

Abstract
Hypothalamic IL1 is suggested to be a critical mediator of the central effects of the adipocyte hormone leptin on energy balance. We hypothesized that IL1 receptor signaling is required for exogenously administered leptin to cause anorexia and weight loss, but not for physiological effects of endogenous leptin signaling on energy balance. To test this hypothesis, we investigated whether chronic hypothalamic over-expression of an IL1 receptor antagonist (AdV-IL1ra) alters food intake and weight gain in normal rats. Our findings demonstrate that impaired IL1 signaling in the CNS did not cause excess weight gain over a period of 11 days (AdV-IL1ra +38.1±4.1g vs. VEH +42.2±5.6g; p=0.6) and caused a slight reduction daily food intake (AdV-IL1ra 29.0±1.1g/day vs. VEH 33.0±1.6g/day; p<0.05). Blocking central IL1 signaling also did not alter the re-feeding response to a prolonged fast, yet was entirely effective in preventing the anorexic effect of exogenously administered leptin (2mg/kg ip, cumulative food intake at 18h AdV-IL1ra 30.5±1.1g vs. VEH 26.4±1.7g, p<0.05) and prevented leptin-induced weight loss (AdV-IL1ra −0.1±1.3g vs. VEH −2.7±1.9g, p<0.05). Together these findings suggest that hypothalamic IL1 signaling is required for the pharmacological effects of leptin administration, but that impaired hypothalamic IL1 signaling does not alter the physiological regulation of energy balance.
Keywords: Inflammation, hypothalamus, appetite, cytokine
Introduction
Following the identification of the adipocyte hormone leptin as a physiological regulator of energy balance, a large body of work has begun to define the critical neuronal pathways that mediate its behavioral and metabolic effects [20,21,25]. Neuronal melanocortin signaling plays a key role since genetic or pharmacological disruption of melanocortin signaling blocks leptin action and invariably leads to obesity [2,3]. Several findings also implicate the pro-inflammatory cytokine IL1β as a mediator of leptin action in the brain. First, leptin-induced anorexia is prevented by intracerebroventricular (icv) administration of an IL1 receptor antagonist [12,29]. In addition, our own studies, as well as those of others, have shown that exogenous leptin administration increases hypothalamic IL1β signaling [10,28] supporting a potential role for IL1β in mediating the CNS effects of leptin. Unlike mutations that disrupt leptin or melanocortin signaling, however, targeted mutation of IL1 receptors do not cause obesity in mice [7] challenging the notion that this signaling pathway plays a physiological role in energy homeostasis.
Based on these observations, we hypothesized that IL1 receptor signaling is required for responses to pharmacological leptin administration, but not for physiological effects of endogenous leptin signaling. To test this hypothesis, we sought to determine whether hypothalamic IL1 signaling is necessary for energy balance under physiological conditions, in addition to its known role in the response to exogenous leptin administration [12,29]. Specifically, we investigated whether chronic hypothalamic over-expression of an IL1 receptor antagonist alters food intake and weight gain in normal rats. Our data demonstrate that impaired IL1 signaling in the CNS does not increase daily food intake or weight gain, nor does it alter the re-feeding response to a prolonged fast, yet is entirely effective in preventing the anorexic effect of exogenously administered leptin.
Results
Effect of adenoviral-induced CNS over-expression of IL1ra on body weight and food intake
Following adenoviral gene transfer surgery all rats were monitored without further intervention for a period of 11 days (Fig. 1 period 1). During this time, weight gain was not different between AdV-IL1ra injected animals and controls (38.1±4.1g vs. 42.2±5.6g; respectively, p=0.6). Cumulative food intake during this time period was slightly lower in AdV-IL1ra injected rats than in control animals (mean 29.0±1.1g/day vs. 33.0±1.6g/day; respectively, p=0.05). Though IL1ra is a secreted protein, following gene transfer of the adenoviral-IL1ra vector into the third ventricle, diffusion of the protein from the third ventricle into key hypothalamic nuclei may be necessary for effects on energy balance. Both weight gain (34±5g) and daily food intake (28.5±1.6g) were no different in animals having received AdV-IL1ra in the third ventricle (3rdV AdV-IL1ra) in comparison to the combined AdV-IL1ra data presented above. Thus chronic adenoviral-mediated CNS over-expression of IL1ra does not cause weight gain, and, if anything, causes a modest reduction in food intake.
Fig. 1.
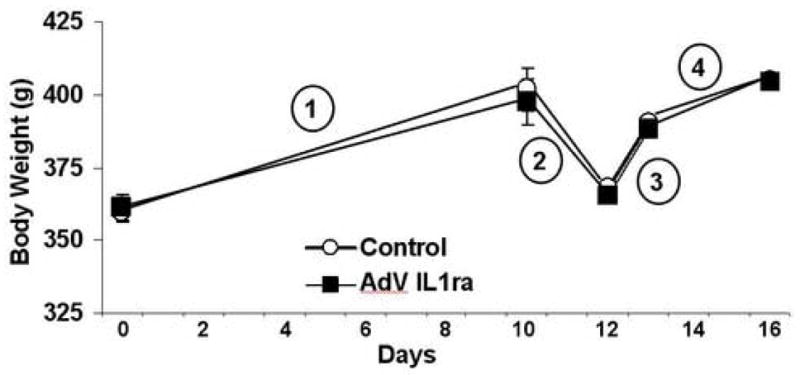
Effect of CNS adenoviral over-expression of IL1ra on body weight. Mean body weight in the AdV-IL1ra group (black square, n=15) and Control group (white circle, n=6) are shown over time. Period 1 demonstrates the weight gain in both groups during the 11 days following adenoviral injection surgery. Period 2 shows the weight loss induced by a 36h fast. Period 3 demonstrates the weight regain following 1 day of re-feeding. Period 4 shows the weight gain following 4d of re-feeding. Data presented are means ± SEM. Statistical analysis by unpaired Student’s t-test.
Effect of adenoviral-induced CNS over-expression of IL1ra on food intake and body weight recovery following a prolonged fast
Since re-feeding following a prolonged fast is, perhaps, the only physiological 5 paradigm in which plasma leptin concentration increases rapidly [11,27], we evaluated whether adenoviral-mediated CNS over-expression of IL1ra alters the feeding response following a fast. Weight loss at the end of the 36h fast was not different between the two groups (AdV-IL1ra rats −32.3±1.1g vs. −34.1±2.2g in Control rats, p=0.4, Fig 1 period 2). Evaluation of food intake during the day prior to the fast was consistent with the mean daily food intake described above (AdV-IL1ra 28.6±0.8g and Control 33.0±1.1g; p=0.01) During re-feeding, food intake in the AdV-IL1ra group was not different from that in Control rats at any of the four time points evaluated (Fig. 2). Weight regain 24h following the end of the fast (AdV-IL1ra 23±1.2g and Control 23±1.7g, p=0.9, Fig. 1 period 3) and body weight 96h after the fast (AdV-IL1ra 404±8.0g and Control 405±8.5g, p=0.9, Fig. 1 period 4) were also equivalent between the two groups. In response to fasting and re-feeding, food intake (2h 10.3±0.4g; 4h 13.5±0.6g; 6h 16.6±0.8g; 8h 22.5±0.8g; 18h 40.4±3.5g) and body weight changes (fasting −32±1.5g, first day of re-feeding 23±1.6g, 4 days post fast 42±6g) were no different in 3rdV AdV-IL1ra relative to the AdV-IL1ra group as a whole. Thus, chronic adenoviral-mediated CNS over-expression of IL1ra does not alter the regulation of food intake or body weight in response to fasting or re-feeding.
Fig. 2.
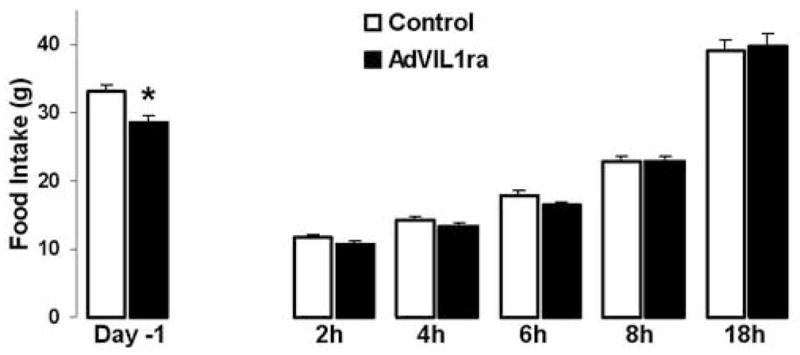
Effect of CNS adenoviral over-expression of IL1ra on food intake. Food intake (g) in two adenoviral injection groups, Control (white bars, n=6) and AdV-IL1ra (black bars, n=10) on the day prior to a 36h fast (Day -1) and at five indicated time points following the initiation of re-feeding. Data presented are means ± SEM. * p<0.05 by unpaired Student’s t-test.
Effect of adenoviral-induced CNS over-expression of IL1ra on leptin-induced anorexia
Previous studies, including our own, have shown that acute icv administration of IL1ra blocks the effect of leptin to reduce food intake [12,29]. Thus, after allowing five days for experimental animals to recover from the fasting/re-feeding protocol, we evaluated whether adenoviral-mediated CNS over-expression of IL1ra protects rats against leptin-induced anorexia. Administration of exogenous leptin (2mg/kg ip) caused significant reductions of food intake in Control rats relative to vehicle injection at all evaluated time points (2h food intake 4.2±0.5g vs. 5.6±0.3g; 4h food intake 7.1±0.9g vs. 9.7±0.9g; and 18h food intake 26.4±1.7g vs. 30.5±1.1g, p<0.05 vs. Vehicle injection for all time points). In contrast, leptin administration had no effect on food intake in the AdV-IL1ra group relative to vehicle injection (2h food intake 6.1±0.7g vs. 5.2±0.4g; 4h food intake 11.5±0.8g vs. 10.4±0.5g; and 18h food intake 30.3±0.9g vs. 29.6±1.7g, all p=ns). In addition, leptin administration induced significant weight loss in Control rats (−2.7±1.9g, p=0.02 vs. Vehicle injection) but not in the AdV-IL1ra group (−0.1±1.3g, p=0.36 vs. Vehicle injection). Similar to our previous results, leptin-induced changes in food intake (2h 5.7±0.9g; 4h 11.4±1.2g; 18h 28.7±2.6g) were no different in 3rdV AdV-IL1ra rats relative to the AdV-IL1ra group as a whole. Thus, chronic adenoviral-mediated over-expression of IL1ra in the CNS prevents the ability of exogenous, pharmacological leptin administration to cause anorexia and weight loss.
Discussion
The current studies were undertaken to determine if impaired IL1β signaling in the CNS alters the ability of leptin to regulate energy balance in a physiological, as well as a pharmacological context. Specifically, we determined whether chronic adenoviral mediated over-expression of an endogenous IL1RI antagonist, IL1ra, in the CNS causes changes in food intake and body weight in both rats fed ad libitum and in response to fasting, and in a pharmacological paradigm of altered leptin signaling. Our results demonstrate that whereas chronic adenoviral-mediated over-expression of IL1ra has no impact on baseline food intake or weight gain in rats, on the amount of weight lost during a fast, or on food intake or weight gain during re-feeding, this intervention, nonetheless, completely prevents the ability of exogenously administered leptin to reduce food intake and body weight.
Understanding the CNS pathways activated by leptin has been a major focus of obesity research in the last decade. Previous studies, including our own, have shown that leptin administration stimulates production of IL1β, an anorectic cytokine, in the hypothalamus [10,28] while conversely, administration of IL1ra into the 3rd cerebral ventricle blocks the effects of exogenously administered leptin on food intake and weight loss [12,29]. However, unlike the proven role of leptin as a physiological regulator of energy balance [24], IL1β signaling in the CNS is principally implicated as a pathological ‘sickness’ signal induced by disease states that trigger inflammation [17,28]. Moreover, unlike leptin- or leptin receptor-deficient mice, mutant animals lacking the IL1β receptor, IL1RI, do not develop obesity [7].
Consistent with these studies, we found that adenoviral-mediated inhibition of IL1β signaling in the CNS does not alter weight gain in normal rats over a period of 11 days, a time period during which blockade of central melanocortin signaling results in dramatic weight gain [4]. This finding suggests that, unlike central melanocortin signaling, the chronic effects of leptin on energy balance are not dependent on IL1β signaling in the brain. If physiological leptin effects were dependent on IL1β signaling one would also expect food intake to increase in animals with CNS over-expression of IL1ra. In contrast, we found that rats with adenoviral-mediated IL1ra expression consumed slightly less food than the control animals. Though preliminary, this finding is consistent with data from a study in which axonal connections to the ARC were severed in rats, which suggested that the effect of IL1β on the isolated ARC was to stimulate food intake [19].
Prolonged fasting decreases circulating leptin levels by 80% or more despite only a small change in fat mass [11]. Upon re-feeding the leptin levels are restored within a 24h period, thus representing perhaps the only physiological paradigm where circulating leptin increases relatively rapidly [11]. Our previous work demonstrated that hypothalamic IL1β decreases with fasting and increases with re-feeding [28] consistent with regulation of hypothalamic IL1β by physiological changes of circulating leptin. The findings from this study demonstrate that adenoviral-mediated over-expression of IL1ra in the CNS does not alter food intake or weight gain during re-feeding following a prolonged fast. Thus, though the leptin surge during re-feeding may stimulate hypothalamic IL1β production, this signaling pathway does not influence re-feeding-induced changes in energy balance.
However, in the present study adenoviral-mediated over-expression of IL1ra in the CNS was able to prevent anorexia and weight loss induced by ip administration of exogenous leptin, as has been shown previously [12,29]. In combination with the findings described above, this data suggests that the CNS signaling pathways activated by pharmacological leptin administration may be fundamentally different from those triggered by physiological excursions of endogenous leptin.
One previous study has attempted to address the role of IL1β in energy balance through chronic icv administration of IL1ra [18]. Like our data, this study suggested that the physiological effects of leptin were not mediated through IL1β, but interpretation of the results was limited by the fact that exogenous administration of leptin was not tested and the low dose of administered IL1ra was not effective in blocking exogenously-administered IL1β, thus the study paradigm may also have been ineffective in preventing endogenous leptin-mediated effects. In contrast, we found that over-expression of IL1ra in the CNS caused the expected inhibition of leptin-mediated anorexia demonstrating that adenoviral expression of IL1ra effectively blocked IL1 signaling. Our previous studies have shown that adenovirus-mediated gene transfer in the CNS can be detected as early as day 3 following adenoviral injections and is maintained for 3–4 weeks [15]. As leptin was administered at the end of our study period (day 21), the degree of CNS IL1ra over-expression at earlier time points when physiological effects of leptin were evaluated would have been more than sufficient to prevent leptin signaling, but failed to do so, suggesting that IL1 signaling is not required for physiological effects of leptin on energy balance.
The mechanism whereby pharmacological effects of leptin are IL1β-dependent remains unknown, and may be related to melanocortin signaling. We have shown that both ip and icv administration of the melanocortin agonist MTII increases IL1β mRNA expression in the mediobasal hypothalamus [29]. Thus IL1β signaling may be downstream of melanocortin signaling. Importantly, physiological changes in leptin appear to alter signaling in only a small subset of POMC neurons in the rostral portion of the ARC [23]. However, leptin receptor expression on POMC cells is wide-spread in the ARC [1] and electrophysiological studies with labeled POMC cells have shown that most POMC cells in the hypothalamus can respond to leptin [5]. This suggests that acute administration, or high concentrations, of leptin may activate POMC cells that involve different ‘downstream’ signaling pathways than those involved during physiological regulation of energy balance. Our previous studies have shown that exogenous administration of leptin (2 mg/kg ip) results in circulating leptin levels 10 fold greater than baseline [28]. Interestingly, one pathological circumstance that causes a rapid increase of endogenous leptin is acute inflammation [8], and it is possible that inflammatory effects of leptin may involve CNS signaling pathways distinct from those involved in the physiological regulation of energy balance. Thus, documented changes in CNS signaling following pharmacological administration of leptin may more accurately reflect the function of leptin as an inflammatory cytokine rather than the effects of leptin as a long-term signal monitoring the adequacy of body fat stores.
In addition to melanocortin and IL1β pathways as potential targets of leptin, the response to exogenously administered leptin may also be dependent on corticotrophin releasing hormone (CRH) and neurotensin (NT)[22,26]. Interestingly, like for IL1β, these two hormones are more commonly associated with stress responses and, in the case of CRH signaling, like for IL1β, mutant models of CRH deficiency do not develop obesity [16]. Thus, it remains possible that pharmacological leptin administration activates multiple ‘stress-related’ pathways which contribute to anorexia and weight loss that are not implicated in the physiological control of energy balance.
In conclusion, IL1β signaling in the CNS is not necessary for acute or chronic effects of endogenous leptin on energy balance, but is necessary for the anorexic effects of exogenously administered leptin. Thus, this study implies that exogenously-administered leptin activates alternate pathways within the CNS that may not be related to physiological energy homeostasis pathways. Therefore, data implicating ‘downstream’ hypothalamic pathways derived from studies using exogenous leptin should be interpreted with caution as pertaining to physiological regulation of energy balance.
Experimental Procedure
Animals
Studies were conducted using adult, male Wistar rats, weighing 300–350g (Charles River Laboratories, Wilmington, MA). All animals were housed individually in a temperature-controlled room (23 ± 2°C) and maintained on a 12h light/dark cycle. All study protocols were approved by the Institutional Animal Care and Use Committee of the University of Washington, Seattle, WA, and were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals. All animals were provided ad libitum access to water and pelleted rodent chow (Test Diet 5012, LabDiet Inc., Richmond, IN), except where otherwise indicated. Acute food intake studies were performed following a 4h fast prior to the onset of the dark cycle.
Adenoviral Injection
The two adenovirus injection protocols were performed as described previously [15] except as noted below. Rats were anesthetized with inhaled isoflurane and placed in a stereotaxic frame (Harvard/American Scientific Institution, Holliston, MA). For targeting of injections bilaterally to the arcuate nucleus (n=6) a 30-g injector was used with the coordinates 3.3mm posterior to bregma, 10.5mm below the surface of the skull and 0.5mm lateral to the midline bilaterally, a technique which has been extensively validated in our laboratory [6,13–15]. Targeting of the 3rd cerebral ventricle (n=9) was accomplished with a single midline injection using a 26-g injector with the tip placed 2.2mm posterior to bregma and 8.5mm below the surface of the skull. Using a syringe injector pump (WPI, Sarasota, FL) recombinant human IL1ra AdV (Ad5RSVIL-1ra 1×10^10pfu/ml, Gene Transfer Vector Core, University of Iowa, Iowa City, IA) was injected at a rate of 100nl/min over 5 min (total volume 500nl) into the arcuate nucleus bilaterally with the needle being withdrawn from the brain 5 min after the end of the injection period. For injections into the 3rd ventricle virus was injected at a rate of 100nl/sec over 20sec with the needle being withdrawn from the brain 5 min after the end of the injection period. Control groups (n=6) received either a 3rd ventricular injection of an adenoviral construct expressing green fluorescent protein (GFP) or were cannulated but received no injection. Preliminary studies where an adenoviral construct expressing GFP was injected using the coordinates and protocol targeting the 3rd ventricle showed maximal adenoviral gene expression in the ependymal cells lining the 3rd ventricle with diminishing, but detectable expression in hypothalamic tissue, lateral ventricles and fourth ventricle, respectively (data not shown). For all outcome measures evaluated in this study no differences were seen between the groups of rats that received AdV-IL1ra injections bilaterally into the arcuate nucleus and rats where AdV-IL1ra was injected into the 3rd ventricle. For this reason, animals from these two groups are combined into a single AdV-IL1ra group for analysis. Similarly, as all outcome measures were equivalent for the AdVGFP (all 3rd icv) and ‘no injection’ control groups these two groups are combined into a single Control group for the results described below.
STUDY PROTOCOLS
Fasting and Re-feeding
The fasting protocol was initiated 11d following adenoviral injections. Rats fasted for 36h and food was returned immediately prior to the onset of the dark cycle. Food intake during re-feeding was evaluated at 2, 4, 6, 8 and 18h of re-feeding. Body weight was evaluated 18h after re-feeding.
Exogenous leptin administration
Five days following the end of the fasting/re-feeding protocol all rats received ip injections of vehicle (0.3ml normal saline) or leptin (2mg/kg, National Hormone and Peptide Program, Harbor-UCLA Medical Center) on two consecutive days immediately prior to the onset of the dark cycle. Food intake was evaluated at 2, 4 and 18h following the onset of the dark cycle. Though serum leptin levels were not determined as part of this study we have previously evaluated serum leptin as part of studies that employed the identical ip leptin administration paradigm. In ~400g Wistar rats the serum leptin 2h into the dark cycle following vehicle injection is 4.4±0.6ng/ml. In comparison, two hours post leptin administration (2mg/kg ip) serum leptin values are 69±12ng/ml [28]. 20h following leptin administration serum leptin levels are no different from vehicle-injected controls as would be expected given published leptin pharmacokinetics in rats [9].
Statistical Methods
Comparisons between group mean values were performed using an unpaired Student’s t-test or using a two-way ANOVA using within subjects comparison for treatment effect and between subjects comparisons for adenoviral gene transfer effect, and the Tukey HSD post-hoc test for multiple comparisons. Statistical analyses were performed using Statistica software (StatSoft Inc., V. 4.1, Tulsa, OK). The null hypothesis of no difference between groups was rejected at p <0.05. All values are presented as the mean ± SEM.
Fig. 3.
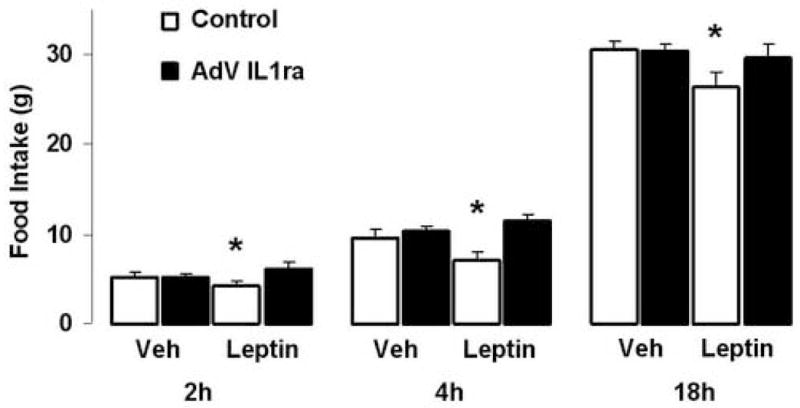
Effect of ip leptin administration in rats with CNS adenoviral over-expression of IL1ra. Food intake (g) in two adenoviral injection groups, Control (white bars, n=6) and AdV-IL1ra (black bars, n=10) at three indicated time points following ip administration of leptin (2mg/kg). Data presented are means ± SEM. * p<0.05 by one-way ANOVA with Tukey HSD post-hoc analysis.
Acknowledgments
We are grateful to A. Cubelo, G. Alkire and C. Leung for their technical assistance. This work was supported by grants from the National Institutes of Health to M.W.S. (P01-DK 068384, DK12829, NS32273), and B.E.W (DK61384), the American Institute of Cancer Research Grant #03B051 (B.E.W.), Pilot & Feasibility Awards from the Clinical Nutrition and Research Unit (P30DK035816) and the Diabetes Endocrinology Research Center (P30DK17047) at the University of Washington (B.E.W.) as well as a grant from the Murdock Charitable Trust (M.W.S.)
Abbreviations
- icv
intracerebroventricular
- IL1ra
Interleukin 1 receptor antagonist
- AdV
adenoviral
- IL1
Interleukin-1
- CNS
central nervous system
- MTII
melanotan II
- POMC
pro-opiomelanocortin
- ARC
arcuate nucleus
- CRH
corticotrophin releasing hormone
- NT
neurotensin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature references
- 1.Baskin DG, Schwartz MW, Seeley RJ, Woods SC, Porte D, Jr, Breininger JF, Jonak Z, Schaefer J, Krouse M, Burghardt C, Campfield LA, Burn P, Kochan JP. Leptin receptor long-form splice-variant protein expression in neuron cell bodies of the brain and co-localization with neuropeptide Y mRNA in the arcuate nucleus. J Histochem Cytochem. 1999;47:353–62. doi: 10.1177/002215549904700309. [DOI] [PubMed] [Google Scholar]
- 2.Benoit S, Schwartz M, Baskin D, Woods SC, Seeley RJ. CNS melanocortin system involvement in the regulation of food intake. Horm Behav. 2000;37:299–305. doi: 10.1006/hbeh.2000.1588. [DOI] [PubMed] [Google Scholar]
- 3.Butler AA. The melanocortin system and energy balance. Peptides. 2006;27:281–90. doi: 10.1016/j.peptides.2005.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cabeza de Vaca S, Hao J, Afroz T, Krahne LL, Carr KD. Feeding, body weight, and sensitivity to non-ingestive reward stimuli during and after 12-day continuous central infusions of melanocortin receptor ligands. Peptides. 2005;26:2314–21. doi: 10.1016/j.peptides.2005.03.041. [DOI] [PubMed] [Google Scholar]
- 5.Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–4. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 6.Gelling RW, Morton GJ, Morrison CD, Niswender KD, Myers MG, Jr, Rhodes CJ, Schwartz MW. Insulin action in the brain contributes to glucose lowering during insulin treatment of diabetes. Cell Metab. 2006;3:67–73. doi: 10.1016/j.cmet.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 7.Glaccum MB, Stocking KL, Charrier K, Smith JL, Willis CR, Maliszewski C, Livingston DJ, Peschon JJ, Morrissey PJ. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J Immunol. 1997;159:3364–71. [PubMed] [Google Scholar]
- 8.Grunfeld C, Zhao C, Fuller J, Pollack A, Moser A, Friedman J, Feingold KR. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. J Clin Invest. 1996;97:2152–7. doi: 10.1172/JCI118653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hill RA, Margetic S, Pegg GG, Gazzola C. Leptin: its pharmacokinetics and tissue distribution. Int J Obes Relat Metab Disord. 1998;22:765–70. doi: 10.1038/sj.ijo.0800656. [DOI] [PubMed] [Google Scholar]
- 10.Hosoi T, Okuma Y, Nomura Y. Leptin regulates interleukin-1beta expression in the brain via the STAT3-independent mechanisms. Brain Res. 2002;949:139–46. doi: 10.1016/s0006-8993(02)02974-8. [DOI] [PubMed] [Google Scholar]
- 11.Kolaczynski JW, Considine RV, Ohannesian J, Marco C, Opentanova I, Nyce MR, Myint M, Caro JF. Responses of leptin to short-term fasting and refeeding in humans: a link with ketogenesis but not ketones themselves. Diabetes. 1996;45:1511–5. doi: 10.2337/diab.45.11.1511. [DOI] [PubMed] [Google Scholar]
- 12.Luheshi GN, Gardner JD, Rushforth DA, Loudon AS, Rothwell NJ. Leptin actions on food intake and body temperature are mediated by IL-1. Proc Natl Acad Sci U S A. 1999;96:7047–52. doi: 10.1073/pnas.96.12.7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morton GJ, Blevins JE, Williams DL, Niswender KD, Gelling RW, Rhodes CJ, Baskin DG, Schwartz MW. Leptin action in the forebrain regulates the hindbrain response to satiety signals. J Clin Invest. 2005;115:703–10. doi: 10.1172/JCI200522081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morton GJ, Gelling RW, Niswender KD, Morrison CD, Rhodes CJ, Schwartz MW. Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2005;2:411–20. doi: 10.1016/j.cmet.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Morton GJ, Niswender KD, Rhodes CJ, Myers MG, Jr, Blevins JE, Baskin DG, Schwartz MW. Arcuate nucleus-specific leptin receptor gene therapy attenuates the obesity phenotype of Koletsky (fa(k)/fa(k)) rats. Endocrinology. 2003;144:2016–24. doi: 10.1210/en.2002-0115. [DOI] [PubMed] [Google Scholar]
- 16.Muglia L, Jacobson L, Dikkes P, Majzoub JA. Corticotropin-releasing hormone deficiency reveals major fetal but not adult glucocorticoid need. Nature. 1995;373:427–32. doi: 10.1038/373427a0. [DOI] [PubMed] [Google Scholar]
- 17.Plata-Salaman CR, Ilyin SE, Gayle D. Brain cytokine mRNAs in anorectic rats bearing prostate adenocarcinoma tumor cells. Am J Physiol. 1998;275:R566–73. doi: 10.1152/ajpregu.1998.275.2.R566. [DOI] [PubMed] [Google Scholar]
- 18.Pu S, Anisman H, Merali Z. Central infusion of interleukin-1 receptor antagonist fails to alter feeding and weight gain. Neuroreport. 2000;11:1699–702. doi: 10.1097/00001756-200006050-00021. [DOI] [PubMed] [Google Scholar]
- 19.Reyes TM, Sawchenko PE. Involvement of the arcuate nucleus of the hypothalamus in interleukin-1-induced anorexia. J Neurosci. 2002;22:5091–9. doi: 10.1523/JNEUROSCI.22-12-05091.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richard D, Baraboi D. Circuitries involved in the control of energy homeostasis and the hypothalamic-pituitary-adrenal axis activity. Treat Endocrinol. 2004;3:269–77. doi: 10.2165/00024677-200403050-00001. [DOI] [PubMed] [Google Scholar]
- 21.Sahu A. Minireview: A hypothalamic role in energy balance with special emphasis on leptin. Endocrinology. 2004;145:2613–20. doi: 10.1210/en.2004-0032. [DOI] [PubMed] [Google Scholar]
- 22.Sahu A, Carraway RE, Wang YP. Evidence that neurotensin mediates the central effect of leptin on food intake in rat. Brain Res. 2001;888:343–347. doi: 10.1016/s0006-8993(00)03107-3. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz MW, Seeley RJ, Woods SC, Weigle DS, Campfield LA, Burn P, Baskin DG. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes. 1997;46:2119–23. doi: 10.2337/diab.46.12.2119. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–71. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 25.Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell. 2001;104:531–43. doi: 10.1016/s0092-8674(01)00240-9. [DOI] [PubMed] [Google Scholar]
- 26.Uehara Y, Shimizu H, Ohtani K, Sato N, Mori M. Hypothalamic corticotropin-releasing hormone is a mediator of the anorexigenic effect of leptin. Diabetes. 1998;47:890–3. doi: 10.2337/diabetes.47.6.890. [DOI] [PubMed] [Google Scholar]
- 27.Wisse BE, Campfield LA, Marliss EB, Morais JA, Tenenbaum R, Gougeon R. Effect of prolonged moderate and severe energy restriction and refeeding on plasma leptin concentrations in obese women. Am J Clin Nutr. 1999;70:321–30. doi: 10.1093/ajcn/70.3.321. [DOI] [PubMed] [Google Scholar]
- 28.Wisse BE, Ogimoto K, Morton GJ, Wilkinson CW, Frayo RS, Cummings DE, Schwartz MW. Physiological regulation of hypothalamic IL-1beta gene expression by leptin and glucocorticoids: implications for energy homeostasis. Am J Physiol Endocrinol Metab. 2004;287:E1107–13. doi: 10.1152/ajpendo.00038.2004. [DOI] [PubMed] [Google Scholar]
- 29.Wisse BE, Ogimoto K, Schwartz MW. Role of hypothalamic interleukin-1beta (IL-1beta) in regulation of energy homeostasis by melanocortins. Peptides. 2006;27:265–73. doi: 10.1016/j.peptides.2005.08.020. [DOI] [PubMed] [Google Scholar]