

Abstract
The endoplasmic reticulum (ER) is the site where proteins enter the secretory pathway. Proteins are translocated into the ER lumen in an unfolded state and require protein chaperones and catalysts of protein folding to attain their final appropriate conformation. A sensitive surveillance mechanism exists to prevent misfolded proteins from transiting the secretory pathway and ensures that persistently misfolded proteins are directed towards a degradative pathway. In addition, those processes that prevent accumulation of unfolded proteins in the ER lumen are highly regulated by an intracellular signaling pathway known as the unfolded protein response (UPR). The UPR provides a mechanism by which cells can rapidly adapt to alterations in client protein-folding load in the ER lumen by expanding the capacity for protein folding. In addition, a variety of insults that disrupt protein folding in the ER lumen also activate the UPR. These include changes in intralumenal calcium, altered glycosylation, nutrient deprivation, pathogen infection, expression of folding-defective proteins, and changes in redox status. Persistent protein misfolding initiates apoptotic cascades that are now known to play fundamental roles in the pathogenesis of multiple human diseases including diabetes, atherosclerosis and neurodegenerative diseases.
Keywords: Endoplasmic reticulum, unfolded protein response, ER, secretory pathway, apoptosis
Introduction
Protein folding is an essential process for protein function in all organisms. As a consequence, all cells have evolved sophisticated mechanisms to ensure proper protein folding occurs and to dispose of irreversibly misfolded proteins. All proteins that transit the secretory pathway in eukaryotic cells first enter the endoplasmic reticulum (ER) where they fold and assemble into multi-subunit complexes prior to transit to the Golgi compartment [1]. ‘Quality control’ is a surveillance mechanism that permits only properly folded proteins to exit the ER en route to other intracellular organelles and the cell surface. Misfolded proteins are either retained within the ER lumen in complex with molecular chaperones or are directed toward degradation through the 26S proteasome in a process called ER-associated degradation (ERAD) or through autophagy.
The efficiency of protein-folding reactions depends on appropriate environmental, genetic and metabolic conditions. Conditions that disrupt protein folding present a threat to cell viability. The ER provides a unique environment that challenges proper protein folding as nascent polypeptide chains enter the ER lumen. A high concentration of partially folded and unfolded proteins predisposes protein folding intermediates to aggregation. Polypeptide binding proteins, such as BiP and GRP94, act to slow protein folding reactions and prevent aberrant interactions and aggregation. The ER lumen is an oxidizing environment so disulfide bond formation occurs. As a consequence, cells have evolved sophisticated machinery composed of several protein disulfide isomerases (PDIs) that are required to ensure proper disulfide bond formation and prevent formation of illegitimate disulfide bonds. The ER is also the primary Ca2+ storage organelle in the cell. Both protein folding reactions and protein chaperone functions require high levels of ER intralumenal calcium. Protein folding in the ER requires extensive amounts of energy and depletion of energy stores prevents proper protein folding. ATP is required for chaperone function, to maintain Ca2+ stores and redox homeostasis, and for ERAD. Finally, proteins that enter the ER lumen are subject to numerous post-translational modifications including N-linked glycosylation, amino acid modifications such as proline and aspartic acid hydroxylation and γ-carboxylation of glutamic acid residues, and addition of glycosylphosphatidylinositol anchors. All these processes are highly sensitive to alterations in the ER luminal environment. As a consequence, innumerable environmental insults alter protein-folding reactions in the ER through mechanisms that include depletion of ER calcium, alteration in the redox status, and energy (sugar/glucose) deprivation. In addition, gene mutations, elevated protein traffic through the ER compartment, and altered post-translational modification all contribute the accumulation of unfolded proteins in the ER lumen.
Accumulation of unfolded protein initiates activation of an adaptive signaling cascade known as the unfolded protein response (UPR). Appropriate adaptation to misfolded protein accumulation in the ER lumen requires regulation at all levels of gene expression including transcription, translation, translocation into the ER lumen, and ERAD. Coordinate regulation of all these processes is required to restore proper protein folding and ER homeostasis [1-6]. Conversely, if the protein folding defect is not resolved, chronic activation of UPR signaling occurs which eventually induces an apoptotic (programmed cell death) response. In this review we summarize the signaling pathways that mediate the UPR, mechanisms that signal cell death, the role of the UPR in mammalian physiology, and the clinical implications of the UPR in health and disease.
Protein folding and quality control in the ER
Protein folding and maturation in vivo is a highly-assisted process. The ER lumen contains molecular chaperones, folding enzymes and quality control factors that assist in folding and trafficking of newly synthesized polypeptides. Nascent polypeptide chains enter the ER lumen through a proteinaceous channel, the Sec 61 translocon complex. The nascent chains of most translocated polypeptides are subject to addition of a preassembled oligosaccharide core (N-acetylglucosamine2-mannose9-glucose3) i.e., Glc3Man9GlcNac2 to selective asparagine (N) residues. N-glycosylation is catalyzed by the oligosaccharyltransferase(OST), a multisubunit enzyme associated with the translocon complex. Subsequently, sequential action by the ER α glucosidases I and II removes the two outermost glucose residues to produce a mono-glucosylated core glycan. The mono-glucosylated glycoprotein can then interact with two homologous ER lectins calnexin (CNX) and calreticulin (CRT) that associate with Erp57, an oxidoreductase that catalyzes disulfide bond formation. Upon release of folding substrates from CNX and/or CRT, the innermost glucose residue is rapidly removed by glucosidase II. Two potential mechanisms by which chaperones may monitor protein folding are through exposed hydrophobic patches or through excessive surface dynamics associated with the non-compact partially-folded state. The protein chaperone BiP binds to the hydrophobic patches exposed on protein folding intermediates. Highly dynamic, non-native deglucosylated glycoproteins are recognized by the ER folding sensor UDP-glucose:glycoprotein glucosyltransferase (UGT1). UGT1 specifically re-glucosylates folding intermediates released from CNX/CRT cycle. Reglucosylation mediates ER retention of immaturely folded glycoproteins so they enter another round of CNX/CRT-assisted folding. Polypeptides that fail to acquire their native transport-competent structure are eventually removed by ER-associated degradation (ERAD). During ERAD, folding-defective proteins are retranslocated to the cytosol, in a process possibly mediated by EDEM and Derlin, and are degraded by the 26S proteasome. Native, properly-folded polypeptides released from CNX/CRT are transported from the ER to the Golgi compartment, in an event possibly assisted by mannose-binding lectins, such as ERGIC-53, VIPL, ERGL [7] (Figure 1).
Figure 1. Protein Trafficking from the ER.
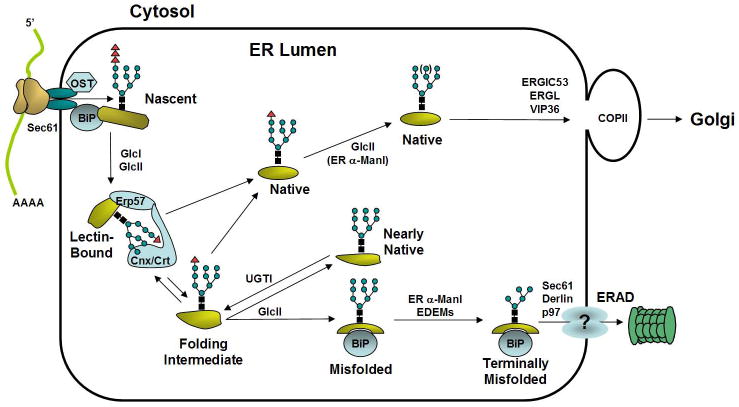
Upon translocation of polypeptides through the Sec61 proteinaceous channel, asparagine residues are frequently modified by covalent addition of a preassembled oligosaccharide core (N-acetylglucosamine2-mannose9-glucose3). This reaction is catalyzed by the oligosaccharyltransferase (OST), a multisubunit complex associated with translocon. To facilitate unidirectional transport through the translocon, nascent polypeptide chains in the ER lumen interact with BiP, a molecular chaperone that binds to exposed hydrophobic residues. Subsequently, rapid deglucosylation of the two outermost glucose residues on the oligosaccharide core structures, mediated by glucosidase I and II (GlcI and GlcII), prepares glycoproteins for association with the ER lectins calnexin and calreticulin. The calnexin/calreticulin-associated oxidoreductase ERp57 facilitates protein folding by catalyzing formation of intra- and inter- molecular disulfide bonds, a rate-limiting step in the protein folding process. Release from calnexin/calreticulin followed by glucosidase II cleavage of the innermost glucose residue prevents further interaction with calnexin and calreticulin. At this point, natively folded polypeptides transit the ER to the Golgi compartment, in a process possibly assisted by mannose-binding lectins, such as ERGIC-53, VIPL, ERGL. As an essential component of protein-folding quality control, non-native polypeptides are tagged for reassociation with calnexin/calreticulin by the UDP-glucose:glycoprotein glucosyltransferase (UGT1) to facilitate their ER retention and prevent anterograde transport. Polypeptides that are folding incompetent are targeted for degradation by retrotranslocation, possibly mediated by EDEM and Derlins, into the cytosol and delivery to the 26S proteosome. Triangles represent glucose residues, squares represent N-acetylglucosamine residues, and circles represent mannose residues.
UPR signaling
In response to ER stress, three ER-localized transmembrane signal transducers are activated to initiate adaptive responses. These transducers are two protein kinases IRE1 (inositol requiring kinase 1) [8,9], and PERK (double stranded RNA-activated protein kinase-like ER kinase) [10] and the transcription factor ATF6 (activating transcription factor 6) [9,11]. These three UPR transducers are constitutively expressed in all known metazoan cells (Fig. 1). IRE1 was the first component of the UPR that was identified, initially in yeast, and is conserved in all eukaryotic cells. The essential and unique properties of IRE1 signaling in the UPR have been conserved in all eukaryotic cells but higher eukaryotes also possess the additional sensors PERK and ATF6 that promote stress adaptation or cell death in a more complex and diverse, yet coordinated manner.
Perk phosphorylates eIF2α to attenuate mRNA translation
The most immediate response to ER stress in metazoan cells is reversibly, transient attenuation of mRNA translation, thereby preventing influx of newly synthesized polypeptides into the stressed ER lumen [12]. This translational attenuation is signaled through PERK-mediated phosphorylation of the eukaryotic translation initiation factor 2 on the alpha subunit (eIF2α) at Ser51. eIF2α phosphorylation inhibits the guanine nucleotide exchange factor eIF2B that recycles the eIF2 complex to its active GTP-bound form. The formation of the ternary translation initiation complex eIF2-GTP-tRNAMet is required for AUG initiation codon recognition and joining of the 60S ribosomal subunit that occurs during initiation phase of polypeptide chain synthesis. Lower levels of active ternary complex result in lower levels of translation initiation [10,13-15]. (Fig.1).
PERK is an ER-associated transmembrane serine/threonine protein kinase. Upon accumulation of unfolded proteins in the ER lumen, PERK dimerization and trans-autophosphorylation leads to activation of its eIF2α kinase function. [10,16]. In addition to translational attenuation, activation of PERK also induces transcription of approximately 1/3 of the UPR-dependent genes [13-15,17]. Although phosphorylation of eIF2α inhibits general translation initiation, it is required for the selective translation of several mRNAs. One fundamental transcription factor for which translation is activated upon PERK-mediated phosphorylation of eIF2α, is the activating transcription factor 4 (ATF4) [13-15,17]. Expression profiling identified that genes encoding amino acid biosynthesis and transport functions, anti-oxidative stress responses, and apoptosis, such as growth arrest and DNA damage 34 (GADD34) and CAAT/Enhancer binding protein (C/EBP) homolgous protein (CHOP/GADD153) [16,18] require PERK, eIF2α phosphorylation, and ATF4 [13-15,17].
Although the majority of PERK signaling is mediated through phosphorylation of eIF2α, studies suggest that the bZiP Cap ‘n’ Collar transcription factor nuclear respiratory factor 2 (NRF2) may also be a substrate for the PERK kinase activity [19]. NRF1 and NRF2 are transcription factors that integrate a variety of responses to oxidative stress. NRF2 is distributed in the cytoplasm through its association with the microtubule-associated protein Keap1 (Kelch-like Ech-associated protein 1). Upon ER stress, PERK phosphorylates NRF2 to promote its dissociation from Keap1, leading to the nuclear accumulation of NRF2. Nrf2−/− cells are sensitive to ER stress-induced apoptosis. NRF2 is a direct PERK substrate and effector of PERK-dependent cell survival [19]. NRF2 binds to the antioxidant response element (ARE) to activate transcription of genes encoding detoxifying enzymes like A1 and A2 subunits of glutathione S-transferase, NAD(P)H:quinone oxidoreductase, γ-glutamylcysteine synthetase, Heme oxygenase-1 (HO-1) and UDP-glucoronosyl transferase [20]. Possibly in a similar manner, NRF1 is localized to the ER membrane and translocates to the nucleus upon ER stress [21]. These data support the notion that PERK phosphorylates multiple substrates to protect cells from oxidative stress. Consistent with this idea, Perk-/- cells accumulate ROS when they are exposed to ER stress [14].
IRE1 initiates nonconventional splicing of XBP1 mRNA
The first component in the UPR pathway was isolated through a genetic screen to identify mutants in UPR signaling in the budding yeast Saccharomyces cerevisiae. In this screen, Ire1p/Ern1p was identified as an ER transmembrane protein kinase that is required for the UPR [3,22]. Subsequently, it was discovered that Ire1p is a bifunctional protein that also has a site-specific endoribonuclease (RNase) activity [3,22]. Under non-stress conditions, Ire1p protein kinase is maintained in an inactive monomeric form through interactions with the protein chaperone Kar2p/BiP. Upon accumulation of unfolded proteins in the ER lumen, Ire1p is released from Kar2p/BiP and undergoes homodimerization and trans-autophosphorylation to activate its RNase activity. The RNase activity of Ire1p cleaves a 252–base intron from mRNA encoding the basic leucine zipper (bZIP)-containing transcription factor Hac1p. This splicing reaction alters the carboxy-terminus of Hac1p to introduce a potent transcriptional activation domain. The protein encoded by spliced HAC1 mRNA binds and activates transcription from the UPR element [UPRE, minimal motif TGACGTG(C/A)] upstream of many UPR target genes [2,23]. In S. cerevisiae, the UPR activates transcription of approximately 381 genes [24], more than 50% of which provide functions in the secretory pathway.
Two mammalian homologues of yeast IRE1 have been identified; IRE1α [25] and IRE1β [26]. IRE1α is expressed in most cells and tissues, with highest levels of expression in the pancreas and placenta [25]. IRE1β expression is prominent only in intestinal epithelial cells [26]. The cleavage specificities of IRE1α and IRE1β are quite similar, thereby suggesting that they do not recognize distinct substrates but rather confer temporal- and tissue- specific expression [27].
Transcriptional analysis of UPR gene targets, such as BiP, GRP94 and calreticulin, identified a mammalian ER stress response element (ERSE, CCAAT(N9)CCACG) that is necessary and sufficient for UPR gene activation [28]. Yoshida et al used a yeast one-hybrid screen to isolate factors that interact with the ERSE. This screen identified one ERSE-binding protein as the bZIP-containing transcription factor XBP1 (X-box binding protein) [28]. Subsequently, several groups using different approaches demonstrated that XBP1 mRNA is a substrate for the endoribonuclease activity of metazoan IRE1 [9,29-31]. Upon activation of the UPR, the IRE1 RNase activity initiates removal of a 26 nucleotide intron from XBP1 mRNA. This splicing reaction creates a translational frameshift to produce a larger form of XBP1 that contains a novel transcriptional activation domain it its C-terminus. Spliced XBP1 is a transcriptional activator that plays a fundamental role activation of wide variety of UPR target genes. Some of the genes identified that require the IRE1/XBP1 pathway are those that encode functions involved in ERAD, such as the ER degradation-enhancing mannosidase-like protein EDEM. Consistent with this observation, cells that are deficient in either IRE1 or XBP1 are defective in ERAD [32](Fig.2).
Figure 2. Signaling the unfolded protein response.
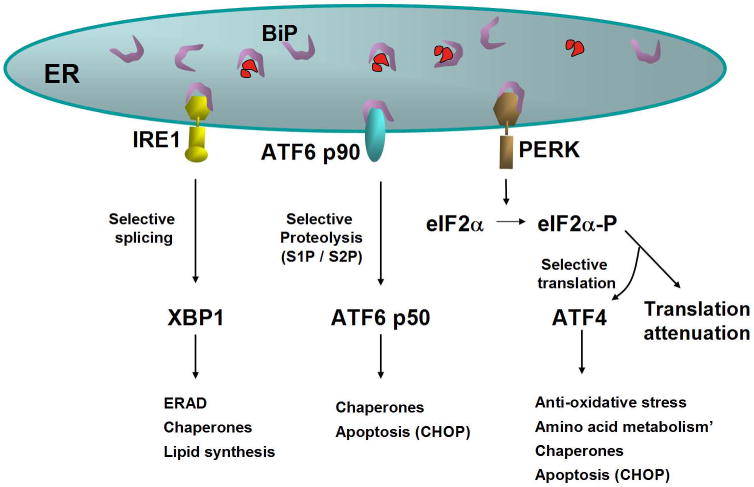
Three proximal sensors IRE1, PERK and ATF6 regulate the UPR through their respective signaling cascades. Under non-stressed conditions, BiP binds to the lumenal domains of IRE1 and PERK to prevent their dimerization. Upon accumulation of unfolded proteins in the ER lumen, IRE1 released from BIP dimerizes to activate its kinase and RNase activities to initiate XBP1 mRNA splicing thereby creating a potent transcriptional activator. Primary targets that require IRE1/XBP1 pathway for induction include genes encoding functions in ERAD. Similarly, ATF6 released from BiP transits to the Golgi compartment where for cleavage by S1P and S2P proteases to yield a cytosolic fragment that migrates to the nucleus to further activate transcription of UPR-responsive genes. Finally, PERK released from BiP dimerizes, autophosphorylates, and phosphorylates eIF2α on Ser 51 leading to general attenuation of translational initiation. Paradoxically, eIF2α phosphorylation induces translation of ATF4 mRNA. The PERK/eIF2α/ATF4 regulatory axis also induces expression of anti-oxidative stress response genes and expression of genes encoding proteins with proapoptotic functions, such as CHOP.
Analysis of gene-deleted mice has provided insight into the physiological roles of IRE1 and XBP1 in mammals. Deletion of Ire1α or Xbp1 in mice creates an embryonic lethality at E11.5-E14 [30,33]. Although deletion of Ire1β had no developmental phenotype, Ire1β-/- mice were susceptible to experimental-induced intestinal colitis [34]. Mice with heterozygous Xbp1 deletion appear normal but develop insulin resistance when fed a high-fat diet [35]. Thus, it was proposed that the UPR might be important in insulin signaling (see below). In addition, both IRE1 and XBP1 have critical roles in B cell differentiation. Antigenic stimulation of mature B lymphocytes activates the UPR and signaling through IRE1-mediated XBP1 mRNA splicing is required to drive B lymphocyte differentiation into plasma cells [29,36-38]. These studies suggest that the IRE1/XBP1 subpathway of the UPR might be required for differentiation of cell types that secrete high levels of protein. This is consistent with a requirement for XBP1 in pancreatic acinar cell development [39].
ATF6–mediated transcriptional activation requires regulated intramembrane proteolysis
The bZiP-containing activating transcription factor 6 (ATF6) was identified as another regulatory protein that, like XBP1, binds to the ERSE1 element in the promoters of UPR–responsive genes [28]. In mammals, there are two alleles of ATF6, ATF6α (90kDa) and ATF6β (110 kDa), both synthesized in all cell types as ER transmembrane proteins. In the unstressed cells, ATF6 is localized at the ER membrane and bound to BiP. In response to ER stress, BiP dissociation permits trafficking of ATF6 to the Golgi complex, where ATF6 is sequentially cleaved by two proteases [40-42]. The serine protease site-1 protease S1P cleaves ATF6 in the luminal domain. The N terminal portion is subsequently cleaved by the metalloprotease site-2 protease S2P [43]. The processed forms of ATF6α and ATF6β translocate to the nucleus and bind to the ATF/cAMP response element (CRE) and to the ER stress responsive element (ERSE-1) to activate target genes [41]. ATF6α and ATF6β both require the presence of the transcription factor CBF (also called NF-Y) to bind ERSEI [40-42]. The proteases S1P and S2P were originally identified for their essential role in processing of the sterol response element binding protein (SREBP) transcription factor, which is activated upon cholesterol deprivation [44] (Fig.1). Over-expression of the cleaved form of ATF6α identified a class of UPR genes that may be transcriptional targets of ATF6α [45]. Since ATF6α and ATF6β have not yet been deleted in model organisms, their precise roles in UPR transcriptional induction remains to be clarified.
Recently, additional bZIP-containing transcription factors that are localized to the ER and regulated by RIP have been identified. CREBH was identified as a liver-specific bZiP transcription factor of the CREB/ATF family with a transmembrane domain that directs localization to the ER [46]. Pro-inflammatory cytokines IL6, 1L-1β and TNFα increase transcription of CREBH to produce an inert protein that is localized to the ER. Upon ER stress, CREBH transits to the Golgi compartment where it is cleaved by S1P and S2P processing enzymes. However, cleaved CREBH does not activate transcription of UPR genes, but rather, induces transcription of a subset of acute phase response genes, such as C-reactive protein and murine Serum Amyloid P component (SAP) in hepatocytes. These studies identified CREBH as a novel ER-localized transcription factor that has an essential role in induction of innate immune response genes and links for the first time ER stress to inflammatory responses [46].
In addition to ATF6 and CREBH, there are additional similarly related factors that are likely regulated through ER stress-induced proteolytic processing, although their physiological roles remain unknown. OASIS (old astrocyte specifically induced substance) and BBF2H7 (BBF2 human homolog on chromosome 7) are cleaved by S1P and S2P in response to ER stress in astrocytes and neurons, respectively [47,48]. Tisp40 (transcript induced in spermiogenesis 40) is cleaved by S1p and S2P to activate transcription of EDEM [49]. These tissue-specific ATF6-like molecules may contribute to the ER stress response. Finally, Luman/LZIP/CREB3 and CREB4 are also two ATF6-like molecules that are cleaved by S1P and S2P to activate UPR transcription, although their cleavage appears to not be activated by ER stress [50-52]. These transcription factors might be activated under conditions other than ER stress to activate transcription of ER chaperones.
BiP is a master regulator of UPR sensor activaton
In non-stressed cells, the luminal domains of IRE1, PERK, and ATF6 are bound to the protein chaperone BiP. In response to stress, unfolded proteins accumulate and bind BiP, thereby sequestering BiP and promoting BiP release from the UPR sensors. When these sensors are bound to BiP, they are maintained in an inactive state [53]. This BiP-mediated negative-regulation model for UPR activation is also supported by the observation that BiP over-expression prevented activation of the UPR upon ER stress [54]. In addition, sufficiently high levels of expression of any protein that binds BiP can activate UPR. In contrast, the accumulation of unfolded proteins that do not bind BiP does not activate the UPR [55,56]. Analysis of the interaction between BiP and ATF6 suggested that this dissociation is not merely a consequence of competition between ATF6 and unfolded protein for binding to BiP, but rather may involve an active ER stress-dependent release of BiP from ATF6 [57]. Recently, based on the x-ray crystal structure of the yeast Ire1p luminal domain, Credle et al. identified a deep, long MHC1-type groove that exists in an Ire1p dimer and proposed that unfolded polypeptides directly bind Ire1p to mediate its dimerization [58]. However, although the x-ray crystal analysis of the human IRE1 luminal domain indicated a similar structure as yeast Ire1p, the MHC1-type groove was not solvent accessible [59]. In addition, the luminal domain was shown to form dimers in vitro in the absence of added polypeptide [59]. These observations bring into question the requirement for peptide binding to the MHC1-type cleft to promote dimerization. It is possible that a combination of BiP binding as well as peptide binding both regulate IRE1 dimerization. Future studies should resolve this issue.
ER Stress-Induced Apoptosis
If the UPR fails to resolve the protein-folding defect, apoptosis is activated. In response to ER stress, apoptosis is signaled through both mitochondrial-dependent and -independent pathways (Fig.2). The ER might actually serve as a site where apoptotic signals are generated and integrated to elicit the death response. Several mechanisms by which apoptotic signals are generated at the ER include: PERK/eIF2α-dependent transcriptional induction of the proapoptotic transcription factor CHOP; Bak/Bax-regulated Ca2+ release from the ER; IRE1-mediated activation of ASK1 (apoptosis signal-regulating kinase 1)/JNK (c-Jun amino terminal kinase); and cleavage and activation of procaspase 12 (Fig.3). Recent evidence also suggests that oxidative stress may provide a significant contributing factor to ER stress-induced apoptosis.
Figure 3. Pathways of ER stress-induced apoptosis.
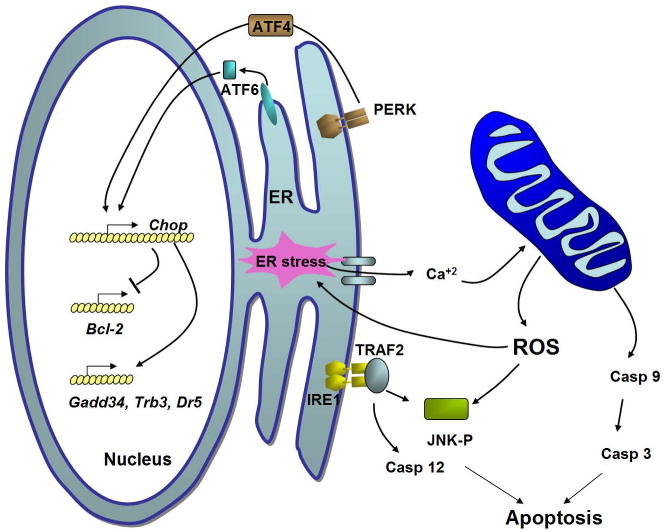
ER stress leads to several redundant pathways for caspase activation that involve mitochondrial- dependent and independent pathways. Activated IRE1 recruits TRAF2 to elicit JNK phosphorylation and activation. Caspase 12 is a murine ER-associated proximal effector in the caspase activation cascade that activates procaspase 9 to cleave procaspase 3, the primary executioner of cell death. A second cell death signaling pathway activated by ER stress is mediated by transcriptional induction of genes encoding proapoptotic functions. Activation of PERK, ATF6 and possibly IRE1 lead to transcriptional induction of CHOP that induces apoptosis possibly through up-regulatng expression of the genes Gadd34, Dr5 and Trb3 or by inhibiting expression of the anti-apoptotic gene Bcl2. Mitochondrial ROS can be generated as a result of ER stress-induced Ca+2 release and depolarization of the inner mitochondrial membrane. Thus, oxidative stress in association of unresolved ER stress contributes to multiple pathways of cell death.
CHOP is a UPR-induced transacription factor that mediates apoptosis
Probably the most significant ER stress-induced apoptotic pathway is mediated through CHOP. CHOP/GADD153 (growth arrest and DNA damage 153) is a bZIP transcription factor that is induced through the ATF6 and PERK UPR pathways [18,60]. Chop−/− cells are protected from ER stress-induced apoptosis [61], indicating the significance of this apoptotic pathway during ER stress. Although the precise mechanism by which CHOP mediates apoptosis in unknown, CHOP activates the transcription of several genes that may potentiate apoptosis. These include GADD34, ERO1, DR5 (death receptor 5), TRB3, and carbonic anhydrase VI. GADD34 encodes a subunit of protein phosphatase 2C that enhances dephosphorylation of eIF2α and promotes protein synthesis [62]. Persistent protein synthesis during periods of ER stress would chronically activate the UPR and initiate cell death pathways. ERO1 encodes an ER oxidase that increases the oxidizing potential of the ER [63]. DR5 encodes a cell surface death receptor that may activate caspase cascades [64]. TRB3 (Tribbles homolog 3) encodes a human ortholog of Drosophila tribble, and TRB3-knock-down cells are resistant to ER stress-induced apoptosis [65]. Carbonic anhydrase VI may decrease the intracellular pH during ER stress [66]. CHOP has also been implicated in repressing transcription of the anti-apoptotic BCL2 protein [67]. However the ability of CHOP to induce ER stress-associated apoptosis has recently been demonstrated to be dependent on the duration of the stress state. Chronic exposure to a mild stress can lead to adaptation by selective attenuation of CHOP expression mediated by degradation of CHOP mRNA and CHOP protein, whereas expression of downstream targets encoding adaptive functions, such as ER chaperones like BiP, is persistent due to long-lived mRNAs and proteins [68].
BCL2-family members regulate ER stress-induced apoptosis
During periods of ER stress, pro-apoptotic members of the Bcl2 family are recruited to the ER surface and activate caspase-12. In contrast, the anti-apoptotic members prevent this recruitment, although the exact relationship between these factors remains unclear. Over-expression of BCL2 family members can prevent ER stress-induced apoptosis. BIM (BCL2-interacting mediator of cell death) translocates from the dynein-rich compartment to the ER membrane and activates caspase-12 in response to ER stress, whereas an antiapoptotic factor, BCL-xL (BCL2-like 1), binds to BIM and inhibits its translocation [69]. Consistent with this notion, BIM knockdown cells are resistant to ER stress-induced death.
BH3 domain (Bcl2-homology domain 3)-only containing pro-apoptotic factors, such as Bax (Bcl2-associated × protein) and Bak (Bcl-2 homologous antagonist/killer), are present at the mitochondrial and ER membranes [70,71]. During ER stress, BAX and BAK oligomerize to possibly permit Ca2+ efflux into the cytoplasm. Increased cytosolic Ca2+ can activate both mitochondrial-dependent and independent caspase cascades [70,71]. In vitro experiments support the idea that the increase in the cytosolic Ca2+ concentration (from micromolar to millimolar) activates the calcium-dependent protease m-Calpain, which subsequently cleaves and activates the ER-resident procaspase-12 to initiate caspase-dependent apoptosis [72]. The Ca2+ released from the ER also enters mitochondria to depolarize the inner membrane, promoting cytochrome c release and activating APAF-1 (apoptosis protease-activating factor 1)/procaspase-9-regulated apoptosis. Thus, BCL2 family members may regulate ER stress-induced apoptotic responses through Ca2+ signaling.
The regulation of BH3-only members of the BCL2 family during ER stress is quite complex. BAX and BAK are required for most forms of apoptosis [73]. Additional BH3 domain-only family members, PUMA (p53 up-regulated modulator of apoptosis) and NOXA (neutrophil NADPH oxidase factor), are up-regulated by p53 during ER stress. In addition, Puma−/− cells and Noxa−/− cells are resistant to ER stress-induced apoptosis [74]. BAX activation during ER stress is inhibited by the ER-localized anti-apoptotic factor BI-1(Bax inhibitor −1). BI-1−/− mice are sensitive to ER stress, whereas mice overexpressing BI-1 are resistant [75]. BIK (BCL2-interacting killer) is an ER-localized pro-apoptotic component that enhances the recruitment of BAX and BAK to the ER membrane [76]. Finally, BAX and BAK associate with IRE1α and potentiate its signaling during ER stress [77].
IRE-dependent activation of JNK mediates ER stress-induced apoptosis
In addition to initiating splicing of XBP1 mRNA, activation of IRE1 signals into the MAP kinase cascade. The IRE1 cytoplasmic domain interacts with the adaptor protein, TRAF2 (tumor necrosis factor receptor-associated factor 2). TRAF2 couples the activation of death receptors at the plasma membrane to activation of Jun kinase (JNK) and stress-activated protein kinase (SAPK) [78]. IRE1 and TRAF2 interact with the mitogen-activated protein kinase kinase kinase, ASK1 (apoptosis signal-regulating kinase 1), which subsequently phosphorylates and activates JNK [79]. Therefore, ER stress-induced JNK activation and apoptosis are reduced in Ire1−/− and Ask1−/− cells. However, this mechanism cannot account for the observation that Traf2−/− cells are more susceptible to ER stress-induced apoptosis [80]. TRAF2 also associates with caspase-12 and regulates its activation [81]. IRE1–TRAF2 activates the transcriptional repressor ATF3 as well, leading to apoptosis [82]. There is also evidence that UPR activation of IRE1 may initiate the extrinsic apoptotic pathway. IRE1 interacts with TNFR1 (tumor necrosis factor receptor 1) to form a complex with TRAF2 and ASK1 to mediate JNK activation. The activation of JNK by ER stress is impaired in Tnfr1−/− cells. In addition, the expression of TNFα is up-regulated by the IRE1 pathway during ER stress [83.,84]. ROS can directly activate ASK1 by disrupting an ASK1 thioredoxin (TDX) complex through oxidation of TDX, and thereby lead to activation of JNK, p38 MAP kinase, and cell death [85]. The Jun activation domain-binding protein (JAB1) may be a feedback regulator since it can interact with IRE1 and inhibit XBP1 mRNA splicing and BiP transcription [86]. Thus, oxidative stress and ER stress may induce cell death by using the same molecular complex consisting of IRE1/TRAF2/ASK1/TDX/JNK. Finally, TNFα can activate the UPR in a ROS-dependent manner. These finding indicate an intricate relationship exists between death receptor signaling, oxidative stress, and activation of the UPR.
ER stress-induces caspase-dependent apoptosis
Caspases 2, 3, 4, 7, 9 and 12 are reported to be involved in ER stress-induced cell death [87-90]. Caspase-12 is associated with the ER membrane, and activated by ER stress, possibly by calpain [91]. In addition, proapoptotic BCL2 family members BAX and BAK colocalize to the ER membrane and function to activate apoptosis through caspase 12 [87-90]. Caspase-12 activates caspase-9, which in turn activates caspase-3 [92] leading to cell death. Caspase-12−/− mice are partially resistant to ER stress-induced apoptosis but sensitive to other death stimuli, suggesting that caspase-12 is a regulator specific to ER stress-induced apoptosis [93]. However, the involvement of caspase-12 in apoptosis of human cells is still open to question, as the human caspase-12 gene contains several inactivating mutations [94]. It is possible that caspase 4 mediates ER stress-induced apoptosis in human cells [90,95].
ER Stress and Oxidative Stress
ROS can be produced in all cellular compartments and ultimately results in protein damage [96]. Furthermore, the exposure of biological systems to various conditions of oxidative stress leads to age-dependent increases in the cellular levels of oxidatively-modified proteins, lipids and nucleic acids, and subsequently predisposes to the development of well-recognized, age-related disorders that cause impaired cognitive function and metabolic integrity [97]. There is accumulating evidence to suggest that protein folding, ER stress and production of ROS are intimately intertwined, however, this area of ER stress is not well explored. Since oxidative protein folding occurs in the ER and perturbations in protein folding can cause deleterious consequences, alterations in redox status or generation of ROS could directly and/or indirectly affect ER homeostasis and protein folding. Elucidating the relationship between oxidative stress and oxidative protein folding represents a major area for future research.
Oxidative stress disrupts ER function
ROS can be produced both as a result of exposure to toxic agents such as irradiation and environmental pollutants, and also as byproducts of oxygen–utilizing enzymatic reactions, such as the mitochondrial respiratory chain, the arachidonic acid pathway, the cytochrome P450 family, glucose oxidase, amino acid oxidases, xanthine oxidase, NADPH/NADPH oxidases, or NO synthases [98,99] (Fig.3A). The electron transport chain produces membrane impermeable superoxide anion and the rate is dependent on the mitochondrial inner membrane potential. In the presence of mitochondrial SOD, superoxide is converted to hydrogen peroxide (H2O2) that can diffuse out of mitochondria into the cytoplasm. In the presence of iron, hydrogen peroxide forms the highly reactive hydroxyl radical (OH•) via the Fenton reaction. The superoxide anion radical (O2-•) also generates other toxic metabolites such as peroxynitrile (ONOO-), hypochlorous acid (HOCl) and singlet oxygen (1O2). Under physiological conditions ROS accumulation is guarded by numerous endogenous antioxidant defense systems that include both enzymatic and non-enzymatic antioxidant mechanisms that can either scavenge ROS or prevent their formation. The enzymatic antioxidant defense mechanisms are mediated through superoxide dismutase (SOD), glutathione peroxidase (GPX), catalase, and thioredoxin reductase. Vitamins provide a non-enzymatic antioxidant defense [100]. Finally, redox homeostasis is contributed by several redox systems including NAD+/NADH, NADP+/NADPH, and oxidized glutathione/reduced glutathione (GSSG/GSH).
Both ER stress and oxidative stress, through ROS generation, can increase leak of Ca2+ from the ER lumen [101-103]. Increases in cytosolic Ca2+ can stimulate mitochondrial ROS production through multiple mechanisms. The mitochondrial electron transport chain generates ROS as a consequence of increased mitochondrial Ca2+ loading. The amount of mitochondrial ROS production primarily reflects the quantity of the ubisemiquinone radical intermediate (QH•), an intermediate in the Q cycle at complex III [104,105]. The level of QH• increases when either complex III is inhibited or with greater flux through the respiratory chain. Ca2+ leak stimulates the TCA cycle, thereby increasing O2 consumption and ROS generation. Ca2+ also stimulates nitric oxide synthase which generates NO• that inhibits complex IV and can thereby enhance ROS production. In addition, Ca2+ opens the permeability transition pore to release cytochrome C from the inner mitochondrial membrane, thereby blocking the respiratory chain at complex III. Finally, Ca2+ -induced permeability transition pore opening may cause leak of GSH from the matrix and as a consequence, deplete reducing equivalents.
High levels of ROS generation within the mitochondria further increases Ca2+ release from the ER. The very close proximity of ER and mitochondria leads to accumulation of Ca2+ near mitochondria, further increasing mitochondrial ROS production and leading to opening of the permeability transition pore [106]. Furthermore, ROS can also feedback to sensitize the Ca2+ release channels at the ER membrane [107,108]. For example, this may occur through ROS or reactive nitrogen species that can oxidize a critical thiol in the ryanodine receptor and cause its inactivation, thereby enhancing Ca2+ release from the sarcoplasmic reticulum [109,110]. As the anti-oxidation potential of the cell diminishes, the vicious cycle of Ca2+ release and ROS production becomes more threatening to cell survival.
Protein folding in the ER causes oxidative stress
The ER is an organelle where proper protein folding and disulfide formation of proteins is dependent on the redox status within the lumen of the ER. In contrast to the cytosol that has a reducing environment, the lumen of the ER is oxidizing with a high ratio of oxidized to reduced glutathione (GSSG/GSH) [111]. Glutathione is a tripeptide (L-γ-glutamyl-L-cysteinyl-glycine) that is synthesized in the cytosol. The cell contains up to 10 mM GSH that is maintained in a reduced form through a cytosolic NADPH-dependent reaction catalyzed by glutathione reductase. Cellular redox homeostasis is maintained by a dynamic interaction between reduced glutathione (GSH) and protein thiols with ROS. Reduced glutathione GSH serves as a major thio-disulfide redox buffer in cells and the ratio of GSH:GSSG is used as an index of the redox state. While the ratio of reduced glutathione to oxidized glutathione is (>50:1) in the cytoplasm, this ratio is (1:1 to 3:1) in the ER lumen [112]. The oxidizing environment of the ER lumen promotes disulfide bond formation. In addition, the greater oxidizing environment of ER was suggested to contribute to the preferred oxidation and inactivation of ER resident proteins, thereby contributing to unfolded protein accumulation [111].
Proteins that transit the secretory pathway frequently require disulfide bond formation for their maturation, stability, and/or function. Mispairing of cysteine residues and formation of inappropriate disulfide bonds prevents proteins from attaining their native conformation and leads to misfolding. Although it is likely that GSH reduces non-native disulfide bonds in misfolded proteins, this is likely not the major pathway utilized in cells. The ER lumen maintains redox conditions that enable a distinct set of folding catalysts to facilitate the formation and isomerization of disulfide bonds [112]. The process of disulfide bond-dependent protein folding is slow due to its dependence on a redox reaction, which requires an electron acceptor. During this folding process a protein may be oxidized to form disulfide bonds, isomerized to allow polypeptide rearrangement, or reduced to allow unfolding and subsequent degradation [113]. The idea that disulfide bond formation is an assisted process in vivo is supported by the discovery of DsbA mutants in Escherichia coli that display compromised disulfide bond formation [114].
In eukaryotes, oxidative protein folding, i.e., disulfide bond formation, is catalyzed by a family of ER oxidoreductases, including PDI (protein disulphide isomerase), ERp57, ERp72, PDIR, PDIp and P5. When chaperone assisted disulfide bond formation occurs, cysteine residues within the PDI active site [-C-X-X-C-] accept two electrons from the polypeptide chain substrate. This electron transfer results in the oxidation of the substrate and the reduction of the PDI active site. Despite the ability of PDI to enhance the rate of disulfide-linked folding, the mechanisms by which the ER disposes of electrons as a result of the oxidative disulfide bond formation has remained an enigma. A number of different factors have been proposed to maintain the oxidizing environment of the ER, including the preferential secretion of reduced thiols and uptake of oxidized thiols, and a variety of different redox enzymes and small molecule oxidants. However, there is a lack of genetic evidence demonstrating that these factors are physiologically important [115-117]. It was believed for many years that the low molecular mass thiol glutathione is responsible for oxidizing the PDI active sites. This was contrary to observations in yeast where depletion of glutathione did not interfere with disulfide bond formation [118,119].
Extensive genetic and biochemical studies using the yeast Saccharomyces cerevisiae have provided detailed insights into the mechanisms underlying oxidative protein folding. A genetic screen in yeast identified a conserved ER-membrane–associated protein Ero1p (ER oxidoreductin 1) [116,120] that plays a role similar to that of the bacterial periplasmic protein DsbB in oxidative folding. The proteins Ero1p and DsbB specifically oxidize a thioredoxin-like protein (PDI in eukaryotes, DsbA in bacteria) that further serves as an intermediate in electron transfer. In both prokaryotes and eukaryotes molecular oxygen serves as the terminal electron acceptor for disulfide bond formation. Ero1p uses a flavin-dependent reaction to pass electrons directly to molecular oxygen. This reaction has the potential to generate ROS that would contribute to cellular oxidative stress. The role of Ero1p in electron transfer suggests that the activity of Ero1p is tightly coupled with the protein-folding load in the ER [121]. In mammals, there are two ERO1 genes hERO1-Lα and hERO1-Lβ [122,123] that differ in their tissue distribution and transcriptional regulation. Only ERO1-Lβ is induced by the UPR [123], whereas ERO1-Lα is induced during hypoxia [124]. Further studies in yeast, and later in metazoan cells, identified a critical role for flavin adenine dinucleotide (FAD) in oxidative protein folding. As Ero1p is a novel FAD-binding protein [121], the FAD requirement in oxidative folding may reflect its function in Ero1p. These studies suggest that the versatile redox molecule FAD functions in disulfide bond formation in the ER lumen.
It has been estimated that approximately 25% of the ROS generated in a cell may result from formation of disulfide bonds in the ER during oxidative protein folding(Fig.3A). Two mechanisms have been proposed for how disulfide bond formation generates ROS. During formation of disulfide bonds, ROS are a byproduct formed as ERO1 and PDI act in concert to transfer electrons from thiol groups in proteins to molecular oxygen. In addition, ROS may be formed as a consequence of the glutathione depletion that occurs as glutathione reduces unstable and improper disulfide bonds. The consumption of GSH would return thiols involved in non-native disulfide bonds to their reduced form so they may again interact with ERO1/PDI1 to be reoxidized. This would generate a futile cycle of disulfide bond formation and breakage in which each cycle would generate ROS and consume GSH (Fig.3B). As a consequence, it is expected that proteins that have multiple disulfide bonds may be more prone to generating oxidative stress.
It is not known whether ROS are generated as a consequence of unfolded protein accumulation in the ER. There several possible mechanisms by which unfolded protein in the ER could lead to ROS production. First, as described above, protein misfolding may be associated with inappropriate pairing and bonding of cysteine residues. A futile cycle of disulfide bond breakage and formation would deplete GSH reducing equivalents, thereby leading to oxidative stress. It order to test this hypothesis, it will be important to determine whether misfolding of a protein that has no disulfide bonds can generate ROS. Alternatively, unfolded protein accumulation in the ER may elicit Ca2+ leak into the cytosol to increase ROS production in mitochondria. Finally, since both protein folding and refolding in the ER lumen are highly energy-dependent processes, ATP depletion as a consequence of protein misfolding could stimulate mitochondrial oxidative phosphorylation to increase ATP production, and consequently increase ROS production.
Future Perspectives
Tremendous progress has been made in understanding the mechanisms underlying the cause of ER stress and cellular adaptive responses. Future studies are required to understand the physiological significance of ER stress and UPR signaling in disease pathogenesis. The relationships between ER stress and apoptosis also remain to be defined. Further studies are also required to elucidate how ER stress and UPR signaling in integrated with other stress signaling pathways, particularly those related to oxidative stress. A greater understanding of the complex interrelationship between protein misfolding and oxidative stress may lead to the development of general pharmacological agents, such as chemical chaperones to improve protein folding and/or antioxidants to reduce oxidative stress, for the treatment of human disease. A coherent mechanistic understanding of the mechanisms and pathways that signal ER stress responses should contribute to the development of more selective and specific-acting therapeutic agents targeted for diseases associated with ER pathologies.
Acknowledgments
R. J. Kaufman is an Investigator of the Howard Hughes Medical Institute and is supported by NIH grants RO1 DK042394 and R01 HL052173.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patil C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol. 2001;13:349–355. doi: 10.1016/s0955-0674(00)00219-2. [DOI] [PubMed] [Google Scholar]
- 3.Mori K, Ma W, Gething MJ, Sambrook J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell. 1993;74:743–756. doi: 10.1016/0092-8674(93)90521-q. [DOI] [PubMed] [Google Scholar]
- 4.Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the Mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- 5.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 6.Wu J, Kaufman RJ. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006;13:374–384. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- 7.Molinari M. N-glycan structure dictates extension of protein folding or onset of disposal. Nat Chem Biol. 2007;3:313–320. doi: 10.1038/nchembio880. [DOI] [PubMed] [Google Scholar]
- 8.Sidrauski C, Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90:1031–1039. doi: 10.1016/s0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- 9.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 10.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. 2000;20:6755–6767. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaufman RJ. Regulation of mRNA translation by protein folding in the endoplasmic reticulum. Trends Biochem Sci. 2004;29:152–158. doi: 10.1016/j.tibs.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 13.Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- 14.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 15.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 16.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 17.Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110:1383–1388. doi: 10.1172/JCI16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- 19.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 21.Wang W, Chan JY. Nrf1 is targeted to the endoplasmic reticulum membrane by an N-terminal transmembrane domain. Inhibition of nuclear translocation and transacting function. J Biol Chem. 2006;281:19676–19687. doi: 10.1074/jbc.M602802200. [DOI] [PubMed] [Google Scholar]
- 22.Cox JS, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87:391–404. doi: 10.1016/s0092-8674(00)81360-4. [DOI] [PubMed] [Google Scholar]
- 23.Mori K, Ogawa N, Kawahara T, Yanagi H, Yura T. mRNA splicing-mediated C-terminal replacement of transcription factor Hac1p is required for efficient activation of the unfolded protein response. Proc Natl Acad Sci U S A. 2000;97:4660–4665. doi: 10.1073/pnas.050010197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 25.Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. Embo J. 1998;17:5708–5717. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niwa M, Sidrauski C, Kaufman RJ, Walter P. A role for presenilin-1 in nuclear accumulation of Ire1 fragments and induction of the mammalian unfolded protein response. Cell. 1999;99:691–702. doi: 10.1016/s0092-8674(00)81667-0. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273:33741–33749. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- 29.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 30.Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, et al. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002;16:452–466. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen X, Ellis RE, Lee K, Liu CY, Yang K, Solomon A, et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development Cell. 2001;107:893–903. doi: 10.1016/s0092-8674(01)00612-2. [DOI] [PubMed] [Google Scholar]
- 32.Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, Mori K. A time-dependent phase shift in the mammalian unfolded protein response. Dev Cell. 2003;4:265–271. doi: 10.1016/s1534-5807(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 33.Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000;14:152–157. [PMC free article] [PubMed] [Google Scholar]
- 34.Bertolotti A, Wang X, Novoa I, Jungreis R, Schlessinger K, Cho JH, et al. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J Clin Invest. 2001;107:585–593. doi: 10.1172/JCI11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 36.Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 37.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 38.Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest. 2005;115:268–281. doi: 10.1172/JCI21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. Embo J. 2005;24:4368–4380. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, et al. Endoplasmic reticulum stress-induced formation of transcription factor complex ERSF including NF-Y (CBF) and activating transcription factors 6alpha and 6beta that activates the mammalian unfolded protein response. Mol Cell Biol. 2001;21:1239–1248. doi: 10.1128/MCB.21.4.1239-1248.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li M, Baumeister P, Roy B, Phan T, Foti D, Luo S, et al. ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol Cell Biol. 2000;20:5096–5106. doi: 10.1128/mcb.20.14.5096-5106.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 44.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 45.Okada T, Yoshida H, Akazawa R, Negishi M, Mori K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J. 2002;366:585–594. doi: 10.1042/BJ20020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 47.Kondo S, Murakami T, Tatsumi K, Ogata M, Kanemoto S, Otori K, et al. OASIS, a CREB/ATF-family member, modulates UPR signalling in astrocytes. Nat Cell Biol. 2005;7:186–194. doi: 10.1038/ncb1213. [DOI] [PubMed] [Google Scholar]
- 48.Kondo S, Saito A, Hino S, Murakami T, Ogata M, Kanemoto S, et al. BBF2H7, a novel transmembrane bZIP transcription factor, is a new type of endoplasmic reticulum stress transducer. Mol Cell Biol. 2007;27:1716–1729. doi: 10.1128/MCB.01552-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nagamori I, Yabuta N, Fujii T, Tanaka H, Yomogida K, Nishimune Y, et al. Tisp40, a spermatid specific bZip transcription factor, functions by binding to the unfolded protein response element via the Rip pathway. Genes Cells. 2005;10:575–594. doi: 10.1111/j.1365-2443.2005.00860.x. [DOI] [PubMed] [Google Scholar]
- 50.Liang G, Audas TE, Li Y, Cockram GP, Dean JD, Martyn AC, et al. Luman/CREB3 induces transcription of the endoplasmic reticulum (ER) stress response protein Herp through an ER stress response element. Mol Cell Biol. 2006;26:7999–8010. doi: 10.1128/MCB.01046-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Raggo C, Rapin N, Stirling J, Gobeil P, Smith-Windsor E, O'Hare P, et al. Luman, the cellular counterpart of herpes simplex virus VP16, is processed by regulated intramembrane proteolysis. Mol Cell Biol. 2002;22:5639–5649. doi: 10.1128/MCB.22.16.5639-5649.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stirling J, O'Hare P. CREB4, a transmembrane bZip transcription factor and potential new substrate for regulation and cleavage by S1P. Mol Biol Cell. 2006;17:413–426. doi: 10.1091/mbc.E05-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 54.Morris JA, Dorner AJ, Edwards CA, Hendershot LM, Kaufman RJ. Immunoglobulin binding protein (BiP) function is required to protect cells from endoplasmic reticulum stress but is not required for the secretion of selective proteins. J Biol Chem. 1997;272:4327–4334. doi: 10.1074/jbc.272.7.4327. [DOI] [PubMed] [Google Scholar]
- 55.Ng DT, Watowich SS, Lamb RA. Analysis in vivo of GRP78-BiP/substrate interactions and their role in induction of the GRP78-BiP gene. Mol Biol Cell. 1992;3:143–155. doi: 10.1091/mbc.3.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Graham KS, Le A, Sifers RN. Accumulation of the insoluble PiZ variant of human alpha 1-antitrypsin within the hepatic endoplasmic reticulum does not elevate the steady-state level of grp78/BiP. J Biol Chem. 1990;265:20463–20468. [PubMed] [Google Scholar]
- 57.Shen J, Snapp EL, Lippincott-Schwartz J, Prywes R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol Cell Biol. 2005;25:921–932. doi: 10.1128/MCB.25.3.921-932.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2005;102:18773–18784. doi: 10.1073/pnas.0509487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, et al. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc Natl Acad Sci U S A. 2006;103:14343–14348. doi: 10.1073/pnas.0606480103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992;6:439–453. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- 61.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004;279:45495–45502. doi: 10.1074/jbc.M406933200. [DOI] [PubMed] [Google Scholar]
- 65.Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. Embo J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sok J, Wang XZ, Batchvarova N, Kuroda M, Harding H, Ron D. CHOP-Dependent stress-inducible expression of a novel form of carbonic anhydrase VI. Mol Cell Biol. 1999;19:495–504. doi: 10.1128/mcb.19.1.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Morishima N, Nakanishi K, Tsuchiya K, Shibata T, Seiwa E. Translocation of Bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J Biol Chem. 2004;279:50375–50381. doi: 10.1074/jbc.M408493200. [DOI] [PubMed] [Google Scholar]
- 70.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 71.Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, et al. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol. 2003;162:59–69. doi: 10.1083/jcb.200302084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001;15:1481–1486. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem. 2006;281:7260–7270. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- 75.Chae HJ, Kim HR, Xu C, Bailly-Maitre B, Krajewska M, Krajewski S, et al. BI-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress. Mol Cell. 2004;15:355–366. doi: 10.1016/j.molcel.2004.06.038. [DOI] [PubMed] [Google Scholar]
- 76.Mathai JP, Germain M, Shore GC. BH3-only BIK regulates BAX, BAK-dependent release of Ca2+ from endoplasmic reticulum stores and mitochondrial apoptosis during stress-induced cell death. J Biol Chem. 2005;280:23829–23836. doi: 10.1074/jbc.M500800200. [DOI] [PubMed] [Google Scholar]
- 77.Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- 78.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 79.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mauro C, Crescenzi E, De Mattia R, Pacifico F, Mellone S, Salzano S, et al. Central role of the scaffold protein tumor necrosis factor receptor-associated factor 2 in regulating endoplasmic reticulum stress-induced apoptosis. J Biol Chem. 2006;281:2631–2638. doi: 10.1074/jbc.M502181200. [DOI] [PubMed] [Google Scholar]
- 81.Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem. 2001;276:13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- 82.Zhang C, Kawauchi J, Adachi MT, Hashimoto Y, Oshiro S, Aso T, et al. Activation of JNK and transcriptional repressor ATF3/LRF1 through the IRE1/TRAF2 pathway is implicated in human vascular endothelial cell death by homocysteine. Biochem Biophys Res Commun. 2001;289:718–724. doi: 10.1006/bbrc.2001.6044. [DOI] [PubMed] [Google Scholar]
- 83.Yang Q, Kim YS, Lin Y, Lewis J, Neckers L, Liu ZG. Tumour necrosis factor receptor 1 mediates endoplasmic reticulum stress-induced activation of the MAP kinase JNK. EMBO Rep. 2006;7:622–627. doi: 10.1038/sj.embor.7400687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol. 2006;26:3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tobiume K, Saitoh M, Ichijo H. Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. J Cell Physiol. 2002;191:95–104. doi: 10.1002/jcp.10080. [DOI] [PubMed] [Google Scholar]
- 86.Oono K, Yoneda T, Manabe T, Yamagishi S, Matsuda S, Hitomi J, et al. JAB1 participates in unfolded protein responses by association and dissociation with IRE1. Neurochem Int. 2004;45:765–772. doi: 10.1016/j.neuint.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 87.Cheung HH, Lynn Kelly N, Liston P, Korneluk RG. Involvement of caspase-2 and caspase-9 in endoplasmic reticulum stress-induced apoptosis: a role for the IAPs. Exp Cell Res. 2006;312:2347–2357. doi: 10.1016/j.yexcr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 88.Dahmer MK. Caspases-2, -3, and -7 are involved in thapsigargin-induced apoptosis of SH-SY5Y neuroblastoma cells. J Neurosci Res. 2005;80:576–583. doi: 10.1002/jnr.20471. [DOI] [PubMed] [Google Scholar]
- 89.Di Sano F, Ferraro E, Tufi R, Achsel T, Piacentini M, Cecconi F. Endoplasmic reticulum stress induces apoptosis by an apoptosome-dependent but caspase 12-independent mechanism. J Biol Chem. 2006;281:2693–2700. doi: 10.1074/jbc.M509110200. [DOI] [PubMed] [Google Scholar]
- 90.Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tan Y, Dourdin N, Wu C, De Veyra T, Elce JS, Greer PA. Ubiquitous calpains promote caspase-12 and JNK activation during endoplasmic reticulum stress-induced apoptosis. J Biol Chem. 2006;281:16016–16024. doi: 10.1074/jbc.M601299200. [DOI] [PubMed] [Google Scholar]
- 92.Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- 93.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 94.Fischer H, Koenig U, Eckhart L, Tschachler E. Human caspase 12 has acquired deleterious mutations. Biochem Biophys Res Commun. 2002;293:722–726. doi: 10.1016/S0006-291X(02)00289-9. [DOI] [PubMed] [Google Scholar]
- 95.Kim SJ, Zhang Z, Hitomi E, Lee YC, Mukherjee AB. Endoplasmic reticulum stress-induced caspase-4 activation mediates apoptosis and neurodegeneration in INCL. Hum Mol Genet. 2006;15:1826–1834. doi: 10.1093/hmg/ddl105. [DOI] [PubMed] [Google Scholar]
- 96.Brookes PS, Darley-Usmar VM. Role of calcium and superoxide dismutase in sensitizing mitochondria to peroxynitrite-induced permeability transition. Am J Physiol Heart Circ Physiol. 2004;286:H39–46. doi: 10.1152/ajpheart.00742.2003. [DOI] [PubMed] [Google Scholar]
- 97.Stadtman ER. Importance of individuality in oxidative stress and aging. Free Radic Biol Med. 2002;33:597–604. doi: 10.1016/s0891-5849(02)00904-8. [DOI] [PubMed] [Google Scholar]
- 98.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 99.Finkel T. Reactive oxygen species and signal transduction. IUBMB Life. 2001;52:3–6. doi: 10.1080/15216540252774694. [DOI] [PubMed] [Google Scholar]
- 100.Brigelius-Flohe R, Banning A, Schnurr K. Selenium-dependent enzymes in endothelial cell function. Antioxid Redox Signal. 2003;5:205–215. doi: 10.1089/152308603764816569. [DOI] [PubMed] [Google Scholar]
- 101.Lizak B, Czegle I, Csala M, Benedetti A, Mandl J, Banhegyi G. Translocon pores in the endoplasmic reticulum are permeable to small anions. Am J Physiol Cell Physiol. 2006;291:C511–517. doi: 10.1152/ajpcell.00274.2005. [DOI] [PubMed] [Google Scholar]
- 102.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 103.Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal. 2006;8:1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- 104.Eckert A, Keil U, Kressmann S, Schindowski K, Leutner S, Leutz S, et al. Effects of EGb 761 Ginkgo biloba extract on mitochondrial function and oxidative stress. Pharmacopsychiatry. 2003;36 1:S15–23. doi: 10.1055/s-2003-40449. [DOI] [PubMed] [Google Scholar]
- 105.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 106.Jacobson J, Duchen MR. Mitochondrial oxidative stress and cell death in astrocytes--requirement for stored Ca2+ and sustained opening of the permeability transition pore. J Cell Sci. 2002;115:1175–1188. doi: 10.1242/jcs.115.6.1175. [DOI] [PubMed] [Google Scholar]
- 107.Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32:235–249. doi: 10.1016/s0143416002001823. [DOI] [PubMed] [Google Scholar]
- 108.Viner RI, Williams TD, Schoneich C. Nitric oxide-dependent modification of the sarcoplasmic reticulum Ca-ATPase: localization of cysteine target sites. Free Radic Biol Med. 2000;29:489–496. doi: 10.1016/s0891-5849(00)00325-7. [DOI] [PubMed] [Google Scholar]
- 109.Favero TG, Zable AC, Abramson JJ. Hydrogen peroxide stimulates the Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. J Biol Chem. 1995;270:25557–25563. doi: 10.1074/jbc.270.43.25557. [DOI] [PubMed] [Google Scholar]
- 110.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 111.van der Vlies D, Makkinje M, Jansens A, Braakman I, Verkleij AJ, Wirtz KW, et al. Oxidation of ER resident proteins upon oxidative stress: effects of altering cellular redox/antioxidant status and implications for protein maturation. Antioxid Redox Signal. 2003;5:381–387. doi: 10.1089/152308603768295113. [DOI] [PubMed] [Google Scholar]
- 112.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 113.Chakravarthi S, Bulleid NJ. Glutathione is required to regulate the formation of native disulfide bonds within proteins entering the secretory pathway. J Biol Chem. 2004;279:39872–39879. doi: 10.1074/jbc.M406912200. [DOI] [PubMed] [Google Scholar]
- 114.Bardwell JC, McGovern K, Beckwith J. Identification of a protein required for disulfide bond formation in vivo. Cell. 1991;67:581–589. doi: 10.1016/0092-8674(91)90532-4. [DOI] [PubMed] [Google Scholar]
- 115.Carelli S, Ceriotti A, Cabibbo A, Fassina G, Ruvo M, Sitia R. Cysteine and glutathione secretion in response to protein disulfide bond formation in the ER. Science. 1997;277:1681–1684. doi: 10.1126/science.277.5332.1681. [DOI] [PubMed] [Google Scholar]
- 116.Frand AR, Kaiser CA. Two pairs of conserved cysteines are required for the oxidative activity of Ero1p in protein disulfide bond formation in the endoplasmic reticulum. Mol Biol Cell. 2000;11:2833–2843. doi: 10.1091/mbc.11.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Frand AR, Kaiser CA. The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol Cell. 1998;1:161–170. doi: 10.1016/s1097-2765(00)80017-9. [DOI] [PubMed] [Google Scholar]
- 118.Jessop CE, Chakravarthi S, Watkins RH, Bulleid NJ. Oxidative protein folding in the mammalian endoplasmic reticulum. Biochem Soc Trans. 2004;32:655–658. doi: 10.1042/BST0320655. [DOI] [PubMed] [Google Scholar]
- 119.Cuozzo JW, Kaiser CA. Competition between glutathione and protein thiols for disulphide-bond formation. Nat Cell Biol. 1999;1:130–135. doi: 10.1038/11047. [DOI] [PubMed] [Google Scholar]
- 120.Pollard MG, Travers KJ, Weissman JS. Ero1p: a novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol Cell. 1998;1:171–182. doi: 10.1016/s1097-2765(00)80018-0. [DOI] [PubMed] [Google Scholar]
- 121.Tu BP, Weissman JS. Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol. 2004;164:341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cabibbo A, Pagani M, Fabbri M, Rocchi M, Farmery MR, Bulleid NJ, et al. ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J Biol Chem. 2000;275:4827–4833. doi: 10.1074/jbc.275.7.4827. [DOI] [PubMed] [Google Scholar]
- 123.Pagani M, Fabbri M, Benedetti C, Fassio A, Pilati S, Bulleid NJ, et al. Endoplasmic reticulum oxidoreductin 1-lbeta (ERO1-Lbeta), a human gene induced in the course of the unfolded protein response. J Biol Chem. 2000;275:23685–23692. doi: 10.1074/jbc.M003061200. [DOI] [PubMed] [Google Scholar]
- 124.Gess B, Hofbauer KH, Wenger RH, Lohaus C, Meyer HE, Kurtz A. The cellular oxygen tension regulates expression of the endoplasmic oxidoreductase ERO1-Lalpha. Eur J Biochem. 2003;270:2228–2235. doi: 10.1046/j.1432-1033.2003.03590.x. [DOI] [PubMed] [Google Scholar]