

Abstract
Capping of the pre-mRNA 5′ end by addition a monomethylated guanosine cap (m7G) is an essential and the earliest modification in the biogenesis of mRNA. The reaction is catalyzed by three enzymes: triphosphatase, guanylyltransferase, and (guanine N-7) methyltransferase. Whereas this modification occurs co-transcriptionally in most eukaryotic organisms, trypanosomatid protozoa mRNAs acquire the m7G cap by trans-splicing, which entails the transfer of the capped spliced leader (SL) from the SL RNA to the mRNA. Intriguingly, the genomes of all trypanosomatid protozoa sequenced to date possess two distinct proteins with the signature motifs of guanylyltransferases: TbCGM1 and the previously characterized TbCE1. Here we provide biochemical evidence that TbCgm1 is a capping enzyme. Whereas RNAi-induced downregulation of TbCe1 had no phenotypic consequences, we found that TbCGM1 is essential for trypanosome viability and is required for SL RNA capping. Furthermore, consistent with co-transcriptional addition of the m7G cap, chromatin immunoprecipitation revealed recruitment of TbCgm1 to the SL RNA genes.
Keywords: mRNA capping, guanylyltransferase, cap 4 modification, trans-splicing, SL RNA, RNAi
1. Introduction
In all eukaryotic cells analyzed so far, including Trypanosoma brucei, addition of the m7G cap represents the earliest event in the modification of the 5′ end of RNA polymerase II (pol II) transcripts [1–3]. Capping occurs co-transcriptionally when the RNA has achieved a chain length of about 30 nucleotides (nt). Formation of the m7GpppN cap entails three enzymatic reactions [4]. The 5′ triphosphate end is first hydrolyzed by RNA 5′ triphosphatase to a 5′ diphosphate, which is then capped with GMP by RNA guanylyltransferase. Finally, the guanosine moiety of the GpppN cap is methylated by RNA (guanine-N7) methyltransferase. Each of the cap-forming activities is essential in budding yeast [5–7].
Even though eukaryotes depend on the same three enzymatic reactions for m7G capping, the physical organization of the capping apparatus is quite diverse in different organisms [4,8,9]. A two-component capping enzyme system consisting of a bifunctional triphosphatase-guanylyltransferase polypeptide and a separate methyltransferase polypeptide is present in metazoans and plants. In contrast, fungi and the microsporidia Encephalitozoon cuniculi have a three component system with separate triphosphatase, guanylyltransferase and methyltransferase polypeptides. The protozoa Plasmodium falciparum [10] and Giardia lamblia [11] have a yeast-like machinery. Extensive mutational analysis and crystal structures revealed that RNA guanylyltransferases are related to DNA and RNA ligases [4,8,9] and are structurally and mechanistically conserved among all eukaryotic species and many viruses. Guanylyltransferases catalyze the nucleotidyl transfer through a covalent enzyme-(lysyl-N)-GMP intermediate and they are defined by a set of six conserved signature motifs [12]. On the other hand, RNA triphosphatases fall into two mechanistically and structurally distinct families. Triphosphatases of fungi, microsporidia, protozoa, including T. brucei, and Chlorella virus belong to the family of metal-dependant NTP phosphohydrolases defined by a unique active site tunnel architecture [13,14], whereas metazoan and plant triphosphatases are part of the cysteine phosphatase superfamily [15,16]. The crystal structure of the (guanine-N7) methyltransferase from the E. cuniculi highlighted the known biochemical specificity of the enzyme [17] and completed the catalogue of protein structures of the three enzymes required for m7GpppN cap formation.
In most eukaryotic organisms m7G capping is restricted to nascent pol II transcripts. In contrast, studies in trypanosomatid protozoa have highlighted a peculiar aspect in the mode of cap formation, because this modification is not only present on the pol II-transcribed spliced leader (SL) RNA, but is also found on a specific subset of pol III transcripts, namely the U1, U2, and U4 snRNAs and the U3 snoRNA. Since the most abundant pol III transcripts (tRNAs, 5S rRNA and 7SL RNA) are not capped, this raises numerous questions about the specificity and regulation of the capping mechanism. Furthermore, mRNA cap addition occurs by post-transcriptional trans-splicing, which entails the transfer of the 39-nt long SL sequence, including the cap structure, from the SL RNA to the 5′ end of all mRNAs [18–20]. The SL RNA cap is unique to trypanosomatids and contains a hypermodified cap 4 structure, which is derived from methylation of seven sites within the first four nucleotides of the SL RNA resulting in the structure: m7guanosine-ppp-N6,N6,2′-O-trimethyladenosine-p-2′-O-methyladenosine-p-2′-O-methylcytosine-p-N3,2′-O-methyluridine [21]. Recently, two 2′-O-methyltransferases required for cap 4 biogenesis have been identified and shown to be intriguingly similar to vaccinia virus VP39 [22–25]. In addition, the cap 4 structure in T. brucei is specifically recognized by an unusual nuclear cap binding complex that contains three novel and essential subunits only present in trypanosomatids [26]. Taken together these observations underscore the uniqueness of the capping apparatus of trypanosomatid protozoa.
Our initial characterization of the enzymatic machinery involved in m7G capping in trypanosomatids identified a polypeptide in Crithidia fasciculata and T. brucei (Ce1) with structural and biochemical properties characteristic of GTP:RNA guanylyltransferases [27]. Whereas the carboxy-terminal half of Ce1 displays the six signature motifs characteristic of guanylyltransferases, the amino-terminal half contains a domain of unknown function with no resemblance to any other domain associated with capping enzymes. In the course of these studies, we noted a second polypeptide in C. fasciculata and T. brucei extracts of approximately 116 kDa, capable of forming a covalent protein-GMP complex, which is indicative of a capping enzyme. However, the characterization of this polypeptide was not pursued further. More recently, a RNA triphosphatase (TbCet1) was characterized in T. brucei, which is structurally and mechanistically related to the fungal metal-dependent phosphohydrolases [28], and recombinant T. brucei Cmt1 was shown to be a (guanine-N7) methyltransferase [29]. In addition, data-base mining identified a second candidate T. brucei capping enzyme, TbCgm1, with a predicted N-terminal guanylyltransferase domain and a C-terminal methyltransferase domain [29]. While this manuscript was in preparation, the Ho laboratory further reported that TbCgm1 caps the SL RNA [30].
Here, we have used RNA interference-induced down regulation, biochemical properties and chromatin immunoprecipitation to examine the in vivo function of the two T. brucei capping enzymes TbCe1 and TbCgm1 and how they interface with the unusual capping of both pol II and pol III transcripts.
2. Materials and methods
2.1. Plasmids, transfections and RNAi
PCR products encompassing nt 1 - 894 of the N-terminal domain of TbCE1, and nt 180 - 1,180 of the guanylyltransferase domain of TbCGM1 were inserted in the sense and antisense orientation separated by a stuffer fragment into plasmid pLEW100 [31] and transfected into the procyclic T. brucei strain 29.13.6 [32], which harbors integrated genes for T7 RNA polymerase and the tetracycline repressor. The cells were selected with phleomycin and cloned in 96-well plates. Clonal cell lines were induced by adding tetracycline to a concentration of 10 μg/ml. A PCR-based approach [33] was used to introduce a BB2 epitope tag at the N-terminus of TbCe1 and TbCgm1 and to replace the second allele with the blasticidin (BSR) resistant gene.
To produce recombinant guanylyltransferase in E. coli, nt 1- 2,355 of TbCGM1, corresponding to amino acids 1 - 785, were cloned in between the NotI and XhoI restriction sites of the T7 RNA polymerase-based expression vector pET-44a (Novagen). The lysine residue in motif I was changed to alanine by a two-step PCR procedure and the mutation was verified by DNA sequencing.
2.2. RNA analysis
RNA extraction and Northern blot analysis were performed as described [34]. In Figure 3B, 4B and 4C, the SL RNA was primer extended with oligonucleotide Y-21, complementary to nt 40 to 60, and in Figure 4D SLIN22, complementary to nt 110 to 131, was used. U2, U3 and U6 snRNAs were primed with oligonucleotide U2B-17, complementary to nt 46 to 62, U3-PE, complementary to nt 96 to 123, and U6-22, complementary to nt 19 to 40, respectively.
Fig. 3.

TbCE1 downregulation by RNAi. (A) Total RNA isolated from uninduced (lanes 1–5) or TbCE1-RNAi cells induced for 8 days (lanes 6–10) was immunoprecipitated with anti-m7G and anti-TMG antibodies and the input (I), supernatant (S) and pellet (P) fractions were assayed by primer extension of the U2 snRNA (U2) and U3 snoRNA (U3). (B) The TbCE1 RNAi cell line was induced with tet for the indicated number of days (lanes 2–4) and total RNA was assayed by primer extension with an SL intron-specific primer. The position of fully-modified (fm SL) and hypomodified (hm SL) SL RNA is indicated. RNA isolated from sinefungin-treated cells served as a control for hypomodified SL RNA (lane 1). A U6 snRNA-specific primer was included in the reactions to control for RNA amounts and quality.
Fig. 4.

TbCGM1 depletion affects SL RNA capping. (A) Total RNA isolated from uninduced (lanes 1–5) or TbGM1-RNAi cells induced for 2.5 days (lanes 6–10) was immunoprecipitated with anti-m7G and anti-TMG antibodies and the input (I), supernatant (S) and pellet (P) fractions were assayed by primer extension of the U2 snRNA (U2) and U3 snoRNA (U3). (B) The TbCGM1 RNAi cell line was induced with tet for the indicated number of days (lanes 2–5) and total RNA was assayed by primer extension with an SL intron-specific primer. The position of fully-modified (fm SL) and hypomodified (hm SL) SL RNA is indicated. RNA isolated from sinefungin-treated cells served as a control for hypomodified SL RNA (lane 1). A U6 snRNA-specific primer was included in the reactions to control for RNA amounts and quality. (C) Total RNA isolated from uninduced cells (lanes 1–3) or from cells depleted of TbCgm1 for 2.5 days (lanes 4–6) was immunoprecipitated with anti-m7G antibodies and the input (I), supernatant (S) and pellet (P) fractions were assayed by primer extension with an SL intron-specific primer. A U6-snRNA-specific primer was included in all the reactions to control for immunoprecipitation specificity (U6). (D) Primer extension of total RNA with an oligonucleotide complementary to nt 110 to 131 of the SL RNA. The position of the Y-structure intermediate (Y) and full-length SL RNA (SL) is indicated. RNA isolated from sinefungin-treated cells was used as a control for trans-splicing inhibition (lane 1). An oligonucleotide complementary to the U2 snRNA was included as a loading and quality control (U2).
Total RNA was subjected to immunoprecipitations with rabbit polyclonal antibodies against the m7G cap (a kind gift of Dr. F. Richards) and mouse monoclonal antibodies against the 2,2,7-trimethylguanosine cap (Calbiochem). RNA samples were analyzed by primer extension.
Uninduced and TbCGM1 RNAi-induced procyclic T. brucei cells were permeabilized with lysolecithin (L-α-lysophosphatidylcholine, palmitoyl, Sigma) and RNA was synthesized for 15 minutes in the presence of [α-32P]GTP or [α-32P]ATP as described [3,35,36]. Total RNA, extracted with TRIZOL reagent, was separated on 6% polyacrylamide - 7M urea gels, the SL RNA was excised and eluted in water. Tobacco acid pyrophosphatase (TAP) cleavage and nuclease P1 digestion were done essentially as described [3] and the digestion products were chromatographed on Avicel cellulose thin layer plates, developed with isobutyric acid/concentrated NH4OH/H2O, 66:1:33 (v/v/v).
2.3. Other procedures
GMP binding [27] and ChIP was done as previously described [37].
3. Results
3.1. The guanylyltransferase domain of TbCgm1 binds GMP
During a search of the T. brucei databases for additional components of the capping machinery, we and others [29] found a predicted protein, TbCgm1, of 116,820 Da (GeneDB accession # Tb927.7.2080) with convincing homologies to RNA (guanine-N7) methyltransferases in the C-terminal half of the protein. Further domain and motif searches revealed that the N-terminal half has the characteristics of a guanylyltransferase domain with the six peptide motifs that are absolutely conserved in the superfamily of capping enzymes and DNA ligases and constitute the active site for GMP binding and nucleotidyl transfer [4,29]. Northern blot, 5′ end RACE and sequencing of cDNA clones established the authenticity of this gene in the T. brucei genome and determined the structure of the mRNA 5′ end (data not shown).
The identification of a second putative guanylyltransferase domain in a eukaryotic organism is unique. However, this finding can be rationalized by our earlier observation that T. brucei whole cell extracts have two polypeptides of 67 and 116 kDa capable of forming a covalent GMP-enzyme complex characteristic of capping enzymes [27]. Whereas the 67 kDa-polypeptide (TbCe1) was shown to have a functional guanylyltransferase domain, the larger polypeptide was not pursued further. In view of a predicted molecular weight of 117 kDa and the presence of a putative N-terminal guanylyltransferase domain, TbCgm1 was a likely candidate for a second GMP-binding enzyme. To test whether TbCGM1 encoded a protein that can bind GMP in a covalent fashion, we initially expressed full-length TbCgm1 in Escherichia coli with several different expression vectors, which did not result in the production of sufficient quantities of recombinant protein in a soluble form to perform enzymatic tests. Thus, we generated a construct containing the first 785 amino acids of TbCgm1 fused at the N-terminus to the E. coli NusA protein. A 142 kDa protein corresponding to the fusion protein was detectable by SDS-PAGE in soluble extracts of IPTG-induced bacteria (upper panel in Fig. 1, lane 2). The recombinant protein formed an SDS-stable nucleotide-protein adduct (lower panel in Fig. 1, lane 2), which was not evident in a sample expressing only the NusA protein (lane 1). Importantly, the protein lost the ability to bind GMP, when the predicted catalytic lysine residue (amino acid 127) of motif I was mutated to alanine (lane 3), providing evidence that TbCgm1 has a functional guanylyltransferase domain.
Fig. 1.
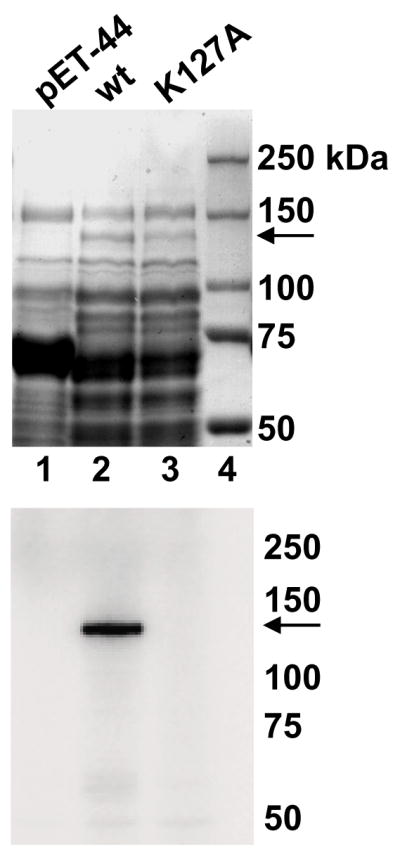
The guanylyltransferase domain of TbCgm1 binds GMP. Soluble protein lysates from bacteria expressing the NusA protein (pET-44, lane 1), amino acids 1 - 785 of TbCgm1 fused to the NusA protein (wt, lane 2), and a fusion protein with the indicated mutation (K127A, lane 3) were fractionated by SDS-PAGE and stained with Coomassie blue dye (upper panel) or tested for GMP binding (lower panel, an autoradiograph is shown). The sizes (in kilodaltons) of marker proteins are shown on the right and the position of the NusA-TbCgm1 fusion proteins is indicated by an arrow.
3.2. TbCgm1 is encoded by an essential gene
To begin to investigate the in vivo function of the two T. brucei polypeptides containing guanylyltransferase domains, we used RNA interference (RNAi) to downregulate the respective mRNAs. To do this, nt 1 - 894 of TbCE1 and nt 180 - 1,180 of TbCGM1 were inserted in the sense and antisense orientation separated by a spacer fragment into plasmid pLEW100 [38]. This cassette, driven by a tetracycline (tet) inducible ribosomal promoter, was stably integrated in the non-transcribed rDNA spacer region and clonal cell lines were established. Upon RNAi induction with tet, production of double-stranded RNA and degradation of the target mRNA was verified by Northern blot (data not shown). In addition, in extracts prepared from CE1-RNAi cell lines induced for 24 hr GMP-enzyme complex formation was reduced to undetectable levels (Fig. 2A), whereas in CGM1-RNAi cell lines the Cgm1 protein was severely decreased, albeit still detectable after 24 hr (Fig. 2B). Importantly, the results of Figures 2A and B demonstrated the specificity of the downregulation of Ce1 or Cgm1 by the corresponding RNAi construct. Next, we monitored cell growth over a period of 4 days (Fig. 2C). Compared to wild-type and uninduced cells, downregulation of TbCE1 had no noticeable effect on cell viability. In fact, maintaining the induction for over 10 days did not reveal any detectable change in growth characteristics. In contrast, monitoring cell growth during TbCGM1 RNAi induction revealed that the growth rate declined between day 2 and day 3 and eventually the cells died (Fig. 2C), indicating that TbCGM1 is essential for viability.
Fig. 2.

RNAi-induced downregulation of TbCE1 and TbCGM1. (A) Two independent clonal cell lines expressing TbCE1 dsRNA were grown in the absence (lanes 1 and 3) or presence (lanes 2 and 4) for 24 hr and total cell extracts were assayed for enzyme-GMP complex formation. (B) Same assay as in panel (A), except that TbCGM1 was downregulated (lanes 2 and 4). (C) Cell densities were measured 24, 48, 72, 90 and 96 hr after induction of RNAi with tetracycline in a clonal cell line expressing double-stranded RNA for either TbCE1 or TbCGM1. Since the growth curves of uninduced cells were essentially identical to wild-type cells (wt), they are represented as one.
3.3. Depletion of TbCE1 or TbCGM1 does not affect capping of the U2 snRNA and U3 snoRNA
Similar to other eukaryotes, the trypanosomatid U1, U2 and U4 snRNAs and the U3 snoRNA contain a 2,2,7-trimethylguanosine (TMG) cap [39–41]. Little is known about the sequence of events leading to a TMG cap in trypanosomatids, but it is presumed that these RNA molecules are initially modified by an m7G cap [42,43]. Thus, in a first set of experiments, we asked whether TbCE1 or TbCGM1 downregulation had an effect on cap formation of the U2 snRNA or the U3 snoRNA. TbCE1-RNAi cells were induced with tet for 8 days, RNA was prepared and assayed by immunoprecipitation with anti-m7G and anti-TMG antibodies followed by primer extension analysis (Fig. 3A). As expected, both U2 and U3 RNAs were affinity selected by anti-TMG antibodies from RNA prepared from uninduced cells (Fig. 3A, lane 5), whereas these RNAs were not detectable in the anti-m7G immunoprecipitate (lane 3). A similar result was obtained with RNA isolated from TbCE1-RNAi cells induced for 8 days (lanes 8 and 10). Furthermore, there was no detectable difference between uninduced TbCGM1-RNAi cells and cells induced for 2.5 days (Fig. 4A, compare lanes 2 to 5 with lanes 7 to 10), indicating that cap formation on the U2 snRNA and U3 snoRNA appeared normal under conditions where TbCe1 and TbCgm1 protein amounts were limiting.
3.4. TbCgm1 is involved in SL RNA capping
We next investigated whether RNAi-induced downregulation of TbCE1 or TbCGM1 affected cap formation of the pol II-transcribed SL RNA. RNA was prepared from TbCE1-RNAi cells induced for 2.5 and 8 days and assayed by primer extension analysis of the SL RNA (Fig. 3B). As shown previously [22,44] and evident in Figure 3B (lane 2), primer extension analysis at low deoxyribonucleotide triphosphate concentrations gives rise to a characteristic primer extension stop at the 5′ end of the SL RNA indicative of a fully-methylated cap 4 structure (indicated as fm SL). In contrast, hypomethylated SL RNA (hm SL) prepared from cells grown in the presence of the methylation inhibitor sinefungin, gives rise to a series of bands characteristic of SL RNA with a partially methylated cap 4 (Fig. 3B, lane 1). TbCE1 downregulation for 8 days did not affect the accumulation or methylation status of the SL RNA, as assayed by primer extension analysis (Fig. 3B, compare lanes 2 and 4). In contrast, following 2 and 2.5 days of induction of TbCGM1 RNAi, the accumulation of the SL RNA was reduced to 76% and 36%, respectively, as compared to uninduced cells (Fig. 4B). In addition, a clear defect in cap 4 formation was evident with fully-modified SL RNA reduced to 34% and 13% at day 2 and 2.5, respectively, and a concomitant appearance of longer extension products indicative of SL RNA carrying a hypomethylated cap 4 (lanes 4 and 5).
To address the apparent effect of TbCGM1 downregulation on the SL RNA cap formation in more detail, we first used immunoprecipitation with anti-m7G antibodies (Fig. 4C). Whereas the majority of the fully-modified SL RNA was affinity selected from RNA isolated from uninduced cells (lane 3), after 2.5 days of induction the hypomethylated SL RNA was predominantly found in the supernatant (compare lanes 5 and 6). In contrast, the small amount of fully-modified SL RNA present after 2.5 days of induction was immunoprecipitated with anti-m7G antibodies (lane 6). Thus, the epitope recognized by the antibody was not present in a considerable proportion of steady-state SL RNA accumulating during downregulation of Cgm1, suggesting a defect in the m7G cap.
Next, we turned to the analysis of newly-synthesized RNA in permeabilized T. brucei cells. Uninduced and TbCGM1-RNAi cells induced for 1 and 2.5 days were permeabilized with lysolecithin and incubated for 15 min with transcription cocktail containing [α-32P]GTP [35]. Using the same number of cells in the permeabilization procedure, there was no obvious difference in total RNA synthesis in the induced cells, as compared to uninduced cells (data not shown). The SL RNA was then gel purified and digested with tobacco acid pyrophosphatase (TAP), which released the cap nucleotide from the SL RNA by hydrolyzing the pyrophosphate bond. The digestion products were separated by thin layer chromatography (TLC) and analyzed by phosphorimaging (Fig. 5A). This treatment released 1.5% of the radioactive label in the SL RNA from uninduced cells in the form of m7G (lane 1). This value did not change after 1 day of TbCGM1 depletion (lane 2, 1.5%), but was reduced to 0.8% following 2.5 days of induction (lane 3). In uninduced (lane 1) and 1 day-induced cells (lane 2) 0.8% of the radioactive label was released by TAP in the form of GMP, indicative of a cap without the methyl group at the N7 position of guanosine. This value increased to 1.4% in the sample isolated from cells induced for 2.5 days (lane 3). In addition, there was a new spot (1.5% of the radioactive label) that did not correspond to any of the available standards (labeled X1 in Fig. 5A). Even two-dimensional TLC analysis (data not shown) failed to give us clues to the possible nature of the X1 spot. However, the above data were in agreement with the immunoprecipitation results with anti-m7G antibodies and further indicated that upon CGM1 silencing there was an increase in the proportion of SL RNA bearing an unmethylated cap structure.
Fig. 5.

Newly-synthesized RNA labeled with [α-32P]GTP or [α-32P]ATP was prepared in permeabilized cells, the spliced leader RNA was gel purified and analyzed by tobacco acid pyrophosphatase and P1 digestion. In both panels, the SL RNA was isolated from uninduced cells (lane 1), or from cells depleted for TbCGM1 for 1 day (lane 2) or 2.5 days (lane 3). (A) The cap nucleotide was visualized by digesting [α-32P]GTP-labeled SL RNA with tobacco acid pyrophosphatase and TLC separation. The positions of nonradioactive m7Gp and Gp markers (visualized with UV light) are indicated. (B) [α-32P]ATP-labeled SL RNA was treated with nuclease P1 and separated by TLC. Two different exposure times are shown. The positions of nonradioactive AMP, ADP and ATP markers (visualized with UV light) are indicated.
To further define the consequences of TbCgm1 depletion on the SL RNA cap, permeabilized cells were labeled with [α-32P]ATP in order to monitor the status of the adenosine at position 1, i.e. the initiating nucleotide, of the SL RNA. The gel purified SL RNA was digested with nuclease P1 (Fig. 5B) that generates 5′nucleotide monophosphates, but does not cleave pyrophosphate bonds, i.e. it will not cleave the triphosphate bridge linking m7G to the adenosine at position 1. As expected, in the sample prepared from uninduced cells the majority of the label (96%) comigrated with AMP (lane 1). The next prominent spot (3%, designated X2) did not correspond to any of the available standards, but decreased over the induction period and was 50% less abundant after 2.5 days. P1 digestion of SL RNA labeled with [α-32P]GTP generated an identical spot (data not shown) and therefore we speculate that this represents a dinucleotide derived from the cap structure. In addition, 0.7% of the radioactive label was associated with ATP, which most likely originates from the 5′ end of primary SL transcripts. Following 1 and 2.5 days of TbCgm1 depletion the level of ATP did not change significantly. A fourth spot was identified as ADP. Whereas SL RNA from uninduced cells had 0.2% of the radioactivity associated with ADP, downregulation of TbCGM1 for 1 and 2.5 days resulted in a gradual increase of ADP reaching 0.5% after 2.5 days. The most likely explanation for this result is that TbCgm1 depletion gave rise to an increase in diphosphate-terminated RNA, i.e. capping of these transcripts was blocked after the triphosphatase step, which removed the terminal phosphate from the primary transcript. Thus, taken together these observations provided supporting evidence that TbCgm1 is required for capping of the SL RNA.
3.5. Trans-splicing utilization of the SL RNA is reduced in TbCgm1-depleted cells
Since the m7G cap may be one of the determinants for utilization of the SL RNA in trans-splicing, we examined trans-splicing activity in TbCgm1-depleted cells (Fig. 4D). In the RNA derived from uninduced cells (lane 2), primer extension analysis produced cDNA corresponding to full-length SL RNA, as well as a primer extension stop mapping at the 5′ end of the SL intron, which is mostly diagnostic of Y-structures (SL RNA intron-pre-mRNA branched intermediates that are formed during the first step of trans-splicing). Y-structures are not detectable in cells treated with sinefungin (lane 1), a drug that blocks trans-splicing in trypanosomes [45]. By this analysis we found that TbCGM1 silencing reduced the accumulation of Y-structures by 35% after 2.5 days of induction (Fig. 4D, lane 5).
3.6. Recruitment of TbCgm1 to the SL RNA genes
To provide independent evidence that TbCgm1 is involved in the maturation of the SL RNA, we monitored the in vivo association of this polypeptide with the SL RNA genes using chromatin immunoprecipitation (ChIP). To this end, we first generated a T. brucei cell line expressing solely an epitope-tagged version of TbCgm1. We deleted one allele by homologous recombination with a PCR-generated cassette encoding the blasticidin resistance gene and then introduced an N-terminal BB2 epitope in the second allele at its original chromosomal locus. This approach ensured that the epitope-tagged version of the protein was functional and expressed at levels comparable to the endogenous protein. In addition, we generated a cell line solely expressing BB2 epitope-tagged TbCe1. We have previously shown by ChIP that pol II and the transcription factor TRF4 are recruited to the SL RNA genes [37]. Using the same protocol, we found TbCgm1 specifically associated with the SL RNA genes (Fig. 6, lane 6), but not with an upstream control region (lane 3). In contrast, we did not detect recruitment of TbCe1 to the SL RNA genes (lane 12). This result corroborated our RNAi and permeable cell experiments and further supported the conclusion that TbCgm1 plays a role in capping of the SL RNA.
Fig. 6.

TbCgm1 is recruited to the SL RNA genes in vivo. Chromatin immunoprecipitations were performed with antibodies directed against the BB2 epitope tag present on TbCgm1 (lanes 3 and 6) or TbCe1 (lanes 9 and 12), or with no antibodies as a control (lanes 2, 5, 8 and 11). PCR amplification of the immunoprecipitates was done with primers upstream of the SL promoter (nt −608 to −530) or within the promoter (nt −56 to +82). Numbering is with respect to the SL RNA transcription start site. An aliquot of the total input is also shown (lanes 1, 4, 7 and 10).
4. Discussion
Eukaryotes rely on three enzymatic reactions for the formation of the m7GpppN cap, namely the sequential action of RNA 5′ triphosphatase, RNA guanylyltransferase and RNA (guanine-N7) methyltransferase. Although the physical organization of the capping apparatus is quite distinct in different taxa [4,8,9], each enzymatic activity can be assigned to a single polypeptide or domain. In contrast, a distinct and novel genetic organization is emerging in trypanosomatids, namely the presence of two guanylyltransferase domains: one (TbCe1) fused to an N-terminal domain of unknown function [27] and a second (TbCgm1) to a C-terminal (guanine-N7) methyltransferase [29,30]. In addition, there is a separate RNA triphosphatase, which is mechanistically related to the fungal enzymes [28,46], and a separate (guanine-N7) methyltransferase [29]. Although some of these polypeptides have been characterized biochemically and shown to be involved in m7G capping, the challenge remains to assign in vivo functions. Here we have a taken a first step towards this goal and analyzed the functional consequences of TbCe1 and TbCgm1 downregulation. Surprisingly, RNAi downregulation of Ce1 had no phenotypic consequences, precluding further investigations by this route. Nevertheless, we do not interpret this result to mean that TbCe1 is not needed for capping in trypanosomes. Since RNAi does not generate a “knockout”, but rather downregulates mRNA levels, the most likely explanation is that the residual amount of TbCe1 protein was sufficient for cell growth. Indeed, so far we have not been able to generate a “classical” TbCE1 knockout strain, where both alleles of the gene are replaced with an antibiotic resistance marker, indicating that the gene is likely to be essential. Nevertheless, the in vivo function of TbCe1 remains to be determined.
As shown by the presence of diagnostic motifs [29] and by our mutagenesis analysis, TbCgm1 has the hallmarks and the activity of an RNA guanylyltransferase. In contrast to the negative results obtained with RNAi downregulation of CE1, silencing of CGM1 had severe consequences both on cell viability and SL RNA capping: between 2 and 3 days after RNAi induction the cells stopped growing demonstrating that Cgm1 function is essential for cell viability. This result is similar to what has been observed in the yeast S. cerevisiae, where it was originally shown that Ceg1, the gene encoding the capping enzyme, is essential [7]. During the time interval when cell growth begins to slow down, we observed several anomalies with regard to the SL RNA. First, the steady-state level of the SL RNA decreased substantially. This effect could be due to the instability of SL RNA molecules without or with unmethylated cap and/or could be the result of the inhibition of trans-splicing, which is likely to cause havoc in cells. Second, there was accumulation of molecules bearing a hypomodified cap 4 structure. The explanation for this finding most likely lies in the requirement of the m7G cap for the biogenesis of the cap 4 structure. Indeed, in vaccinia virus the steps in the biosynthesis of the cap must occur in a unique sequence, i.e. the modification of the nucleotide adjacent to the cap requires that the m7G cap be in place [47]. Third, during CGM1 silencing the proportion of SL RNA bearing a methylated cap substantially decreased, as assessed by antibody immunoprecipitation of steady-state SL RNA and by TAP digestion of newly–synthesized SL RNA. Lastly, P1 digestion and TLC analysis showed that there was accumulation of newly-synthesized SL RNA molecules with an ADP terminus. In summary, our results showed that Cgm1 depletion results in defects in both the addition of GMP and the methylation of the N7 position of guanine. On the basis of our analyses, we propose that the guanylyltransferase domain of TbCgm1 is responsible for adding GMP to diphosphate terminated SL RNA. Activity assays with a recombinant protein corroborated the proposed function of this domain. Furthermore, the observation that the relative amount of m7G cap decreased, as a consequence of TbCgm1 depletion, with a concomitant increase of the unmethylated GMP cap, provides evidence that this polypeptide is also required for the methylation step of the capping reaction. So far, we have not been able to produce the putative C-terminal (guanine-N7) methyltransferase domain in a recombinant form to test its proposed enzymatic activity. While this manuscript was in preparation, the Ho laboratory reported that recombinant TbCgm1 is a bifunctional capping enzyme capable of transferring GMP to the SL RNA via covalent enzyme-GMP intermediate [30]. In addition, it was shown that TbCgm1 methylates the guanine N-7 position of GpppN-terminated RNA. Taken together, these results are in agreement with TbCgm1 being responsible for capping of the SL RNA.
We have previously shown that SL RNA transcripts as short as 30 nucleotides are capped, consistent with a co-transcriptional mechanism for m7G capping addition [3]. This model is further strengthened by our ChIP experiments presented here, which revealed an in vivo association of TbCgm1 with the SL RNA genes. What remains to be explored is how the capping enzyme is recruited to the transcriptional machinery. Since the SL RNA genes are transcribed by pol II [48], the mechanism is most likely similar to that described in yeast and mammals, where there is a physical interaction of the capping enzyme with the CTD of the largest subunit of pol II [49,50].
The presence of two guanylyltransferase domains in trypanosomatid protozoa was unexpected and so far, to the best of our knowledge, has not been reported in any other eukaryotic organism. It is important to bear in mind that capping in these organisms not only occurs on pol II transcripts, namely the SL RNA, but also on a specific subset of pol III transcripts. One attractive possibility is that TbCe1 is involved in capping of a subset of pol III transcripts. How TbCe1 may be directed to the U1, U2, U3 and U4 snRNAs, but not to other more abundant pol III transcripts, is at present a matter of speculation. Since pol III is common to all these genes, other possibilities to consider are that this specificity is achieved either by a gene-specific transcription factor(s), by RNA determinants or by a combination of both mechanisms.
Acknowledgments
We thank Andrei Alexandrov for help and discussion with the TLC analysis. This study received support from National Institutes of Health Grant AI43594 to C.T.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rasmussen EB, Lis JT. In vivo transcriptional pausing and cap formation on three Drosophila heat shock genes. Proc Natl Acad Sci U S A. 1993;90:7923–7. doi: 10.1073/pnas.90.17.7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hagler J, Shuman S. A freeze-frame view of eukaryotic transcription during elongation and capping of nascent mRNA. Science. 1992;255:983–6. doi: 10.1126/science.1546295. [DOI] [PubMed] [Google Scholar]
- 3.Mair G, Ullu E, Tschudi C. Cotranscriptional cap 4 formation on the Trypanosoma brucei spliced leader RNA. J Biol Chem. 2000;275:28994–9. doi: 10.1074/jbc.M004193200. [DOI] [PubMed] [Google Scholar]
- 4.Shuman S. Structure, mechanism, and evolution of the mRNA capping apparatus. Prog Nucleic Acid Res Mol Biol. 2001;66:1–40. doi: 10.1016/s0079-6603(00)66025-7. [DOI] [PubMed] [Google Scholar]
- 5.Shibagaki Y, Itoh N, Yamada H, Nagata S, Mizumoto K. mRNA capping enzyme. Isolation and characterization of the gene encoding mRNA guanylytransferase subunit from Saccharomyces cerevisiae. J Biol Chem. 1992;267:9521–8. [PubMed] [Google Scholar]
- 6.Mao X, Schwer B, Shuman S. Yeast mRNA cap methyltransferase is a 50-kilodalton protein encoded by an essential gene. Mol Cell Biol. 1995;15:4167–74. doi: 10.1128/mcb.15.8.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsukamoto T, Shibagaki Y, Imajoh-Ohmi S, Murakoshi T, Suzuki M, Nakamura A, Gotoh H, Mizumoto K. Isolation and characterization of the yeast mRNA capping enzyme beta subunit gene encoding RNA 5′-triphosphatase, which is essential for cell viability. Biochem Biophys Res Commun. 1997;239:116–22. doi: 10.1006/bbrc.1997.7439. [DOI] [PubMed] [Google Scholar]
- 8.Shuman S. What messenger RNA capping tells us about eukaryotic evolution. Nat Rev Mol Cell Biol. 2002;3:619–25. doi: 10.1038/nrm880. [DOI] [PubMed] [Google Scholar]
- 9.Shuman S, Lima CD. The polynucleotide ligase and RNA capping enzyme superfamily of covalent nucleotidyltransferases. Curr Opin Struct Biol. 2004;14:757–64. doi: 10.1016/j.sbi.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Ho CK, Shuman S. A yeast-like mRNA capping apparatus in Plasmodium falciparum. Proc Natl Acad Sci U S A. 2001;98:3050–5. doi: 10.1073/pnas.061636198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hausmann S, Altura MA, Witmer M, Singer SM, Elmendorf HG, Shuman S. Yeast-like mRNA capping apparatus in Giardia lamblia. J Biol Chem. 2005;280:12077–86. doi: 10.1074/jbc.M412063200. [DOI] [PubMed] [Google Scholar]
- 12.Hakansson K, Doherty AJ, Shuman S, Wigley DB. X-ray crystallography reveals a large conformational change during guanyl transfer by mRNA capping enzymes. Cell. 1997;89:545–53. doi: 10.1016/s0092-8674(00)80236-6. [DOI] [PubMed] [Google Scholar]
- 13.Lima CD, Wang LK, Shuman S. Structure and mechanism of yeast RNA triphosphatase: an essential component of the mRNA capping apparatus. Cell. 1999;99:533–43. doi: 10.1016/s0092-8674(00)81541-x. [DOI] [PubMed] [Google Scholar]
- 14.Gong C, Shuman S. Chlorella virus RNA triphosphatase: mutational analysis and mechanism of inhibition by tripolyphosphate. J Biol Chem. 2002;13:13. doi: 10.1074/jbc.M200532200. [DOI] [PubMed] [Google Scholar]
- 15.Takagi T, Moore CR, Diehn F, Buratowski S. An RNA 5′-triphosphatase related to the protein tyrosine phosphatases. Cell. 1997;89:867–73. doi: 10.1016/s0092-8674(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 16.Changela A, Ho CK, Martins A, Shuman S, Mondragon A. Structure and mechanism of the RNA triphosphatase component of mammalian mRNA capping enzyme. EMBO J. 2001;20:2575–86. doi: 10.1093/emboj/20.10.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fabrega C, Hausmann S, Shen V, Shuman S, Lima CD. Structure and mechanism of mRNA cap (guanine-N7) methyltransferase. Mol Cell. 2004;13:77–89. doi: 10.1016/s1097-2765(03)00522-7. [DOI] [PubMed] [Google Scholar]
- 18.Murphy WJ, Watkins KP, Agabian N. Identification of a novel Y branch structure as an intermediate in trypanosome mRNA processing: evidence for trans splicing. Cell. 1986;47:517–25. doi: 10.1016/0092-8674(86)90616-1. [DOI] [PubMed] [Google Scholar]
- 19.Sutton RE, Boothroyd JC. Evidence for trans splicing in trypanosomes. Cell. 1986;47:527–35. doi: 10.1016/0092-8674(86)90617-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laird PW, Zomerdijk JC, de Korte D, Borst P. In vivo labelling of intermediates in the discontinuous synthesis of mRNAs in Trypanosoma brucei. EMBO J. 1987;6:1055–62. doi: 10.1002/j.1460-2075.1987.tb04858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bangs JD, Crain PF, Hashizume T, McCloskey JA, Boothroyd JC. Mass spectrometry of mRNA cap 4 from trypanosomatids reveals two novel nucleosides. J Biol Chem. 1992;267:9805–15. [PubMed] [Google Scholar]
- 22.Arhin GK, Li H, Ullu E, Tschudi C. A protein related to the vaccinia virus cap-specific methyltransferase VP39 is involved in cap 4 modification in Trypanosoma brucei. RNA. 2006;12:53–62. doi: 10.1261/rna.2223406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arhin GK, Ullu E, Tschudi C. 2′-O-methylation of position 2 of the trypanosome spliced leader cap 4 is mediated by a 48 kDa protein related to vaccinia virus VP39. Mol Biochem Parasitol. 2006;147:137–9. doi: 10.1016/j.molbiopara.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 24.Zamudio JR, Mittra B, Zeiner GM, Feder M, Bujnicki JM, Sturm NR, Campbell DA. Complete cap 4 formation is not required for viability in Trypanosoma brucei. Eukaryot Cell. 2006;5:905–15. doi: 10.1128/EC.00080-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hall MP, Ho CK. Functional characterization of a 48 kDa Trypanosoma brucei cap 2 RNA methyltransferase. Nucleic Acids Res. 2006;34:5594–602. doi: 10.1093/nar/gkl573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Tschudi C. Novel and essential subunits in the 300-kilodalton nuclear cap binding complex of Trypanosoma brucei. Mol Cell Biol. 2005;25:2216–26. doi: 10.1128/MCB.25.6.2216-2226.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silva E, Ullu E, Kobayashi R, Tschudi C. Trypanosome capping enzymes display a novel two-domain structure. Mol Cell Biol. 1998;18:4612–9. doi: 10.1128/mcb.18.8.4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho CK, Shuman S. Trypanosoma brucei RNA triphosphatase. Antiprotozoal drug target and guide to eukaryotic phylogeny. J Biol Chem. 2001;276:46182–6. doi: 10.1074/jbc.M108706200. [DOI] [PubMed] [Google Scholar]
- 29.Hall MP, Ho CK. Characterization of a Trypanosoma brucei RNA cap (guanine N-7) methyltransferase. RNA. 2006;12:488–97. doi: 10.1261/rna.2250606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takagi Y, Sindkar S, Ekonomidis D, Hall MP, Ho CK. Trypanosoma brucei encodes a bifunctional capping enzyme essential for cap 4 formation on the spliced leader RNA. J Biol Chem. 2007;282:15995–6005. doi: 10.1074/jbc.M701569200. [DOI] [PubMed] [Google Scholar]
- 31.Djikeng A, Shen S, Tschudi C, Ullu E. Analysis of gene function in Trypanosoma brucei using RNA interference. In: Melville S, editor. Parasite Genomics Protocols. Vol. 270. Humana Press Inc; 2004. pp. 287–97. [DOI] [PubMed] [Google Scholar]
- 32.Wirtz E, Leal S, Ochatt C, Cross GA. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol. 1999;99:89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
- 33.Arhin GK, Shen S, Ullu E, Tschudi C. A PCR-based method for gene deletion and protein tagging in Trypanosoma brucei. Methods Mol Biol. 2004;270:277–86. doi: 10.1385/1-59259-793-9:277. [DOI] [PubMed] [Google Scholar]
- 34.Djikeng A, Shi H, Tschudi C, Ullu E. RNA interference in Trypanosoma brucei: cloning of small interfering RNAs provides evidence for retroposon-derived 24–26-nucleotide RNAs. RNA. 2001;7:1522–30. [PMC free article] [PubMed] [Google Scholar]
- 35.Ullu E, Tschudi C. Permeable trypanosome cells as a model system for transcription and trans-splicing. Nucleic Acids Res. 1990;18:3319–26. doi: 10.1093/nar/18.11.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ullu E, Tschudi C. Trans splicing in trypanosomes requires methylation of the 5′ end of the spliced leader RNA. Proc Natl Acad Sci U S A. 1991;88:10074–8. doi: 10.1073/pnas.88.22.10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruan JP, Arhin GK, Ullu E, Tschudi C. Functional characterization of a Trypanosoma brucei TATA-binding protein-related factor points to a universal regulator of transcription in trypanosomes. Mol Cell Biol. 2004;24:9610–8. doi: 10.1128/MCB.24.21.9610-9618.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi H, Djikeng A, Mark T, Wirtz E, Tschudi C, Ullu E. Genetic interference in Trypanosoma brucei by heritable and inducible double-stranded RNA. RNA. 2000;6:1069–76. doi: 10.1017/s1355838200000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Djikeng A, Ferreira L, D’Angelo M, Dolezal P, Lamb T, Murta S, Triggs V, Ulbert S, Villarino A, Renzi SUE, Tschudi C. Characterization of a candidate Trypanosoma brucei U1 snRNA gene. Mol Biochem Parasitol. 2001;113:91–7. doi: 10.1016/s0166-6851(00)00384-4. [DOI] [PubMed] [Google Scholar]
- 40.Tschudi C, Richards FF, Ullu E. The U2 RNA analogue of Trypanosoma brucei gambiense: implications for a splicing mechanism in trypanosomes. Nucleic Acids Res. 1986;14:8893–903. doi: 10.1093/nar/14.22.8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mottram J, Perry KL, Lizardi PM, Luhrmann R, Agabian N, Nelson RG. Isolation and sequence of four small nuclear U RNA genes of Trypanosoma brucei subsp. brucei: identification of the U2, U4, and U6 RNA analogs. Mol Cell Biol. 1989;9:1212–23. doi: 10.1128/mcb.9.3.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gunzl A, Bindereif A, Ullu E, Tschudi C. Determinants for cap trimethylation of the U2 small nuclear RNA are not conserved between Trypanosoma brucei and higher eukaryotic organisms. Nucleic Acids Res. 2000;28:3702–9. doi: 10.1093/nar/28.19.3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tschudi C, Ullu E. Unconventional rules of small nuclear RNA transcription and cap modification in trypanosomatids. Gene Expr. 2002;10:3–16. [PMC free article] [PubMed] [Google Scholar]
- 44.Mandelboim M, Estrano CL, Tschudi C, Ullu E, Michaeli S. On the role of exon and intron sequences in trans-splicing utilization and cap 4 modification of the trypanosomatid Leptomonas collosoma SL RNA. J Biol Chem. 2002;277:35210–8. doi: 10.1074/jbc.M201910200. [DOI] [PubMed] [Google Scholar]
- 45.McNally KP, Agabian N. Trypanosoma brucei spliced-leader RNA methylations are required for trans splicing in vivo. Mol Cell Biol. 1992;12:4844–51. doi: 10.1128/mcb.12.11.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gong C, Martins A, Shuman S. Structure-function analysis of Trypanosoma brucei RNA triphosphatase and evidence for a two-metal mechanism. J Biol Chem. 2003;278:50843–52. doi: 10.1074/jbc.M309188200. [DOI] [PubMed] [Google Scholar]
- 47.Barbosa E, Moss B. mRNA(nucleoside-2′-)-methyltransferase from vaccinia virus. Characteristics and substrate specificity. J Biol Chem. 1978;253:7698–702. [PubMed] [Google Scholar]
- 48.Gilinger G, Bellofatto V. Trypanosome spliced leader RNA genes contain the first identified RNA polymerase II gene promoter in these organisms. Nucleic Acids Res. 2001;29:1556–64. doi: 10.1093/nar/29.7.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cho EJ, Takagi T, Moore CR, Buratowski S. mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev. 1997;11:3319–26. doi: 10.1101/gad.11.24.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCracken S, Fong N, Rosonina E, Yankulov K, Brothers G, Siderovski D, Hessel A, Foster S, Program AE, Shuman S, Bentley DL. 5′-Capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes Dev. 1997;11:3306–18. doi: 10.1101/gad.11.24.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]