

Abstract
Vaccine immunogens derived from the envelope glycoproteins of the human immunodeficiency virus type 1 (HIV-1) that elicit broad neutralizing antibodies remains an elusive goal. The highly conserved 30 amino acid membrane proximal external region (MPER) of HIV gp41 contains the hydrophobic epitopes for two rare HIV-1 broad cross-reactive neutralizing antibodies, 2F5 and 4E10. Both these antibodies possess relatively hydrophobic HCDR3 loops and demonstrate enhanced binding to their epitopes in the context of the native gp160 precursor envelope glycoprotein by the intimate juxtaposition of a lipid membrane. The Hepatitis B surface antigen (HBsAg) S1 protein forms nanoparticles that can be utilized both as an immunogenic array of the MPER and to provide the lipid environment needed for enhanced 2F5 and 4E10 binding. We show that recombinant HBsAg particles with MPER (HBsAg-MPER) appended at the C-terminus of the S1 protein are recognized by 2F5 and 4E10 with high affinity compared to positioning the MPER at the N-terminus or the extracellular loop (ECL) of S1. Addition of C-terminal hydrophobic residues derived from the HIV-1 Env transmembrane region further enhances recognition of the MPER by both 2F5 and 4E10. Delipidation of the HBsAg-MPER particles decreases 2F5 and 4E10 binding and subsequent reconstitution with synthetic lipids restores optimal binding. Inoculation of the particles into small animals raised cross-reactive antibodies that recognize both the MPER and HIV-1 gp160 envelope glycoproteins expressed on the cell surface; however, no neutralizing activity could be detected. Prime:boost immunization of the HBsAg-MPER particles in sequence with HIV envelope glycoprotein proteoliposomes (Env-PLs) did not raise neutralizing antibodies that could be mapped to the MPER region. However, the Env-PLs did raise anti-Env antibodies that had the ability to neutralize selected HIV-1 isolates. The first generation HBsAg-MPER particles represent a unique means to present HIV-1 envelope glycoprotein neutralizing determinants to the immune system.
Introduction
The human immunodeficiency virus type 1 (HIV-1) envelope glycoproteins, gp120 and gp41 are derived from a heavily glycosylated precursor protein, gp160. The exterior envelope glycoprotein, gp120, constitutes the receptor binding domain and undergoes a series of entry-related conformational changes, first upon interaction with the primary receptor, CD4, and subsequently with the co-receptor, CCR5 or CXCR4 (Alkhatib et al., 1996; Berson et al., 1996; Choe et al., 1996; Deng et al., 1996; Doranz et al., 1996; Dragic et al., 1996; Feng et al., 1996). The transmembrane glycoprotein, gp41, constitutes the trimerization and membrane fusion domain that mediates virus-to-cell membrane fusion and entry of the viral genetic material into target cells. The conserved, membrane proximal region (MPER) of the Env gp41 ectodomain, consists of approximately 30 amino acid proximal to the viral membrane, ending at Lys 683 (HXBc2 numbering system) immediately upstream of the transmembrane domain. The MPER is highly conserved across various strains of HIV and selected hydrophobic residues in the MPER were shown to be important for viral entry (Salzwedel, West, and Hunter, 1999). The membrane proximal heel of the HIV envelope ectodomain is the target of two of the most broadly reactive anti-HIV-1 antibodies identified to date, 2F5 and 4E10 (Muster et al., 1993; Stiegler et al., 2001). Most attempts as immunogens to re-elicit 2F5 or 4E10-like antibodies, using variety of different contexts, have met with limited success (Coeffier et al., 2000; Eckhart et al., 1996; Ernst et al., 1998; Ho, MacDonald, and Barber, 2002; Liang et al., 1999; Muster et al., 1994; Xiao et al., 2000) and summarized in (Ofek et al., 2004). To understand the atomic-level details of 2F5 recognition, the structure of 2F5 with gp41 peptides corresponding to its epitope was solved (Ofek et al., 2004). The structure revealed that the 2F5 antibody bound to only one face of an extended peptide in a cleft between the heavy and light chain, and that this face was much more charged than the unbound hydrophobic face. We observed that the hydrophobic residues at the tip of the 2F5 CDR3 loop directly adjacent to the peptide comprised a contiguous hydrophobic surface. Modeling of the 2F5-bound 17-mer peptide in the context of the virus and surrounding gp41 sequences suggested an antibody-antigen interaction proximal to the viral membrane. To confirm the involvement of lipid in 2F5 antibody-epitope recognition, we carried out a biochemical analysis with solid phase Env-containing proteoliposomes either possessing or lacking a reconstituted membrane. The binding of 2F5 as well as that of another membrane-proximal binding antibody, 4E10, could be enhanced almost two orders of magnitude by the presence of lipid membrane. The solved structure of 4E10 bound to its epitope revealed helical nature of the epitope to which 4E10 binds and implicated lipid proximity of both antibody and antigen (Brunel et al., 2006; Cardoso et al., 2005). These structural and biochemical studies suggested that conformational restriction, steric occlusion of the antibody unbound faces of the epitope and the inclusion of the lipid in the immunogen should be considered in designing immunogens with the potential to elicit 2F5- and 4E10-like antibodies (Ofek et al., 2004; and reviewed in Phogat and Wyatt, 2007; Phogat, Wyatt, and Karlsson Hedestam, 2007).
Viral B-cell epitopes that are presented in rigid, highly repetitive, paracrystalline form were shown to induce neutralizing antibodies that help to clear virus (Zinkernagel et al., 1996). Furthermore, the arrayed B-cell epitopes were recognized as foreign and induced strong B-cell activation to produce protective neutralizing antibodies against surface antigens in several pathogenic viral models (Bachmann et al., 1997; Bachmann and Zinkernagel, 1997; Zinkernagel et al., 2001). Historically, viral vaccines have been live-attenuated or chemically inactivated forms of the virus. The exception to these traditional vaccines are few: the particulate Hepatitis B virus (HBV) vaccine and the similarly particulate human papilloma virus (HPV) vaccine (Brown et al., 2004; Fife et al., 2004; Frazer, 2004; Koutsky et al., 2002) but hopefully recombinant, protein-based vaccines will increase in number. Such vaccines are generally considered safer and less expensive for large scale manufacture (Pantaleo and Koup, 2004; Plotkin, 2003). The vaccine against HBV makes use of particle-forming hepatitis B surface antigen protein, S1, and for HPV, the L1 protein. Both of these proteins self-assemble to form viral-like particles (VLPs). Initially, the HBV surface antigen particles contain plasma-membrane derived lipid constituents that surround the membrane-spanning HBsAg proteins. Assembly of viral antigens into polyvalent VLPs, rather than as free antigens, enhanced the vaccine potency up to 1000-fold for the HBsAg (Cabral et al., 1978; Kirnbauer et al., 1992) and glycoprotein G of rabies vaccine (Piza et al., 2002). The HBV and HPV VLPs were shown to be recognized by dendritic cells (DCs), traffic intracellularly and to induce neutralizing antibodies and CD4+/CD8+ T cell activation in a manner comparable to native virions (Frazer, 2004; Lowe et al., 1997; Schirmbeck, Bohm, and Reimann, 1996).
Previously, recombinant HBsAg particles containing HIV gp120 envelope glycoproteins fused with the S1 protein and the malarial vaccine RTS,S were studied as potential vaccine candidates (Berkower et al., 2004; Heppner et al., 2005). The RTS,S malarial vaccine is in clinical trials and recently was shown to be protective against experimental challenge with P.falciparum sporozoite (Stoute et al., 1997). Here we have investigated the use of the immunogenic HBsAg particulate platform to array the conserved, neutralization-sensitive MPER region of HIV-1.
Results
Design of the HBsAg-MPER fusion constructs
Previous structural and biochemical studies suggested that to elicit 2F5- and 4E10-like antibodies, the membrane context of the respective epitopes was a critical element to be incorporated into immunogen design. And, since peptide or antigenic arrays were shown to enhance B cell responses, we sought to express the MPER “miniproteins” in a Hepatitis B surface antigen S1 protein particulate format (Fig 1). Although not crystallized, the predicted secondary structure of the S1 protein is shown as a model in Fig 1. The S1 protein monomers spontaneously associate into cysteine-linked dimers and eventually to highly cysteine-crosslinked, lipid-containing nanoparticles approximately 22 nm in diameter as determined by electron microscopy (Berkower et al., 2004; Mangold et al., 1997; McAleer et al., 1984; see Fig 1 for approximate positions of the intermolecular cysteine residues). Previously, the multi array, lipid-containing HepB surface antigen platform has demonstrated enhanced immunogenicity and protective responses when presented as a fusion protein with sequences derived from a malarial protein (Heppner et al., 2005)
Fig 1.

Schematic representation of the HIV-1 gp41, the MPER, the Hepatitis B S1 protein and the HBsAg-MPER recombinant particles. The top bar diagram displays the overall organization of the HIV gp41 transmembrane glycoprotein, which contains the MPER between amino acids 656 to 683 (fusion peptide (fp); N-heptad (heptad repeat 1); S-S (cysteine-linked immunodominant region); C-heptad (heptad repeat 2); transmembrane region (tm). In expanded section below, the MPER residues are shown. The residues in contact with 2F5 or 4E10 in the crystal structures are “boxed” in red or purple and the core residues defined in the original peptide mapping of the epitopes are italicized (Cardoso et al., 2005; Ofek et al., 2004). From the crystal structures of peptide with antibody, the 2F5 peptide is depicted as an extended loop (in red) and the 4E10 epitope as an alpha helix (in purple; Cardoso et al., 2005; Ofek et al., 2004). In the next panel, the MPER is schematically shown fused to either the N- or C-terminus of the hepatitis B surface antigen S1 membrane-spanning protein as indicated by the ½ arrowheads. The S1 protein is in blue and the predicted alpha helices and beta strands are shown (from Mahoney FJ, Kane M. Hepatitis B Vaccine. In Plotkin, Orenstein [ed] Vaccines 3rd edition, 1999). The center full arrowhead indicates the approximate position where the MPER was inserted into the S1 extracellular loop. An “SS” marks the approximate positions of the five S1 protein cysteine residues involved in intermolecular dimerization and oligomeric particle formation. The black asterisk indicates where additional hydrophobic C-terminal residues were added to the MPER in selected constructs. Directly below is a schematic diagram of an HBsAg-MPER particle containing an array of an (theoretical) 180 S1-C-terminal MPER fusion sequences per particle (not to scale).
The first sets of constructs (Fig 1 and 2A) were designed to append the MPER to the S1 proteins at three distinct positions, the C-terminus, the N-terminus and the immunodominant ECL region. We chose each of these positions as they were exposed but relatively close to the membrane bilayer and each might enhance presentation of MPER elements for different reasons. In the context of the HIV-1 envelope glycoprotein, the 4E10 epitope lies proximal to the TM, which is more analogous to the N-terminal positioning. In the ECL, we reasoned that since this region is immunodominant it is likely well-exposed and therefore the MPER might be better presented in this context. Positioning the MPER at the C-terminus would constrain the 2F5 epitope-proximal portion of the MPER in a manner similar to its orientation in gp160. However, at the C-terminus the 4E10 epitope region would remain free of anchorage by the hydrophobic TM residues found in its natural gp160 context. Therefore, we added HIV-1 gp160 transmembrane region sequences of varying length (5, 10, 15, or 22 amino acids) to the C-terminus of the 4E10 epitope (Fig 1 and 2B) to better mimic the natural 4E10 epitope configuration. In other instances, to better mimic possible but unknown natural environs of the MPER in the functional spike, gp41 sequences upstream of the 2F5 epitope were included and for another subset of constructs we added a trimerization motif derived from T4 bacteriophage (foldon), in an attempt present the MPER region as a trimer on the HBsAg particles (see Fig 2C).
Fig 2.
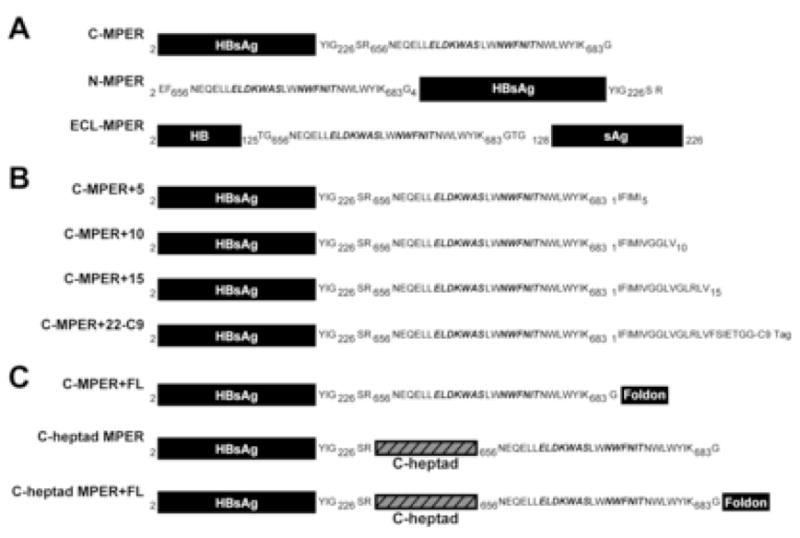
Schematic representation of the construct design using the Hepatitis B surface antigen S1 protein as a carrier molecule. A) The MPER was appended at the N-terminus, C-terminus and the immunodominant extracellular loop of the HepB S1 gene; B) different lengths of the hydrophobic HIV transmembrane region was cloned at the C-terminus of MPER to potentially enhance 4E10 recognition; C) the portion of the the gp41 extracellular domain beginning just after the gp41 second heptad repeat through the MPER ending at the lysine residue at position 683 was appended at the C-terminus of the S1 protein; additionally; we attempted to oligomerize the MPER by appending a foldon trimerization motif as shown.
Partial purification of the recombinant HBsAg-MPER variants
To assess particle production of the designed HBsAg-MPER fusion constructs, individual plasmids were transfected into HEK293 cells. Following expression of the S1 or S1-MPER proteins, cell supernatants were harvested and, in parallel, the cells were treated with lysis buffer to determine the best source for isolation of the HBsAg particles. To detect the presence of recombinant particles, either concentrated cell supernatants or cell lysates were fractionated by sucrose density gradient centrifugation and analyzed by ELISA (before and after sucrose density fractionation) with the HBsAg-specific antibodies. The HIV-1 Gag particles were used as a negative control for antibody and epitope specificity. The presence of the MPER in the particles was confirmed using the 2F5 antibody (Fig 3A). By this analysis, most constructs generated particles with the exception of those containing the C-terminal C9 tag, heptad sequences N-terminal to the MPER and the C-terminal foldon sequences. Further analysis of the HBsAg particles containing the MPER at either the N- or C-terminus was done by Western blotting and recognition of the particles by both 2F5 and antisera specific for the HBsAg S1 protein confirmed that MPER sequences were present in the particles (Fig 3B). When produced from the mammalian cells, the S1-MPER proteins appeared as an approximate 27 kDa doublet; the two bands likely due to differences in glycosylation. The purified yeast-produced HBsAg standard migrated as a 24 kDa monomer and displayed a faint dimer band and high order oligomers (Fig 3B and not shown).
Fig 3.

Antigenic, biochemical and EM analysis of the variant HepB-MPER expression constructs. A) Shown in tabular +/− format is the ELISA analysis of the cell lysates/supernatants derived from the expression constructs (first column) for recognition by the HepBsAg antibody (second column). In the next two columns are ELISA results following sucrose pelleting of lysate for 2F5 to determine if particles were detected in the pellets (third column). HIV Gag particles and HepBsAg commercially available particles were used as controls; B) The partially purified HepB-MPER particles were resolved side-by-side with commercial yeast-purified HBsAg particles on SDS gels followed by Western blotting. As shown by Western (left), the yeast particles displayed a band at 24kDa, the mammalian-expressed HepB-MPER particles show a ~27kDa band and a slightly higher glycosylated form. The HepBsAg C-MPER and N-MPER particles were recognized by 2F5 but not the HBsAg particles and all three particle types were recognized by the HBsAg antibody. C) EM analysis of cells transfected with HbsAg-MPER DNA showed that the predominant compartment for accumulation of the MPER particles was in the rough endoplasmic reticulum.
Following determination that the HBsAg C-MPER protein formed particles, the C-MPER particle were visualized by electron microscopy to see whether they form small ~ 22nm particles similar to the HBsAg particles. The HEK293 cells transfected with C-MPER construct were negatively stained and electron micrographs showed that they form small spherical particles that predominantly accumulate in the rough endoplasmic reticulum inside the cell (Fig 3C).
Antigenic and biophysical analysis of the recombinant HBsAg-MPER variants
To evaluate the positioning of the MPER sequences in the context of S1 protein, S1–MPER particles containing the MPER at either the N- or C-terminus of HBsAg or at the tip of the extracellular loop (ECL) of the S1 protein. The MPER sequences placed either at the N-terminus or the ECL of HBsAg were not well-recognized by either 2F5 or 4E10 antibody, perhaps indicating that the MPER was not proximal to the lipid bilayer. When the MPER sequences were placed at the C-terminus, however, strikingly different recognition pattern of the isolated HBsAg-MPER particles by both 2F5 and 4E10 antibodies was observed (Fig 4A, C-MPER). We interpret the dramatic increase in recognition of the C-MPER particles relative to the N-MPER or ECL-MPER particles by both 2F5 and 4E10 to indicate a much higher affinity for their cognate epitopes in this context. Because the 4E10 binding to the C-MPER particles was slightly less than that compared to 2F5, different lengths of sequences derived from the HIV-1 TM region (5, 10, 15 and 22-C9) were inserted at the C-terminus of the C-MPER construct. The rationale was to potentially enhance 4E10 recognition if the hydrophobic TM-derived residues might insert into the lipid bilayer. Appending the 15 hydrophobic residues from the TM increasing after the C-MPER (C-MPER+15) had the greatest impact on recognition by both the 2F5 and 4E10 antibodies (Fig 4B). Lesser increases in 2F5 and 4E10 recognition were observed by addition of 5 and 10 residues from the hydrophobic TM region (not shown).
Fig 4.

Binding of 2F5 and 4E10 to HepBsG-MPER variants by ELISA. A) the binding 2F5 and 4E10 to HBsAg-MPER particles containing MPER sequences at the N-terminus (N-MPER), the C-terminus (C-MPER) and the extracellular loop (ECL-MPER), B) shows binding of 2F5, 4E10 to N, C-terminus and Extracellular loop HBsAG-MPER particles, and C) the binding of 2F5 and 4E10 to C-MPER particles compared to those containing an additional 15 hydrophobic, C-terminal residues derived from the HIV transmembrane region that lies immediately proximal to the MPER.
Following the observation that the C-terminal positioning of the MPER formed particles (C-MPER) and were well recognized by 2F5 and 4E10, we generated recombinant baculovirus for large-scale production of particles in Hi5 cells. Following expression and sucrose density gradient centrifugation, the partially purified HepB particles were subjected further to CsCl density fractionation. The density of the C-MPER particles on the cesium chloride density gradient was 1.18, which is similar to the density of 1.2reported previously for the wild-type HBsAg particles (Berkower et al., 2004). A representative gradient analysis is shown in Fig 5A. This material was relatively homogeneous by SDS gels and was used for subsequent delipidation and immunogenicity experiments. Following additional hydrophobic interaction chromatography, SDS gel analysis followed by Commassie-blue staining showed a major single protein species migrating at approximately 27 kDa (Fig 5B).
Fig 5.

Analysis of the HepB-MPER particles by density gradient centrifugation and gel electrophoresis. A) A CsCl density gradient of the HepBsAg-MPER particles. Gradient fractions were collected and analyzed for the presence of the MPER by ELISA. The MPER particles displayed a density of 1.18 which very close to the density of 1.2 displayed by the HepB particles. B) An SDS gel of the commercial HBsAg particles compared to the sucrose pelleted, CsCl banded, and reverse phase purified C-MPER particles.
Delipidation and lipid reconstitution of the HBsAg-MPER particles to determine affects on 2F5 and 4E10 recognition
The yeast-expressed HBsAg particles used in the current vaccine are subjected to series of drastic changes in pH (cycles of low and high) and treated with detergent during the purification process to remove lipids and other cellular components. Therefore, we tested if a similar process would affect 2F5/4E10 recognition. As expected from our previous analysis of the 2F5/4E10 binding in the context of proteoliposomes (Ofek et al., 2004), delipidation reduces the binding of the two antibodies to HBsAg-MPER particles (Fig 6). Therefore we asked if lipid reconstitution would restore recognition of 2F5/4E10 to the MPER particles. Both 2F5 (Fig 6, left) and 4E10 antibodies bound with high relative affinity to the baculovirus-purified HBsAg-C-MPER particles and removal of lipids significantly decreased the binding of these antibodies. On reconstitution using the synthetic lipids, DOPC:DOPS (7:3), the binding was fully restored (Fig 6), confirming that both 2F5 and 4E10 binding is enhanced by the presence of lipids. However, recognition by these antibodies is not dependent upon native membrane lipids as reconstituted synthetic lipid bilayers nearly fully restored recognition by both antibodies, consistent with previous observations (Ofek et al., 2004). These data suggest that it will be feasible to make large quantities of the particles in baculovirus systems or in yeast. The particles can then be purified free of cellular components and then reconstituted with synthetic lipids to present the MPER for optimal recognition by both the 2F5 and 4E10 antibodies.
Fig 6.

ELISA binding of 2F5 and 4E10 to HepB MPER particles before and after delipidation and following lipid reconstitution. Left, the binding of 2F5 to unmodified HepB-MPER particles (closed squares), to HepB-MPER particles depleted of lipids (open squares) and to HepB-MPER particles possessing a reconstituted synthetic lipid bilayer (solid triangles). Right, the binding of 4E10 to the particle preps is shown.
HBsAg C-MPER particles raise antibodies that bind to MPER and to HIV-1 envelope glycoproteins
Female Balb/C mice were immunized by the intramuscular route, at intervals of three weeks with HBsAg, HBsAg C-MPER particles or plasmid DNAs expressing these proteins. All the immunogens raised significant endpoint titer antibodies against the commercially available yeast HBsAg particles as determined by ELISA. Endpoint titers of 1:50,000 were observed for mice immunized with HBsAg purified particles or HBsAg DNA, 1:15,000 for HBsAg C-MPER particles or C-MPER encoding plasmid DNA. In parallel, ELISA analysis of the sera for recognition of peptides containing the 2F5 or 4E10 epitopes showed that after four or five inoculations did sera from HBsAg-MPER-particle-immunized mice reacted to the 2F5 or 4E10 peptides. Relatively low titers of 1:1,000 were observed. Similarly, three DNA inoculations followed by two boosts with the HBsAg-MPER particles elicited high-titer antibodies directed toward the HBsAg and low-titer antibodies against the MPER constructs. Sera from mice inoculated four times with the HBsAg C-MPER particles were further analyzed for binding to the MPER expressed on the cell surface by FACS. The sera displayed an end titer similar to that observed by peptide ELISA (Fig 7, left). Importantly, the same sera elicited by the C-MPER showed cross reactivity to the primary envelope HIV envelope glycoproteins JR-FL, YU2 and ADA expressed on the cell surface with roughly equivalent end titers (Fig 7, right and not shown). The binding of the mice sera to the MPER and HIV Env on the cell surface was at least equivalent to the binding seen with 2F5 to these molecules (data not shown)
Fig 7.

FACS-based binding analysis of mouse anti-sera elicited by HepBsAg and HepB sAg-MPER particles. A) Binding to the MPER expressed on the cell surface of HEK 293 cells; B) Binding of the same anti-sera to both cleavage-competent JR-FL gp160 (+) and cleavage-defective JR-FL gp160 (−) expressed on the cell surface. Similar patterns of binding were obtained for YU2 gp160 expressed on the cell surface (not shown).
In preliminary analysis, there was no detectable neutralizing activity in the mouse sera, but due to both limits of available sera and non-specific affects of mouse sera on HIV entry, immunogenicity analysis of the particles was performed in rabbits. Based upon the ability of HBsAg-MPER particles to elicit cross-reactive antibodies to the HIV-1 Env, we used the particles by themselves and in prime-boost regimens with native ADA gp160 presented on proteoliposomes (ADA Env-PLs; (Grundner et al., 2002) in rabbits to evaluate the ability of these novel reagents to elicit neutralizing antibodies.
Prime:Boost Immunization with the HBsAg-MPER particles and ADA Env-PLs in rabbits
The immunization protocol consisted of two priming inoculations followed by subsequent boosting inoculations at weeks 8 and 12 following the second “prime” (Table 1). We tested the hypothesis that by prime:boosting with immunogens that shared in common the MPER only, we might better focus the antibody response to this region and thereby increase MPER-directed neutralizing antibodies. As can be seen in Table 1, we tested prime:boosting in both directions and included appropriate controls. After the 4th inoculation, sera were collected 7 to 10 days post-injection and were assessed for binding to gp120, HBsAg particles and to MPER peptides by ELISA and subsequently for HIV-1 neutralizing reactivity. All rabbits in groups immunized with envelope containing proteoliposomes (ADA EnvPLs) had high endpoint titer anti-gp120 antibodies (reciprocal dilutions >10^5). Animal groups immunized with either wild type or recombinant HBsAg particles had high anti-HepB endpoint titers (reciprocal dilutions of 50,000 – 100,000). Endpoint titers against the MPER were relatively low, (reciprocal dilutions of 1,000 to 3,000).
Table 1.
Immunogenicity with selected immunogens and elicite neutralization activity against selected pseudotyped HIV-1 isolatesa
| Group | Immunogenb | Immunogen | Rabbits | HXBc2 | SF162 | MN | MW965 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Prime Wk 0, 4 | Boost Wk 8, 12 | IDs | Post 2 | Post 4 | Post 2 | Post 4 | Post 2 | Post 4 | Post 2 | Post 4 | |
| IC50 | |||||||||||
| I | Blank beadsc | Blank beads | 1 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 |
| 2 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | |||
| II | HBsAg particles | HBsAg particles | 3 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 |
| 4 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | |||
| III | HBsAg particles | ADA Env-PLs | 5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 |
| 6 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | |||
| IV | HBsAg MPER particles | HBsAg MEPR particles | 7 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 |
| 8 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | |||
| 9 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | |||
| 10 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | |||
| V | HBsAg MPER particles | ADA Env-PLs | 11 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | 49 |
| 12 | <5 | <5 | <5 | 35 | <5 | 1428 | <5 | 449 | |||
| 13 | <5 | <5 | <5 | <5 | <5 | 6 | <5 | 16 | |||
| 14 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | 45 | |||
| VI | ADA Evn-PLs | HBsAg MPER particles | 15 | <5 | <5 | <5 | <5 | <5 | 20 | <5 | <5 |
| 16 | <5 | <5 | <5 | <5 | <5 | <5 | 13 | <5 | |||
| 17 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | |||
| 18 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | |||
| VII | ADA Env-PLs | HBsAg particles | 19 | <5 | <5 | 16 | <5 | <5 | <5 | 112 | <5 |
| 20 | <5 | <5 | 30 | <5 | <5 | <5 | 166 | 154 | |||
| VIII | ADA Env-PLs | ADA Env-PLs | 21 | <5 | 11 | <5 | 18 | <5 | 3310 | <5 | 345 |
| 22 | <5 | <5 | <5 | <5 | <5 | 9 | 100 | 269 | |||
| 23 | <5 | <5 | 18 | 432 | <5 | 4107 | 99 | 886 | |||
| 24 | <5 | <5 | <5 | <5 | <5 | 3597 | <5 | 346 | |||
| 25 | <5 | <5 | <5 | 58 | 1631 | 1809 | 74 | 841 | |||
| 26 | 116 | 1485 | 418 | 513 | 520 | 818 | 258 | 470 | |||
| IX | HBsAg MPER-20 particle | ADA Env-PLs | 27 | <5 | <5 | <5 | <5 | <5 | 391 | <5 | 137 |
| 28 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | |||
| 29 | <5 | <5 | <5 | 59 | <5 | 30821 | <5 | 85 | |||
| 30 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | 28 | |||
| X | HBsAg MPR-20 particles | HBsAg MPER-20 particles | 31 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 |
| 32 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | |||
| 33 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | |||
| 34 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | |||
| 35 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | |||
| 36 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | <5 | |||
Shown are neutralization titers from a single-round neutralization assay with sera after the second and fourth inoculations against the indicated isolates. The values represent reciprocal serum dilutions to achieve 50 % (IC50) neutralization.
All immunogens were combined with CpG+Alum as described in the Methods.
The immunogens of blank beads and unmodified Hepatitis B surface antigen particles (HBsAg) were used a negative controls for either prime or boost as indicated.
To assess neutralizing activity in the sera, the pre-immune and immune serum obtained post 2nd and 4th immunization were diluted and tested in a single-round “TZM” neutralization assay (Mascola et al., 2002; Shu et al., 2007) against the clade B molecular clones HXBc2, SF162, MN and the clade C molecular clone MW965 (Table 1). The sera were tested to determine the titer that resulted in 50% neutralization (IC50; Table 1) or 80% neutralization (IC80; not shown). Neutralization was only observed when the rabbits were immunized with ADA Env-PL either as a prime or a boost; we did not observe any neutralization with the recombinant HBsAg particles, or MPER-HBsAg particles either alone or as a prime or a boost. The anti-Env antibodies showed significant neutralization of the relatively neutralization-sensitive MN and MW965 pseudoviruses and in some cases, neutralization of HXBc2. Mapping studies were performed using selected neutralizing sera pre-incubated with peptides capable of inhibiting V3-directed neutralization as previously described (Dey et al., 2007; Li et al., 2006)(Li et al; Dey et al) or MPER-directed neutralization (Li et al, manuscript in preparation). While there was an indication of some V3-directed neutralization activity in some of the Env-PL elicited sera, we could not detect the presence of any anti-MPER neutralizing antibodies as defined by the MPER peptide inhibition assay using the HXBc2 isolate (not shown).
Discussion
Most attempts to elicit MPER-directed neutralizing antibodies have failed completely or met with limited success at eliciting broadly neutralizing antibodies (summarized in (Ofek et al., 2004)). Here we present a rational design of immunogens to present the MPER on the particulate hepatitis B surface antigen platform that has been used successfully to elicit protective antibodies against hepatitis B infection. The hepatitis B particles form spontaneously from the multi-spanning hepatitis S1 protein. We present data regarding several HBsAg S1-MPER fusion constructs, their ability to form particles, recognition of the particles by the MPER-specific antibodies 2F5 and 4E10, the affect of lipid on recognition by these antibodies and an initial immunogenicity analysis of sera from mice and rabbits immunized with selected HBsAg-MPER constructs. We show that the recombinant HBsAg particles with the MPER appended at the C-terminus of the S1 protein bind 2F5 and 4E10 with the highest affinity compared to the positioning of MPER at N-terminus or the extracellular loop of S1. Positioning the MPER at the C-terminus of the S1 protein may provide proximity or an orientation to the membrane to enhance antibody recognition compared to positions at either the N-terminus or the ECL. Alternatively, coupling of the MPER to the S1 protein at these two positions may occlude antibody recognition in some unanticipated manner not predicted by modeled secondary structure of the S1 protein (see Fig 1). The 2F5 epitope is in close proximity to the membrane in C-MPER construct but on the native gp160 or gp120/gp41 Env, the 4E10 epitope is immediately upstream of the membrane. In a rational attempt to further improve 4E10 binding in the C-MPER particles, we appended hydrophobic residues from the HIV-1 Env transmembrane to the C-terminal region of the C-MPER. Addition of the hydrophobic TM residues improved relative binding affinity of both the2F5 and 4E10 antibodies, presumably due the presence of hydrophobic context immediately following the hydrophobic 4E10 epitope. Perhaps the additional hydrophobic residues permit a more intimate association of the epitope with the lipid bilayer. This is consistent with previous data presenting HIV gp160 in a proteoliposome context (Ofek et al., 2004) as well as delipidation/reconstitution experiments presented here.
The data suggest that specific cellular lipids were not required for binding rather a hydrophobic context was needed, as described for 2F5 and 4E10 binding on the proteoliposome (Ofek et al., 2004). This further suggests that these particles can be made in yeast following similar processes used for the HepB vaccine, and at a final step, a lipid bilayer can be reconstituted using synthetic lipids.
In this study, we describe our initial attempts to test the hypothesis that MPER arrayed on HBsAg particles will enhance immunogenicity of this relatively hydrophobic and membrane-proximal region. To this end, we have immunized mice with C-terminal MPER HBsAg particles and observed that relatively low titer MPER-directed antibodies were elicited. Although the relatively low titer was disappointing, perhaps this result is not so surprising given that the two immunodominant and relatively well-exposed HepB-specific epitopes are present in the particles. Future studies deleting these regions, or masking them with N-linked glycans may improve the relative immunogenicity of this region.
Encouragingly and importantly, the HBsAg C-MPER particles elicited sera that showed cross-reactive binding to a primary isolate-derived HIV-1 envelope glycoprotein expressed on the cell surface. We feel that this is a substantial improvement over other presentations of the MPER, For example, when we coupled a peptide containing the structurally defined 2F5 epitope to KLH and immunized rabbits, we did elicited antibodies that could recognize the free peptide but not antibodies capable of cross recognition of native, cell-surface gp160 (not shown). This suggests that the MPER in the particle format presents itself in a qualitatively different form than the free peptide, although in this format both the non-neutralizing and neutralizing faces of the 2F5 and 4E10 epitopes might be accessible to the immune system. The data do, in part, alleviate some concerns that the MPER region is not immunogenic due to mimicry of self-structures such as cardiolipin (Haynes et al., 2005), but does not rule out this as a potential immune dampening mechanism. It is possible that cloaking of the MPER in self-lipid renders it poorly immunogenic or poorly accessible on the virus to most MPER-directed antibodies. The elicitation of antibodies directed toward the MPER may require the utilization of selected MPER constructs in concert with native gp160 trimers in a heterologous prime:boost strategy to immuno-focus the humoral antibody response on relevant MPER conformations as suggested previously (Ofek et al., 2004). As an initial test of this hypothesis, we performed prime:boost immunization in rabbits using both the HBsAg -MPR and Env-PL immunogens. As show in Table 1, we observed neutralization only when the Env-PLs were one component of the immunization regimen. Also the most potent neutralization was observed when the Env-PLs were immunized four times consecutively (Group VIII animals). Since we observed less potent immunization when the MPER-HBsAg particles were a component of the regimen, the data indicate that the prime:boost strategy did not preferentially elicit MPER-directed neutralizing antibodies. As previously reported, the Env-PLs raised anti-Env antibodies that had the ability to neutralize some HIV isolates (Grundner et al., 2002), but no neutralization directed toward the MPER could be detected. Likely the neutralization elicited here by the Env-PLs is also gp120-directed and not against the MPER, consistent with the inability of peptides from this region to inhibit neutralization of the HXBc2 isolate (not shown). The data again emphasize that the MPER region is weakly immunogenic relative to elements of gp120 and that presentation of the correct MPER conformation to the immune system is difficult. Perhaps the use of more potent adjuvants along with presentation of the elusive relevant conformations of the MPER would better elicit MPER-directed neutralizing antibodies.
Materials and Methods
Construction of HBsAg–MPER variants
The synthetic S-gene HBsAg (Berkower et al., 2004) amino acid 2 to 226 was used as a scaffold to implant the membrane proximal regions of HIV-1 gp41 at the N-terminus, C-terminus or the extra-cellular loop of the HBsAg. S1-gene was PCR-amplified from the vector pGEM using the forward primer 5′ GGAGCTCGTCGACAGCAA3′ and reverse primer 5′GCTCTA GACCCGATGTAGACCCA 3′ to introduce Sal I site at the 5′ and Xba I site at the 3′ end of the gene. The PCR-amplified product was cloned in to pCMV/R vector at the Sal I and Xba I sites. Variants of gp41 sequences containing the MPER were PCR-amplified using codon-optimized HIV-1 YU2 gp160 or JRFLgp160 constructs as the template. Primer pairs used are listed in Table 2. The MPER was positioned at the C-terminus of S1 gene (C-MPER) and at the N-terminus after the 2nd and 3rd amino acids (E and F, respectively), of the HBsAg S1 sequence (N-MPER). An Age I site was created in the extra cellular loop of the S1 gene by replacing amino acids P126 and A127 with T and G substitutions. Following the G residue, the MPER codons (with a 3 amino acid linker GTG at the C-terminus of the MPER codons) were cloned at the Age I site to place it in the extra cellular loop of S1 sequences (ECL-MPER). The second sets of constructs were generated to introduce different lengths of HIV-1 transmembrane region after the lysine 683 of the MPER, 1IFIMI5 for C-MPER-5, 1IFIMIVGGLV10 for C-MPER-10, 1IFIMIVGGLVGLRLV15 for C-MPER-15 and 1IFIMIVGGLVGLRLVFSIETGG 22 TETSQVAPA – C9 tag for C-MPER-22-C9 in order to further stabilize and orient the 4E10 epitope. The final set of constructs was generated with HIV-1 gp41 region, C-heptad and or the MPER at the C-terminus of HBsAg. Between the HBsAg and the gp41 region, a two amino acids S and R were introduced and following the K at residue 683, a glycine was placed immediately before the stop codon. The T4 fibritin trimerization domain, foldon, was also introduced in two of the constructs to see the effect of trimerization on recombinant HBsAg particle production and recognition of 2F5 and 4E10.
Table 2.
Oligonucleotides used for generating HBsAg-MPER constructs
| Primer Name | Primer Sequence |
|---|---|
| SAg-Forward | 5′ GGAGCTCGTCGA CAGCAA 3′ |
| SAg-Reverse | 5′ GC TCT AGA CCC GA T GTA CAC CCA 3′ |
| MPER Forward | 5′ GC TCT AGA AAC GAG CAG GAG CTG CTG 3′ |
| MPER Reverse | 5′ CGC GGA TCC TCA CCC CTT GAT GTA CCA CAG CCA CTT 3′ |
| MPER-Foldon Rev | 5′ CGC GGA TCC TCA ATG GTG ATG GTG ATG GTG GGG 3′ |
| C-heptad-MPER Forward | 5′ GC TCT AGA GCC GTG GAG CGG TAC CTG 3′ |
| MPER-Tm5 Reverse | 5′ CTCGGATCCTCAAATCATGATGAAAATCTTGAT 3′ |
| MPER-Tm10 Reverse | 5′ CTCGGATCCTCACACCAGGCCACCAACAAT 3′ |
| MPER-Tm15 Reverse | 5′ CTCGGATCCTCACACCAGCCTCAGGCCCAC 3′ |
| MPER-Tm23-C9 Reverse | 5′ CTCGGATCCTCAGGCGGGCGC 3′ |
| AgeI Forward | 5′ CCCTGCAAGACCTGCACC ACCACCGGTCAGGGCAACTCCAAGTTCCCC 3′ |
| AgeI reverse | 5′ GGGGAACTTG GAGTTGCCCT GACCGGTGGT GGTGCAGGTC TTGCAGGG 3′ |
| MPER AgeI Forward | 5′ GGC ACC GGT AAC GAG CAG GAG CTG CTG 3′ |
| MPER AgeI Reverse | 5′ GGC ACC GGT CCC CTT GAT GTA CCA CAG CCA CTT 3′ |
| MPERSAG Forward | 5′ AGC GAA TTC AAC GAG CAG GAG CTG CTG 3′ |
| MPER SAG Reverse | 5′ CGC GGA TCC TCA CCC GA T GTA CAC CCA 3′ |
| SAGMPER RI forward | 5′ CAG GAA GCC GGA GGT GATGAA CCC CTT GAT GTA CCA CAG CCA CTT 3′ |
| SAG MPER RI Reverse | 5′ AAG TGG CTG TGG TAC ATC AAG GGG TTC ATC ACC TCC GGC TTC CTG 3′ |
Transient transfections, particle production and analysis
All the constructs were transfected in HEK293T cells. One day prior to transfection, 8 million HEK 293T cells in DMEM, 10 % FBS, 1 % penicillin-streptomycin (pen-strep) were seeded in a 150 mm tissue culture dish. The cells were transfected with the plasmids encoding recombinant HBsAg-MPER and MPER variants or wild-type HBsAg using Fugene6 (Roche) at a ratio of DNA:Fugene6 1:3 and 10 ug total DNA plate. Four days after transfection, cells and supernatant were collected. Supernatant was concentrated using Centricon Plus-80 100 kDa Biomax membrane (Millipore, Billerica, MA) to 25 mls. The cells were lysed by resuspending it in 10 ml of 1 X PBS and sonication for 1 min at 20 Hz every 10 sec using a probe sonicator and the cell lysate was clarified by centrifugation at 15,000 rpm in an Eppendorf centrifuge for 15 mins. Cell lysates were loaded on a 20% sucrose cushion (20% sucrose in PBS) and centrifuged at 23,000 rpm for 16 hr (Surespin rotor, Sorvall). The partially purified VLPs were resuspended in PBS and analyzed by ELISA or Western blotting. To further purify the VLPs, the particles were loaded onto a 10–40% (wt/wt) CsCl step gradient (in PBS) and centrifuged at 22h at 36000 rpm (TV-860 rotor; Sorvall), and a 500μl fractions were taken from the bottom of the tube. ELISA identified the fractions containing VLPs. The positive fractions were, desalted, concentrated, washed with PBS using Amicon YM-100 filter (Millipore).
EM analysis of the particles
The HEK293 cells transfected with C-terminus HBsAG-MPER DNA were collected 24 hrs after transfection and washed with PBS. Approximately 3 × 106 cells were fixed in 2% paraformaldehyde-2% glutaraldehyde in 0.10M cacodylate buffer. Thin sections were negatively stained with 2% methylamine tungstate and analyzed by electron microscopy at a magnification of 100,000X. For cell-free particles, the fractions from the CsCl gradient were selected by optical density and the particles contained in those fractions were negatively stained with 2% methylamine tungstate and analyzed by electron microscopy.
Recombinant baculovirus particle production
The HBsAg-MPER sequence was PCR-amplified from pCMV/R HBsAg-MPER construct and ligated into the pFastbac plasmid between the Sal I and Spe I restriction sites. The pFastbac sAg-MPER plasmid was transformed into competent cells containing full-length bacmid DNA (Invitrogen), to generate recombinant bacmid. Two positive bacmids were sent to ATG Laboratories, Inc., in Eden Prairie, MN for titer and production of recombinant baculovirus in Sf9 cells. For particle production, the Hi5 or Sf8 cells were infected at a multiplicity of infection of four and. the cell pellets were harvested at about 48 hours following infection. Cells were lysed by sonication and the lysate was layered onto sucrose gradients. After sedimentation for 2 hrs at 27,000 rpm in an SW28 rotor, fractions were collected from the bottom. Each fraction was assayed for MPER content, using monoclonal antibody 2F5. Particles were further purified either by CsCl gradient centrifugation for immunogenicity as described above or on a Macro-prep methyl HIC column (from Bio Rad) for SDS gel analysis. For purification by the hydrophobic methyl Macro-prep method, particles in the pooled sucrose fractions were suspended in 0.01 M sodium phosphate buffer, pH 6.8 with 0.6 M ammonium sulfate and 0.05% CHAPSO (Anagrade), washed in the same buffer and eluted with 0.2 M ammonium sulfate.
ELISA analysis of the HBsAg-MPER particles
To determine the presence of HBsAg, HBsAg-MPER and MPER variants VLPs in the preparations, ELISAs were performed as follows. For direct ELISA, the particles were adsorbed onto a high-protein-binding microwell plate (Corning) for 2hrs, then incubated with blocking buffer (bb; PBS with 2% dry milk). After one wash with PBS/0.2% Tween-20, anti-HBsAg antibody NE3 or NF5 (Aldevron) was added to each well in serial dilution and incubated at 37C for 1hr. After three washes with PBS/0.2% Tween-20, a secondary Anti-Mouse-IgG-HRP antibody (Sigma) was added in washing buffer at a 1:5000 dilution for 1 h at 37C. Following three washes, the ELISAs were developed with 100 μl TMB Peroxidase substrate (KPL). The reaction was stopped by adding 100 μl 1 M HCl to each well. The optical density at 450 nm was read on a microplate reader (Molecular Devices).
For sandwich ELISA, 500 nanograms of the HBsAg-specific mouse monoclonal antibody NE3 (Aldevron) was adsorbed onto each well overnight at 4°C. The next day, following incubation of bb, the particles were resuspended in PBS and 100 ul of the suspension was added to each well and incubated 37°C for 2 hr. After one wash with PBS/0.2% Tween-20, either the antibody 2F5 or 4E10 (kindly provided by H Katinger) or HIVIgG (NIH AIDS Reagent Repository Program) was added to each well as a serial dilution and incubated at 37°C for 1hr. After three washes with PBS/0.2% Tween-20, a secondary Anti-human-IgG-HRP antibody (Jackson Immuno Research labs) was added in washing buffer at a 1:5000 dilution for 1 h at 37°C. Following three washes, the ELISAs were developed as described above.
For competition ELISA, all the steps similar to sandwich ELISA were performed except that the peptide NEQELLELDKWASLWN was mixed along with 2F5 diluted and incubated at 37C for 1hr. To determine the effect of lipid on antibody binding, the Baculovirus expressed C-terminus MPER particles were treated with high and low pH and in PBS containing 1% CHAPSO and then diafiltered to remove the lipids and concentrate the particles. To a portion of the delipidated particles, the lipids DOPC:DOPS (7:3) (Avanti Polar lipids, Inc) were added, the samples was dialyzed against PBS and all particle preparations were analyzed by sandwich ELISA.
Inoculations
BalB/c mice were inoculated by the intramuscular route at intervals of 3 weeks with 5 μg of HBsAg or HBsAg-MPER particles in the presence of alum or 50 μg of HBsAg, HBsAg-MPER, HBsAg-MPER+15 DNA followed by HBsAg or HBsAg-MPER particle/alum boost. Animals were inoculated up to a total of five times. Sera was collected 7–10 days after each inoculation and analyzed either by ELISA, cell surface staining by FACS or in selected instances by a neutralization assay.
For the prime:boost immunization strategy, rabbits were immunized by two priming immunizations at weeks 0 and 4, followed by two boosting immunizations at weeks 8 and 12. The dose of inoculation of the ADA Env-PLs was approximately 20 to 30 ug of envelope glycoproteins as estimated by SDS gels using a defined volume of Env-PL bead suspension. Inoculation was by the intradermal route and CpG plus alum was used as adjuvant for all the immunogens.
Fluorescence-activated cell sorting (FACS) staining at the cell surface
FACS staining was performed as previously described (Koch et al., 2003; Pancera and Wyatt, 2005). Forty-eight hours following transfection, the cells were harvested and washed in FACS buffer (PBS, 5% fetal bovine serum, 0.02% azide) and stained with sera from mice immunized with HBsAg or HBsAg-MPER. The polyclonal antibodies were washed in FACS buffer and anti-mouse-FITC (SIGMA) at a 1:200 dilution was added for 30 minutes and then washed to remove unbound secondary antibody. The stained cells were analyzed by FACS on a Beckman Coulter Calibur Instrument.
Virus Neutralization Assay
Single round virus neutralization was performed as described earlier (Li et al., 2006; Shu et al., 2007). In brief, HIV-1 was pseudotyped with selected envelope glycoproteins by the cotransfection of an env expressor and viral genomic DNA with a deletion of Env into 293T cells. The MN Env plasmid was provided by David Montefiori, MW965 was obtained for the NIH AIDS Research and Regent Program. Following the production of pseudotyped virus, a luciferase-based neutralization assay was performed as previously described. Briefly, TZM-bl cells expressing CD4, CXCR4, and CCR5 were used for HIV-1 infection. These target cells contain Tat-responsive reporter genes for firefly luciferase and the Escherichia coli β-galactosidase gene under the regulatory control of the HIV-1 long terminal repeat. The level of HIV-1 infection was quantified by measuring relative light units (RLU) of luminescence, which is directly proportional to the amount of virus input. The assays were performed using a 96-well microtiter plate format with 10,000 TZM-bl cells per well. For neutralization assays, each pseudotyped virus stock was diluted to a level that produced approximately 100,000 to 500,000 RLU. The percentage of virus neutralization by each immune serum sample was derived by calculating the reduction in RLUs in the test wells compared to the RLUs in the wells containing preimmune serum from the corresponding animal. To control for nonspecific neutralization in protein-immunized rabbits, sera from two animals immunized with BSA were analyzed. All serum samples were also assayed for neutralizing activity against a pseudovirus expressing the amphotropic murine leukemia virus envelope to test for non-HIV-1-specific plasma effects (Mascola et al., 2002). To obtain IC50 and IC80 data, fivefold serial dilution of immune sera were incubated with viruses before infection of target cells. Antiserum dose-response curves were fit with a nonlinear function, and the IC50 and IC80 for the corresponding virus was calculated by a least-squares regression analysis.
Acknowledgments
We would like to thank Adhuna Phogat for help with expression of selected constructs and thanks to Brenda Hartman and Michael Cichanowski for assistance with the graphics. These studies were supported by the NIAID intramural research program, the International AIDS Vaccine Initiative and the Bill and Melinda Gates Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272(5270):1955–8. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- Bachmann MF, Kalinke U, Althage A, Freer G, Burkhart C, Roost H, Aguet M, Hengartner H, Zinkernagel RM. The role of antibody concentration and avidity in antiviral protection. Science. 1997;276(5321):2024–7. doi: 10.1126/science.276.5321.2024. [DOI] [PubMed] [Google Scholar]
- Bachmann MF, Zinkernagel RM. Neutralizing antiviral B cell responses. Annu Rev Immunol. 1997;15:235–70. doi: 10.1146/annurev.immunol.15.1.235. [DOI] [PubMed] [Google Scholar]
- Berkower I, Raymond M, Muller J, Spadaccini A, Aberdeen A. Assembly, structure, and antigenic properties of virus-like particles rich in HIV-1 envelope gp120. Virology. 2004;321(1):75–86. doi: 10.1016/j.virol.2003.12.017. [DOI] [PubMed] [Google Scholar]
- Berson JF, Long D, Doranz BJ, Rucker J, Jirik FR, Doms RW. A seven-transmembrane domain receptor involved in fusion and entry of T-cell-tropic human immunodeficiency virus type 1 strains. J Virol. 1996;70(9):6288–95. doi: 10.1128/jvi.70.9.6288-6295.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DR, Fife KH, Wheeler CM, Koutsky LA, Lupinacci LM, Railkar R, Suhr G, Barr E, Dicello A, Li W, Smith JF, Tadesse A, Jansen KU. Early assessment of the efficacy of a human papillomavirus type 16 L1 virus-like particle vaccine. Vaccine. 2004;22(21–22):2936–42. doi: 10.1016/j.vaccine.2003.11.059. [DOI] [PubMed] [Google Scholar]
- Brunel FM, Zwick MB, Cardoso RM, Nelson JD, Wilson IA, Burton DR, Dawson PE. Structure-function analysis of the epitope for 4E10, a broadly neutralizing human immunodeficiency virus type 1 antibody. J Virol. 2006;80(4):1680–7. doi: 10.1128/JVI.80.4.1680-1687.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral GA, Marciano-Cabral F, Funk GA, Sanchez Y, Hollinger FB, Melnick JL, Dreesman GR. Cellular and humoral immunity in guinea pigs to two major polypeptides derived from hepatitis B surface antigen. J Gen Virol. 1978;38(2):339–50. doi: 10.1099/0022-1317-38-2-339. [DOI] [PubMed] [Google Scholar]
- Cardoso RM, Zwick MB, Stanfield RL, Kunert R, Binley JM, Katinger H, Burton DR, Wilson IA. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity. 2005;22(2):163–73. doi: 10.1016/j.immuni.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85(7):1135–48. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- Coeffier E, Clement JM, Cussac V, Khodaei-Boorane N, Jehanno M, Rojas M, Dridi A, Latour M, El Habib R, Barre-Sinoussi F, Hofnung M, Leclerc C. Antigenicity and immunogenicity of the HIV-1 gp41 epitope ELDKWA inserted into permissive sites of the MalE protein. Vaccine. 2000;19(7–8):684–93. doi: 10.1016/s0264-410x(00)00267-x. [DOI] [PubMed] [Google Scholar]
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381(6584):661–6. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- Dey B, Pancera M, Svehla K, Shu Y, Xiang SH, Vainshtein J, Li Y, Sodroski J, Kwong PD, Mascola JR, Wyatt R. Characterization of human immunodeficiency virus type 1 monomeric and trimeric gp120 glycoproteins stabilized in the CD4-bound state: antigenicity, biophysics, and immunogenicity. J Virol. 2007;81(11):5579–93. doi: 10.1128/JVI.02500-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996;85(7):1149–58. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381(6584):667–73. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- Eckhart L, Raffelsberger W, Ferko B, Klima A, Purtscher M, Katinger H, Ruker F. Immunogenic presentation of a conserved gp41 epitope of human immunodeficiency virus type 1 on recombinant surface antigen of hepatitis B virus. J Gen Virol. 1996;77 (Pt 9):2001–8. doi: 10.1099/0022-1317-77-9-2001. [DOI] [PubMed] [Google Scholar]
- Ernst W, Grabherr R, Wegner D, Borth N, Grassauer A, Katinger H. Baculovirus surface display: construction and screening of a eukaryotic epitope library. Nucleic Acids Res. 1998;26(7):1718–23. doi: 10.1093/nar/26.7.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272(5263):872–7. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- Fife KH, Wheeler CM, Koutsky LA, Barr E, Brown DR, Schiff MA, Kiviat NB, Jansen KU, Barber H, Smith JF, Tadesse A, Giacoletti K, Smith PR, Suhr G, Johnson DA. Dose-ranging studies of the safety and immunogenicity of human papillomavirus Type 11 and Type 16 virus-like particle candidate vaccines in young healthy women. Vaccine. 2004;22(21–22):2943–52. doi: 10.1016/j.vaccine.2003.11.058. [DOI] [PubMed] [Google Scholar]
- Frazer IH. Prevention of cervical cancer through papillomavirus vaccination. Nat Rev Immunol. 2004;4(1):46–54. doi: 10.1038/nri1260. [DOI] [PubMed] [Google Scholar]
- Grundner C, Mirzabekov T, Sodroski J, Wyatt R. Solid-phase proteoliposomes containing human immunodeficiency virus envelope glycoproteins. J Virol. 2002;76(7):3511–21. doi: 10.1128/JVI.76.7.3511-3521.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes BF, Fleming J, St Clair EW, Katinger H, Stiegler G, Kunert R, Robinson J, Scearce RM, Plonk K, Staats HF, Ortel TL, Liao HX, Alam SM. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science. 2005;308(5730):1906–8. doi: 10.1126/science.1111781. [DOI] [PubMed] [Google Scholar]
- Heppner DG, Jr, Kester KE, Ockenhouse CF, Tornieporth N, Ofori O, Lyon JA, Stewart VA, Dubois P, Lanar DE, Krzych U, Moris P, Angov E, Cummings JF, Leach A, Hall BT, Dutta S, Schwenk R, Hillier C, Barbosa A, Ware LA, Nair L, Darko CA, Withers MR, Ogutu B, Polhemus ME, Fukuda M, Pichyangkul S, Gettyacamin M, Diggs C, Soisson L, Milman J, Dubois MC, Garcon N, Tucker K, Wittes J, Plowe CV, Thera MA, Duombo OK, Pau MG, Goudsmit J, Ballou WR, Cohen J. Towards an RTS,S-based, multi-stage, multi-antigen vaccine against falciparum malaria: progress at the Walter Reed Army Institute of Research. Vaccine. 2005;23(17–18):2243–50. doi: 10.1016/j.vaccine.2005.01.142. [DOI] [PubMed] [Google Scholar]
- Ho J, MacDonald KS, Barber BH. Construction of recombinant targeting immunogens incorporating an HIV-1 neutralizing epitope into sites of differing conformational constraint. Vaccine. 2002;20(7–8):1169–80. doi: 10.1016/s0264-410x(01)00441-8. [DOI] [PubMed] [Google Scholar]
- Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc Natl Acad Sci U S A. 1992;89(24):12180–4. doi: 10.1073/pnas.89.24.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M, Pancera M, Kwong PD, Kolchinsky P, Grundner C, Wang L, Hendrickson WA, Sodroski J, Wyatt R. Structure-based, targeted deglycosylation of HIV-1 gp120 and effects on neutralization sensitivity and antibody recognition. Virology. 2003;313(2):387–400. doi: 10.1016/s0042-6822(03)00294-0. [DOI] [PubMed] [Google Scholar]
- Koutsky LA, Ault KA, Wheeler CM, Brown DR, Barr E, Alvarez FB, Chiacchierini LM, Jansen KU. A controlled trial of a human papillomavirus type 16 vaccine. N Engl J Med. 2002;347(21):1645–51. doi: 10.1056/NEJMoa020586. [DOI] [PubMed] [Google Scholar]
- Li Y, Svehla K, Mathy NL, Voss G, Mascola JR, Wyatt R. Characterization of antibody responses elicited by human immunodeficiency virus type 1 primary isolate trimeric and monomeric envelope glycoproteins in selected adjuvants. J Virol. 2006;80(3):1414–26. doi: 10.1128/JVI.80.3.1414-1426.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Munshi S, Shendure J, Mark G, 3rd, Davies ME, Freed DC, Montefiori DC, Shiver JW. Epitope insertion into variable loops of HIV-1 gp120 as a potential means to improve immunogenicity of viral envelope protein. Vaccine. 1999;17(22):2862–72. doi: 10.1016/s0264-410x(99)00125-5. [DOI] [PubMed] [Google Scholar]
- Lowe RS, Brown DR, Bryan JT, Cook JC, George HA, Hofmann KJ, Hurni WM, Joyce JG, Lehman ED, Markus HZ, Neeper MP, Schultz LD, Shaw AR, Jansen KU. Human papillomavirus type 11 (HPV-11) neutralizing antibodies in the serum and genital mucosal secretions of African green monkeys immunized with HPV-11 virus-like particles expressed in yeast. J Infect Dis. 1997;176(5):1141–5. doi: 10.1086/514105. [DOI] [PubMed] [Google Scholar]
- Mangold CM, Unckell F, Werr M, Streeck RE. Analysis of intermolecular disulfide bonds and free sulfhydryl groups in hepatitis B surface antigen particles. Arch Virol. 1997;142(11):2257–67. doi: 10.1007/s007050050240. [DOI] [PubMed] [Google Scholar]
- Mascola JR, Louder MK, Winter C, Prabhakara R, De Rosa SC, Douek DC, Hill BJ, Gabuzda D, Roederer M. Human immunodeficiency virus type 1 neutralization measured by flow cytometric quantitation of single-round infection of primary human T cells. J Virol. 2002;76(10):4810–21. doi: 10.1128/JVI.76.10.4810-4821.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAleer WJ, Buynak EB, Maigetter RZ, Wampler DE, Miller WJ, Hilleman MR. Human hepatitis B vaccine from recombinant yeast. Nature. 1984;307(5947):178–80. doi: 10.1038/307178a0. [DOI] [PubMed] [Google Scholar]
- Muster T, Guinea R, Trkola A, Purtscher M, Klima A, Steindl F, Palese P, Katinger H. Cross-neutralizing activity against divergent human immunodeficiency virus type 1 isolates induced by the gp41 sequence ELDKWAS. J Virol. 1994;68(6):4031–4. doi: 10.1128/jvi.68.6.4031-4034.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muster T, Steindl F, Purtscher M, Trkola A, Klima A, Himmler G, Ruker F, Katinger H. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J Virol. 1993;67(11):6642–7. doi: 10.1128/jvi.67.11.6642-6647.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofek G, Tang M, Sambor A, Katinger H, Mascola JR, Wyatt R, Kwong PD. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J Virol. 2004;78(19):10724–37. doi: 10.1128/JVI.78.19.10724-10737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancera M, Wyatt R. Selective recognition of oligomeric HIV-1 primary isolate envelope glycoproteins by potently neutralizing ligands requires efficient precursor cleavage. Virology. 2005;332(1):145–56. doi: 10.1016/j.virol.2004.10.042. [DOI] [PubMed] [Google Scholar]
- Pantaleo G, Koup RA. Correlates of immune protection in HIV-1 infection: what we know, what we don’t know, what we should know. Nat Med. 2004;10(8):806–10. doi: 10.1038/nm0804-806. [DOI] [PubMed] [Google Scholar]
- Phogat S, Wyatt R. Rational modifications of HIV-1 envelope glycoproteins for immunogen design. Curr Pharm Des. 2007;13(2):213–27. doi: 10.2174/138161207779313632. [DOI] [PubMed] [Google Scholar]
- Phogat S, Wyatt RT, Karlsson Hedestam GB. Inhibition of HIV-1 entry by antibodies: potential viral and cellular targets. J Intern Med. 2007;262(1):26–43. doi: 10.1111/j.1365-2796.2007.01820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piza AT, Pieri KM, Lusa GM, Caporale GM, Terreran MT, Machado LA, Zanetti CR. Effect of the contents and form of rabies glycoprotein on the potency of rabies vaccination in cattle. Mem Inst Oswaldo Cruz. 2002;97(2):265–8. doi: 10.1590/s0074-02762002000200022. [DOI] [PubMed] [Google Scholar]
- Plotkin SA. Vaccines, vaccination, and vaccinology. J Infect Dis. 2003;187(9):1349–59. doi: 10.1086/374419. [DOI] [PubMed] [Google Scholar]
- Salzwedel K, West JT, Hunter E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J Virol. 1999;73(3):2469–80. doi: 10.1128/jvi.73.3.2469-2480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmbeck R, Bohm W, Reimann J. Virus-like particles induce MHC class I-restricted T-cell responses. Lessons learned from the hepatitis B small surface antigen. Intervirology. 1996;39(1–2):111–9. doi: 10.1159/000150482. [DOI] [PubMed] [Google Scholar]
- Shu Y, Winfrey S, Yang ZY, Xu L, Rao SS, Srivastava I, Barnett SW, Nabel GJ, Mascola JR. Efficient protein boosting after plasmid DNA or recombinant adenovirus immunization with HIV-1 vaccine constructs. Vaccine. 2007;25(8):1398–408. doi: 10.1016/j.vaccine.2006.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiegler G, Kunert R, Purtscher M, Wolbank S, Voglauer R, Steindl F, Katinger H. A potent cross-clade neutralizing human monoclonal antibody against a novel epitope on gp41 of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 2001;17(18):1757–65. doi: 10.1089/08892220152741450. [DOI] [PubMed] [Google Scholar]
- Stoute JA, Slaoui M, Heppner DG, Momin P, Kester KE, Desmons P, Wellde BT, Garcon N, Krzych U, Marchand M. A preliminary evaluation of a recombinant circumsporozoite protein vaccine against Plasmodium falciparum malaria. RTS,S Malaria Vaccine Evaluation Group. N Engl J Med. 1997;336(2):86–91. doi: 10.1056/NEJM199701093360202. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Zhao Y, Lu Y, Chen YH. Epitope-vaccine induces high levels of ELDKWA-epitope-specific neutralizing antibody. Immunol Invest. 2000;29(1):41–50. doi: 10.3109/08820130009105143. [DOI] [PubMed] [Google Scholar]
- Zinkernagel RM, Bachmann MF, Kundig TM, Oehen S, Pirchet H, Hengartner H. On immunological memory. Annu Rev Immunol. 1996;14:333–67. doi: 10.1146/annurev.immunol.14.1.333. [DOI] [PubMed] [Google Scholar]
- Zinkernagel RM, LaMarre A, Ciurea A, Hunziker L, Ochsenbein AF, McCoy KD, Fehr T, Bachmann MF, Kalinke U, Hengartner H. Neutralizing antiviral antibody responses. Adv Immunol. 2001;79:1–53. doi: 10.1016/S0065-2776(01)79001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]