

Abstract
Mutations in the transcriptional repressor methyl CpG binding protein 2 (MeCP2) are responsible for most cases of Rett Syndrome (RS), a severe neurodevelopmental disorder characterized by developmental regression, minimal speech, seizures, postnatal microcephaly and hand sterotypies. Absence of the maternal copy of ubiquitin protein ligase 3A (UBE3A) results in Angelman syndrome, also a severe developmental disorder that shares some clinical features with RS. As MeCP2 regulates gene expression, this has led to the hypothesis that MeCP2 may regulate UBE3A expression; however, there are conflicting reports regarding the expression of Ube3a in MeCP2 null mutant mice. We have generated a novel MeCP2 mutant knock-in mouse with the mutation R168X, one of the most common mutations in patients with RS. These mice show features similar to RS, including hypoactivity, forelimb stereotypies, breathing irregularities, weight changes, hind limb atrophy, and scoliosis. The male mice experience early death. Analysis of Ube3a mRNA and protein levels in the Mecp2R168X male mice showed no significant difference in expression compared to their wildtype littermates.
Keywords: Rett Syndrome, Angelman Syndrome, MeCP2, UBE3A, mouse model
1. INTRODUCTION1
Rett syndrome (RS) is a severe neurodevelopmental disorder characterized by apparently normal initial development followed by slowing of development and head growth(Hagberg et al., 1983; Hagberg et al., 1985; Hagberg et al., 2002; Rett, 1966). Purposeful hand skills are lost and replaced by characteristic stereotypies. Spoken language is lost. Patients may also develop seizures, breathing irregularities, sleep disturbance, autistic symptoms and scoliois. Mutations or deletions in methyl-CpG-binding protein 2 (MECP2) located at Xq28 are detectable in 96% of patients with RS (Amir et al., 1999; Moretti and Zoghbi, 2006). A small percentage of RS patients with early onset seizures have mutations in cyclin dependent kinase-like 5 (CDKL5) (Evans et al., 2005; Scala et al., 2005; Tao et al., 2004). CDKL5 may play a role in phosphorylation and regulation of MeCP2 (Mari et al., 2005).
Because MeCP2 binds to methyl CpG dinucleotides, an early leading hypothesis was that it serves as a global repressor of transcription. However, transcriptional profiling studies have failed to reveal a global de-repression of transcription in the setting of MeCP2 deficiency (Tudor et al., 2002). An alternate hypothesis that MeCP2 instead regulates transcription in a highly specific and selective manner has thus been raised. Strong evidence for this hypothesis is the finding that MeCP2 regulates BDNF in a calcium and phosphorylation dependent manner (Chen et al., 2003; Zhou et al., 2006).
Angelman syndrome (AS) is an imprinting disorder caused by a decrease in or loss of function of the maternal copy of ubiquitin protein ligase E3A (UBE3A) located at 15q11q13 (Kishino et al., 1997; Magenis et al., 1987). This protein, unlike MeCP2, is not a regulator of gene transcription but is involved in the ubiquitination pathway, which targets specific proteins for degradation. AS patients exhibit profound speech deficits, gait ataxia, seizures, characteristic EEG, postnatal acquired microcephaly, sleep disturbance, and an unusually happy demeanor with propensity to paroxysms of laughter (Angelman, 1965; Clayton-Smith and Laan, 2003; Williams et al., 1995). Several of these features are in common with RS, including the speech deficits, acquired microcephaly, sleep disturbance, and seizures. These similarities suggest that UBE3A could be a target for regulation by MeCP2. However, recent studies designed to test this hypothesis yielded conflicting results (Jordan and Francke, 2006; Makedonski et al., 2005; Samaco et al., 2005).
The study by Samaco et al looked at two lines of mutant Mecp2 adult mice, as well as post mortem brain tissue from RS patients (Samaco et al., 2005). One mouse line studied, Mecp2tm1.1Jae, was constructed with an exon 3 deletion and characterized as functionally Mecp2 null (Chen et al., 2001). The other mouse line studied was Mecp2tm1.1Bird, which is Mecp2 null (Guy et al., 2001). They reported a significant reduction in expression of both UBE3A RNA and protein products. Next, Makedonski et al investigated newborns of one of the same mouse lines studied by Samaco et al (Mecp2tm1.1Bird) as well as post mortem brain tissue from RS patients and a lymphoblast cell line from an RS patient (Makedonski et al., 2005). They also reported reductions in UBE3A RNA and protein expression. Jordan and Francke then reported studies on both mouse lines, Mecp2tm1.1Jae and Mecp2tm1.1Bird, at 3 and 21 days of age (Jordan and Francke, 2006). However, in direct contrast to the previous two reports, no significant changes in Ube3a RNA or protein expression were detected. This lack of consensus prompted us to investigate Ube3a expression in a novel Mecp2 mutant mouse line designed in our laboratory.
2. RESULTS
Mecp2R168X mice show RS features
We designed a line of Mecp2 mutant mice, which have the most common mutation associated with RS, R168X, knocked-in the mouse gene (Mecp2R168X)(Bienvenu and Chelly, 2006). This sequence change replaces a codon for arginine with a stop codon. Using site directed mutagenesis, we altered the AGA sequence coding for arginine at codon 168 to a TGA coding for stop (Fig 1A). The mutant transcript is transcribed, and is easily detectable by RT-PCR (Fig. 1B). The amplicon was confirmed to be Mecp2 by sequencing, which also confirmed the presence of the mutation in the mouse line. The mutant transcript is relatively stable compared to the WT transcript (Fig 1C). The Mecp2R168X mice do not express full length wild type Mecp2(Fig. 1D). These Mecp2 mutant mice differ from the mice previously used to study the impact of Mecp2 on Ube3a expression, which had null mutations.
Figure 1. Mecp2R168X mutant mice.
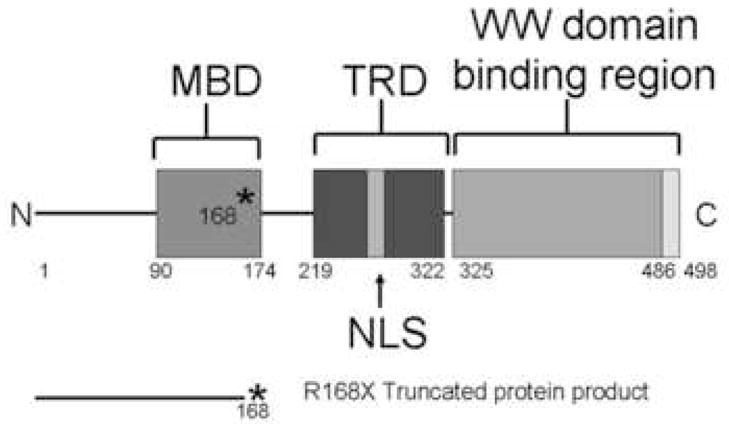
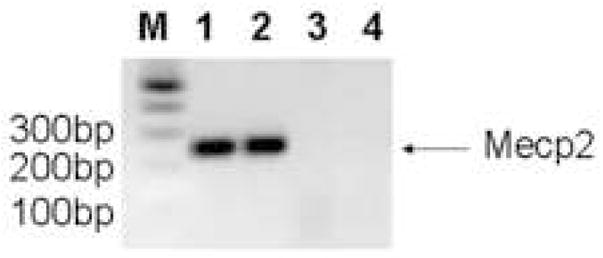
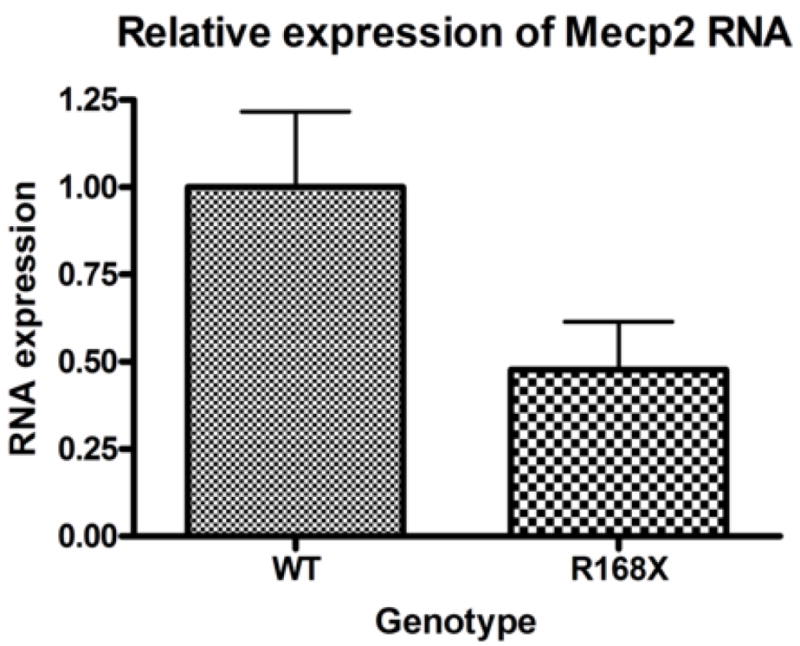
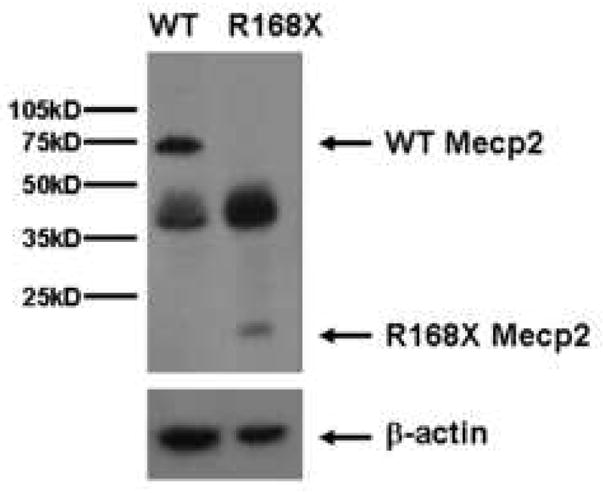
(A) An A to T point mutation was engineered to create a premature stop codon in place of an arginine at codon 168. (B) Confirmation of the mutant transcript by RTPCR. RTPCR was performed with primers flanking the mutation using cDNA reverse transcribed from WT mouse RNA (lane 1), cDNA reverse transcribed from mutant mouse RNA (lane 2), WT mouse RNA (lane 3), and mutant mouse RNA (lane 4). The expected 238bp product was detected in lanes 1 and 2 and confirmed as MeCP2 by sequencing. Larger bands are produced from genomic DNA in lanes 3 and 4 due to the presence of intron 3. (C) The Mecp2R168X mutant RNA is relatively stabled compared to the WT transcript. Error bars show standard error of the mean. (D) Mecp2R168X mutant mice (right lane) express a small protein which may be a prematurely truncated mutant MeCP2 protein, but do not express the full length WT protein. The WT mice (left lane) express the full length protein.
Male hemizygotes are more severely affected than the female heterozygotes. The males have a shortened lifespan of 85.8 ± 24.2 days (± SD; n=43) (Fig 2A). We observed forelimb stereotypies, hindlimb atrophy, hypoactivity, and breathing irregularities. The males showed great variability in weight as compared to the wildtype littermates that became more accentuated with age (Fig 2B and 2C). While some of the affected males were similar in weight to their unaffected littermates, one subset showed failure to thrive (Fig. 2D) and another showed excessive weight gain (Fig. 2E). By 7 weeks they showed significant hindlimb clasping (Fig. 2F and 2G) and breathing irregularities.
Figure 2. Characterization of Mecp2R168X mutant mice.
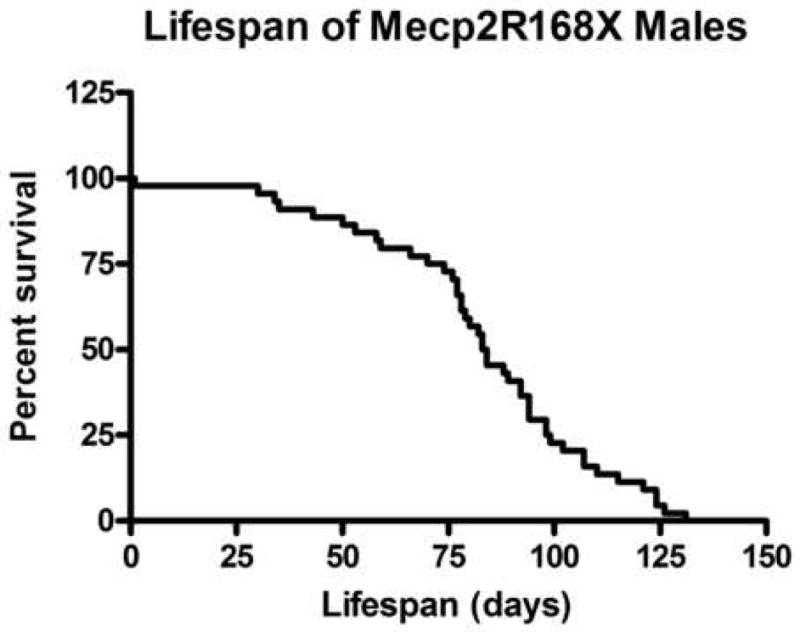
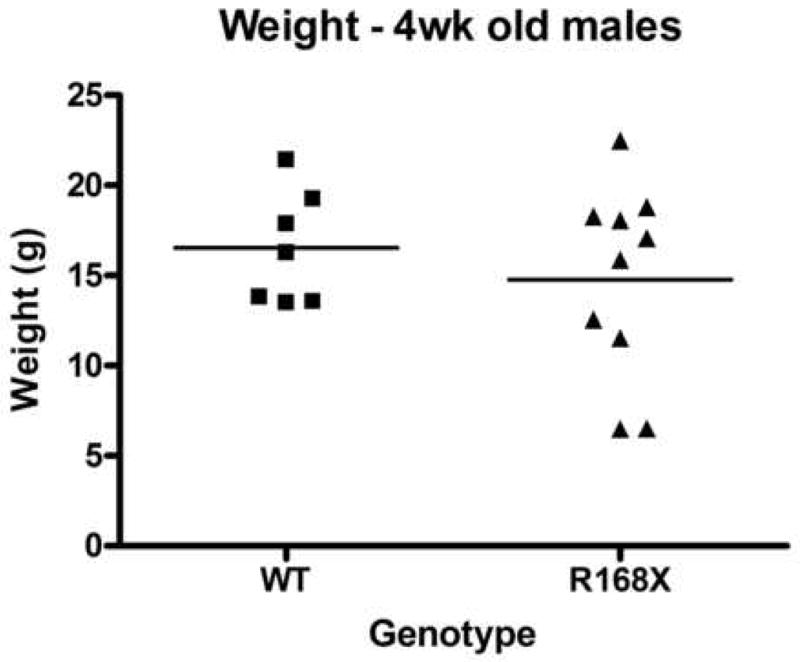
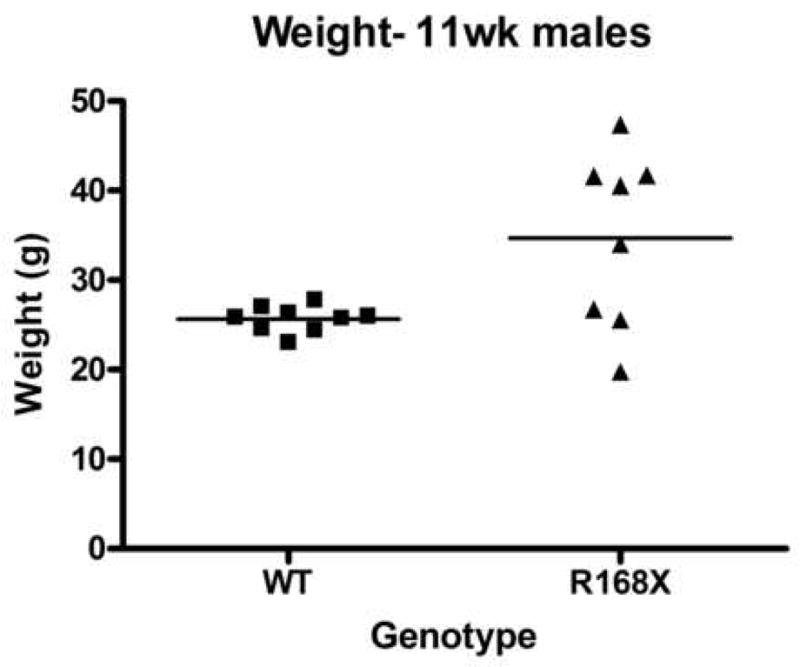
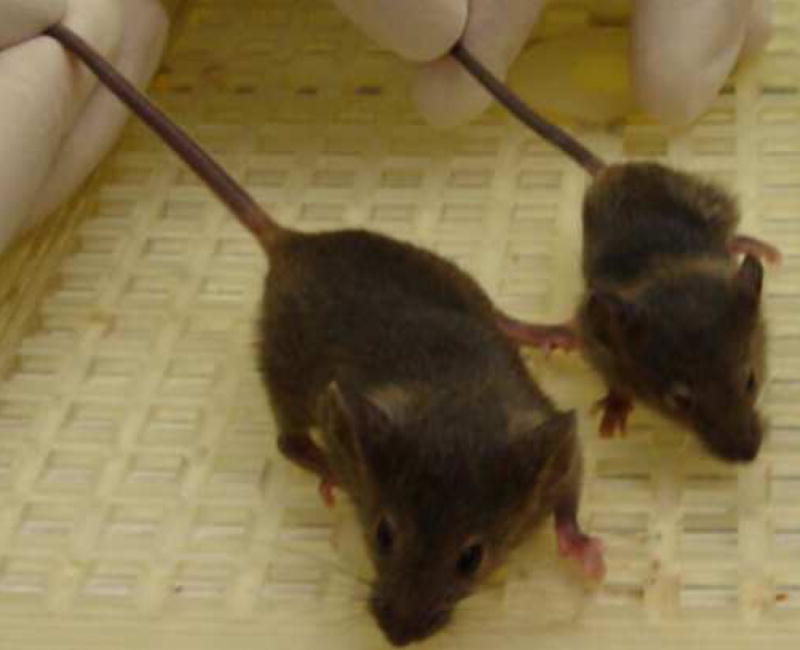
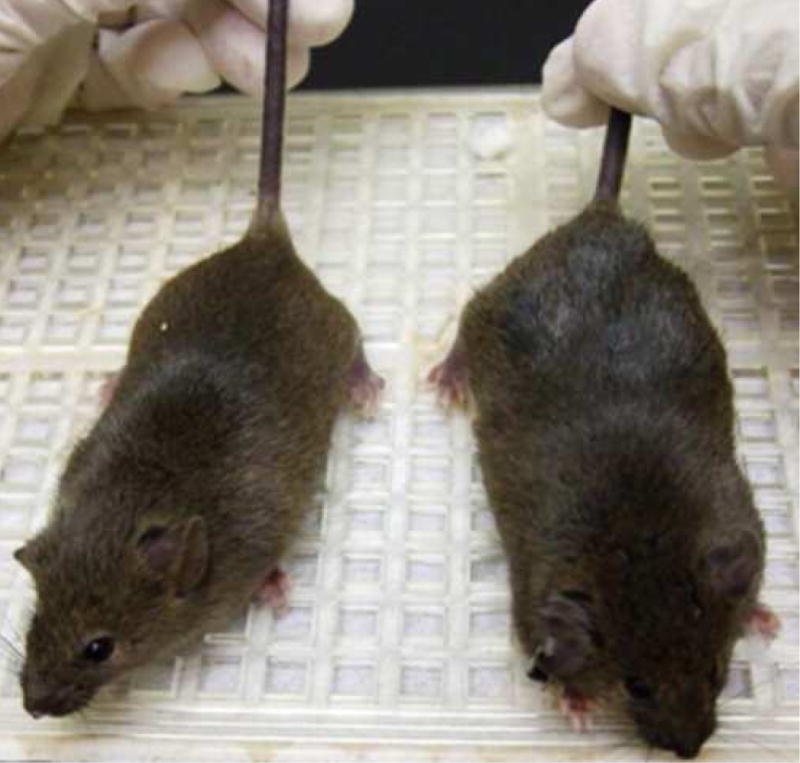
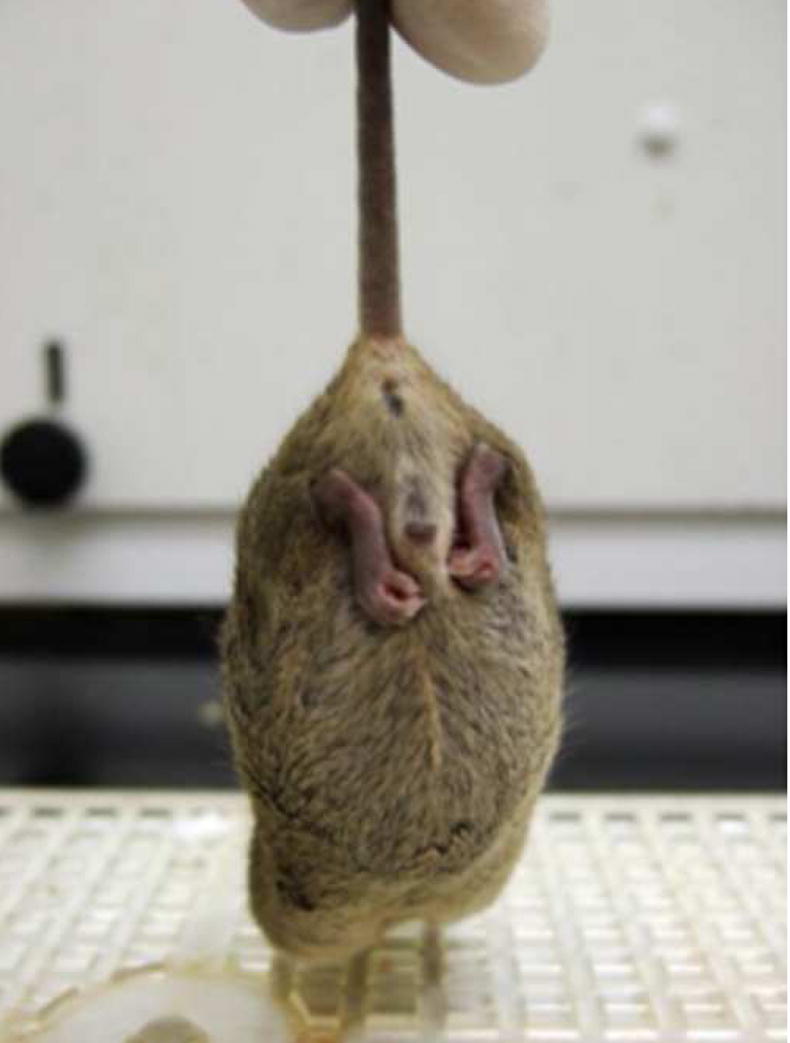
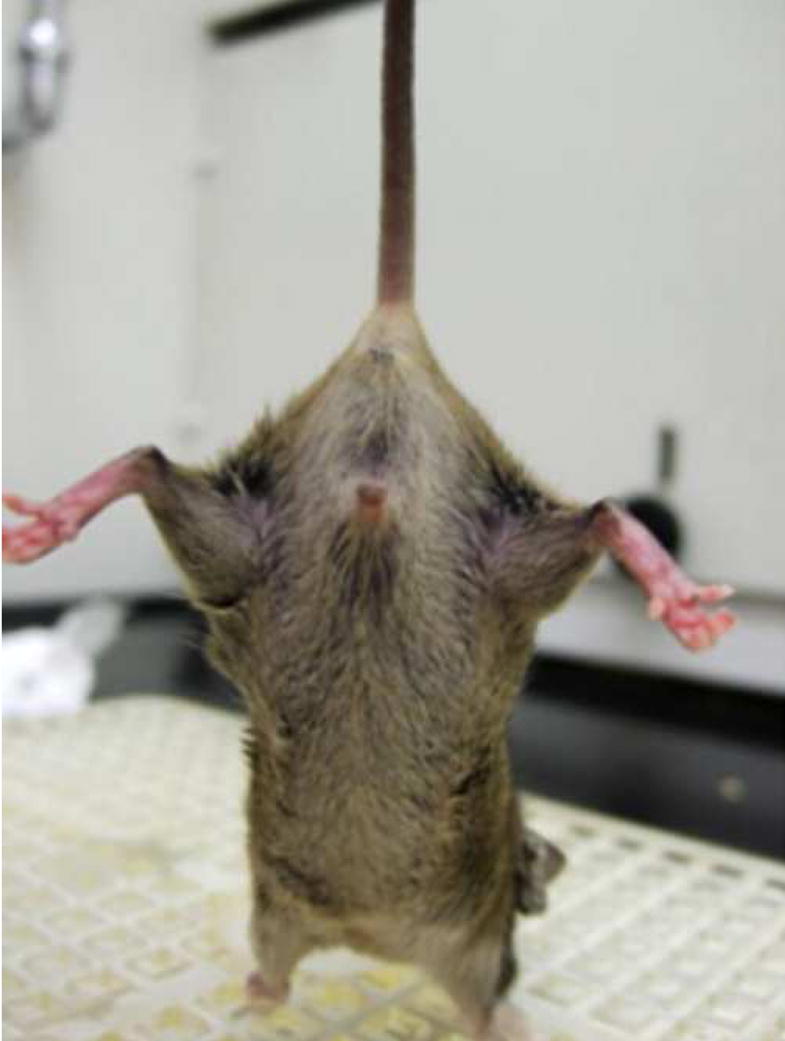
(A) Lifespan of the Mecp2R168X mutant mice is shortened, with a median lifespan of 83.5 days (n=45). (B) At 4 weeks of age, greater variance in the weight distribution of the affected males was apparent, with numerous low weight mice. (C) In the mice surviving to 11 weeks, weight variance was also apparent, but many more mice were obese. (D) At 3 weeks, an affected hemizygous Mecp2R168X male (mouse on the right) had difficulty gaining weight while his WT littermate thrived. (E) At 7 weeks, an affected hemizygous Mecp2R168X male (mouse on the right) showed greater weight gain his WT littermate. (F) 7 week affected male showing hindlimb clasping. (G) 7 week old WT male littermate showing normal function of hindlimbs.
The female heterozygotes showed significant symptoms by approximately 6 months. Lifespan was much longer than that of the males, however, with multiple females surviving more than 1 year. They also showed significant hindlimb clasping and breathing irregularities starting after 6 months of life.
Ube3a and Ube3a-antisense transcript levels are similar in Mecp2R168X mice and wild type (WT) mice
We collected cortical brain tissue samples from seven male Mecp2R168X mice and seven male WT littermates between the ages of six and seven weeks. In this age range, the male mice are severely affected. Using real-time quantitative PCR (qRT-PCR), Ube3a mRNA expression levels showed no significant difference in the affected mice as compared to their WT littermates (Fig. 2A). We also tested expression levels of the Ube3a antisense transcript (Ube3a-ATS) using the same seven pairs of mice (Fig. 2B). UBE3A is imprinted in brain, and so its transcription occurs from only one allele(Rougeulle et al., 1997; Vu and Hoffman, 1997). When UBE3A is not transcribed, an alternate transcript, UBE3A-antisense (UBE3A-ATS), is produced from the antisense strand(Chamberlain and Brannan, 2001; Rougeulle et al., 1998; Runte et al., 2001). Disruption of the maternal Ube3a transcript in murine brain correlates with elevated expression of Ube3a-ATS(Landers et al., 2005). Expression levels between the same WT and the Mecp2R168X groups again showed no significant difference.
Ube3a protein product levels are similar in Mecp2R168X mice and WT mice
Cortical brain tissue samples from the same seven male Mecp2R168X mice and seven WT littermates were also used to analyze Ube3a protein expression. The samples were analyzed by Western blot (Fig. 3A) and quantified by densitometry relative to a reference protein, β-actin (Fig. 3B). We did not detect any significant difference between the affected males and their WT littermates. In fact, both groups had virtually identical expression levels.
Figure 3. Ube3a and Ube3a-ATS mRNA expression.

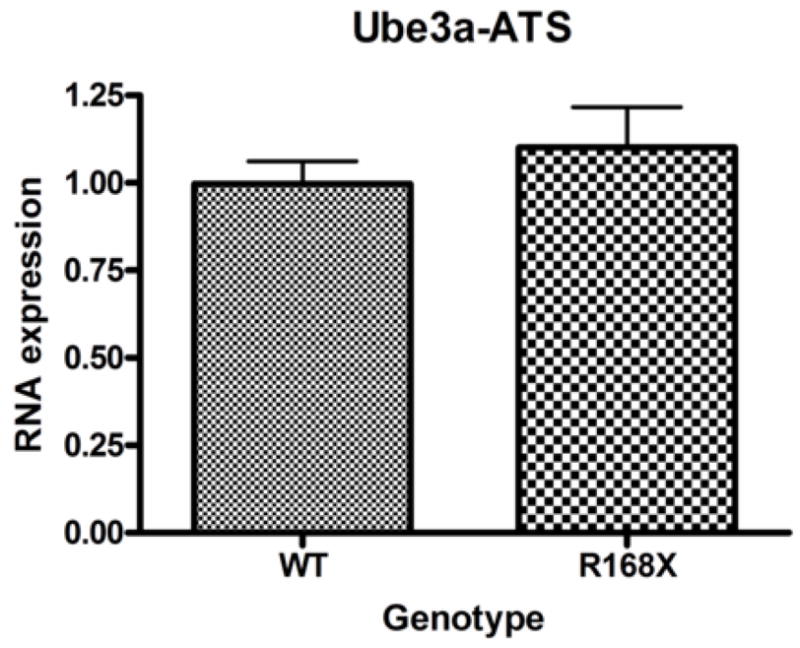
Male hemizygous Mecp2R168X mice no significant difference in Ube3a (A) or Ube3a-ATS (B) mRNA expression in cortical tissue as compared to their WT littermates when analyzed by real time qRT-PCR. Error bars show standard error of the mean.
3. DISCUSSION
Conceptually, MeCP2 regulation of UBE3A is an attractive hypothesis, given the similarities between RS and AS. However, the literature is far from a clear consensus. Two groups using mouse models and human post-mortem brain tissue observed decreases in UBE3A RNA and protein in the setting of MeCP2 deficiency(Makedonski et al., 2005; Samaco et al., 2005), while a third group found no change using the same two mouse models as the previous two groups(Jordan and Francke, 2006).
We chose to probe this question using our novel model, which is unique in that it carries a common Mecp2 mutation found in patients with RS. Study of such mutant models with common mutations knocked-in can greatly enrich the information gained by knock-out models. These models may more closely mimic actual disease states and may help to provide information on genotype-phenotype correlations within heterogeneous diagnostic categories. It is possible that some Mecp2 mutations may have dominant negative activity, which could lead to a more complex picture than simple loss of function.
Our studies showed no significant differences between Ube3a mRNA and protein levels between the WT and Mecp2R168X groups. We chose to study males at the 6 to 7 weeks of age because the males are more severely affected than the females, and this is the most affected age range among the males, just prior to death. We hypothesized that if there were a difference in Ube3a expression, it would be best demonstrated in this most severely affected group of mice.
Mecp2 function, however, has recently been demonstrated to be strikingly dynamic(Zhou et al., 2006). Mecp2 is phosphorylated following neuronal activation, which could then lead to very specific time limited changes in gene expression. One could hypothesize that Mecp2 might truly impact Ube3a expression, but only at discrete and specific times. Mouse model studies, or even post-mortem brain tissue, may have difficulty detecting such time specific effects. Further studies on neurons in culture may better test the hypothesis that Mecp2 regulates Ube3a. Alternatively, the effect could be for a short time at a very specific age or developmental point, including the prenatal period. Our data at this time, however, do not support the hypothesis.
4. EXPERIMENTAL PROCEDURE
Mouse lines
Site directed mutagenesis was performed using the Transformer Site-Directed Mutagenesis Kit (CLONTECH). Sequence alteration was confirmed by sequencing (Massachusetts General Hospital DNA Core Facility). The targeting construct containing a Neomycin resistance (neo) cassette flanked by lox P sites and the mutant sequence was electroporated onto 129SvJ embryonic stem cells. The neo cassette was cloned from pLITMUS-28 (New England Biolabs). Neomycin-resistant colonies were isolated, expanded and characterized by PCR using MeCP2 and Neo cassette specific primers (sequences available upon request). Cells with the correct recombination were injected into C57BL/6 blastocytes and transferred to pseudopregnant females. Chimeric mice were bred with 129S6/SvEv Tac mice. The F1 mice with germ line transmission of R168X were crossed with Cre Recombinase-expressing mice to remove the neo cassette, yielding Mecp2R168X mice. The mice used in the experiments described here were back crossed through a minimum of 10 generations. Care and handling of the mice was conducted in concordance with Institutional Animal Care and Use Committee approved protocols.
RT-PCR and Real-time qRT-PCR
Mouse brain cortex samples were harvested and immediately placed in RNAlater (Ambion). Total RNA was isolated from cortex tissue using the RNeasy Mini Kit (Qiagen) and reverse transcribed to cDNA using the Transcriptor First Strand cDNA synthesis kit (Roche Diagnostics) following manufacturers’ protocols. qRT-PCR was then performed on a MiniOpticon MJ Mini Personal Thermal Cycler (Bio-Rad) using the following conditions: 95°C for 3 minutes, followed by 36 cycles of 95°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec. All samples were processed in triplicate. The primer sequences were as follows: MeCP2 forward (ACA GCG GCG CTC CAT TAT C), MeCP2 reverse (CCC AGT TAC CGT GAA GTC AAA A), Ube3a forward (GCG AGC AGC TGC AAA GCA TCT AAT), Ube3a reverse (AGC TTG CTC CTT TCT TGG AGG GAT), Ube3a-ATS forward (TTG TAT ACA GGA AGC TAA TGG GG)(Landers et al., 2005), Ube3a-ATS reverse (CAA AAG TTT ACA AAT AAA TAA TGT TCC)(Landers et al., 2005), β-actin forward (AGT GTG ACG TTG ACA TCC GTA), and β-actin reverse (GCC AGA GCA GTA ATC TCC TTC T). The fold change in expression was calculated using the standard curve method with β-actin as a reference control gene. All qRT-PCR products were analyzed using gel electrophoresis on 3% agarose, as well as by melting curve analysis. All samples were observed to have a single product with the expected length in gel electrophoresis, and the melting curve analysis showed a single peak.
Western blot analysis
Mouse brain cortex samples were harvested and immediately placed on dry ice, then stored at −80°C until use. The tissue was disrupted by sonication in lysis buffer (Sigma C3228). Protein was quantified with a bicinchoninic assay (Pierce), and 10μg was resolved by polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. Western blots were tested as indicated with a β-actin specific antibody (Abcam ab8227), a Ube3a specific antibody (Abcam ab10488) or a Mecp2 N-terminal specific antibody (Sigma M7443). Secondary antibodies were goat anti-mouse peroxidase conjugate (Sigma A0168) or goat anti-rabbit peroxidase conjugate (Upstate 12–348) as appropriate. All samples were tested at least in duplicate.
Figure 4. Ube3a protein expression.
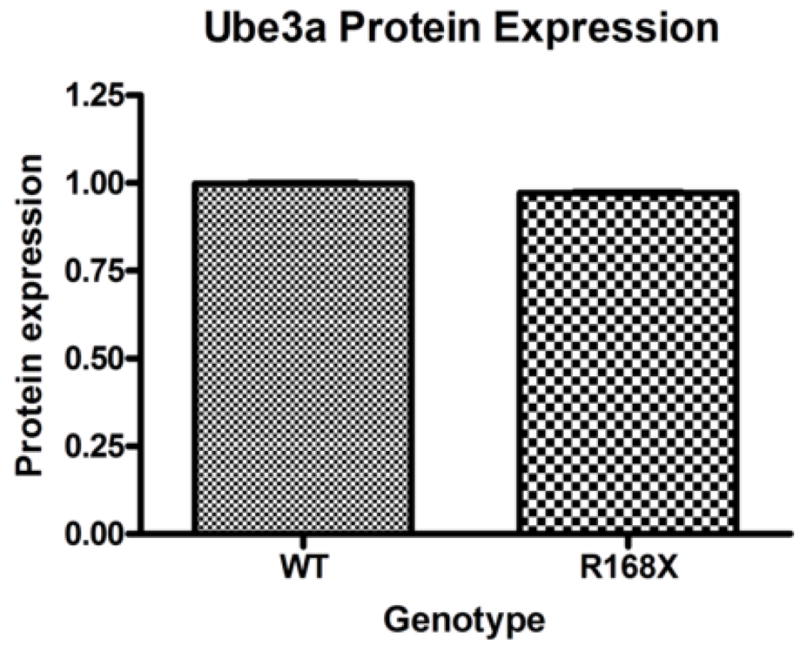
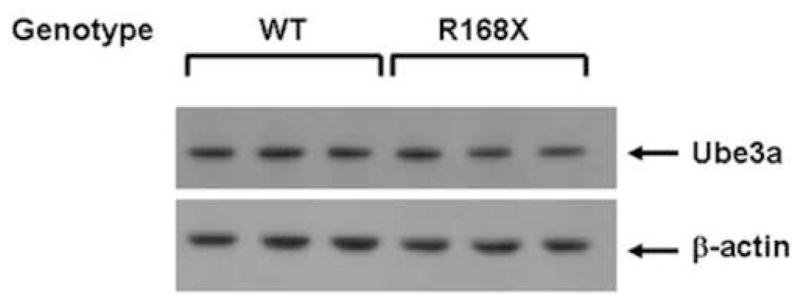
(A) Male hemizygous Mecp2R168X mice showed no change in Ube3a protein expression in cortical tissue as compared to their WT littermates when analyzed by Western blot. Error bars show standard error of the mean. (B) Western blot showing representative samples from 3 pairs of mice.
Acknowledgments
Funding for this study was through NIH grant MH-572901 and.
Footnotes
Abbreviations: AS – Angelman Syndrome; MeCP2 - methyl CpG binding protein 2; qRT-PCR – quantitative real-time reverse transcriptase polymerase chain reaction; RS – Rett Syndrome; UBE3A - ubiquitin protein ligase 3A; UBE3A-ATS – UBE3A antisense transcript
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Angelman H. “Puppet children.” A report of three cases. Dev Med Child Neurol. 1965;7:681–8. doi: 10.1111/j.1469-8749.2008.03035.x. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet. 2006;7:415–26. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- Chamberlain SJ, Brannan CI. The Prader-Willi syndrome imprinting center activates the paternally expressed murine Ube3a antisense transcript but represses paternal Ube3a. Genomics. 2001;73:316–22. doi: 10.1006/geno.2001.6543. [DOI] [PubMed] [Google Scholar]
- Chen RZ, et al. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–31. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Chen WG, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–9. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Clayton-Smith J, Laan L. Angelman syndrome: a review of the clinical and genetic aspects. J Med Genet. 2003;40:87–95. doi: 10.1136/jmg.40.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JC, et al. Early onset seizures and Rett-like features associated with mutations in CDKL5. Eur J Hum Genet. 2005 doi: 10.1038/sj.ejhg.5201451. [DOI] [PubMed] [Google Scholar]
- Guy J, et al. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–6. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Hagberg B, et al. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann Neurol. 1983;14:471–9. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- Hagberg B, et al. Rett syndrome: criteria for inclusion and exclusion. Brain Dev. 1985;7:372–3. doi: 10.1016/s0387-7604(85)80048-6. [DOI] [PubMed] [Google Scholar]
- Hagberg B, et al. An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol. 2002;6:293–7. doi: 10.1053/ejpn.2002.0612. [DOI] [PubMed] [Google Scholar]
- Jordan C, Francke U. Ube3a expression is not altered in Mecp2 mutant mice. Hum Mol Genet. 2006;15:2210–5. doi: 10.1093/hmg/ddl146. [DOI] [PubMed] [Google Scholar]
- Kishino T, et al. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–3. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- Landers M, et al. Maternal disruption of Ube3a leads to increased expression of Ube3a-ATS in trans. Nucleic Acids Res. 2005;33:3976–84. doi: 10.1093/nar/gki705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magenis RE, et al. Is Angelman syndrome an alternate result of del(15)(q11q13)? Am J Med Genet. 1987;28:829–38. doi: 10.1002/ajmg.1320280407. [DOI] [PubMed] [Google Scholar]
- Makedonski K, et al. MeCP2 deficiency in Rett syndrome causes epigenetic aberrations at the PWS/AS imprinting center that affect UBE3A expression. Hum Mol Genet. 2005 doi: 10.1093/hmg/ddi097. [DOI] [PubMed] [Google Scholar]
- Mari F, et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet. 2005;14:1935–46. doi: 10.1093/hmg/ddi198. [DOI] [PubMed] [Google Scholar]
- Moretti P, Zoghbi HY. MeCP2 dysfunction in Rett syndrome and related disorders. Curr Opin Genet Dev. 2006;16:276–81. doi: 10.1016/j.gde.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Rett A. [On a unusual brain atrophy syndrome in hyperammonemia in childhood] Wien Med Wochenschr. 1966;116:723–6. [PubMed] [Google Scholar]
- Rougeulle C, et al. An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat Genet. 1998;19:15–6. doi: 10.1038/ng0598-15. [DOI] [PubMed] [Google Scholar]
- Rougeulle C, et al. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat Genet. 1997;17:14–5. doi: 10.1038/ng0997-14. [DOI] [PubMed] [Google Scholar]
- Runte M, et al. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum Mol Genet. 2001;10:2687–700. doi: 10.1093/hmg/10.23.2687. [DOI] [PubMed] [Google Scholar]
- Samaco RC, et al. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. 2005;14:483–92. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scala E, et al. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J Med Genet. 2005;42:103–7. doi: 10.1136/jmg.2004.026237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet. 2004;75:1149–54. doi: 10.1086/426460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudor M, et al. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci U S A. 2002;99:15536–41. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TH, Hoffman AR. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet. 1997;17:12–3. doi: 10.1038/ng0997-12. [DOI] [PubMed] [Google Scholar]
- Williams CA, et al. Angelman syndrome: consensus for diagnostic criteria. Angelman Syndrome Foundation. Am J Med Genet. 1995;56:237–8. doi: 10.1002/ajmg.1320560224. [DOI] [PubMed] [Google Scholar]
- Zhou Z, et al. Brain-Specific Phosphorylation of MeCP2 Regulates Activity-Dependent Bdnf Transcription, Dendritic Growth, and Spine Maturation. Neuron. 2006;52:255–69. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]