, Hoberg JE, Wilkinson AS, Rumble JM, Csomos RA, Komarck CM, et al. COMMD proteins: a novel family of structural and functional homologs of Murr1. J Biol Chem. 2005;280:22222–22232. doi: 10.1074/jbc.M501928200. [DOI] [PubMed] [Google Scholar]
- 14.De Bie P, Van De Sluis B, Burstein E, Duran KJ, Berger R, Duckett CS, et al. Characterization of COMMD protein-protein interactions in NF-kB signalling. Biochem J. 2006 doi: 10.1042/BJ20051664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller S, Janin J, Lesk AM, Chothia C. Interior and surface of monomeric proteins. J Mol Biol. 1987;196:641–656. doi: 10.1016/0022-2836(87)90038-6. [DOI] [PubMed] [Google Scholar]
- 16.Contreras-Moreira B, Jonsson PF, Bates PA. Structural context of exons in protein domains: implications for protein modelling and design. J Mol Biol. 2003;333:1045–1059. doi: 10.1016/j.jmb.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 17.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 18.Stebbins CE, Galan JE. Modulation of host signaling by a bacterial mimic: structure of the Salmonella effector SptP bound to Rac1. Mol Cell. 2000;6:1449–1460. doi: 10.1016/s1097-2765(00)00141-6. [DOI] [PubMed] [Google Scholar]
- 19.Rice LM, Brunger AT. Crystal structure of the vesicular transport protein Sec17: Implications for SNAP function in SNARE complex assembly. Mol Cell. 1999;4:85–95. doi: 10.1016/s1097-2765(00)80190-2. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H, Saebra MC, Deisenhofer J. Crystal structure of Rab geranylgeranyltransferase at 2.0 A resolution. Structure. 2000;8:241–251. doi: 10.1016/s0969-2126(00)00102-7. [DOI] [PubMed] [Google Scholar]
- 21.Walsh STR, Cheng H, Bryson JW, Roder H, DeGrado WF. Solution structure and dynamics of a de novo designed three-helix bundle protein. Proc Natl Acad Sci USA. 1999;96:5486–5491. doi: 10.1073/pnas.96.10.5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider JP, Lombardi A, DeGrado WF. Analysis and design of three-stranded coiled coils and three-helix bundles. Fold Des. 1998;3:R29–R40. doi: 10.1016/S1359-0278(98)00011-X. [DOI] [PubMed] [Google Scholar]
- 23.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup, execution, and analysis of Poisson-Boltzmann electrostatic calculations. Nucleic Acid Research. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacobs MD, Harrison SC. Structure of an IkBa/NFkB complex. Cell. 1998;95:749–758. doi: 10.1016/s0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- 26.Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkBa/NF-kB complex reveals mechanisms of NF-kB inactivation. Cell. 1998;95:759–770. doi: 10.1016/s0092-8674(00)81699-2. [DOI] [PubMed] [Google Scholar]
- 27.Chen FE, Huang D-B, Chen Y-Q, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kB bound to DNA. Nature. 1998;391:410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- 28.Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, et al. Structure of the Cul1-Rbx1-Skp1-Fbox(Skp2) SCF ubiquitin ligase complex. Nature. 2002;416:703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- 29.Sun C, Cai M, Maedows RP, Xu N, Gunasekera AH, Herrmann J, et al. NMR structure and mutagenesis of the third Bir domain of the inhibitor of apoptosis protein XIAP. J Biol Chem. 2000;275:33777–33781. doi: 10.1074/jbc.M006226200. [DOI] [PubMed] [Google Scholar]
- 30.Walker JM, Huster D, Ralle M, Morgan CT, Blackburn NJ, Lutsenko S. The N-terminal metal-binding site 2 of the Wilson’s disease protein plays a key role in the transfer of copper from Atox1. J Biol Chem. 2004;279:15376–15384. doi: 10.1074/jbc.M400053200. [DOI] [PubMed] [Google Scholar]
- 31.DeLaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 32.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francsico: 2002. [Google Scholar]
- 33.Bax A, Grzesiek S. Methodological advances in protein NMR. Acc Chem Res. 1993;26:131–138. [Google Scholar]
- 34.Yamazaki T, Forman-Kay JD, Kay LE. Two-dimensional NMR experiments for correlating 13Cb and 1Hd/e chemical shifts of aromatic residues in 13C-labeled proteins via scalar couplings. J Am Chem Soc. 1993;115:11054–11055. [Google Scholar]
- 35.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 36.Linge JP, Habeck M, Rieping W, Nilges M. ARIA: automated NOE assignment and NMR structure calculation. Bioinformatics. 2003;19:315–316. doi: 10.1093/bioinformatics/19.2.315. [DOI] [PubMed] [Google Scholar]
- 37.Laskowski RA, Rullmann JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 38.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 39.Delano WL. The PyMOL molecular graphics system. DeLano Scientific; San Carlos, CA: 2002. [Google Scholar]
- 40.Thompson JD, Higgins DG, Gibson TJ. CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.