

Abstract
Growth hormone (GH) and insulin-like growth factor-I (IGF-I) are important physiologic regulators of growth, body composition, and kidney function. Perturbations in the GH-IGF-I axis are responsible for many important complications seen in chronic kidney disease (CKD), such as growth retardation and cachectic wasting, as well as disease progression. Recent evidence suggests that CKD is characterized by abnormalities in GH and IGF-I signal transduction and the interaction of these pathways with those that involve other molecules such as ghrelin, myostatin, and the suppressor of cytokine signaling (SOCS) family. Further understanding of GH/IGF pathophysiology in CKD may lead to the development of therapeutic strategies for these devastating complications, which are associated with high rates of mortality and morbidity.
Keywords: Growth hormone, Insulin-like growth factors, Chronic kidney disease, Ghrelin, Myostatin, Suppressor of cytokine signaling
Introduction
Chronic kidney disease (CKD) is a common disorder, involving as much as 11% of the general population [1]. Patients with CKD often exhibit a common final pathophysiology, despite differences in the precipitating pathologies that cause loss of kidney function. Hormonal imbalance is a common phenomenon in CKD and results in widespread complications in patients. The growth hormone (GH)-insulin-like growth factor (IGF)-I axis has important physiological functions in maintaining normal growth and body composition as well as kidney function. In children with CKD, growth retardation is a significant complication and occurs even though serum GH levels are normal or even elevated, reflecting a state of acquired GH resistance [2-5]. Treatment with recombinant GH can, in part, overcome this resistant state [4]. Furthermore, there is a suggestion that there may also be IGF-I resistance in CKD that additionally contributes to the pathogenesis of growth retardation [5,6]. Perturbations in hypothalamic signaling pathways involving leptin and ghrelin, a ligand of the GH secretagogue receptor, may contribute to cachexia and wasting in CKD [7-10]. Tissue myostatin/IGF-I imbalance further contributes to sarcopenia [11]. The GH-IGF-I axis may also be involved in the progression of CKD [12]. The purpose of this review is to provide an update on the perturbations of GH-IGF-I axis in CKD and their role in the pathobiology of growth retardation, wasting, and progression.
The GH-IGF-I axis in modulating growth and organ development
The outline of the GH-IGF-I axis was first promulgated 50 years ago, when Salmon and Daughaday performed an elegant series of rodent studies that demonstrated the existence of a circulating factor that was responsible for the effects of GH on sulfation activity, a surrogate measure of longitudinal bone growth [13]. This putative factor was termed somatomedin, since it was postulated to mediate the somatotropic effects of GH, with the implicit suggestion that GH did not itself exert direct effects on somatic growth. Contemporary studies had independently identified a growth-promoting factor that displayed insulin-like activity that was not affected by anti-insulin antibodies, termed “non-suppressible insulin-like activity”, or NSILA. Subsequent characterization of protein and cDNA structures revealed that somatomedin C (as somatomedin was subsequently designated to distinguish it from other putative activities termed somatomedins A and B) and NSILA were identical to each other and to the protein designated IGF-I [14]. A distinct growth factor, termed multiplication-stimulating activity or MSA, was first characterized from buffalo rat liver (BRL)-3A cells, and was subsequently shown to be identical to a related protein, insulin-like growth factor-II (IGF-II), defined by protein and cDNA sequences.
GH action is mediated through its binding to the GH receptor (GHR), a transmembrane protein that, upon ligand-induced dimerization, recruits intracellular JAK tyrosine kinases, specifically JAK2, which phosphorylate the GH receptor on tyrosine residues [15]. The phosphorylated GH receptor serves as a docking site for members of the signal transducer and activator of transcription (STAT) family, in particular STATs 1, 3, and 5, which are then tyrosine phosphorylated by the GHR-associated JAK kinase. Upon phosphorylation, the STATs dimerize and translocate to the nucleus, where they regulate the expression of target genes responsible for GH action, including the IGF-I gene.
IGF-I and IGF-II function principally through their activation of the IGF-I receptor (IGF-IR), a transmembrane tyrosine kinase structurally related to the insulin receptor [16]. The IGF-IR is widely expressed, and through its coupling to signaling cascades such as the PI3K and MAPK (ERK) pathways, controls cellular proliferation, survival, and differentiation. IGF-II also exhibits affinity for a specific splice variant of the insulin receptor, through which it can also exert growth effects. Additionally, IGF-II, but not IGF-I, binds with high affinity to the IGF-II receptor, which is identical to the cation-independent mannose-6-phosphate receptor (M6PR). This IGF-II/M6PR does not have signal transduction capability, and is thought to function primarily as a clearance receptor in IGF action by controlling the extracellular levels and bioavailability of IGF-II. Finally, IGF-I and IGF-II interact with a series of IGF-binding proteins (IGFBPs) in the circulation and the extracellular fluid, which increase IGF half-life and can both positively and negatively modulate interaction with the IGF-IR and, therefore, biological activity.
The “somatomedin hypothesis” proposed that GH released from the pituitary under the influence of hypothalamic hormones such as GH-releasing hormone (GHRH) acted in an endocrine fashion on hepatic GH receptors to trigger the synthesis and release of IGF-I from the liver, which itself then acted in an endocrine fashion to stimulate bone growth and other effects in other tissues (Fig. 1). Subsequent studies demonstrated that extrahepatic tissues also produce IGF-I, although, in rodents at least, the liver is the predominant source of circulating IGF-I. It has also been shown that GH has IGF-I-independent effects that influence growth. Thus, the current version of the somatomedin hypothesis includes both direct and indirect effects of GH on somatic growth, with the latter being mediated through IGF-I produced by the liver as well as other tissues [17].
Fig. 1.
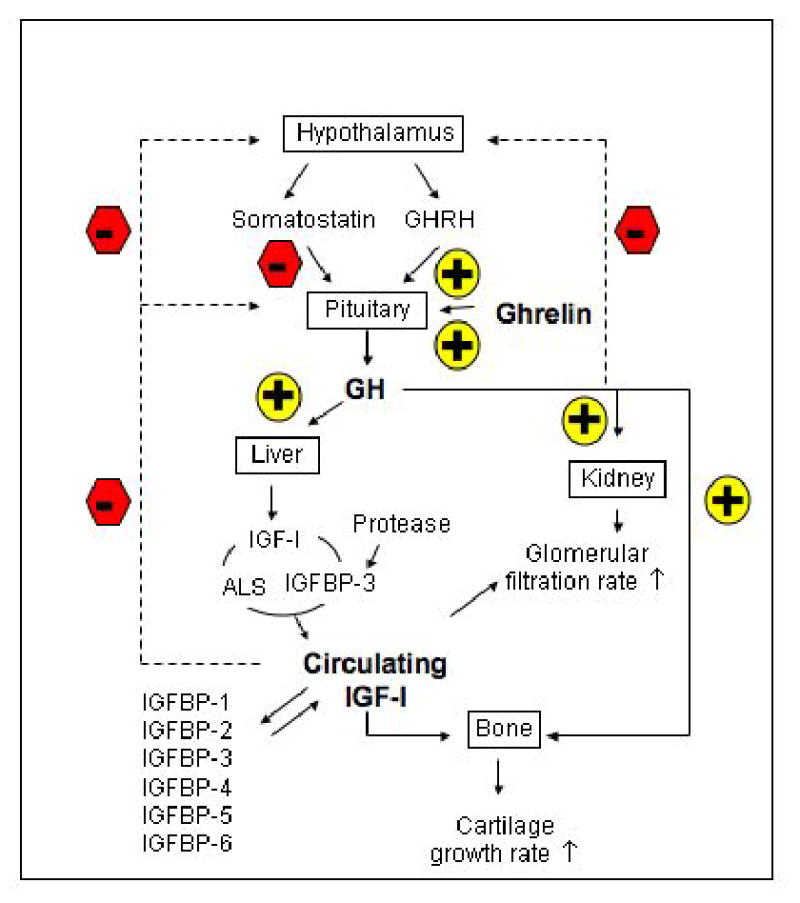
The normal growth hormone (GH) and insulin-like growth factor-I (IGF-I) axis in modulating growth and body composition. The synthesis and release of GH from pituitary are modulated by the hypothalamic hormones such as GH-releasing hormone (GHRH) and somatostatin, which in turn are modulated by feedback (dashed lines) from blood GH and IGF-I levels. Ghrelin also stimulates the release of GH. Circulating GH acts on many organs to stimulate the production of IGF-I. Liver is the major source of blood IGF-I. Most of the circulating IGF-I is bound to IGF-binding protein-3 (IGFBP-3) in a ternary complex with acid-labile subunit (ALS). A smaller fraction of the total IGF-I is bound to the five other IGFBPs. A small fraction of the total IGF-I in blood is in a bioactive-free fraction. In the kidney, IGF-I increases renal plasma flow and glomerular filtration rate. GH also affects many organs such as cartilage.
As would be predicted, defects in GH and IGF action in humans and experimental animals have effects on both overall growth and the development of specific tissues. GH-deficient and GHR mutant humans and rodents exhibit decreased growth, and GH excess in GH transgenic mice or acromegalic humans causes somatic overgrowth [18-23]. IGF-I excess in humans has only been seen to date in acromegalics secondary to elevated GH levels, while the hemihypertrophy seen in Beckwith-Wiedemann Syndrome is thought to be, in many cases, caused by increased expression of IGF-II. IGF-I, IGF-II, and IGF-IR-deficient knockout mice are growth-retarded at birth, although this is thought to be a deficit in GH-independent, but still IGF-mediated, growth [24,25]. Rare IGF-I-deficient and IGF-IR mutant humans also exhibit growth retardation [26].
Effects of GH and IGFs on specific tissues have been seen in analyses of transgenic and knockout mouse models. While GH-over-expressing mice exhibit increased kidney size and hypertrophy [27], IGF-I over-expression results in hyperplasia [28] and increased glomerular size [27,29] indicative of distinct effects of GH and IGF-I on kidney growth or development. IGF-II over-expression also results in an overall increase in kidney size [30,31]. IGF-I-deficient animals display particular muscle hypoplasia [24], and IGF-I excess results in skeletal muscle hypertrophy [32]. Interestingly, IGF-I excess increases fat mass [29], while IGF-II excess decreases fat mass [33,34].
GH and IGF-I and kidney function
The GH/IFGF-I system is present in the kidney and is important to kidney structure and function (Fig. 1). GH receptors, IGF-I and IGF-II, as well as IGF-I and IGF-II receptors and IGFBPs are normally expressed in the adult (rat) kidney, suggesting a role for GH and IGF in regulating kidney function. GH may increase renal hemodynamics and filtration rate, and glomerular filtration rate (GFR) and renal plasma flow rates are elevated in patients with acromegaly. GH increases GFR with a delay of many hours up to a day, consistent with induction of IGF-I synthesis. IGF-I increases GFR acutely when given pharmacologically. Endogenous IGF-I may contribute to the physiologic regulation of GFR [12].
GH undergoes glomerular ultrafiltration and tubular absorption and lysosomal degradation. Under normal conditions, small amounts of GH are excreted in the urine. Due to their low molecular mass (∼7.6 kDa), the glomerular ultrafiltration of IGF-I and IGF-II should occur at great rates. However, since <95% of the circulating IGFs are sequestered in complexes with IGFBPs, principally a 150-kDa ternary complex of IGF-I or II, IGFBP-3, and the acid-labile subunit (ALS) (∼150 and ∼45 kDa), ultrafiltration of these protein complexes occurs at very low rates [12].
GH and IGF-I circulating levels in CKD
CKD is associated with several derangements in the GH/IGF-I axis, as illustrated in Fig. 2. Circulating levels of GH are a balance between the production rate and metabolic clearance. GH synthesis may be reduced, normal, or even increased in CKD [35]. GH secretion is normal or low in late pre-pubertal and pubertal children with CRF [36]; whereas GH secretion rates are increased with increased GH burst frequency in pre-pubertal children and in adults with end-stage renal disease (ESRD) [36-38]. The release of GH in response to GHRH is increased in adults [39], whereas, in children, the mid-to-late adolescent increase in GH production fails to occur in CKD [36]. This decreased GH production is a consequence of attenuated GRH expression and increased somatostatin release [40,41]. Acidosis may also suppress GH secretion. The metabolic clearance rate of GH is obviously decreased in advanced CKD [42]. Overall, plasma GH levels are normal or increased in patients with CKD.
Fig. 2.
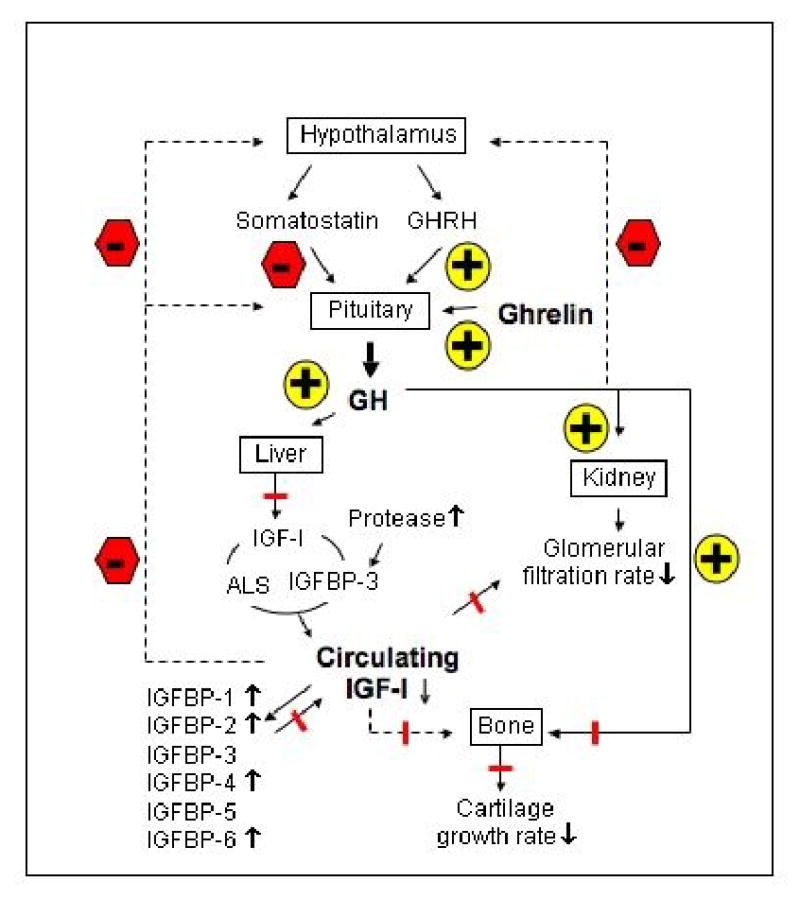
Deranged GH and IGF-I axis in chronic kidney disease (CKD). The GH/IGF-I axis in CKD is changed markedly compared with the normal axis, as illustrated in Fig.1. In CKD, the circulating levels of GH are not reduced while circulating levels IGF-I are decreased. Furthermore, there is reduced effectiveness of endogenous GH and IGF-I, which probably plays a major role in reducing linear bone growth. The reduced effectiveness of endogenous IGF-I is likely due to decreased levels of free, bioactive IGF-I as levels of circulating inhibitory IGFBP1, 2, 4 and 6 are increased. In addition, less IGF-I is circulating in the complex with ALS and IGF-BP3 as the result of increased proteolysis of IGF-BP3. In sum, these lead to decreased IGF-I receptor activation and a decreased feedback to the hypothalamus and pituitary. Low free IGF-I and high IGFBP levels probably contribute to a reduced renal function and lead to a reduced stature. The direct effects of GH on bone, which are not fully understood, also are blunted. Blunt-ended line represents inhibition.
In children with advanced CKD, serum IGF-I levels are normal or low [3], while, in adult patients, serum IGF-I levels are reduced only in cachectic subjects [5]. The rate of IGF-I production is reduced in uremic children [43], but appears to be normal in adults with ESRD [44,45]. Basal IGF-I mRNA expression is reduced in some but not all tissues. Reduced IGF-I mRNA expression has been described in liver, muscle, and growth plates [46,47], but not in heart and kidney. This reduction in tissue IGF-I mRNA appears to be nutritionally mediated [46].
GH-IGF-I insensitivity and growth failure in CKD
Growth retardation is a significant complication in children with CKD and occurs even though serum GH levels are normal or even elevated, reflecting a state of acquired GH resistance. An important recently described cause of GH resistance in uremia is a defect in the GHR-JAK2-STAT signal transduction pathway [48,49]. In CRF rats, resistance to GH was evident from the diminished response to treatment with supramaximal doses of recombinant GH with respect to body growth, and both hepatic and skeletal muscle IGF-I gene expression, compared with control animals ingesting the same quantity of food [48,49]. This attenuated response could not be attributed to a defect at the level of the GHR, as GHR binding, GHR protein levels, and the distribution of the receptor between the cytoplasmic and plasma membrane compartments were unaltered in liver, as were the GHR protein levels in skeletal and cardiac muscle. Nevertheless, despite normal GHR levels, there were striking abnormalities in GH signaling. Phosphorylation of JAK2 and of the downstream proteins phosphorylated by this kinase, namely the GHR, STAT5, STAT3, and STAT1, was impaired. In liver, STAT5 phosphorylation was depressed by 50%–75%. In all three tissues examined, nuclear phosphorylated STAT levels were reduced, indicating that nuclear translocation was impaired. Since activation of STAT5b is required for normal growth and IGF-I gene expression [50,51], this defect in JAK2-STAT5 phosphorylation contributes to the GH resistance and growth retardation in CKD.
Insensitivity to IGF-I is an important cause of GH resistance, and this appears to arise because of the accumulation of circulating IGFBPs that are normally cleared through the kidney. Serum IGFBP-1, -2, -4, and -6 levels are elevated, as are immunoreactive IGFBP-3 levels, although the latter is largely the result of the accumulation of immunoreactive fragments with reduced IGF-I affinity; intact IGFBP-3 levels are not elevated [52-55]. The bioavailability of IGF-I in CKD is compromised because of increased levels of IGFBP-1, -2, -4, and -6 [48-50]. Furthermore, as a result of an increased proteolysis of IGFBP-3, less IGF-I circulates as the 150-kDa ternary complex. Conversely, IGFBP-3 (29-kD) fragments increase, and these have less affinity for IGF-I. Consequently, effective delivery of IGF-I to its sites of action is reduced. These binding proteins form high-affinity complexes with IGF-I and IGF-II and thus reduce bioavailability. It is also conceivable that tissue IGF-I resistance may arise because of altered local IGFBP production and accumulation. Furthermore, there is evidence of other circulating inhibitors of growth in uremia. Sera from uremic rats were separated into two different fractions, high (>10 kDa, containing IGFBPs) and low (<10 kDa, containing free IGF) molecular weight fractions. Both fractions decreased the growth of control chondrocytes up to 40% compared with sera from control rats with normal renal function. Thus, unidentified small molecular weight uremic serum factors, other than IGFBPs, may also contribute to growth retardation [56].
Furthermore, there may be end-organ resistance to GH and IGF-I in CKD. Growth responses of chondrocytes from uremic rats were reduced compared with control chondrocytes when grown in normal rat serum. Growth responses of uremic chondrocytes were reduced to GH and IGF-I specifically, since growth responses to fibroblast growth factor and transforming growth factor were normal [57]. A study of uremic rats showed a reduction in GHR density in tibial growth plate [58] and low levels of mRNA for hepatic GH receptor [59]. In addition, there is evidence from animal studies that IGF-I resistance also arises from tissue insensitivity caused by a post-receptor defect in IGF-I-mediated signal transduction [60], although the site of the abnormality is controversial [61]. Ding et al. [60] studied IGF-IR isolated from skeletal muscle of rats with CRF and reported an increase in IGF-IR number and a decrease in IGF-I-activated receptor autophosphorylation and tyrosine kinase activity that could not be attributed to IGFBP activity. In contrast, Tsao et al. [61], also studying IGF-I-activated signaling in skeletal muscle of uremic rats, found that IGF-IR levels, receptor autophosphorylation, and tyrosine kinase activity were unaltered and suggested the possibility of a more-distal defect in signal transduction. The reason for the disparity between these studies of IGF-IR signaling is not readily apparent and awaits further study.
Ghrelin in CKD
Ghrelin, first described by Kojima et al. [62], is a peptide of 28 amino acids that stimulates GH release from the pituitary. Ghrelin, a natural ligand of the GH secretagogue receptor 1a (GHS-R1a), is secreted into the bloodstream primarily from the stomach and degraded by kidney. Apart from the stomach, lower amounts of ghrelin have been detected in many other tissues, including the hypothalamus, pituitary, lung, bowel, pancreas, kidney, testis, immune cells, and placenta [63]. Ghrelin stimulates GH-releasing activity in both animals and humans [62,64]. It is more potent than GRH, stimulating GH release in a dose-dependent manner.
The stimulatory effects of ghrelin on food intake and meal appreciation suggest that ghrelin could be an effective treatment for anorexic CKD patients [10]. The potential use of ghrelin for the treatment of anorexia in CRF patients was demonstrated in a recent report. Wynne et al. have tested the effects of subcutaneous ghrelin administration on CRF patients with mild to moderate malnutrition. Single-dose (3.6 nmol/kg) subcutaneous ghrelin administration significantly increased the mean absolute energy intake in CRF patients as compared with those patients on saline placebo. When expressed as proportional energy increase for each individual patient, ghrelin administration resulted in immediate doubling of energy intake. The immediate increase in energy intake in those patients was followed by a trend toward increased energy intake over the following 24 hours. In addition, there was no subsequent compensatory reduction in energy intake over the following 3 days, which would negate any potential therapeutic benefit of subcutaneous ghrelin administration [65].
The therapeutic potential of ghrelin in CKD remains to be exploited. Other studies have demonstrated that the modest short-term weight gain produced by ghrelin administration is primarily due to body fat gain, with very little beneficial effects on lean body tissues. Central or peripheral administration of ghrelin significantly increased food intake and body weight gain in rodents; however, the increase in body weight is caused by an augmentation in fat mass without changes in longitudinal skeletal growth and with a decrease in lean mass. Theander-Carrillo et al. studied the effects of central ghrelin administration on insulin sensitivity and body composition in experimental CKD. During the 6-day experimental period, daily central ghrelin infusion (3 nmol per day) increased food intake in ad libitum-fed rats compared with saline-infused ad libitum-fed controls. Ghrelin treatment increased food efficiency (calculated as the ratio of body weight gain to cumulative food intake measured). Chronic central ghrelin infusion did not alter total energy expenditure or spontaneous physical activity but resulted in a significant increase in the respiratory quotients, indicative of elevated adiposity of those rodents. Body weight gain in ghrelin-treated rats was related to the gain of fat mass, as determined by NMR imaging, without any change in lean body mass [66].
Myostatin and IGF-I balance in muscle mass regulation in CKD
Myostatin (also known as growth/differentiation factor-8) is a member of the transforming growth factor-β family that plays an essential role in the regulation of skeletal muscle mass. Myostatin is expressed initially in the myotomal compartment of developing somites and continues to be expressed in muscle throughout development and in adult animals. Mice with a targeted disruption of the myostatin gene display a marked increase in muscle mass, up to three times normal size, as a result of a combination of muscle fiber hyperplasia and hypertrophy [67]. IGF-I is essential for embryonic and postnatal development of skeletal muscle and other tissues. Mice engineered to lack IGF-I or the IGF-IR were significantly smaller than their control littermates and had reduced muscle tissue, whereas transgenic mice with targeted overexpression of IGF-I to muscle showed increased muscle mass [68]. Mak and Rotwein proposed that the balance between the local expression of myostatin and IGF-I represents a yin-and-yang system regulating muscle mass in CKD [11]. Sun and colleagues showed that uremia and exercise have opposing effects in affecting this balance of myostatin and IGF-I in the pathogenesis of uremic wasting [69].
Myostatin appears to induce muscle wasting by activating the ubiquitin proteolytic system through an NF-κB-independent pathway (Fig. 3). Myostatin mRNA is increased and IGF-I mRNA is decreased in skeletal muscles of uremic mice. Cheung et al. recently showed that AgRP administration almost completely normalized these uremic molecular pertubations and they were accompanied by the improvement in weight gain as well as lean bldy mass as measured by DEXA. The myostatin and IGF-I gene expression changes were similar in white adipose tissue in uremic mice in which there was a loss of fat mass after induction of CKD. Following improvement in adipose tissue mass induced by AgRP, there was normalization of IGF-I but not myostatin gene expression [70]. This was not surprising, since IGF-I signaling is associated with adipogenesis [71], whereas there is no current information on the role of myostatin in adipose tissue.
Fig. 3.
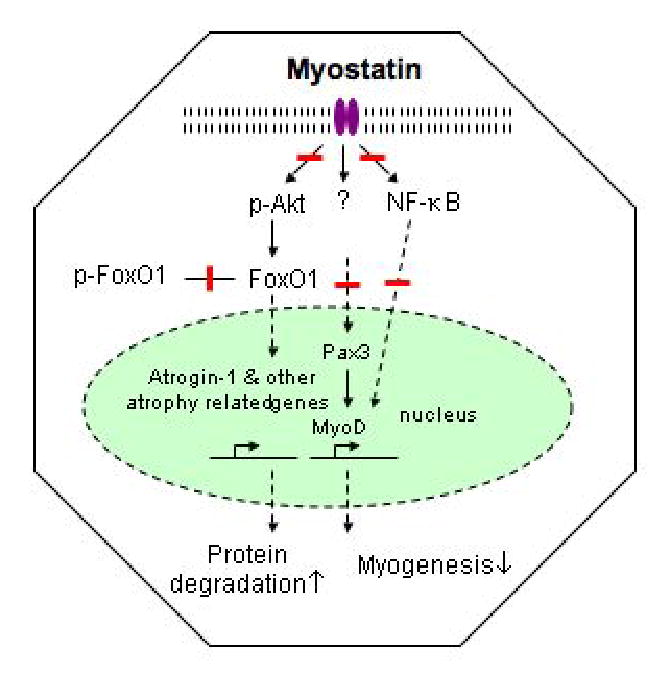
Molecular mechanism of myostatin in muscle mass regulation in CKD. Myostatin appears to induce muscle wasting independent of NF-κB pathway. Increased levels of myostatin blocks myogenesis by downregulating pax3 and myoD expression. In addition, myostatin upregulates proteolysis by phosphorylating FOXO1 through the inhibition of the PI3K/AKT signaling pathway. Blunt-ended line represents inhibition.
Suppressor of cytokine signaling (SOCS) genes in CKD
A growing body of evidence suggests that SOCS proteins act in the GH and IGF-I signaling pathways. Mice lacking SOCS-2 exhibit accelerated postnatal growth resulting in adult mice that are 1.3 to 1.5 times the size of normal mice. This growth enhancement in the SOCS-2 null mice is dependent on STAT-5b [72]. Schaefer et al. showed that CKD causes a post-receptor defect in GH signal transduction characterized by impaired phosphorylation and nuclear translocation of GH-activated STAT proteins, which is possibly mediated, at least in part, by over-expression of SOCS proteins (Fig. 4). Specifically, these investigators showed that impaired phosphorylation of STAT5b is accompanied by a 2-fold increase in SOCS-2 in uremic animals [48]. In skeletal muscle and liver, GH produced an exaggerated increase in SOCS-2 mRNA levels, while, in liver, the basal SOCS3 mRNA levels were elevated. If this increase in gene expression is accompanied by an increase in protein expression, this would provide a potential explanation for the depressed signal transduction. An acquired defect in GHR-JAK-2-STAT signaling with upregulation of SOCS expression has also been described in inflammatory conditions. This may be relevant to uremic GH resistance, as patients with ESRD and malnutrition often have underlying subclinical chronic inflammation [73]. Sun et al. studied skeletal muscle GH resistance in CKD and showed that there was a modest but significant increase in C-reactive protein levels [49]. In the same study, skeletal muscle protein tyrosine phosphatase activity was increased significantly. If this reflects the activity of phosphatases involved in the dephosphorylation of proteins in the JAK2-STAT pathway, then this could be another cause of the attenuated transduction of the GH signal along this pathway.
Fig. 4.
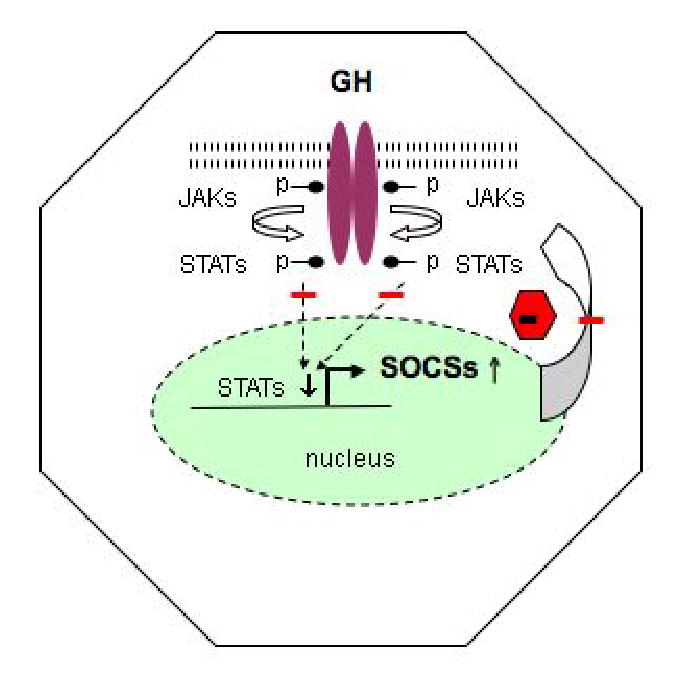
Janus kinase (JAK), signal transducers and activators of the transcription (STAT) and suppressor of cytokine signaling (SOCS) pathways in CKD. Binding of GH to the GH receptor activates JAK2, which in turn activates downstream signaling pathway, including STAT1, 3, 5a and 5b. The phosphorylated STATs dimerize, translocate into the nucleus and bind to specific DNA sequences and induce target genes including SOCS. In turn, these SOCS proteins inhibit JAK/STAT signal transduction, forming a negative feedback loop. The SOCS proteins bind to the phosphorylated receptors or JAKs and inhibit signal transduction by suppressing JAK activity or by competing with the STATs for receptor docking sites or by targeting the interacting signaling proteins for proteasomal degradation. Growth retardation and muscle wasting in CKD may arise through several mechanisms such as decreased phosphorylation of the GHR, JAK2, and STAT5 in liver and muscle. Nuclear translocation of phosphorylated STAT5 is reduced. As STAT5b is essential for the transcription of IGF-I and IGF-I is an important stimulator of body growth, this signaling defect may presumably contribute to the impaired GH-induced body growth retardation and muscle wasting in CKD. One mechanism that may contribute to the impaired JAK2-STAT phosphorylation involves the SOCS molecules. In CKD, levels of hepatic SOCS2 and SOCS3 and in muscle SOCS2 mRNA are increased. Blunt-ended line represents inhibition.
Cheung et al. recently confirmed and extended these results in the literature. SOCS-2 gene expression was increased significantly in the skeletal muscles in uremic animals fed ad libitum and intracranial AgRP administration in pair-fed uremic animals resulted in almost normalization of SOCS-2 expression as well as IGF-I expression in skeletal muscles [70]. The mechanism accounting for the increase in SOCS gene expression levels in uremia, where GH-activated JAK2-STAT signaling is impaired, is unclear. One possible mechanism could involve the action of inflammatory cytokines that stimulate signal transduction via other members of the Janus kinase family such as JAK1, JAK3, and Tyk2, which may be unaffected in renal failure. Another possible mechanism for the increase in SOCS expression might involve GH signaling through a non-STAT-mediated GH-activated pathway. Finally, it should be borne in mind that uremia affects several biological processes, including gene transcription and stability and cross-talk between the action of several hormones and cytokines [73].
GH and IGF-I in progression of CKD
The GH-IGF-I system has been implicated in CKD progression [74]. In aging rats, there is a correlation between the severity of glomerulosclerosis and plasma GH levels [75]. Moreover, administration of GH to rats with puromycin-aminoglycoside-induced CKD accelerates the development of glomerulosclerosis [76]. Furthermore, GH administration accelerates glomerulosclerosis and CKD progression in partially nephrectomized rats [52]. Mice transgenic for GH develop mesangial proliferation and progressive glomerular sclerosis [77]. On the other hand, GH-deficient rats are protected from glomerular sclerosis after partial nephrectomy [78].
There is evidence that IGF-I may contribute to the development of glomerular sclerosis and interstitial fiborsis. In cultured rat glomerular mesangial cells, IGF-I stimulates type I and type IV collagen expression [79]. IGF-I infustion increases matrix proteins such as fibronectin, laminin, heparan sulfate proteoglycan, and type II and type IV collagen in rat kidneys [80]. In rats with subtotal nephrectomy, IGF-I administration augments glomerular procollagen IV mRNA [81]. Exogenous IGF-I to rats with reduced renal mass causes renal hypertrophy, similar to the extent induced by a high-protein diet. However, the degree of glomerular sclerosis and the increase in glomerular type I and type IV collagen is less in rats with IGF-I infusion compared with rats fed a high-protein diet [82]. IGF-I administration to female rats resulted in increased renal growth and hyperplasia, but there was no evidence of premature glomerular sclerosis [83]. In rodents, and probably even more so in humans, the contribution of IGF-I to the development of glomerular sclerosis and progression of CKD, if any, may be rather modest.
Acknowledgments
Supported by grant K24 DK59574 from the National Institutes of Health to RHM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mak RH. Chronic kidney disease in children: state of the art. Pediatr Nephrol. 2006 doi: 10.1007/s00467-006-0360-7. EPUB Nov 4. [DOI] [PubMed] [Google Scholar]
- 2.Kaskel F. Chronic renal disease: A growing problem. Kidney Int. 2003;64:1141–1151. doi: 10.1046/j.1523-1755.2003.00194.x. [DOI] [PubMed] [Google Scholar]
- 3.Tonshoff B, Blum WF, Mehls O. Derangements of the somatotropic hormone axis in chronic renal failure. Kidney Int. 1997;S58:S106–S113. [PubMed] [Google Scholar]
- 4.Wuhl E, Schaefer F. Effects of growth hormone in patients with chronic renal failure: experience in children and adults. Horm Res. 2002;3:35–38. doi: 10.1159/000066480. [DOI] [PubMed] [Google Scholar]
- 5.Mak RH, Pak Y. End-organ resistance to growth hormone and IGF-I in epiphyseal chondrocytes of uremic rats. Kidney Int. 1996;50:400–406. doi: 10.1038/ki.1996.329. [DOI] [PubMed] [Google Scholar]
- 6.Ding H, Gao XL, Hirschberg R, Vadgama JV, Kopple JD. Impaired actions of insulin-like growth factor I on protein synthesis and degradation in skeletal muscle of rats with chronic renal failure, Evidence for a postreceptor defect. J Clin Invest. 1996;97:1064–1075. doi: 10.1172/JCI118499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mak RH, Cheung W, Cone RD, Marks DL. Leptin and inflammation-associated cachexia in chronic kidney disease. Kidney Int. 2006;69:794–797. doi: 10.1038/sj.ki.5000182. [DOI] [PubMed] [Google Scholar]
- 8.Mak RH, Cheung W, Cone RD, Marks DL. Orexigenic and anorexigenic mechanisms in the control of nutrition in chronic kidney disease. Pediatr Nephrol. 2005;20:427–431. doi: 10.1007/s00467-004-1789-1. [DOI] [PubMed] [Google Scholar]
- 9.Mak RH, Cheung W, Cone RD, Marks DL. Energy homeostasis and cachexia in chronic kidney disease. Pediatr Nephrol. 2006;21:1807–1814. doi: 10.1007/s00467-006-0194-3. [DOI] [PubMed] [Google Scholar]
- 10.Mak RH, Cheung W, Purnell J. Ghrelin in chronic kidney disease: too much or too little. Perit Dial Int. 2007;27:51–55. [PubMed] [Google Scholar]
- 11.Mak RH, Rotwein P. Myostatin and insulin-like growth factors in uremic sarcopenia: the yin and yang in muscle mass regulation. Kidney Int. 2006;70:410–412. doi: 10.1038/sj.ki.5001622. [DOI] [PubMed] [Google Scholar]
- 12.Feld S, Hirschberg R. Growth hormone, the insulin-like growth factor system and the kidney. Endocrinol Rev. 1996;17:423–477. doi: 10.1210/edrv-17-5-423. [DOI] [PubMed] [Google Scholar]
- 13.Salmon WD, Jr, Daughaday WH. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J Lab Clin Med. 1957;49:825–836. [PubMed] [Google Scholar]
- 14.Klapper DG, Svoboda ME, van Wyk JJ. Sequence analysis of somatomedian-C: confirmation of identity with insulin-like growth factor-I. Endocrinology. 1983;112:2215–2217. doi: 10.1210/endo-112-6-2215. [DOI] [PubMed] [Google Scholar]
- 15.Pilecka I, Whatmore A, van Huijsduijnen RH, Destenaves B, Clayton P. Growth hormone signaling: sprouting links between pathways, human genetics and therapeutic options. Trends Endocrinol Metab. 2007;18:12–18. doi: 10.1016/j.tem.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 16.LeRoith D, Werner H, Beitner-Johnson D, Roberts CT., Jr Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16:143–163. doi: 10.1210/edrv-16-2-143. [DOI] [PubMed] [Google Scholar]
- 17.Liu JL, LeRoith D. Insulin-like growth factor I is essential for postnatal growth in response to growth hormone. Endocrinology. 1999;140:5178–7184. doi: 10.1210/endo.140.11.7151. [DOI] [PubMed] [Google Scholar]
- 18.Snell GD. Dward, A new mendelian recessive character of the house mouse. Proc Natl Acad Sci USA. 1929;15:733–734. doi: 10.1073/pnas.15.9.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sornson MW, Wu W, Dasen JS, Flynn SE, Norman DJ, O'Connell SM, Gukovsky I, Carrière C, Ryan AK, Miller AP, Zuo L, Gleiberman AS, Andersen B, Beamer WG, Rosenfeld MG. Pituitary lineage determination by the Prophet of Pit-1 homeodomain factor defective in Ames dwarfism. Nature. 1996;385:327–333. doi: 10.1038/384327a0. [DOI] [PubMed] [Google Scholar]
- 20.Liu JL, Coschigano KT, Robertson K, Lipsett M, Guo Y, Kopchick JJ, Kumar U, Liu YL. Disruption of growth hormone receptor gene causes diminished pancreatic islet size and increased insulin sensitivity in mice. Am J Physiol Endocrinol Metab. 2004;287:E405–E413. doi: 10.1152/ajpendo.00423.2003. [DOI] [PubMed] [Google Scholar]
- 21.Coschigano KT, Holland AN, Riders ME, List EO, Flyvbjerg A, Kopchick JJ. Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased life span. Endocrinology. 2003;144:3799–3810. doi: 10.1210/en.2003-0374. [DOI] [PubMed] [Google Scholar]
- 22.Palmiter RD, Brinster RL, Hammer RE, Trumbauer ME, Rosenfeld MG, Bimberg NC, Evans RM. Dramatic growth of mice that develop from eggs microinjected with metallothionein-growth hormone fusion genes. Nature. 1982;300:611–615. doi: 10.1038/300611a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmiter RD, Norstedt G, Gelinas RE, Hammer RE, Brinster RL. Metallothionein-human GH fusion genes stimulate growth of mice. Science. 1983;222:809–814. doi: 10.1126/science.6356363. [DOI] [PubMed] [Google Scholar]
- 24.Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 25.DeChiara TM, Efstratiadis A, Robertson EJ. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature. 1990;345:78–90. doi: 10.1038/345078a0. [DOI] [PubMed] [Google Scholar]
- 26.Woods KA, Camacho-Hubner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med. 1996;335:1363–1367. doi: 10.1056/NEJM199610313351805. [DOI] [PubMed] [Google Scholar]
- 27.Doi T, Striker LJ, Gibson CC, Agodoa LYC, Brinster RL, Striker GE. Glomerular lesions in mice transgenic for growth hormone and insulin-like growth factor-I. I. Relationship between increased glomerular size and mesangial sclerosis. Am J Pathol. 1990;137:541–552. [PMC free article] [PubMed] [Google Scholar]
- 28.Mathews LS, Hammer RE, Behringer RR, D'Ecole AJ, Bell GI, Brinster RL, Palmiter RD. Growth enhancement of transgenic mice expressing human insulin-like growth factor I. Endocrinology. 1988;123:2827–2833. doi: 10.1210/endo-123-6-2827. [DOI] [PubMed] [Google Scholar]
- 29.Quaife CJ, Mathews LS, Pinkert CA, Hammer RE, Brinster RL, Palmiter RD. Histopathology associated with elevated levels of growth hormone and insulin-like growth factor I in transgenic mice. Endocrinology. 1989;124:40–48. doi: 10.1210/endo-124-1-40. [DOI] [PubMed] [Google Scholar]
- 30.Wolf E, Kramer R, Blum WF, Foll J, Brem G. Consequences of postnatally elevated insulin-like growth factor-II in transgenic mice: endocrine changes and effects on body and organ growth. Endocrinology. 1994;135:1877–1886. doi: 10.1210/endo.135.5.7525257. [DOI] [PubMed] [Google Scholar]
- 31.Blackburn A, Schmitt A, Schmidt P, Wanke R, Hermanns W, Brem G, Wolf E. Actions and interactions of growth hormone and insulin-like growth factor-II: body and organ growth of transgenic mice. Transgenic Res. 1997;6:213–222. doi: 10.1023/a:1018494108654. [DOI] [PubMed] [Google Scholar]
- 32.Coleman ME, DeMayo F, Yin KC, Lee HM, Geske R, Montgomery C, Schwartz RJ. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem. 1995;270:12109–12116. doi: 10.1074/jbc.270.20.12109. [DOI] [PubMed] [Google Scholar]
- 33.Rogler CE, Yang D, Rossetti L, Donohoe J, Alt E, Chang CJ, Rosenfeld R, Neely K, Hintz R. Altered body composition and increased frequency of diverse malignancies in insulin-like growth factor-II transgenic mice. J Biol Chem. 1994;269:13779–13784. [PubMed] [Google Scholar]
- 34.Ward A, Bates P, Fisher R, Richardson L, Graham CF. Disproportionate growth in mice with Igf-2 transgenes. Proc Natl Acad Sci U S A. 1994;91:10365–10369. doi: 10.1073/pnas.91.22.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feneberg R, Schaefer F, Veldhuis JD. Neuroendocrine adaptations in renal disease. Pediatr Nephrol. 2003;18:492–497. doi: 10.1007/s00467-003-1160-y. [DOI] [PubMed] [Google Scholar]
- 36.Schaefer F, Veldhuis JD, Stanhope R, Jones J, Scharer K. Alterations in growth hormone secretion and clearance in peripubertal boys with chronic renal failure and after renal transplantation. Cooperative Study Group of Pubertal development in Chronic Renal Failure. J Clin Endocrinol Metab. 1994;78:1298–1306. doi: 10.1210/jcem.78.6.8200929. [DOI] [PubMed] [Google Scholar]
- 37.Tonshoff B, Veldhuis JD, Heinrich U, Mehls O. Deconvolution analysis of spontaneous nocturnal growth hormone secretion in prepubertal children with preterminal chronic renal failure and with end-stage renal disease. Pediatr Res. 1995;37:86–93. doi: 10.1203/00006450-199501000-00017. [DOI] [PubMed] [Google Scholar]
- 38.Veldhuis JD, Iranmanesh A, Wilkowski MJ, Samojlik E. Neuroendocrine alterations in the somatotropic and lactotropic axes in uremic men. Eur J Endocrinol. 1994;131:489–498. doi: 10.1530/eje.0.1310489. [DOI] [PubMed] [Google Scholar]
- 39.Ramirez G, Bercu BB, Bittle PA, Ayers CW, Ganguly A. Response to growth hormone-releasing hormone in adult renal failure patients on hemodialysis. Metabolism. 1990;39:764–768. doi: 10.1016/0026-0495(90)90114-r. [DOI] [PubMed] [Google Scholar]
- 40.Garcia E, Santos F, Rodriguez J, Martinez V, Rey C, Veldhuis J, Krieg RJ. Impaired secretion of growth hormone in experimental uremia: relevance of caloric deficiency. Kidney Int. 1997;52:648–653. doi: 10.1038/ki.1997.378. [DOI] [PubMed] [Google Scholar]
- 41.Metzger DL, Kerrigan JR, Krieg RJ, Chan JC, Rogol AD. Alterations in the neuroendocrine control of growth hormone secretion in the uremic rat. Kidney Int. 1993;43:1042–1048. doi: 10.1038/ki.1993.146. [DOI] [PubMed] [Google Scholar]
- 42.Haffner D, Schaefer F, Girard J, Ritz E, Mehls O. Metabolic clearance of recombinant human growth hormone in health and chronic renal failure. J Clin Invest. 1994;93:1163–1171. doi: 10.1172/JCI117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blum WF. Insulin-like growth factors (IGFs) and IGF binding proteins in chronic renal failure: evidence for reduced secretion of IGFs. Acta Paediatr Scand. 1991;S379:24–31. doi: 10.1111/j.1651-2227.1991.tb12039.x. [DOI] [PubMed] [Google Scholar]
- 44.Fouque D, Peng SC, Kopple JD. Pharmacokinetics of recombinant human insulin-like growth factor-1 in dialysis patients. Kidney Int. 1995;47:869–875. doi: 10.1038/ki.1995.130. [DOI] [PubMed] [Google Scholar]
- 45.Rabkin R, Fervenza FC, Maidment H, Ike J, Hintz R, Liu F, Bloedow DC, Hoffman AR, Gesundheit N. Pharmacokinetics of insulin-like growth factor-1 in advanced chronic renal failure. Kidney Int. 1996;49:1134–1140. doi: 10.1038/ki.1996.164. [DOI] [PubMed] [Google Scholar]
- 46.Tonshoff B, Powell DR, Zhao D, Durham SK, Coleman ME, Domene HM, Blum WF, Baxter RC, Moore LC, Kaskel FJ. Decreased hepatic insulin-like growth factor (IGF)-I and increased IGF binding protein-1 and -2 gene expression in experimental uremia. Endocrinology. 1997;138:938–946. doi: 10.1210/endo.138.3.4977. [DOI] [PubMed] [Google Scholar]
- 47.Hanna JD, Santos F, Foreman JW, Chan JC, Han VK. Insulin-like growth factor-I gene expression in the tibial epiphyseal growth plate of growth hormone-treated uremic rats. Kidney Int. 1995;47:1374–1382. doi: 10.1038/ki.1995.193. [DOI] [PubMed] [Google Scholar]
- 48.Schaefer F, Chen Y, Tsao T, Nouri P, Rabkin R. Impaired JAK-STAT signal transduction contributes to growth hormone resistance in chronic uremia. J Clin Invest. 2001;108:467–475. doi: 10.1172/JCI11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun D, Zheng Z, Tummala P, Oh J, Schaefer F, Rabkin R. Chronic uremia attenuates growth hormone induced signal transduction in skeletal muscle. J Am Soc Nephrol. 2004;15:2630–2636. doi: 10.1097/01.ASN.0000139492.36400.6C. [DOI] [PubMed] [Google Scholar]
- 50.Woefle J, Billiard J, Rotwein P. Acute control of insulin-like growth factor-I gene transcription by growth hormone through STAT5b. J Biol Chem. 2003;278:22696–22702. doi: 10.1074/jbc.M301362200. [DOI] [PubMed] [Google Scholar]
- 51.Woefle J, Rotwein P. In vivo regulation of growth-hormone stimulated gene transcription by STAT5b. Am J Physiol Endocrinol Metab. 2003;286:E393–E401. doi: 10.1152/ajpendo.00389.2003. [DOI] [PubMed] [Google Scholar]
- 52.Tonshoff B, Blum WF, Wingen AM, Mehls O. Serum insulin-like growth factors and IGF binding proteins 1,2, and 3 in children with chronic renal failure: relationship to height and glomerular filtration rate. The European Study Group for Nutritional Treatment of Chronic Renal Failure in Childhood. J Clin Endocrinol Metab. 1995;80:2684–2691. doi: 10.1210/jcem.80.9.7545697. [DOI] [PubMed] [Google Scholar]
- 53.Ulinski T, Mohan S, Kiepe D, Blum WF, Wingen AM, Mehls O, Tönshoff B. Serum insulin-like growth factor binding protein (IGFBP)-4 and IGFBP-5 in children with chronic renal failure: relationship to growth and glomerular filtration rate. The European Study Group for Nutritional Treatment of Chronic Renal Failure in Childhood. German Study Group for Growth Hormone Treatment in Chronic Renal Failure. Pediatr Nephrol. 2000;14:589–97. doi: 10.1007/s004670000361. [DOI] [PubMed] [Google Scholar]
- 54.Powell DR, Liu F, Baker BK, Hintz RL, Durham SK, Brewer ED, Frane JW, Tonshoff B, Mehls O, Wingen AM, Watkins SL, Hogg RJ, Lee PD. Insulin-like growth factor-binding protein-6 levels are elevated in serum of children with chronic renal failure: a report of the Southwest Pediatric Nephrology Study Group. J Clin Endocrinol Metab. 1997;82:2978–2984. doi: 10.1210/jcem.82.9.4215. [DOI] [PubMed] [Google Scholar]
- 55.Powell DR, Liu F, Baker BK, Hintz RL, Kale A, Suwanichkul A, Durham SK. Effect of chronic renal failure and growth hormone therapy on the insulin-like growth factors and their binding proteins. Pediatr Nephrol. 2000;14:579–583. doi: 10.1007/s004670000349. [DOI] [PubMed] [Google Scholar]
- 56.Mak RH, Chang SL, Pak YK. Growth impairment of primary chondrocyte cells by serum of rats with chronic renal failure. Exp Mol Med. 2004;36:243–250. doi: 10.1038/emm.2004.33. [DOI] [PubMed] [Google Scholar]
- 57.Mak RH, Pak YK. End-organ resistance to growth hormone and IGF-I in epiphyseal chondrocytes of uremic rats. Kidney Int. 1996;50:400–406. doi: 10.1038/ki.1996.329. [DOI] [PubMed] [Google Scholar]
- 58.Tonshoff B, Eden S, Weiser E, Carlsson B, Robinson IC, Blum WF, Mehls O. Reduced hepatic growth hormone (GH) receptor gene expression and increased plasma GH binding protein in experimental uremia. Kidney Int. 1994;45:1085–1092. doi: 10.1038/ki.1994.145. [DOI] [PubMed] [Google Scholar]
- 59.Edmondson SR, Baker NL, Oh J, Kovacs G, Werther GA, Mehls O. Growth hormone receptor abundance in tibial growth plates of uremic rats: GH/IGF-I treatment. Kidney Int. 2000;58:62–70. doi: 10.1046/j.1523-1755.2000.00141.x. [DOI] [PubMed] [Google Scholar]
- 60.Ding H, Gao XL, Hirschberg R, Vadgama JV, Kopple JD. Impaired actions of insulin-like growth factor I on protein synthesis and degradation in skeletal muscle of rats with chronic renal failure. Evidence for a postreceptor defect. J Clin Invest. 1996;97:1064–1075. doi: 10.1172/JCI118499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsao T, Fervenza F, Friedlaender M, Chen Y, Rabkin R. Effect of prolonged uremia on insulin-like growth factor-I receptor autophosphorylation and tyrosine kinase activity in kidney and muscle. Exp Nephrol. 2002;10:285–292. doi: 10.1159/000063703. [DOI] [PubMed] [Google Scholar]
- 62.Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 63.Kojima M, Kangawa K. Ghrelin: structure and function. Physio Rev. 2005;85:495–522. doi: 10.1152/physrev.00012.2004. [DOI] [PubMed] [Google Scholar]
- 64.Arvat E, Di Vito L, Broglio F, Papotti M, Muccioli G, Dieguez C, Casanueva FF, Deghenghi R, Camanni F, Ghigo E. Preliminary evidence that ghrelin, the natural GH secretagogue (GHS)-receptor ligand, strongly stimulates GH secretion in humans. J Endocrinol Invest. 2000;23:493–495. doi: 10.1007/BF03343763. [DOI] [PubMed] [Google Scholar]
- 65.Wynne K, Giannitsopoulou K, Small CJ, Patterson M, Frost G, Ghatei MA, Brown EA, Bloom SR, Choi P. Subcutaneous ghrelin enhances acute food intake in malnourished patients who receive maintenance peritoneal dialysis: a randomized, placebo-controlled trial. J Am Soc Nephrol. 2005;16:2111–2118. doi: 10.1681/ASN.2005010039. [DOI] [PubMed] [Google Scholar]
- 66.Theander-Carrillo C, Wiedmer P, Cettour-Rose P, Nogueiras R, Perez-Tilve D, Pfluger P, Castaneda TR, Muzzin P, Schurmann A, Szanto I, Tschop MH, Rohner-Jeanrenaud F. Ghrelin action in the brain controls adipocyte metabolism. J Clin Invest. 2006;116:1983–1993. doi: 10.1172/JCI25811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee SJ. Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol. 2004;20:61–86. doi: 10.1146/annurev.cellbio.20.012103.135836. [DOI] [PubMed] [Google Scholar]
- 68.Kuninger DT, Rotwein PS. IGF action and skeletal muscle. In: LeRoith D, Zumkeller W, Baxter R, editors. Insulin-like growth factors, Landes Bioscience. Kluwer Academic/Plenum; New York: 2003. pp. 235–243. [Google Scholar]
- 69.Sun DF, Chen Y, Rabkin R. Work-induced changes in skeletal muscle IGF-I and myostatin gene expression in uremia. Kidney Int. 2006;70:453–459. doi: 10.1038/sj.ki.5001532. [DOI] [PubMed] [Google Scholar]
- 70.Cheung W, Mak RH. Melanocortin signaling modulation ameliorates cachexia in chronic kidney disease: peripheral mechanism. Kidney Int. submitted. [Google Scholar]
- 71.Fleenor D, Arumugam R, Freemark M. Growth hormone and prolactin receptors in adipogenesis: STAT activation, suppressors of cytokine signaling, and regulation of insulin-like growth factor I. Horm Res. 2006;66:101–110. doi: 10.1159/000093667. [DOI] [PubMed] [Google Scholar]
- 72.Greenhalgh CJ, Bertolino P, Asa SL, Metcalf D, Corbin JE, Adams TE, Davey HW, Nicola NA, Hilton DJ, Alexander WS. Growth enhancement in suppressor of cytokine 2 (SOCS-2)-deficient mice is dependent on signal transducer and activator of transcription 5b (STAT5b) Mol Endocrinol. 2002;16:1394–1406. doi: 10.1210/mend.16.6.0845. [DOI] [PubMed] [Google Scholar]
- 73.Rabkin R, Sun DF, Chen Y, Tan J, Schaefer F. Growth hormone resistance in uremia, a role for impaired JAK/STAT signaling. Pediatr Nephrol. 2005;20:313–318. doi: 10.1007/s00467-004-1713-8. [DOI] [PubMed] [Google Scholar]
- 74.Mak RH, Cheung WW. Transforming growth factors and insulin-like growth factors in chronic kidney disease. J Organ Dysfunction. 2007 in press. [Google Scholar]
- 75.Goya RG, Castelleto L, Sosa YE. Plasma levels of growth hormone correlate with the severity of pathologic changes in the renal structure of aging rats. Lab Invest. 1991;64:29–34. [PubMed] [Google Scholar]
- 76.Trachtman H, Futterweit S, Schwob N, Maesaka J, Vladerrama E. Recombinant growth hormone excerbates chronic puromycin aminoglycoside nephropathy in rats. Kidney Int. 1993;44:1281–1288. doi: 10.1038/ki.1993.380. [DOI] [PubMed] [Google Scholar]
- 77.Doi T, Striker LJ, Kimata K, Peten EP, Yamada Y, Striker GE. Glomerulosclerosis in mice transgenic for growth hormone: Increased mesangial extracellular matrix is correlated with kidney mRNA levels. J Exp Med. 1991;173:1287–1290. doi: 10.1084/jem.173.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Allen DB, Fogo A, El-Hayek R, Langhough R, Friedman AL. Effects of prolonged growth hormone administration in rats with chronic renal insufficiency. Pediatr Res. 1992;31:406–410. doi: 10.1203/00006450-199204000-00020. [DOI] [PubMed] [Google Scholar]
- 79.Yoshida H, Mitarai T, Kitamura M, Suzuki T, Ishikawa H, Fogo A, Sakai O. The effect of selective growth hormone defect in the progression of glomerulosclerosis. Am J Kidney Dis. 1994;23:302–312. doi: 10.1016/s0272-6386(12)80988-7. [DOI] [PubMed] [Google Scholar]
- 80.Feld SM, Hirschberg R, Artishevsky A, Nast C, Alder SG. Insulin-like growth factor-I induces mesangial proliferation and increases mRNA and secretion of collagen. Kidney Int. 1995;48:45–51. doi: 10.1038/ki.1995.265. [DOI] [PubMed] [Google Scholar]
- 81.Haylor J, Johnson T, El-Nahas M. Renal matrix protein mRNA elevated by insulin-like growth factor-I. J Am Soc Nephrol. 1995;6:896. abstract. [Google Scholar]
- 82.Hirschberg R, Nast C. Glomerular hypertrophy and segmental glomerular sclerosis are not linked to subtotally nephrectomized rats. J Am Soc Nephrol. 1992;3:739. abstract. [Google Scholar]
- 83.Mehls O, Irzynjec T, Ritz E, Eden S, Kovacs G, Klaus G, Floege J, Mall G. Effects of rhGH and rhIGF-I on renal growth and morphology. Kidney Int. 1993;44:1251–1258. doi: 10.1038/ki.1993.376. [DOI] [PubMed] [Google Scholar]