

SUMMARY
The benefits of endurance exercise on general health make it desirable to identify orally active agents that would mimic or potentiate the effects of exercise to treat metabolic diseases. Although certain natural compounds, such as reseveratrol, have endurance-enhancing activities, their exact metabolic targets remain elusive. We therefore tested the effect of pathway-specific drugs on endurance capacities of mice in a treadmill running test. We found that PPARβ/δ agonist and exercise training synergistically increase oxidative myofibers and running endurance in adult mice. Because training activates AMPK and PGC1α, we then tested whether the orally active AMPK agonist AICAR might be sufficient to overcome the exercise requirement. Unexpectedly, even in sedentary mice, 4 weeks of AICAR treatment alone induced metabolic genes and enhanced running endurance by 44%. These results demonstrate that AMPK-PPARδ pathway can be targeted by orally active drugs to enhance training adaptation or even to increase endurance without exercise.
Keywords: PPARδ, AMPK, exercise, running endurance, skeletal muscle, genetic re-programming, oxidative metabolism
INTRODUCTION
Skeletal muscle is an adaptive tissue composed of multiple myofibers that differ in their metabolic and contractile properties including oxidative slow-twitch (type I), mixed oxidative/glycolytic fast-twitch (type IIa) and glycolytic fast-twitch (type IIb) myofibers (Pette et al., 2000; Fluck et al., 2003). Type I fibers preferentially express enzymes that oxidize fatty acids, contain slow isoforms of contractile proteins and are more resistant to fatigue than glycolytic fibers. Type II fibers preferentially metabolize glucose and express the fast isoforms of contractile proteins. Endurance exercise training triggers a remodeling program in skeletal muscle that progressively enhances performance in athletes such as marathon runners, mountain climbers and cyclists. This involves change in metabolic programs and structural proteins within the myofiber that alter the energy substrate utilization and contractile properties that act to reduce muscle fatigue (Pette et al., 2000; Fluck et al., 2003). Training based adaptations in the muscle are linked to increases in the expression of genes involved in the slow-twitch contractile apparatus, mitochondrial respiration and fatty acid oxidation (Holloszy et al., 1984; Booth et al., 1991; Schmitt et al., 2003; Yoshioka et al., 2003; Mahoney et al., 2005a and b; Siu et al., 2004; Garnier et al., 2005; Short et al., 2005; Timmons et al., 2005). These adaptations that improve performance, can also protect against obesity and related metabolic disorders (Wang et al., 2004; Koves et al., 2005). Moreover, skeletal muscles rich in oxidative slow-twitch fibers are resistant to muscle wasting (Minnaard et al., 2005).
Given the numerous benefits of exercise on general health, identification of orally active agents that mimic or potentiate the genetic effects of endurance exercise is a long standing, albeit elusive medical goal. High doses of certain natural extracts such as resveratrol can improve endurance (Lagouge et al 2006). The aerobic effects of resveratrol are thought to dependent on activation of SIRT1-PGC1α coactivator complex in skeletal muscle. However, the downstream transcriptional factor(s) targeted by SIRT1/PGC1α in mediating these effects are not known. More importantly, both SIRT1/PGC1α and resveratrol each activate multiple targets and thus whether there is a specific signaling pathway that can be selectively activated by a synthetic drug to improve endurance is not known.
Exercise training activates a number of transcriptional regulators as well as serine/threonine kinases in skeletal muscles that contribute to metabolic re-programming (Bassel-Duby and Olson, 2006). We and others previously identified a critical role for PPARβ/δ (henceforth referred to as PPARδ) in transcriptional regulation of skeletal muscle metabolism (Dressel et al., 2003; Luquet et al., 2003; Schuler et al., 2006; Wang et al., 2004). Over-expression of a constitutively active PPARδ (VP16-PPARδ) in skeletal muscles of transgenic mice pre-programs an increase in oxidative muscle fibers, enhancing running endurance by nearly 100% in untrained adult mice (Wang et al., 2004). One of the best understood serine/threonine kinases is AMP-activated protein kinase (AMPK), a master regulator of cellular and organismal metabolism whose function is conserved in all eukaryotes (Hardie, 2007). In mammals, AMPK has been shown to contribute to glucose homeostasis, appetite, and exercise physiology (Andersson et al., 2004; Hardie, 2007; Kubota et al., 2007; Mu et al, 2001; Minokoshi et al., 2004; Thomson et al., 2007). These observations raise the question as to whether synthetic PPARδ or AMPK agonists can re-program established fiber specification in adult muscle toward an overt endurance phenotype. We have found that the PPARδ agonist GW1516 (shown to be bioactive in humans, Sprecher et al., 2007) enables mice to run 60-75% longer and further than the non-treated controls only when combined with exercise training. This ‘super-endurance phenotype’ is linked to a transcriptional boost provided by exercise-activated AMPK resulting in a novel endurance gene signature. A more critical role of AMPK in the super-endurance phenotype is revealed in our unexpected finding that, the orally active AMPK agonist AICAR is sufficient as a single agent to improve running endurance by nearly 45% in non-exercised mice. Together these results provide new insights into the pharmacological malleability of muscle performance.
RESULTS
GW1516 increases muscle gene expression but not endurance in sedentary mice
To examine whether treatment with PPARδ ligands alone can re-program the muscle transcriptome and endurance capacity, wild-type C57Bl/6J age matched cohorts were treated with vehicle or GW1516 for 4 weeks. QPCR analysis of selective target genes confirmed that drug treatment induced oxidative metabolic biomarkers such as uncoupling protein 3 (Ucp3), muscle carnitine palmitoyl transferase I (mCPT I, Cpt 1b) and pyruvate dehydrogenase kinase 4 (Pdk4) (Figure 1A). These changes in gene expression were detected as early as 4 days post-treatment as well as with drug concentrations ranging from 2-5 mg/kg/day. Moreover, in all our gene expression studies, maximal effects of PPARδ activation were detected in pre-dominantly fast-twitch (quadricep and gastrocnemius) but not slow-twitch (soleus) muscles (data not shown). In primary muscle cells cultured from wild type and PPARδ null mice (Chawla et al., 2003; Man et al., 2007), we confirmed that the induction of oxidative genes by GW1516 is mediated via selective activation of PPARδ in skeletal muscles (Supplementary Figure S1 A-C). Moreover, this is similar to the expression changes found in the same genes in muscles expressing the constitutively active VP16-PPARδ transgene (Wang et al., 2004) (Figure 1A), supporting the concept that pharmacological activation of PPARδ is sufficient to initiate an oxidative response in adult skeletal muscle. To determine the functional effects of ligand, age and weight matched cohorts of treated and control mice were subjected to an endurance treadmill performance test before (week 0) and after (week 5) treatment. Curiously, running performance was unchanged by GW1516 treatment (Figure 1B). Furthermore, long-term drug treatment of up to 5 months also did not change running endurance (data not shown). These results indicate that pharmacologic activation of the PPARδ genetic program in adult C57Bl/6J mice is insufficient to promote a measurable enhancement of treadmill endurance.
Figure 1. Synthetic PPARδ activation in mice.
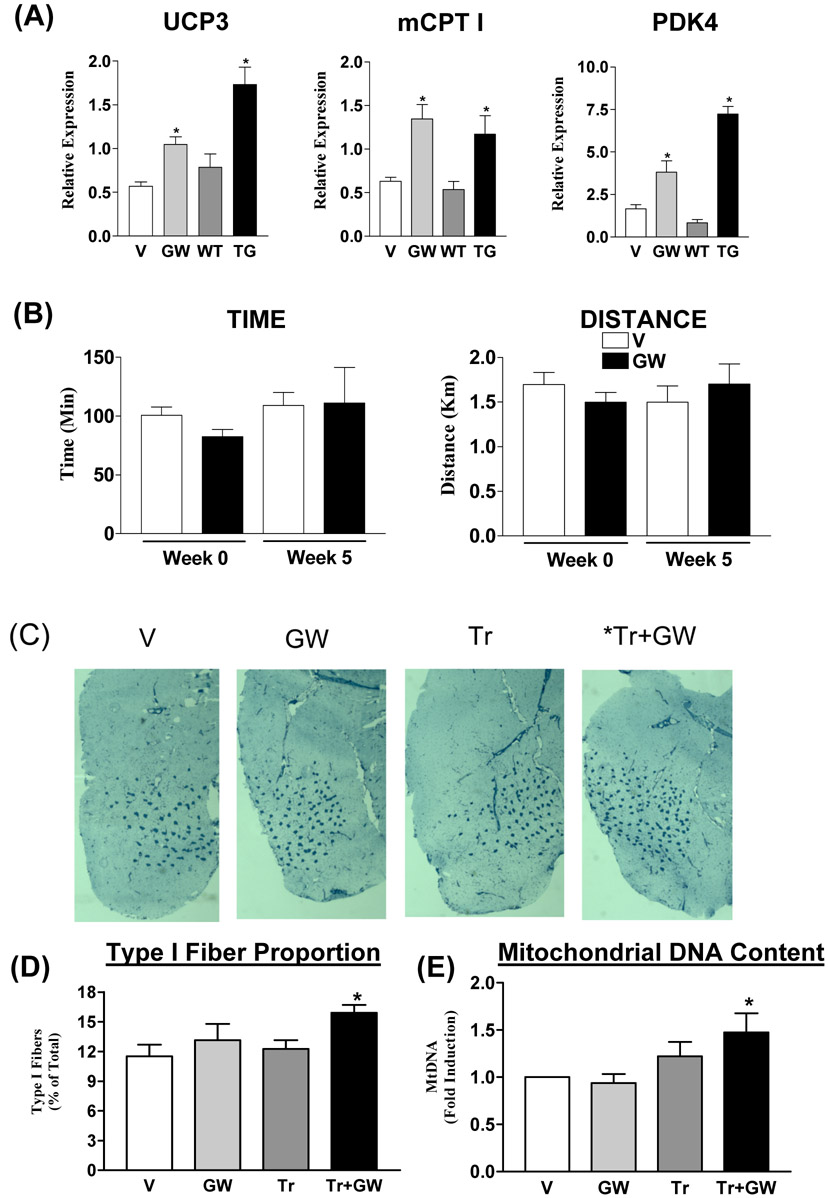
(A) Relative gene expression levels of Ucp3, Cpt 1 and Pdk4 in quadriceps isolated from vehicle (V) and GW1516 (GW)-treated wild type mice as well as from muscle VP16-PPARδ transgenic (Wang et al., 2004) (TG) and non-transgenic (WT) littermates. Data is presented as mean±SEM (N=4-9). * Indicates statistically significant differences between GW and V groups or TG and WT groups (p<0.05, Unpaired student’s t test). (B) Running endurance in sedentary mice. Endurance was tested in V (open bars) and GW (black bars)-treated wild type mice before (Week 0) and after (Week 5) treatment. Data is represented as mean±SD (N=6). (C) Representative meta-chromatically stained frozen gastrocnemius cross-sections from vehicle-treated sedentary (V), GW1516-treated sedentary (GW), vehicle-treated exercised (Tr) and GW-treated exercised (Tr+GW) mice. Type I fibers are stained dark blue. (D) Type I fiber quantification (N=3). (E) Fold change in mitochondrial DNA to nuclear DNA ratio (N=9). Data in (D) and (E) are presented as mean±SEM. * Indicates statistical differences between V and indicated groups (p<0.05, One Way ANOVA; post hoc: Dunnett’s Multiple Comparison Test).
GW1516 remodels skeletal muscle in exercise-trained mice
Since endurance exercise remodels the skeletal muscle to progressively alter performance (Holloszy et al., 1984; Booth et al., 1991; Schmitt et al., 2003; Yoshioka et al., 2003; Mahoney et al., 2005a and b; Siu et al., 2004; Garnier et al., 2005; Short et al., 2005; Timmons et al., 2005), we speculated whether co-administration of GW1516 in the context of exercise training might enhance anticipated changes in fiber type composition and mitochondrial biogenesis. The effect of GW1516 and exercise on fiber type composition was determined using meta-chromatic staining of cryo-sections of the gastrocnemius (Wang et al., 2004). As expected from the results of the running performance in Figure 1B, there was no significant difference in the proportion of type I fibers between vehicle and GW1516-treated sedentary mice (Figure 1C). In contrast, in trained mice, GW1516 increased the proportion of type I fibers (by ∼38%) compared to the vehicle-treated sedentary mice (Figure 1C and 1D). In addition to its effects on the fiber type, exercise training increases skeletal muscle mitochondrial biogenesis, which was measured as a function of mitochondrial DNA expression levels using quantitative real time PCR (QPCR). Similar to type I fiber changes, mitochondrial DNA expression was not changed by drug alone but was increased by approximately 50% with the combination of exercise and GW1516 treatment (Figure 1E).
The effects of GW1516 treatment and exercise, singly or in combination, on components of the oxidative metabolism of fatty acids were further analyzed by measuring the gene expression levels of selective biomarkers for fatty acid β-oxidation. As expected, we found that previously examined genes such as Ucp3, Cpt 1b and Pdk4 were up-regulated by GW1516 but showed no further induction with exercise (Figure 2A). Unexpectedly, we discovered a second set of genes that show no response to exercise or drug alone but are robustly induced by the combination. This intriguing response profile includes a series of genes involved in the regulation of fatty acid storage [such as steroyl-CoA-desaturase (Scd1), fatty acyl coenzyme A synthase (FAS, Fasn) and serum response element binding protein 1c (SREBP1c) (Srebf1c)] and fatty acid uptake [such as the fatty acid transporter (FAT) (Cd36) and lipoprotein lipase (Lpl)] (Figs. 2B, 2C and 3).
Figure 2. Gene and protein expression in quadriceps.

Relative gene expression levels of (A) FAO (Ucp3, Cpt 1b, Pdk4), (B) fatty acid storage (Scd1, Fasn, srebf1c) and (C) fatty acid uptake (Cd36, Lpl) biomarkers in quadriceps from V, GW, Tr and Tr+GW groups. Data is presented as mean±SEM (N=9) mice. * Indicates statistically significant difference between V and indicated groups (p<0.05, One Way ANOVA; post hoc: Dunnett’s Multiple Comparison Test). (D) Protein expression levels of oxidative biomarkers (myoglobin, UCP3, CYCS, SCD1) and loading control (tubulin) in quadriceps (N=3).
Figure 3. Running endurance and gene signature in exercise trained mice.

Running endurance was tested in V (open bars) and GW- (black bars) treated mice before (Week 0) and after (Week 5) exercise training. Running endurance is depicted as time (A) and distance (B) that animals in each group ran. Data is represented as mean±SD (N=6). ***Indicates statistically significant difference between V and GW-treated exercised mice (p<0.001) (One Way ANOVA; post hoc: Tukey’s Multiple Comparison Test). (C) Venn diagram comparing GW, Tr and Tr+GW target genes identified in microarray analysis of quadriceps (N=3). The selection criteria used a p<0.05 on Bonferroni’s multiple comparison test. (D) Classification of target genes in Tr+GW mice. (E) Relative expression of 48 unique TR+GW target genes in GW, TR, TR+GW and VP16-PPARδ muscles. Each condition is represented by data from two samples (each sample is pooled from three mice). (Color scheme for fold change is provided).
We also measured the protein levels of selective oxidative biomarkers including myoglobin, UCP3, cytochrome c (CYCS) and SCD1. In each case, a more robust up-regulation of protein expression was found by combining exercise and GW1516 treatment relative to either drug or exercise alone (Figure 2D). Altered triglycerides are one way to assess changes in muscle oxidative capacity. Triglyceride levels were unchanged in vehicle or GW1516-treated sedentary mice, but showed a striking increase with exercise. In contrast, this increase was completely reversed by GW1516 treatment presumably due to enhanced fat utilization (Supplementary Figure S1 D).
GW1516 and exercise training synergistically increase running endurance
As described above, although GW1516 treatment alone induces wide spread genomic changes associated with oxidative metabolism, it fails to increase running endurance. On the other hand, drug treatment in conjunction with exercise produces an enriched remodeling program that includes a series of transcriptional and post-translational adaptations in the skeletal muscle. This suggests that exercise training serves as a key trigger to unmask a cryptic set of PPARδ target genes leading us to re-examine the ability of the drug to modulate endurance. Indeed, the same dose and duration of GW1516 treatment that previously failed to alter performance, when paired with 4 weeks of exercise training, increases running time by 68% and running distance by 70% over vehicle-treated trained mice (Figure 3A and 3B, compare Week 5). It is also important to note that comparison of running time and distance before (week 0) and after (week 5) exercise and drug treatment revealed a 100% increment in endurance capacity for individual mice, underscoring the robustness of the combination paradigm (Figure 3A and 3B). Finally, it is noteworthy that the combined effects of GW1516 and exercise reduces the epididymal fat to body weight ratio as well as fat cross-sectional area in these mice (Supplementary Figure S1 E, F) suggesting the broader systemic effects of this protocol.
PPARδ agonist and exercise establish an endurance gene signature
To dissect the mechanism underlying the super-endurance phenotype, we conducted a comprehensive study of the muscle transcriptome induced either by ligand, exercise or the combination, which produced three overlapping networks of 96, 113 and 130 genes, respectively (Figure 3C). Approximately 50% of the target genes were common between GW1516 and exercise demonstrating that PPARδ activation partially mimics exercise. To our surprise, combined GW1516 treatment and exercise established a unique gene expression pattern that was neither an amalgamation nor a complete overlap of the individual interventions (Figure 3C). This signature included 48 new target genes (Supplementary Table S1) not regulated by either GW1516 or exercise alone while excluding 74 genes regulated by GW1516 or exercise (selective genes are listed in Supplementary Table S3). The majority of the genes in the GW1516-exercise signature were induced (108/130); the components of which are described in Figure 3D. While the largest gene subclass (32% of genes) was linked to positive regulation of aerobic capacity, additional pathways implicated in muscle remodeling and endurance were also represented in the signature (see Supplementary Table S2 for detailed description). It is noteworthy that comparative expression analysis of the 48 exclusive genes of the endurance signature (but not of either intervention alone) revealed a striking similarity to ‘untrained’ VP16-PPARδ transgenic mice (Figure 3E). This observation confirms the primary dependence of the 48 genes on PPARδ and points to the possibility that exercise-generated signals may function to synergize PPARδ transcriptional activity to levels comparable to transgenic over-expression.
AMPK-PPARδ interaction in transcriptional regulation
What might be the molecular interface between mechanical exercise and PPARδ transcription? Exercise training is known to activate multiple kinases; among which, AMPK has profound effects on skeletal muscle gene expression and oxidative metabolism (Chen et al., 2003, Reznick et al., 2006). Indeed, mice defective for AMPK signaling in muscle exhibit reduced capacity for voluntary running (Mu et al, 2001; Thomson et al., 2007). As previously observed (Durante et al., 2002; Frøsig et al., 2004), we found increased AMPK activation in the quadriceps of exercised mice relative to the sedentary controls (Figure 4A). Furthermore and unexpectedly, AMPK is constitutively activated in muscles of VP16-PPARδ transgenic mice in absence of exercise or drug (Figure 4B). In contrast, in our experiments GW1516 treatment alone does not activate AMPK in either sedentary or exercise trained muscles, as previously suggested by some (Tareda et al., 2006), but not by others (Kramer et al., 2007). Taken together these results strongly suggest that the ability to promote endurance in mice is associated with activation of both AMPK and PPARδ.
Figure 4. Synergistic regulation of muscle gene expression by PPARδ and AMPK.

(A) and (B) represent AMPK activation by exercise and VP16-PPARδ over-expression, respectively, in skeletal muscle. (C) Comparison of Tr+GW and AI+GW dependent gene signatures in quadriceps (N=3). The selection criteria used is similar to one used in Figure 3C. (D) Classification of 52 targets that were common to Tr+GW and AI+GW gene signatures. Expression of Scd1 (E), ATP citrate lyase (Acly) (F), HSL (Lipe) (G), Fabp3 (H), Lpl (I) and Pdk4 (J) transcripts in quadriceps of mice treated with vehicle (V), GW1516 (GW, 5mg/kg/day), AICAR (AI, 250 mg/kg/day) and the combination of the two drugs (GW+AI) for 6 days. Data is presented as mean±SEM (N=6). * Indicates statistically significant difference between V and indicated groups (p<0.05, One Way ANOVA; post hoc: Dunnett’s Multiple Comparison Test).
According to this hypothesis selective co-activation of AMPK and PPARδ would induce gene expression changes that mimic those triggered by combined exercise and PPARδ as well as VP16-PPARδ over-expression. To investigate this possibility, we compared the transcriptional changes induced in skeletal muscle by combined exercise and GW1516 treatment with that of combined AMPK activator (the cell permeable AMP analog AICAR) and GW1516 treatment. It is noteworthy that simultaneous GW1516 and AICAR treatment created a unique gene expression signature in the quadriceps of untrained C57Bl/6J mice (Supplementary Figure S2) that shares 40% of the genes with that of combined GW1516 treatment and exercise (Figure 4C). Classification of the 52 genes common to the two signatures (Figure 4D, listed in Supplementary Table S4) revealed that the majority of the targets were linked to oxidative metabolism. Quantitative expression analysis of selective oxidative genes by QPCR showed that several of these biomarkers including Scd1, ATP citrate lyase (Acly), hormone sensitive lipase (HSL) (Lipe), muscle fatty acid binding protein (mFABP, Fabp3), Lpl and Pdk4 were induced in a synergistic fashion by GW1516 and AICAR in the quadriceps (Figure 4E-4J). It is also noteworthy that all of the above genes were induced in quadriceps of untrained VP16-PPARδ mice where AMPK is constitutively active (Supplementary Figure S1 G). Collectively, these results show that interaction between AMPK and PPARδ substantially contributes to re-programming of the skeletal muscle transcriptome during exercise.
AMPK increases transcriptional activation by PPARδ
The above described pathway crosstalk raised the possibility that AMPK directly regulates the transcriptional activity of PPARδ in skeletal muscles. An analysis of the effects of GW1516 and AICAR on gene expression in primary muscle cells isolated from wild type and PPARδ null mice revealed that synergism is completely dependent on PPARδ and lost in the null cells (Figure 5A-D). These observations show that AMPK enhances a subset of ligand-dependent PPARδ transcriptional targets in a cell-autonomous fashion.
Figure 5. AMPK-PPARδ interaction.

(A-D) Expression of metabolic genes in wild type and PPARδ null primary muscle cells treated with V, GW, AI and GW+AI for 24 hr. In (E-F, J), AD293 cells were transfected with PPARδ+RXRα+Tk-PPRE along with control vector, AMPK α1, α2 and/or PGC1α as indicated. (E) Induction of basal PPARδ transcriptional activity by AMPK α1 or α2. (F) Dose-dependent induction of PPARδ transcriptional activity is enhanced by AMPKα1 (closed circle) or AMPK α2 (closed square) compared to control (open triangle). In (G-I, K), AD293 cells were transfected and processed as indicated. (G-H) Representative blot showing co-immunoprecipitation of transfected (G) or endogenous (H) AMPK with Flag-PPARδ. (I) Metabolic p32 labeling of PPARδ in AD293 cells transfected as described. (J) Synergistic regulation of basal (V) and ligand (GW) dependent PPARδ transcriptional activity by AMPK α2 subunit and PGC1α. (K) Co-immunoprecipitation of PPARδ but not AMPK α2 subunit with Flag-PGC1α. Data in (A)-(D) (n = 6), (E), and (J) (n = 3-4) are presented as mean ± SEM, and * indicates statistical significance (p < 0.05, one-way ANOVA; post hoc: Dunnett’s multiple comparison test).
To more directly examine this connection, we utilized reporter gene expression assays. Co-transfection of either catalytic AMPK α1 or α2 subunits but not control vector with PPARδ increased the basal (Figure 5E) and GW1516-dependent transcriptional activity (Figure 5F) of PPARδ in inducing a PPRE-driven reporter gene in AD293 cells. It should be noted that AMPK over-expression or GW1516 treatment did not change reporter activity in transfections excluding the PPARδ expression vector (data not shown) negating the possibility of an effect via RXR. Additionally, in AD293 cells co-transfected with Flag-PPARδ and with either catalytic AMPK α1 or α2 subunits, we discovered that each of the AMPK subunits co-immunoprecipitated as a complex with Flag-PPARδ (Figure 5G). Furthermore, Flag-PPARδ co-immunoprecipitated endogenous AMPKα subunits from AD293 cells confirming a tight physical interaction between the nuclear receptor and the kinase (Figure 5H). Despite this association, AMPK failed to increase PPARδ phosphorylation. In vivo orthophosphate labeling of PPARδ in AD 293 cells in the presence or absence of either AMPK alpha isoform under the same conditions where AMPK promotes PPARδ-dependent transcription revealed no change in overall PPARδ phosphorylation (Figure 5I). These data suggest PPARδ phosphorylation is not increased by AMPK in vivo. However, co-transfection of AMPKα2 and co-activator PGC1α (a previously reported direct substrate of AMPK) cooperatively interact to further induce both the basal and ligand-dependent transcriptional activity of PPARδ (Figure 5J). Strikingly, we did not detect physical interaction between Flag-PGC1α and AMPK (Figure 5K), though both independently interacted with PPARδ. Collectively, these observations suggest that AMPK may be present in a transcriptional complex with PPARδ where it can potentiate receptor activity via direct protein-protein interaction and/or by phosphorylating co-activators such as PGC1α.
Pharmacologic AMPK activation increases running endurance in untrained mice
Our findings show that pharmacologic activation of PPARδ in adult mice can increase running endurance only in conjunction with exercise signals. The central role for AMPK in this process is especially underscored by the observations that it is both robustly stimulated by exercise as well as is constitutively active in muscles of VP16-PPARδ transgenic mice that exhibit endurance without exercise. Further, AMPK can integrate multiple transcriptional programs by interacting not only with PPARδ but also other transcriptional regulators of metabolism (e.g. PGC1α, PPARα) (Hong et al., 2003; Leff, 2003; Bronner et al., 2004; Jäger et al., 2007). This raises the interesting question as to whether chemical activation of AMPK is sufficient to increase running endurance without exercise.
To test this idea we treated C57B/6J mice with AICAR (500mg/kg/day) for 4 weeks. AICAR increased phosphorylation of AMPK α subunit and acetyl CoA carboxylase (ACC) as well as increased expression of UCP3 in quadriceps confirming effective activation of AMPK signaling (Figure 6A). Interestingly, 4 weeks of drug treatment decreased epididymal fat mass to body weight ratio and increased oxygen consumption without changing body weight (Figure 6B-E), supporting the speculation that AICAR may positively regulate endurance. Indeed, in a treadmill endurance test ACIAR-treated mice ran longer (∼23%) and further (∼44%) compared to vehicle-treated mice revealing that increase in endurance can be achieved without exercise (Figure 6F). Furthermore, global gene expression analysis of quadriceps revealed that AICAR treatment alone up-regulated a set of 32 genes linked to oxidative metabolism (Figure 6G and Supplementary Table S5). Notably, 30 of these 32 genes were also up-regulated in VP16-PPARδ transgenic mice suggesting that stimulation of oxidative genes by AMPK may depend on PPARδ (Supplementary Table S5). To test this possibility, we utilized wild type and PPARδ null primary muscle cells. Treatment of wild type primary cells with AICAR (for 72 hr) increased expression of key oxidative biomarker genes (Scd1, fasn (FAS), Ppargc 1a, Pdk4) (Figure 6H). In contrast, AICAR failed to increase the expression of the above genes in PPARδ null cells, demonstrating the requirement of the receptor for transcriptional effects of AMPK on oxidative genes.
Figure 6. AICAR increases running endurance.

In A-F, C57Bl/6J mice were treated with vehicle (open bars/thin lines) or AICAR (500mg/kg/day, 4 weeks) (closed bar/thick lines). (A) Representative immunoblots showing levels of UCP3, phospho-acetyl CoA carboxylase (ACC), phospho-AMPK and total-AMPK in quadriceps. (B) Average body weight. (C) Percent epididymal fat mass to body weight ratio. (D) Oxygen consumption rates (mg/kg/hr) measured over 12 hr period. (E) Data in (D) is represented as AUC. (F) Running endurance measured as a function of time (upper panel) and distance (lower panel). (G) Representative oxidative genes induced by AICAR treatment (250 mg/kg/day, 6 days). (H) Expression of oxidative biomarkers (Scd1, Fasn, Ppargc1a, Pdk4) in wild type and PPARδ null primary myoblast treated with vehicle (open bar) or AICAR (closed bar) for 72 hr. (I) Model depicting exercise/AMPK-PPARδ interaction in re-programming muscle genome. Data in (B) and (C) (n = 10), (D) and (E) (n = 4), (F) (n = 15-20), and (H) (n = 9) are presented as mean ± SEM, and * indicates statistical significance (p < 0.05, unpaired student’s t test).
DISCUSSION
In this study, we show that the AMP-mimetic AICAR can increase endurance in sedentary mice by genetically reprogramming muscle metabolism in a PPARδ-dependent manner. We also found that a PPARδ agonist in combination with exercise synergistically induces fatigue resistant type I fiber specification and mitochondrial biogenesis ultimately enhancing physical performance. These changes correlate with an unexpected but interesting establishment of a muscle endurance gene signature that is unique to the drug-exercise paradigm. Such a signature is an outcome of molecular crosstalk and perhaps a physical association between exercise-activated AMPK and PPARδ. These findings identify a novel pharmacologic strategy to re-program muscle endurance by targeting AMPK-PPARδ signaling axis with orally active ligands.
Transgenic over-expression as well as knockout studies have identified PPARδ and AMPK as key regulators of type I fiber specification and endurance adaptations during exercise (Mu et al, 2001; Röckl et al., 2007; Thomson et al., 2007; Wang et al., 2004). Whether and how these endogenously expressed regulators can be targeted to re-program adult muscle without exercise has been a subject of unresolved speculation. We found that the AMPK activator AICAR increased oxygen consumption and endurance in untrained adult mice in part by stimulating PPARδ-dependent oxidative genes. Despite a demonstrated role for PPARδ in endurance, 5 week treatment with a potent and selective agonist failed to alter either fiber type composition or endurance revealing that direct and pharmacologic activation of PPARδ is insufficient to enhance running performance. In contrast, transgenic over-expression of activated PPARδ at birth pre-programs the nascent myofibers to trans-differentiate into slow-twitch fibers, thus imparting a high basal endurance capacity to adult transgenic mice. Apparently, once fiber type specification is complete in adults the potential plasticity of muscle to synthetic activation of a single transcriptional pathway is constrained. Along these lines, the unexpected yet successful re-programming of endurance in untrained adults with synthetic AMP-mimetic might be linked to the ability of AMPK to simultaneously target multiple transcriptional programs governed by its substrates such as PGC1α, PPARα and PPARδ triggering a genetic effect akin to exercise (Hong et al., 2003; Leff, 2003; Bronner et al., 2004; Jäger et al., 2007).
Interestingly, the recalcitrance of adult skeletal muscle endurance to manipulation by PPARδ agonist alone is relieved by combining drug treatment with exercise. Indeed, this strategy generates an endurance gene signature that is unique from either paradigm alone reflecting a crosstalk between exercise and PPARδ signaling (Supplementary Table S2). While exercise activates a cascade of signaling events, we feel AMPK is central to this genetic adaptation for several reasons. First, AMPK is a metabolic sensor that detects low ATP levels (such as occur during exercise) and in turn increases oxidative metabolism (Mu et al., 2001, Reznick et al., 2006). Second, long term effects of AMPK are in part mediated via regulation of gene expression (Reznick et al., 2006). Third, exercise induces activation and nuclear import of AMPK, where it can potentially interact with transcription factors (this study and McGee et al., 2003). And finally, transgenic mice defective for AMPK activation exhibit reduced voluntary exercise (Mu et al, 2001; Thomson et al., 2007), making it an attractive exercise cue that modulates receptor signaling.
The notion that exercise-activated AMPK interacts with PPARδ in regulating gene expression is supported by our demonstration that AMPK associates with PPARδ and dramatically increases basal and ligand-dependent transcription via the receptor. Despite physical interaction, we found that AMPK does not induce PPARδ phosphorylation in metabolic labeling studies. Interestingly, AMPK and its previously reported substrate PGC1α synergistically increased PPARδ transcription, suggesting indirect regulation of receptor function by AMPK via co-regulator modification. Nevertheless, we cannot rule out the possible regulation of PPARδ by AMPK via direct protein-protein interaction. Indeed, regulation of other transcription factors by AMPK via similar mechanisms has been previously demonstrated (Hong et al., 2003; Leff, 2003; Bronner et al., 2004). A physiological validation of AMPK-PPARδ interaction comes from our observation that GW1516 and AICAR (AMPK activator) synergistically induces several endurance related genes in wild type but not in PPARδ null primary muscle cells. More importantly, treatment of animals with AICAR and GW1516 creates a gene signature in skeletal muscle that replicates up to 40% of the genetic effects of combined exercise and GW1516 treatment. Notably, the shared genes between the two profiles are linked to oxidative metabolism, angiogenesis and glucose sparing, pathways that are directly relevant to muscle performance (Figure 4D, listed in Supplementary Table S4).
While not all genes regulated by either exercise (data not shown) or exercise-PPARδ interaction (non-overlapping signature, Figure 4D) are AMPK dependent, two key findings assign a critical role for the kinase in promoting endurance compared to other known exercise signals (Bassel-Duby and Olson, 2006, Goodyear et al., 1996, Lagouge et al., 2006). First, AMPK is constitutively active in VP16-PPARδ transgenic muscles that exhibit endurance without exercise. Second, AMPK activation by AICAR was sufficient to increase running endurance without additional exercise signals. Strikingly, majority of the oxidative genes (30 out of 32) up-regulated by AICAR are active in super-endurance VP16-PPARδ mice and perhaps are the core set of genes required to improve muscle performance. Interestingly, AICAR failed to induce oxidative gene expression in PPARδ null muscle cells, indicting the requirement of PPARδ, at least for regulation of oxidative metabolism by AMPK. Collectively, these findings demonstrate a molecular partnership between AMPK and PPARδ in re-programming skeletal muscle transcriptome and endurance (Figure 6I) that can be readily exploited by orally active AMPK drugs to replace exercise.
In humans, endurance exercise leads to physiological adaptations in the cardio-pulmonary, endocrine and neuromuscular systems (Jones et al., 2000; Lucia et al., 2001). While our current investigation focused on skeletal muscle, extra-muscular effects of PPARδ, AMPK and exercise may also contribute to increased endurance. Although, potentiation of extra-muscular adaptations by PPARδ and AMPK agonists remain to be studied, we found that drug treatment can reduce epididymal fat mass, possibly conferring additional systemic benefits. It is noteworthy that PPARδ is important for normal cardiac contractility as well as for the endocrine function of adipose tissue (Wang et al., 2003; Cheng et al., 2004). Similarly, the activation of AMPK by metformin is thought to mediate its ability to lower blood glucose levels (Shaw et al, 2005). In addition to increasing performance in athletes, exercise has beneficial effects in a wide range of patho-physiological conditions such as respiratory disorders, cardiovascular abnormalities, type 2 diabetes and cancer risk. Therefore, understanding the effects of exercise on normal physiology as well as identifying pharmaceutically targetable pathways that can boost these effects is crucial. In this study, we revealed that synthetic PPARδ activation and exercise or more importantly AMPK activation alone, provides a robust transcriptional cue that re-programs the skeletal muscle genome and dramatically enhances endurance. We believe that the strategy of re-organizing the preset genetic imprint of muscle (as well as other tissues) using exercise mimetic drugs has therapeutic potential in treating certain muscle diseases such as wasting and frailty as well as obesity where exercise is known to be beneficial.
EXPERIMENTAL PROCEDURES
Exercise training and drug treatment
Male C57B/6J mice (8 wks old) were randomly divided into 4 cohorts comprising (i) vehicle-treated & sedentary (V), (ii) GW1516-treated & sedentary (GW), (iii) vehicle-treated & exercise trained (Tr) and (iv) GW1516-treated & exercise trained (Tr+GW) (N=9). Mice in all groups were acclimated to moderate treadmill running (10 m/min for 15 min) every other day for 1 week. After acclimation, basal running endurances for the 4 groups were determined using a treadmill running test, where the speed was gradually increased from 0 to 15 m/min and then maintained constant until exhaustion (Week 0). Following the initial test, the mice in the exercise groups were subjected to 4 weeks (5 days/week) of exercise training. The mice were trained on a treadmill inclined at 5 degrees, with progressively increasing intensity and time. At the end of 4 weeks, all exercise-trained mice were running for 50 min/day at 18 m/min. During the 4 weeks, mice from both the sedentary and trained groups were either treated with vehicle or GW1516 (5 mg/kg/day). At the end of the drug treatment and/or training protocol (Week 5) 6 mice per group were subjected to the running test. Three mice in each group were not subjected to treadmill test to confirm that changes observed in the skeletal muscle were not due to the acute run, but related to the exercise training. It should be noted that the above interventions do not affect body weight and food intake in mice (data not shown).
In another study, male C57B/6J mice (8 wks old) were treated with GW1516 (5 mg/kg/day, oral gavage), AICAR (250 mg/kg/day, i.p.) or the combination of the two drugs for 6 days for gene expression analysis. Additionally, C57B/6J mice (8 wks old) were also treated with AICAR (500mg/kg/day, i.p.) for 4 weeks for treadmill running tests.
Tissue collection
The animals were euthanized by carbon dioxide asphyxiation 72 hrs after the last bout of exercise. Gastrocnemius and quadriceps were isolated, frozen and stored at -80°C until further analysis. In GW1516/AICAR study, quadriceps were similarly collected on the 6th day 4 hr after drug treatment.
Meta-chromatic staining and histology
Cryo-sectioning of frozen gastrocnemius and meta-chromatic ATPase staining was performed as previously described (Wang et al., 2003; Wang et al., 2004).
Gene and protein expression analysis
RNA was extracted from gastrocnemius or quadriceps using Trizol and analyzed for gene expression using real time quantitative PCR. Protein homogenates were prepared from quadriceps and analyzed by western blotting with myoglobin (Dako), UCP3 (Affinity Bioreagents), CYCS (Santacruz), SCD1 (Santacruz), tubulin (Sigma), phospho-, total-AMPK α and phospho-ACC antibodies (Cell Signaling).
Microarray Analysis
Genome-wide analysis was performed in quadriceps from V, GW, Tr and Tr+GW mice and from V, GW, AICAR and AICAR+GW mice as well as from wild type and VP16-PPARδ transgenic mice. Preparation of in vitro transcription products, oligonucleotide array hybridization, and scanning were performed by using Affymetrix high-density oligonucleotide array mouse genome 430A 2.0 chips according to Affymetrix protocols. To minimize discrepancies due to variables, the raw expression data were scaled by using Affymetrix MICROARRAY SUITE 5.0 software, and pairwise comparisons were performed. The trimmed mean signal of all probe sets was adjusted to a user-specified target signal value (200) for each array for global scaling. No specific exclusion criteria were applied. Additional analysis was performed by using the freeware program BULLFROG 7 (Zapala et al., 2002) and the Java-based statistical tool VAMPIRE (Hsiao et al, 2004).
Cell culture and transfection experiments
Primary muscle cells were isolated from wild type and PPARδ null mice as previously described (Rando and Blau, 1994). These cells were treated with drugs as described in the figure legends. AD 293 cells were cultured in DMEM containing 10% serum and penicillin/streptomycin cocktail. Transfections with CMX-Flag, CMV-myc, pTAP, CMX-Flag PPARδ, pTAP-PPARδ, CMX-Tk-PPRE, CMX-βGAL, CMV-myc-hAMPK (α1 and α2 subunits) or CMX-Flag PGC1α were performed using Lipofectamine 2000. Skeletal muscle C2C12 cells were cultured in DMEM containing 20% serum and penicillin/streptomycin cocktail. For differentiation, cells at 80% confluence were switched to a differentiation medium (DMEM + 2% serum) for 4 days to obtain differentiated myotubules. Drug treatments are described in figure legends.
Immuno-precipitation and western blotting
Flag-PPARδ or Flag-PGC1α was immuno-precipitated from cell lysates with Anti-Flag conjugated agarose beads (Sigma). For co-immunoprecipitation experiments SDS was excluded from the lysis buffer. Western blotting was performed with rabbit Anti-Flag, AMPK α subunit or PPARδ antibodies. For metabolic labeling, transfected AD 293 cells were treated with p32 for 2 hr before immunoprecipitation.
Data Analysis
Data was analyzed using either one way ANOVA with an appropriate post hoc test for comparison of multiple groups, or using unpaired student’s t-test for comparison between two groups as described in figure legends.
The global gene expression data presented in this manuscript has been deposited in the NCBI Gene Expression Omnibus under the GEO series accession number GSE11805.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs H. Cho, G.D. Barish and C.H. Zhang for valuable suggestions and discussion of the manuscript; and S. Ganley and E. Ong for administrative assistance. R.M.E is an investigator of the Howard Hughes Medical Institute at the Salk Institute for Biological Studies and March of Dimes Chair in Molecular and Developmental Biology. V.A.N. is supported by a Ruth L. Kirschstein National Research Service Award from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (AR053803-03). This work was supported by the Howard Hughes Medical Institute, Hilblom Foundation, and National Institutes of Health (HD027183 and DK057978).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, Carling D, Small CJ. AMP-activated protein kinase plays a role in the control of food intake. J. Biol. Chem. 2004;279:12005–8. doi: 10.1074/jbc.C300557200. [DOI] [PubMed] [Google Scholar]
- Booth FW, Thomason DB. Molecular and cellular adaptation of muscle in response to exercise: perspectives of various models. Physiol. Rev. 1991;71:541–85. doi: 10.1152/physrev.1991.71.2.541. [DOI] [PubMed] [Google Scholar]
- Bronner M, Hertz R, Bar-Tana J. Kinase-independent transcriptional co-activation of peroxisome proliferator-activated receptor alpha by AMP-activated protein kinase. Biochem. J. 2004;384:295–305. doi: 10.1042/BJ20040955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassel-Duby R, Olson EN. Signaling pathways in skeletal muscle remodeling. Annu. Rev. Biochem. 2006;75:19–37. doi: 10.1146/annurev.biochem.75.103004.142622. [DOI] [PubMed] [Google Scholar]
- Chen ZP, Stephens TJ, Murthy S, Canny BJ, Hargreaves M, Witters LA, Kemp BE, McConell GK. Effect of exercise intensity on skeletal muscle AMPK signaling in humans. Diabetes. 2003;52:2205–12. doi: 10.2337/diabetes.52.9.2205. [DOI] [PubMed] [Google Scholar]
- Chawla A, Lee CH, Barak Y, He W, Rosenfeld J, Liao D, Han J, Kang H, Evans RM. PPARdelta is a very low-density lipoprotein sensor in macrophages. Proc. Natl. Acad. Sci. U S A. 2003;100:1268–73. doi: 10.1073/pnas.0337331100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, Evans RM, Schneider MD, Brako FA, Xiao Y, Chen YE, Yang Q. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 2004;10:1245–50. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- Durante PE, Mustard KJ, Park SH, Winder WW, Hardie DG. Effects of endurance training on activity and expression of AMP-activated protein kinase isoforms in rat muscles. Am. J. Physiol. Endocrinol. Metab. 2002;283:E178–86. doi: 10.1152/ajpendo.00404.2001. [DOI] [PubMed] [Google Scholar]
- Dressel U, Allen TL, Pippal JB, Rohde PR, Lau P, Muscat GE. The peroxisome proliferator-activated receptor beta/delta agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol. Endocrinol. 2003;17:2477–93. doi: 10.1210/me.2003-0151. [DOI] [PubMed] [Google Scholar]
- Fluck M, Hoppeler H. Molecular basis of skeletal muscle plasticity-from gene to form and function. Rev. Physiol. Biochem. Pharmacol. 2003;146:159–216. doi: 10.1007/s10254-002-0004-7. [DOI] [PubMed] [Google Scholar]
- Frøsig C, Jørgensen SB, Hardie DG, Richter EA, Wojtaszewski JF. 5′-AMP-activated protein kinase activity and protein expression are regulated by endurance training in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2004;286:E411–7. doi: 10.1152/ajpendo.00317.2003. [DOI] [PubMed] [Google Scholar]
- Goodyear LJ, Chang PY, Sherwood DJ, Dufresne SD, Moller DE. Effects of exercise and insulin on mitogen-activated protein kinase signaling pathways in rat skeletal muscle. Am. J. Physiol. 1996;271:E403–8. doi: 10.1152/ajpendo.1996.271.2.E403. [DOI] [PubMed] [Google Scholar]
- Garnier A, Fortin D, Zoll J, N’Guessan B, Mettauer B, Lampert E, Veksler V, Ventura-Clapier R. Coordinated changes in mitochondrial function and biogenesis in healthy and diseased human skeletal muscle. FASEB. J. 2005;19:43–52. doi: 10.1096/fj.04-2173com. [DOI] [PubMed] [Google Scholar]
- Holloszy JO, Coyle EF. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J. Appl. Physiol. 1984;56:831–8. doi: 10.1152/jappl.1984.56.4.831. [DOI] [PubMed] [Google Scholar]
- Hong YH, Varanasi US, Yang W, Leff T. AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J. Biol. Chem. 2003;278:27495–501. doi: 10.1074/jbc.M304112200. [DOI] [PubMed] [Google Scholar]
- Hsiao A, Worrall DS, Olefsky JM, Subramaniam S. Variance-modeled posterior inference of microarray data: detecting gene-expression changes in 3T3-L1 adipocytes. Bioinformatics. 2004;20:3108–3127. doi: 10.1093/bioinformatics/bth371. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell. Biol. 2007;8:774–85. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Jones AM, Carter H. The effect of endurance training on parameters of aerobic fitness. Sports. Med. 2000;29:373–86. doi: 10.2165/00007256-200029060-00001. [DOI] [PubMed] [Google Scholar]
- Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. U S A. 2007;104:12017–22. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koves TR, Li P, An J, Akimoto T, Slentz D, Ilkayeva O, Dohm GL, Yan Z, Newgard CB, Muoio DM. Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J. Biol. Chem. 2005;280:33588–98. doi: 10.1074/jbc.M507621200. [DOI] [PubMed] [Google Scholar]
- Kramer DK, Al-Khalili L, Perrini S, Skogsberg J, Wretenberg P, Kannisto K, Wallberg-Henriksson H, Ehrenborg E, Zierath JR, Krook A. Direct activation of glucose transport in primary human myotubes after activation of peroxisome proliferator-activated receptor delta. Diabetes. 2005;54:1157–63. doi: 10.2337/diabetes.54.4.1157. [DOI] [PubMed] [Google Scholar]
- Kramer DK, Al-Khalili L, Guigas B, Leng Y, Garcia-Roves PM, Krook A. Role of AMP kinase and PPARdelta in the regulation of lipid and glucose metabolism in human skeletal muscle. J. Biol. Chem. 2007;282:19313–20. doi: 10.1074/jbc.M702329200. [DOI] [PubMed] [Google Scholar]
- Kubota N, Yano W, Kubota T, Yamauchi T, Itoh S, Kumagai H, Kozono H, Takamoto I, Okamoto S, Shiuchi T, Suzuki R, Satoh H, Tsuchida A, Moroi M, Sugi K, Noda T, Ebinuma H, Ueta Y, Kondo T, Araki E, Ezaki O, Nagai R, Tobe K, Terauchi Y, Ueki K, Minokoshi Y, Kadowaki T. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell. Metab. 2007;6:55–68. doi: 10.1016/j.cmet.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Lucia A, Hoyos J, Chicharro JL. Physiology of professional road cycling. Sports. Med. 2001;31:325–37. doi: 10.2165/00007256-200131050-00004. [DOI] [PubMed] [Google Scholar]
- Leff T. AMP-activated protein kinase regulates gene expression by direct phosphorylation of nuclear proteins. Biochem. Soc. Trans. 2003;31:224–7. doi: 10.1042/bst0310224. [DOI] [PubMed] [Google Scholar]
- Luquet S, Lopez-Soriano J, Holst D, Fredenrich A, Melki J, Rassoulzadegan M, Grimaldi PA. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB. J. 2003;17:2299–301. doi: 10.1096/fj.03-0269fje. [DOI] [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–22. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Mauriege P, Prud’Homme D, Marcotte M, Yoshioka M, Tremblay A, Despres JP. Regional differences in adipose tissue metabolism between sedentary and endurance-trained women. Am. J. Physiol. 1997;273:E497–506. doi: 10.1152/ajpendo.1997.273.3.E497. [DOI] [PubMed] [Google Scholar]
- Mu J, Brozinick JT, Jr., Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol. Cell. 2001;7:1085–94. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- McGee SL, Howlett KF, Starkie RL, Cameron-Smith D, Kemp BE, Hargreaves M. Exercise increases nuclear AMPK alpha2 in human skeletal muscle. Diabetes. 2003;52:926–8. doi: 10.2337/diabetes.52.4.926. [DOI] [PubMed] [Google Scholar]
- Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferré P, Birnbaum MJ, Stuck BJ, Kahn BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–74. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- Minnaard R, Drost MR, Wagenmakers AJ, van Kranenburg GP, Kuipers H, Hesselink MK. Skeletal Muscle wasting and contractile performance in septic rats. Muscle. Nerve. 2005;31:339–48. doi: 10.1002/mus.20268. [DOI] [PubMed] [Google Scholar]
- Mahoney DJ, Parise G, Melov S, Safdar A, Tarnopolsky MA. Analysis of global mRNA expression in human skeletal muscle during recovery from endurance exercise. FASEB. J. 2005;19:1498–500a. doi: 10.1096/fj.04-3149fje. [DOI] [PubMed] [Google Scholar]
- Mahoney DJ, Tarnopolsky MA. Understanding skeletal muscle adaptation to exercise training in humans: contributions from microarray studies. Phys. Med. Rehabil. Clin. N. Am. 2005;16:859–73b. doi: 10.1016/j.pmr.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Man MQ, Barish GD, Schmuth M, Crumrine D, Barak Y, Chang S, Jiang Y, Evans RM, Elias PM, Feingold KR. Deficiency of PPARbeta/delta in the Epidermis Results in Defective Cutaneous Permeability Barrier Homeostasis and Increased Inflammation. J. Invest. Dermatol. 2007;128:370–377. doi: 10.1038/sj.jid.5701026. [DOI] [PubMed] [Google Scholar]
- Pette D, Staron RS. Myosin isoforms, myosin fiber types and transitions. Microsc. Res. Tech. 2000;50:500–509. doi: 10.1002/1097-0029(20000915)50:6<500::AID-JEMT7>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Rando TA, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J. Cell. Biol. 1994;125:1275–87. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznick RM, Shulman GI. The role of AMP-activated protein kinase in mitochondrial biogenesis. J. Physiol. 2006;574:33–9. doi: 10.1113/jphysiol.2006.109512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röckl KS, Hirshman MF, Brandauer J, Fujii N, Witters LA, Goodyear LJ. Skeletal muscle adaptation to exercise training: AMP-activated protein kinase mediates muscle fiber type shift. Diabetes. 2007;56:2062–9. doi: 10.2337/db07-0255. [DOI] [PubMed] [Google Scholar]
- Schmitt B, Fluck M, Decombaz J, Kreis R, Boesch C, Wittwer M, Graber F, Vogt M, Howald H, Hoppeler H. Transcriptional adaptations of lipid metabolism in tibialis anterior muscle of endurance-trained athletes. Physiol. Genomics. 2003;15:148–57. doi: 10.1152/physiolgenomics.00089.2003. [DOI] [PubMed] [Google Scholar]
- Siu PM, Donley DA, Bryner RW, Always SE. Myogenin and oxidative enzyme gene expression levels are elevated in rat soleus muscles after endurance training. J. Appl. Physiol. 2004;97:277–85. doi: 10.1152/japplphysiol.00534.2004. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–6. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short KR, Vittone JL, Bigelow ML, Proctor DN, Coenen-Schimke JM, Rys P, Nair KS. Changes in myosin heavy chain mRNA and protein expression in human skeletal muscle with age and endurance exercise training. J. Appl. Physiol. 2005;99:95–102. doi: 10.1152/japplphysiol.00129.2005. [DOI] [PubMed] [Google Scholar]
- Schuler M, Ali F, Chambon C, Duteil D, Bornert JM, Tardivel A, Desvergne B, Wahli W, Chambon P, Metzger D. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell. Metab. 2006;4:407–14. doi: 10.1016/j.cmet.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Sprecher DL, Massien C, Pearce G, Billin AN, Perlstein I, Willson TM, Hassall DG, Ancellin N, Patterson SD, Lobe DC, Johnson TG. Triglyceride:high-density lipoprotein cholesterol effects in healthy subjects administered a peroxisome proliferator activated receptor delta agonist. Arterioscler. Thromb. Vasc. Biol. 2007;27:359–65. doi: 10.1161/01.ATV.0000252790.70572.0c. [DOI] [PubMed] [Google Scholar]
- Timmons JA, Larsson O, Jansson E, Fischer H, Gustafsson T, Greenhaff PL, Ridden J, Rachman J, Peyrard-Janvid M, Wahlestedt C, Sundberg CJ. Human muscle gene expression responses to endurance training provide a novel perspective on Duchenne muscular dystrophy. FASEB. J. 2005;19:750–60. doi: 10.1096/fj.04-1980com. [DOI] [PubMed] [Google Scholar]
- Terada S, Wicke S, Holloszy JO, Han DH. PPARdelta activator GW-501516 has no acute effect on glucose transport in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2006;290:E607–11. doi: 10.1152/ajpendo.00430.2005. [DOI] [PubMed] [Google Scholar]
- Thomson DM, Porter BB, Tall JH, Kim HJ, Barrow JR, Winder WW. Skeletal muscle and heart LKB1 deficiency causes decreased voluntary running and reduced muscle mitochondrial marker enzyme expression in mice. Am. J. Physiol. Endocrinol. Metab. 2007;292:E196–202. doi: 10.1152/ajpendo.00366.2006. [DOI] [PubMed] [Google Scholar]
- Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS. Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell. 2003;113:159–70. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- Yoshioka M, Tanaka H, Shono N, Snyder EE, Shindo M, St-Amand J. Serial analysis of gene expression in the skeletal muscle of endurance athletes compared to sedentary men. FASEB. J. 2003;17:1812–9. doi: 10.1096/fj.02-1200com. [DOI] [PubMed] [Google Scholar]
- Zapala, et al. Zapala MA, Lockhart DJ, Pankratz DG, Garcia AJ, Barlow C, Lockhart DJ. Software and methods for oligonucleotide and cDNA array data analysis. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-6-software0001. (2002) SOFTWARE0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.