

Abstract
Endothelin-converting enzyme-2 (ECE-2a), a member of M13 family of zinc metallopeptidases, has previously been shown to process a number of neuropeptides including those derived from prodynorphin, proenkephalin, proSAAS and amyloid precursor protein. ECE-2, unlike ECE-1, exhibits restricted neuroendocrine distribution and acidic pH optimum; it's consistent with a role in the regulation of neuropeptide levels in vivo. Here we report the generation of a three-dimensional (3D) molecular model of ECE-2 using the crystal structure of neprilysin (EC 3.4.24.11) as a template. Based on the predictions made from the molecular model we mutated and tested two residues, Trp 148 and Tyr 563 in the catalytic site. The mutation of Tyr 563 was found to significantly affect the catalytic activity and inhibitor binding. The molecular model was used to virtually screen a small molecule library of 13000 compounds. Among the top-scoring compounds three were found to inhibit ECE-2 with high affinity and exhibited specificity for ECE-2 as compared to neprilysin. Thus the model provides a new useful tool to probe the active site of ECE-2 and design additional selective inhibitors of this enzyme.
Introduction
Neuroendocrine peptides are synthesized as large precursor proteins that undergo multiple posttranslational processing steps to generate bioactive peptides. Most of the peptide precursors are cleaved at specific sites that usually contain multiple basic amino acids that are considered to be the “classical” cleavage sites 1. This cleavage is performed by endoproteases of the subtilisin family of serine proteases, namely prohormone convertases (PCs) 2. In addition a number of neuropetides have been identified that are generated by processing at “non-classical” sites that do not contain basic amino acids. These peptides were identified among the bulk-purified peptides of neuroendocrine tissues and also detected in the brains from mice lacking specific processing enzymes such as CPE 3-6. In addition, an examination of precursor sequences of some endogenous peptides showed that non-classical cleavage is needed in order to generate the active forms of these peptides 7-9.
Members of the metalloprotease family have been implicated in the processing of neuroendocrine peptides at non-classical sites 10. Among them ECE-2 fits the criteria of a neuropeptide processing enzyme. ECE-2 is a member of the neprilysin family of Zn2+ metallopeptidases and shares most of the common features of this family. It is a type II integral membrane protein with a short cytoplasmic tail, transmembrane domain and a large C-terminal domain that contains the active site. ECE-2 shares 37% overall homology with neprilysin (NEP) and contains consensus sequences (such as HExxH and ExxxD) involved in Zn2+ coordination and catalysis, which are highly conserved among NEP family members. Moreover, the ectodomain of ECE-2 includes ten cysteine residues and multiple glycosylation sites that are also conserved among family members 9, 11. ECE-2 cleaves its substrates at the amino-terminal side of aromatic or aliphatic residues and is inhibited by the generic inhibitor of metalloproteases, phosphoramidon, with a nanomolar potency 12-14. ECE-2 converts big endothelin-1 to ET-1 by cleaving the Trp-Val site, a property shared by its close homolog, ECE-112, 13, 15. Both of these enzymes have also been shown to process β-amyloid peptides and modulate their levels in the mouse brain 16, 17. We have recently reported that ECE-2 is able to cleave a number of neuroendocrine peptide precursors in vitro. The analysis of the cleavage sites revealed that ECE-2 displays endopeptidase- or peptidyldicarboxypeptidase-like activity and prefers cleaving at sites containing an aromatic or aliphatic residue with a large branched side chain at the P1' site18. It is likely that ECE-2 is involved in the generation of neuropeptides by processing at these “non-classical” sites.
The physiological functions of ECE-2 or other members of the neprilysin family are not well defined. The structural similarities among NEP family members and the similarities in substrate specificity as well as the overlapping tissue distribution make it difficult to define a distinct role for ECE-2 in neuropeptide processing. A better understanding of the structural architecture of the active site of ECE-2 and its homologs is likely to lead to the delineation of differences in substrate specificity and help us uncover the physiological functions for each of these metalloproteases.
Until recently, the information about the organization of the active site and the residues involved in zinc binding and catalysis of NEP family of enzymes was obtained from multiple mutagenesis studies 19-26. Early studies examining the catalytic mechanisms of these enzymes used models based on the crystal structure of thermolysin (TLN), a bacterial homolog of NEP 27,28. The availability of a high resolution crystal structure of NEP complexed with phosphoramidon 11 has allowed the generation of more reliable 3D molecular models for related enzymes. In this study we describe the generation of the homology model of ECE-2. Studies to test the predictions based on the model revealed that Tyr 563, a non-conserved residue of the consensus motif 561NAYY564 plays a role in the catalytic activity and inhibitor binding. We also describe studies identifying ECE-2 selective inhibitors by virtually docking a library of 13000 small drug-like molecules on to the 3D model of ECE-2; this led to the selection of a subset of the compounds that were further screened biochemically. Here we report the identification of three compounds that inhibit ECE-2 enzyme activity with micromolar potency and display an order of magnitude higher affinity towards ECE-2 as compared to NEP.
Results
Modeling of ECE-2
A comparison of the amino acid sequences between neprilysin and ECE-2 reveals a high degree of homology (37%), which suggests that they share a common origin and could share a common fold. Based on sequence alignment (Figure 1), it is also clear that the residues around the Zn2+ and phosphoramidon binding sites are well conserved between ECE-2 and NEP; thus it is likely that the homology modeling of ECE-2 using neprilysin structure could be very useful in identifying critical residues involved in substrate/inhibitor binding. We built a 3D molecular model of ECE-2 using the coordinates from the reported crystal structure of NEP (PDB code 1DMT) 11 and checked the quality of the modeling using its Ramachandran plot (Figure S1). This revealed that 93% of the residues were within the conventional limits. The superimposition of the ECE-2 model with the crystal structure of NEP shows that the common fold of both enzymes is composed of two largely α-helical domains. These domains form a central cavity, which contains the active site of the enzyme (Figure 2A).
Figure 1.

Alignment of human NEP sequence with the sequence of human ECE-2. Alignment was done using ClustalW multiple sequence alignment program. Three regions that are highly conserved and represent consensus sequences are boxed and the residues participating in zinc binding and coordination as well as in catalysis are shown in bold. The numbers on the right indicate amino acid numbering and the asterisks indicate conserved residues.
Figure 2.
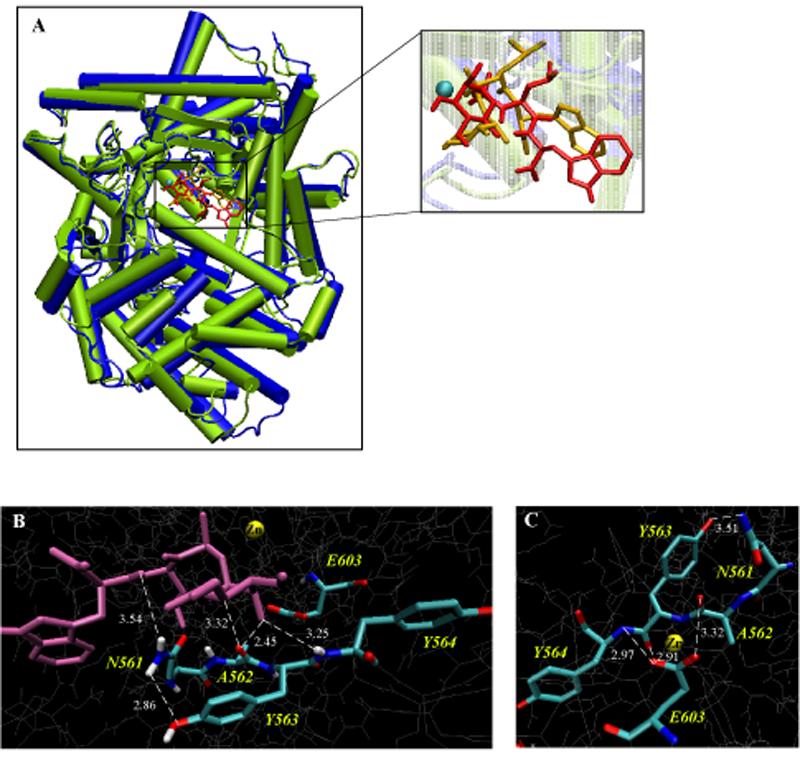
Comparison of the ECE-2 model with the NEP crystal structure. (A) Superimposition of diagrams of the X-ray structure of NEP (blue) and the model of ECE-2 (lime) reveals structural similarity of these two proteins. Cylinders represent α-helices. The inset depicts the position of phosphoramidon in the active site of NEP (yellow) and ECE-2 (red). The position of phosphoramidon in the ECE-2 model was calculated using Autodock (v.3/0/5). The Zn2+ ion is shown as a cyan sphere. (B) 3D representation of ECE-2 active site with docked phosphoramidon (purple) highlighting the binding of the inhibitor to the residues of the 561NAYY564 consensus sequence. (C) 3D representation of the active site of ECE-2 highlighting the residues of the 561NAYY564 consensus sequence that contribute to the positioning of the catalytic Glu 603. The images B and C were generated using VMD software.
Next we took advantage of the fact that both NEP and ECE-2 are inhibited by a common ligand, phosporamidon, and that the crystal structure of NEP was solved with phosphoramidon docked in its active site. We used the homology model to get information on the nature of inhibitor binding to ECE-2. For this we docked phosphoramidon into the active site of the enzyme using AutoDock (v.3/0/5) (Figure 2A insert).
The results from the superimposition of the active site residues of NEP and ECE-2 (with the inhibitor docked) demonstrate that most of the residues are structurally aligned, including the zinc binding residues, H602, H606 and E662, the catalytic E603 of the HExxH motif and the ExxxD consensus sequence. Interestingly, a third consensus sequence that is conserved among the NEP family members, N-A-Ar-Ar (where Ar indicates aromatic residues), exhibits differences in structural alignment. This sequence in NEP consists of 542NAFY545 whereas in ECE-2 it comprises of 561NAYY564. From the crystal structure of NEP it is clear that A543 is hydrogen bound with the peptide amide group of the P1' residue of phosphoramidon and N542 forms two hydrogen bonds with the NH and CO groups of the P2' residue of the inhibitor 11. Our analysis of the active site of ECE-2 reveals that both the N561 and A562 form hydrogen bonds with the inhibitor in a manner similar to that in NEP 11. However, the presence of Y563 (as compared to F544 in NEP) leads to changes in binding properties such as the binding orientation and interaction with phosphoramidon (Figure 2B). In addition, our model indicates that this Y563 participates in a network of hydrogen bonds with several residues including the catalytic E603. Therefore, Y563 could contribute to the positioning and orientation of E603, and thus, is likely to be important for the catalytic activity/inhibitor binding of ECE-2 (Figure 2C).
An additional difference revealed from the comparison of NEP and ECE-2 is in the S2' pocket of the binding site: R110 in NEP is replaced with W148 in ECE-2. The side chains of R102, D107 and R110 form the S2' pocket and provide the space to hold the indole moiety of phosphoramidon. We, therefore, predicted that the presence of Y563 in the position of F544 and W148 in the position of R110 could lead to changes in the binding of the inhibitor and the substrate, since previous studies have shown that phosphoramidon binds to NEP in the substrate binding pocket.
Construction and Expression of Recombinant ECE-2s
In order to test these predictions and to investigate the functional roles of Y563 and W148 residues we performed site-directed mutagenesis. These residues present in the active site of ECE-2 were replaced with the corresponding residues from the NEP sequence: W148R and Y563F. ECE-2 and the mutated enzymes were expressed as soluble secreted proteins in an eukaryotic expression system as described previously 18. To examine if the mutations affected the expression and secretion of proteins, we carried out Western blotting analysis with a polyclonal antiserum generated against the C-terminal of ECE-2 (Figure 3A). A band of equal intensity at ~85kDa (corresponding to ECE-2 lacking transmembrane and N-terminal domains) was detected in all cases thus confirming that the mutations did not affect protein expression levels. Next we subjected the medium containing the recombinant proteins to purification by anion exchange and metal ion affinity chromatography. The ECE-2 activity was measured using the quenched fluorescent peptide substrate, McaBk2 18, 29. The purification efficiency of ECE-2 activity is presented in Table S1. The purified proteins were assessed for enzyme activity and intensity (by Western blot using anti-ECE-2 antibody, Figure 3B). Similar to wild type ECE-2, purified mutant enzymes were able to hydrolyze McaBk2 and the maximal activity was observed at pH 5.5.
Figure 3.

Expression and purification of wild type and mutated ECE-2s. (A) Western blot analysis of medium infected with baculovirus expressing wild type and mutated ECE-2s. The ECE-2 recombinant protein was detected with anti-ECE2 antiserum raised against the C-terminal region of the protein. (B) Western blot analysis of purified proteins. Recombinant protein containing medium was subjected to purification by ion exchange and metal ion chromatography and equal amounts of the resultant fractions were run on SDS-PAGE. Protein was visualized using the anti-ECE-2 antiserum.
Determining the kinetic parameters of recombinant ECE-2s
The enzymatic activity of purified wild type and mutated ECE-2s were analyzed by determining the kinetic parameters (Km and kcat) at pH 5.5 using McaBk2 as a substrate. As shown previously, this substrate is readily hydrolyzed by ECE-2 with a Km of 5.76±0.55µM and kcat of 8.51±1.32min-1 (n=3). The mutant W148R (substitution of Trp 148 to Arg in the S2' pocket of ECE-2) exhibits a slight but significantly increased substrate binding to the enzyme, however this did not have an effect on the rate of hydrolysis, resulting in a kcat/Km ratio comparable to that for the wild type enzyme (Figure 4A and Table 1). In contrast, the mutant Y563F (with the substitution of Tyr 563 to Phe) exhibited no significant alterations in the Km value but showed a significant decrease in the rate of hydrolysis, thus resulting in more than five-fold decrease in kcat/Km ratio (Figure 4A and Table 1) indicating the importance of this residue for the catalytic activity of ECE-2.
Figure 4.
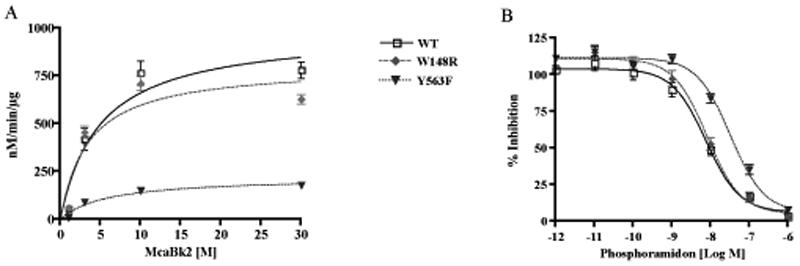
The initial rate of substrate hydrolysis and inhibition by phosphoramidon of wild type (WT) and mutated ECE-2s. (A) The initial rate of substrate hydrolysis (V0) at final concentrations of 0.1-30μM was determined by measuring the appearance of product under initial rate conditions. (V0) values were plotted as a function of substrate concentration ([S]) and fit to the Michaelis-Menten equation. Data represent the mean ±SEM (n=3). (B) Dose-dependent inhibition curve for phosphoramidon was generated by measuring the substrate hydrolysis in the presence of inhibitor at concentrations 1pM to 1µM. Data represent the mean±SEM (n=4).
Table 1.
Kinetic parameters and inhibition of wild type and mutated ECE-2s by phosphoramidona
| Enzymes | Km(µM) | kcat(min-1) |
kcat/Km (mM-1 min-1) |
Ki(nM) |
|---|---|---|---|---|
| ECE-2 WT | 5.76±0.55 | 8.51±1.32 | 1.48±0.23 | 1.56±0.23 |
| W148R | 3.94±0.15* | 6.84±0.57 | 1.74±0.15 | 1.58±0.21 |
| Y563F | 6.60±1.37 | 2.10±0.11** | 0.32±0.02** | 11.07±2.36*** |
The kinetic parameters (Km and kcat) of the purified enzymes were determined from Michaelis-Menten plots. Inhibition of recombinant enzymes by phosphoramidon (Ki) was determined from dose-dependent inhibition curves for a final McaBk2 concentration of 10μM. Data represent the mean±SEM (n=3).
p<0.05,
p<0.005,
p<0.001.
Potency of phosphoramidon to inhibit recombinant ECE-2s
Next we examined the extent to which the substitutions (W148R and Y563F) affected inhibitor binding. For this the potency of phosphoramidon, the generic metalloprotease inhibitor, to inhibit the activity of wild type and mutated ECE-2s was examined. As shown previously, phosphoramidon is a potent inhibitor of ECE-2 activity 12 and inhibits the wild type enzyme with a Ki of 1.56±0.23nM (n=4) (Table 1). We find that the Y563F mutation results in a significant decrease in inhibition as compared to the wild type enzyme (Figure 4B and Table 1). In contrast, the W148R mutation had no significant effect on the inhibitor potency (Table 1). We also tested the sensitivity of mutated enzymes to thiorphan, a specific inhibitor of NEP, which has previously been shown to not inhibit ECE-2 activity 12. Consistent with this, we find that a high concentration of thiorphan (10μM) had no significant effect on the activity of wild type or the mutated ECE-2s (data not shown). Taken together these results show that mutating Y563 affects inhibitor binding and catalysis in agreement with our prediction. Thus by verifying our 3D molecular model of ECE-2, these results suggest that the model could be used to screen for selective inhibitors of ECE-2.
Inhibitor screening
To identify specific inhibitors of ECE-2 we screened a library of ~13000 small, drug-like compounds from a chemical library obtained from ChemBridge. This library was generated by combining three individual libraries; CNS-SetTM, DIVERSet SetTM and MicroFormatsTM. The combined library represents a collection of a diverse set of compounds carefully selected to provide the broadest pharmacophore coverage (and display more than 260,000 unique 3-point pharmacophores). First we subjected the library of molecules to virtual docking onto the 3D model of ECE-2 using the AutoDock program. The compound library was screened by docking the target to the known binding site inside the large cavity of the ECE-2 model. To obtain inhibitors that are specific to ECE-2 we used a two-pronged approach: First, with the help of the Dockres program, we selected top-scoring ligands that dock closest to residues in the binding site specific to ECE-2. Second, we screened the same library by docking to the crystal structure of NEP (without the ligand and crystallographic water but with Zn2+). Those ligands that scored high in docking to both proteins were disregarded. The top-scoring forty compounds that displayed highest binding energy (> -13 kcal/mole-1 on the estimated free-energy scale) to ECE-2 were selected. We then tested if these compounds could inhibit the activity of ECE-2 in vitro. The screening showed that at 10μM concentration two compounds, 5719593 (1) and 5871159 (2) were able to reduce ECE-2 activity by more than 70% (Figure 5A). When these compounds were further characterized using dose-dependent inhibition curves, we found that they displayed IC50 values in the low (~ 6 μM) micro molar range (Figure 5B and C). To test the selectivity of these compounds we measured their inhibitory potency on NEP activity and found that they exhibited ~10 fold lower potency towards NEP as compared to ECE-2 (Figure 5C). To further investigate the nature of this inhibition (competitive vs. non-competitive) of ECE-2 activity by these inhibitors, we determined the rate of McaBk2 hydrolysis by ECE-2 in the presence of increasing concentrations of the substrate. Analysis of the data using Lineweaver-Burk plots revealed that both compounds, 1 and 2, display mixed inhibition (i.e., exhibit features of both competitive and non-competitive inhibitors) (Figure S2). At lower concentrations they exhibit competitive inhibition and increase Km. This is expected since they were selected to bind to the substrate-binding pocket of the ECE-2 model. However at higher concentrations these compounds display mixed, competitive and non-competitive, inhibition since they also decrease the Vmax of the hydrolysis. This is likely due to the binding of the inhibitors to additional sites (other than the active site). Alternatively, at higher concentrations the binding of the inhibitors could alter the conformation of the active site such that it could affect substrate binding. Both of these scenarios would lead to a non-competitive type of inhibition.
Figure 5.
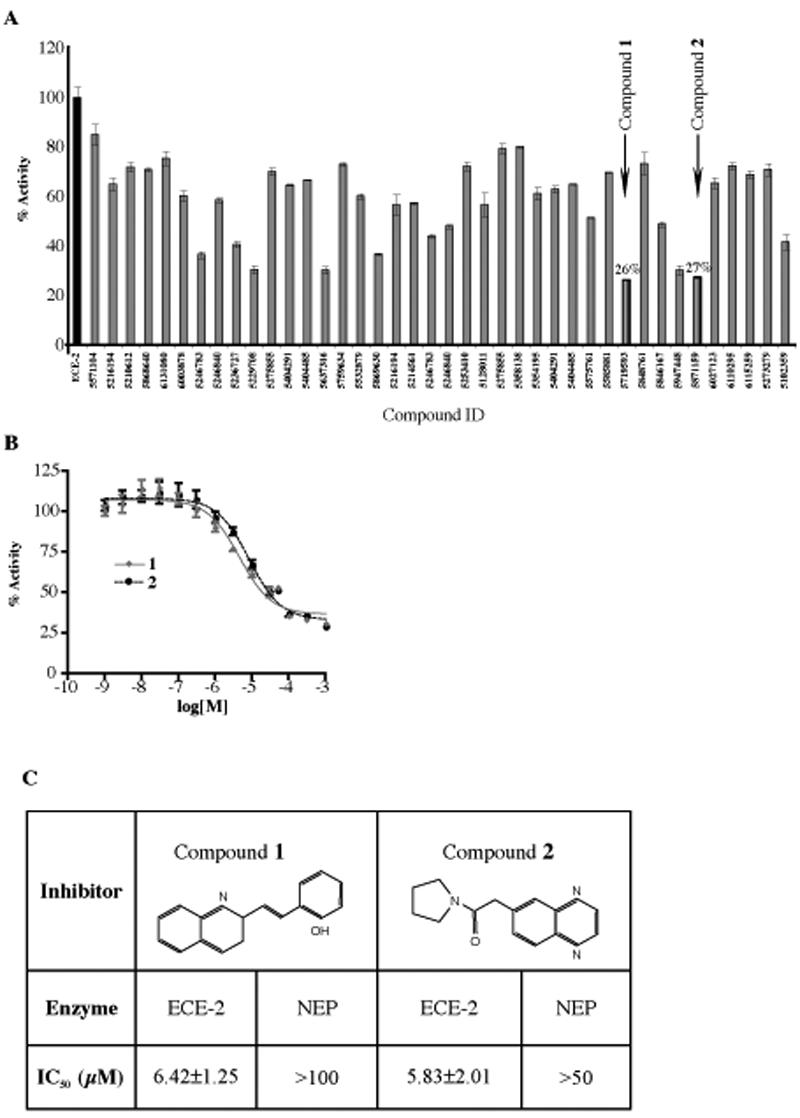
Screening of compounds for the inhibition of ECE-2 activity. (A) The rate of McaBk2 fluorescent substrate hydrolysis by recombinant ECE-2 was measured in the absence and presence of 10μM of each of forty top scoring compounds by initial screening. Data are represented as % activity of ECE-2 without inhibitor. Two most potent compounds, 1 and 2 inhibited ECE-2 activity by more than 70%. (B) Concentration-dependent inhibition curves for selected inhibitor compounds, 1 and 2, were generated by measuring substrate hydrolysis in the presence of inhibitors at concentrations 1nM-1mM. (C) IC50 values for each selected inhibitor compound were determined for ECE-2 and NEP by establishing concentration-dependent inhibition curves for a final McaBk2 concentration of 10μM. Data represent the mean±SEM (n=5).
In order to probe the structures of these two compounds and define parameters important for inhibitor binding, we searched the ZINC database of commercially available compounds for analogs of the two selected compounds 30. Thirty compounds from the database were docked to the active site of our 3D molecular model of ECE-2 using AutoDock software as described earlier. Three analogs of 1 that exhibited highest binding affinity, 3 (S136492), 4 (6634449) and 5 (6636797), were selected for biochemical analysis. Examination of their inhibitory potency revealed 3 to be the most potent (with a low μM potency similar to that of 1). Interestingly the other two compounds exhibited five times reduced potency suggesting that the additional CH3 group in the P1' position plays an important role in the binding and inhibition (Figure 6A and B).
Figure 6.
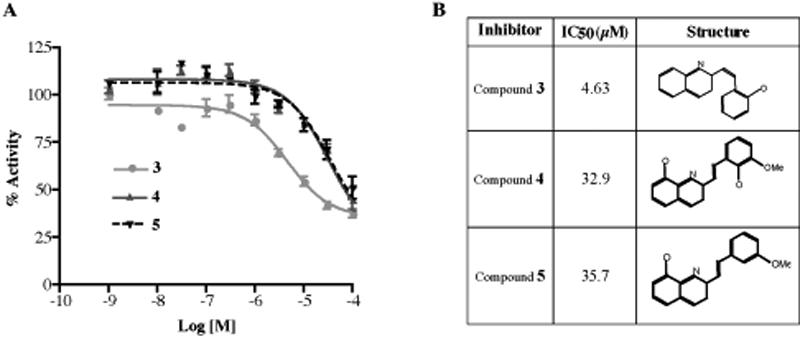
Inhibition of ECE-2 activity by 1 analogs. (A) Concentration-dependent inhibition curves for three selected analogs, 3, 4 and 5 were generated by measuring the substrate hydrolysis in the presence of inhibitors at concentrations 1nM-100μM. (B) IC50 values for each selected inhibitor compound were determined by establishing concentration-dependent inhibition curves for a final McaBk2 concentration of 10μM. Data represent the mean±SEM (n=4).
Thus, using a combination of homology modeling and virtual screening we have been able to identify selective inhibitors of ECE-2. Docking of the inhibitors to the active site of ECE-2 in the molecular model reveals that these compounds bind in the vicinity of Zn2+ atom and form multiple interactions with the residues of the active site involved in substrate binding and catalysis (Figure 7A and B).
Figure 7.
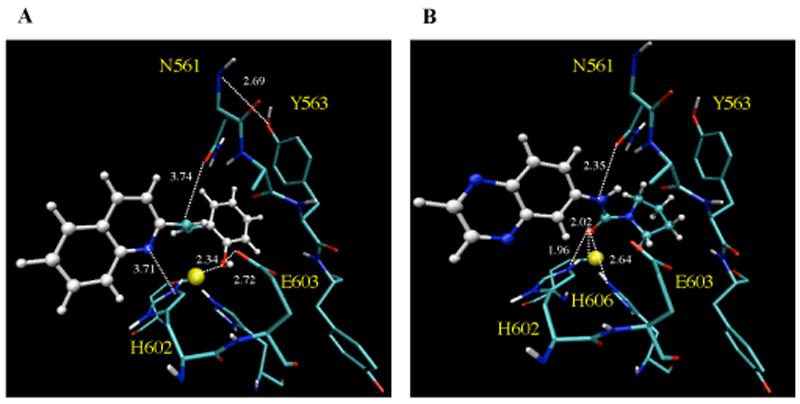
Binding of inhibitor compounds to the molecular model of the active site of ECE-2. The images highlight the binding of 1 (A) and 2 (B) to the H602 and H606 involved in zinc coordination, catalytic E603 and residues of the 541NAYY544 consensus sequence. The images were generated using VMD software.
Discussion
ECE-2 is a recently identified member of M13 family of zinc metallopeptidases of which NEP is the best characterized. Until recently most of the structural information about these enzymes was based on the studies carried out on the bacterial homolog-thermolysin (TLN). Although there is very limited homology between the primary sequences of TLN and NEP, these two enzymes have similar catalytic properties and are inhibited by the same type of molecules such as phosphoramidon and thiorphan. In addition they both contain the highly conserved consensus sequences, HExxH and ExxxD, which contain residues critical for Zn2+ coordination and catalysis. Based on early crystallographic studies Matthews proposed a mechanism for the TLN-catalyzed cleavage of peptides 31. According to this model the two His residues (H142 and H146) together with the E166 and a water molecule are involved in the tetrahedral coordination of the Zn2+ atom. The incoming substrate displaces the water molecule towards the catalytic E143 residue. The negative charge of E143 polarizes the zinc-coordinated water molecule and promotes its nucleophilic attack on the carbonyl carbon of the scissile peptide bond. Structural studies also indicated the importance of H231 and Y157 residues in the stabilization of the transition state.
Later studies in NEP using site-directed mutagenesis have identified corresponding residues involved in zinc coordination, catalysis and substrate binding and confirmed that the proposed catalytic mechanism was also valid for the mammalian enzymes 19-26. Several studies also modeled the structure of NEP, as well as other family members using the crystal structure of TLN as a template. However, the availability of the crystal structure of NEP complexed with phosphoramidon provides a more suitable and reliable template for the modeling of metallopeptidases of this family and has therefore been used to model the structures of several members of M13 family, including endothelin-converting enzyme-1 (ECE-1), Kell blood group protein and neprilysin-2 (NEP-2) 32-34. ECE-1 and ECE-2 share common substrates and approximately 60% sequence homology. The consensus sequences that are conserved among zinc metallopeptidases are identical between these two proteins, indicating significant structural similarities. Therefore the molecular model of ECE-1 could provide considerable information about the architecture of the ECE-2 active site32. However, since the molecular model of ECE-1 was not deposited in the PDB, a detailed comparison of our model with that of ECE-1 was not possible.
There are a number of important physiological differences between ECE-1 and ECE-2, such as the pH dependency and sensitivity to inhibition by phosphoramidon 12. This generic metallopeptidase inhibitor is ~100 less potent towards ECE-1 (IC50=3.5μM) than ECE-2 and NEP (IC50=2-4nM) suggesting that despite a number of similarities, some aspects of catalysis and inhibitor binding to the active site of each of these peptidases could be quite distinct. In addition, the detailed information about the binding pocket of each of these peptidases will be useful to design specific and selective inhibitors of ECE-1 and ECE-2. Because of the involvement of ET in various cardiovascular, renal, pulmonary and central nervous system diseases 35-37 much effort has been put towards developing inhibitors of this pathway as potential therapeutic agents. In this light, inhibitors of ECE, which catalyze a rate-limiting step in ET production, have received a great deal of attention. In addition, double inhibitors of ECE and NEP that would interfere with the production of ET, a vasoconstrictor, as well as with the degradation of atrial natriuretic peptide (ANP), a potent vasorelaxant, have also been designed as potential therapeutics for the treatment of hypertension 38. However, most of these studies did not differentiate between ECE-1 and ECE-2 and focused on the inhibition of ECE-1 activity without addressing the effect of these inhibitors on ECE-2. To date only a trisubstituted quinazoline, PD069185 (6) (Scheme 1) and its analogs, have been tested and reported to be selective for ECE-1 over ECE-2 39.
Scheme 1.
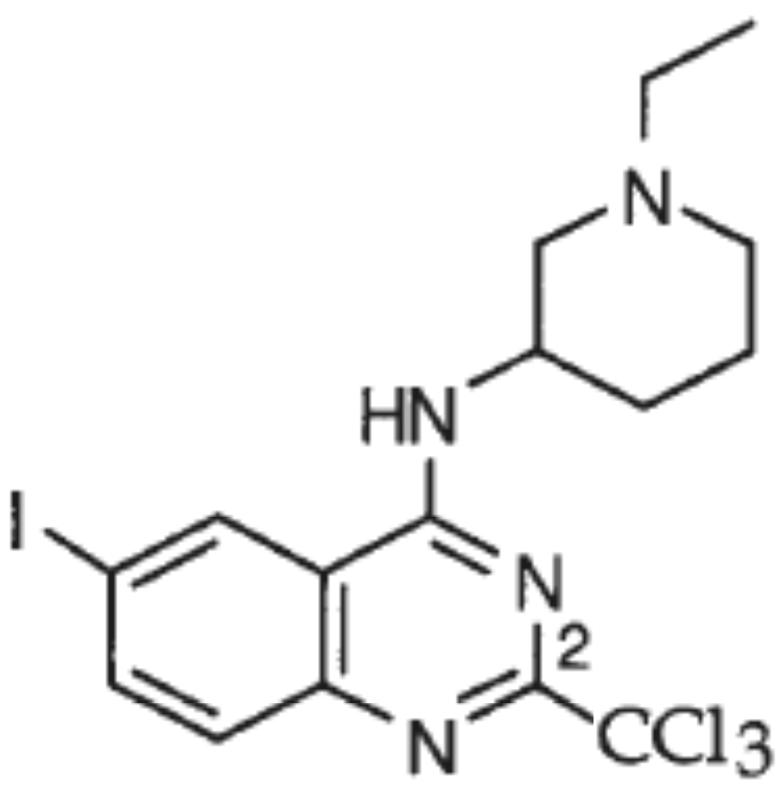
Structure of compound 6 39.
In order to help us identify specific inhibitors of ECE-2, we generated a 3D molecular model of ECE-2 based on the x-ray structure of NEP. A comparison of the active sites of these two enzymes revealed that the residues involved in catalysis as well as the residues in the vicinity of the substrate/inhibitor binding pocket that interact with the substrate are highly conserved. However, we identified two amino acid differences in the active sites of NEP and ECE-2, which could potentially account for pharmacological differences between NEP and ECE-2.
One of the residues, F544 of NEP within the conserved consensus sequence 542NAFY545 is substituted by Y563 in ECE-2 and Y552 in ECE-1. Based on earlier mutagenesis studies as well as crystal structure, both N542 and A543 of NEP were shown to form hydrogen bonds with phosphoramidon 11, 25. Sansom and colleagues 40 also demonstrated the importance of Y552 within this sequence, for the catalytic activity of ECE-1. Although in the ECE-2 model Y563 was not found to directly interact with the phosphoramidon, it was found to hydrogen bond to N561 and thus affect inhibitor binding. Y563 is also in a position to interact with the catalytic E603. While the distance between these two residues suggests only a weak interaction, even a slight movement of the catalytic Glu could have a drastic effect on the catalytic activity of the enzyme. Indeed, we found that the substitution of the Tyr 563 residue with Phe in ECE-2 resulted in approximately six-fold lower catalytic activity for the enzyme and more than sevenfold decrease in inhibitor potency. The fact that this mutation did not influence substrate binding but affected inhibition by the transition state analog-phosphoramidon, leads us to hypothesize that Y563 may be involved in the catalytic activity, and in the stabilization of the transition state of the enzyme, by participating in the positioning and orientation of E603. Considering that this substitution is very conservative, such small, albeit significant change in enzyme properties demonstrate the importance of this residue for the catalytic activity of ECE-2.
Another difference we found in the active site of these two enzymes is that the Arg 110 residue in NEP is substituted by Trp in ECE-2. R110 is one of the residues which along with R102 and D107 form the S2' subsite of NEP. In ECE-2 this subsite is large, similar to NEP, and can accommodate bulky side-chains. The presence of Trp in place of R110 could result in a wider pocket with reduced specificity. We observed that mutation of Trp 148 back to Arg slightly but significantly increased affinity for the substrate however it did not affect the inhibitor potency. The change in affinity may be explained by the fact that the McaBk2 peptide has a Lys residue in the P2' position and its long side chain is held tighter in the S2' pocket with the reduced volume. The more compact Trp side chain of the inhibitor phosphoramidon in the S2' pocket is not affected by this change. Testing additional substrates with different residues in P2' position is needed to address this notion.
After confirming the validity of the 3D molecular model of ECE-2 by site-directed mutagenesis we used the model to screen a library of approximately 13000 small drug-like molecules. Initially the structures of the compounds were docked into the active site of the ECE-2 model. Compounds that displayed highest binding were tested experimentally using in vitro an enzyme assay. In the experimental screening we identified two compounds, 1 and 2, that inhibited ECE-2 activity with micromolar potency and were ~ 10 times more selective for ECE-2 as compared to NEP. We found that these compounds also inhibit ECE-1 activity with an affinity similar to that of ECE-2 (Gagnidze and Devi, unpublished). This is not surprising since the homology between ECE-2 and ECE-1 is higher than that between ECE-2 and NEP and that Y563 of ECE-2 is also conserved in ECE-1. Furthermore, Y563 in ECE-1 has been shown to play a role in the catalytic activity of the enzyme40.
An examination of the docking of these compounds to the active site of ECE-2 revealed that they bind in close proximity to the Zn2+ atom and are in a position to form hydrogen bonds with the His residues that coordinate Zn2+ as well as the catalytic Glu603. Thus they are in a position to affect the hydrolysis of the substrate (Figure 7). In order to identify compounds with higher inhibitory potency towards ECE-2 we searched for analogs of 1 and found three compounds that were commercially available. Two of these three analogs, 4 and 5, were ~5-6 times less effective compared to 1. These two compounds have additional CH3 groups in the P1' position and this may interfere with the stringent specificity of S1' site that is characteristic of these metallopeptidases. An optimal way to increase the potency of these inhibitors would be through chemical optimization using the structures of 1 and 2 as a lead. Additional analyses are also needed to ensure the selectivity of these compounds for ECE-2 and the chemical optimization may be a useful strategy to achieve this goal as well. Information from these studies will be useful in the identification of specific inhibitors of ECE-2 that will serve as hitherto unavailable tools for studies examining the physiological role of this peptidase in vivo.
Conclusions
Endothelin-converting enzyme-2 belongs to the M13 family of Zn2+ metallopeptidases and shares a substantial structural similarity with its homolog neprilysin. We used the crystal structure of NEP to generate a 3D molecular model of ECE-2. Analyses of the architecture of the active site and the binding specificities of the inhibitor-phosphoramidon to the enzyme led us to identify two non-conserved residues that affect the catalytic activity and substrate/inhibitor binding of ECE-2. Tyr 563 of ECE-2 is part of the conserved consensus sequence 561NAYY564 and participates in a network of hydrogen bonds with the catalytic Glu 603. Mutation of Tyr 563 of ECE-2 to Phe results in more than four-fold decrease of kcat of the peptidase. Tyr 563 is also involved in the binding and positioning of the inhibitor-phosphoramidon in the active site of ECE-2 and the substitution of this residue with Phe attenuates the inhibition by phosphoramidon by almost ten-fold. The second non-conserved residue Trp 148 is located in the S2' site of the catalytic cleft of ECE-2 that harbors the indole moiety of phosphoramidon. Substitution of this residue to Arg affected substrate binding without influencing catalytic activity of the enzyme or binding of the inhibitor.
The 3D molecular model of ECE-2 was next used to screen inhibitor compounds for their binding to the active site of the enzyme and a number of selected compounds were tested experimentally. The tests led to the identification of three compounds: 1, 2 and 3 that inhibited ECE-2 activity with IC50 values in the lower micro molar range and displayed higher selectivity towards ECE-2 versus NEP. These compounds can be used in future studies as leads for the development of potent and specific inhibitors of ECE-2 and will aid in the elucidation of the physiological functions of the peptidase.
Experimental Section
Homology Modeling
The model building of ECE-2 using NEP structure (PDB code 1DMT) was achieved using MODELLER 8v2 program 41, 42. The quality of the model was checked using PROCHECK 43, 44 and ProSa 2003 45, 46. A Ramachandran plot generated in PROCHECK with a hypothetical resolution of 3.0 Å, revealed that 93% of the amino acid residues are in the most favorable region. The alignment of two the protein structures was carried out using the 'align' command of pymol which works by doing a sequence alignment and then trying to align the structures to minimize the rmsd between the aligned residues. A rmsd value of 0.36 Å was obtained between ECE-2 and NEP by aligning all atoms against the entire C-alpha backbone. The Z-score, calculated using the software ProSa 2003, showed that the predicted model of ECE-2 was well within the range of a typical native structure.
Molecular Docking
Docking of phosphoramidon to NEP and ECE-2 was achieved using AutoDock v3.0.5 47, 48, which uses a genetic algorithm as a global optimizer combined with energy minimization as a local search method. The starting structure of phosphoramidon was taken from the X-ray structure (PDB code 1DMT) and the partial charges were assigned using Gasteiger-Marsili method using AutoDockTools (http://mgltools.scripps.edu/downloads). Autotors program was used to define torsional degrees of freedom in the ligand and a total of eight torsions were allowed. Binding-site-centered affinity grid maps were generated for NEP with 0.375Å spacing using Autogrid for NEP. The minimization was achieved using Lamarckian genetic algorithm (LGA) and pseudo-Solis and Wets method 49. The grid size was set to 50 x 50 x 60 points, centered on the Zn2+ atom in the crystal structure of the complex. Each LGA job consisted of 200 runs with 270, 000 generations in each run and the maximum number of energy evaluations was set to 5.0 x 106. Resulting docked conformations within 1.0Å rmsd tolerance of each other were clustered together and analyzed using AutoDockTools. The zinc parameters used were, radius, r =1.1 Å, well depth, ε = 0.35 kcal/mol and the charge of +1.0 e.
Inhibitor screening
For the inhibitor screening we used a chemical library from ChemBridge that was generated by combining three individual libraries from CNS-SetTM, DIVERSet SetTM and MicroFormatsTM. The combined library represents a collection of a diverse set of druglike small molecule compounds carefully selected to provide the broadest pharmacophore coverage (and display more than 260,000 unique 3-point pharmacophores). For virtual screening, the AutoDock program (version 3.05 47) was used. The screening was driven by scripts for our cluster of Apple G5 processors running in parallel and the analysis and filtering was done by the program Dockres (URL:http://inka.mssm.edu/~mezei/dockres),
Materials
Internally quenched fluorescent substrate McaBk2 ((7-methoxycoumarin-4-yl) acetyl-RPPGFSAFL-(2,4-dinitrophenyl)), was custom synthesized by Open Biosystems (Huntsville, AL). DEAE-Sepharose fast flow was obtained from Amersham Biosciences AB (Uppsala, Sweden), Talon™ metal affinity resin was purchased from Clontech (Palo Alto, CA). Inhibitor compound were purchased from ChemBridge corporation (San Diego, CA) and are identified by database ID. Compound 3 was purchased from Sigma-Aldrich's Rare Chemical Library (St. Louis, MO).
Cloning and Site-directed Mutagenesis
The cloning of a secreted soluble form of ECE-2 was described previously 18. The same construct was used as a template to introduce the two mutations: W148R and Y563F. Two overlapping regions of the ECE-2 sequence were amplified separately using two flanking oligonucleotides: Forw-5' TCATCATGCCCTAGGGGTC 3' and Rev-5' CTGCAGCGGCCGCGACCTG 3'in combination with two overlapping oligonucleotides containing the mutated residues: W148R: 5' CAGCCTCAGGGACCAAAAC 3'; Y563F: 5'ACAGTGAATGCCTTCTACCTTCCAACT 3'.
The products of the first round of amplification were used as the template for a second PCR with the two flanking oligonucleotides. The final PCR product was digested and cloned into the baculovirus expression vector, pVL1393. The DNA constructs were sequenced to ensure that correct mutations were introduced. The recombinant virus production was carried out as described previously 18.
Expression and Purification of Soluble ECE-2s
Sf9 insect cells growing in Baculo Gold Max-XP serum-free insect cell medium (BD Biosciences, San Jose, CA) were co-transfected with recombinant wildtype and mutated ECE-2 cDNA in pVL1393 and BaculoGold viral DNA (BD Biosciences) using the calcium phosphate procedure. After culturing for 5 days, 100-500 μl of medium were used to infect 3×105 Sf9 cells in a 25-cm2 flask. Then 1 ml of the medium containing amplified virus was used to infect 3×105 Sf9 cells in 5ml of medium in 25-cm2 flasks. After 3 days of infection, the medium was collected and cleared by centrifugation at 20,000 × g for 10 min, and the resulting supernatant was frozen at -80°C until use. Wild type and mutated ECE-2-containing conditioned media was concentrated using Amicon ultrafiltration membrane YM100 (Millipore Corp., Bedford, MA), and purified using DEAE-Sepharose anion exchange column and Talon™-Sepharose Co2+ affinity resin column as described in Supporting Information. Fractions containing ECE-2 activity were subjected to SDS-PAGE and visualized by Western blot. The amount of purified recombinant mutant ECE-2 proteins used in all subsequent experiments, were normalized to that of wild type ECE-2.
Western Blotting
The expression of wild type and mutated ECE-2s in the conditioned media from the baculovirus infected Sf9 cells was analyzed by Western blotting. 25 μg of total protein were separated by sodium dodecyl sulfate polyacrylamide gel-electrophoresis, transferred onto a nitrocellulose membrane and blotted with a polyclonal antiserum generated against the C-terminal 16 amino acids of ECE-2 (1:1000). This antiserum has been previously characterized and is able to recognize the ~85kDa truncated form of ECE-2 secreted from baculovirus cells 18. There was no significant difference in the expression levels of wild type and mutated ECE-2 recombinant proteins.
Assay for ECE-2 Activity
Enzyme activity was assayed using the synthetic quenched fluorescent substrate, McaBk2. The assay was carried out at 37°C with 10μM McaBk2 in 0.2M sodium acetate buffer, pH 5.5, containing the detergent C12E8 (octaethylene glycol dodecyl ether, EMD Chemicals, Inc., San Diego, CA) unless otherwise indicated. Substrate hydrolysis was monitored on a Fluoromax plate reader with excitation at 335nm and emission at 440nm, and initial velocity was determined.
Determination of Kinetic Constants for ECE-2
The enzymatic parameters of the wild type and mutated ECE-2s were determined in a microtiter plate, by monitoring the rate of hydrolysis of a synthetic substrate, McaBk2, in sodium acetate buffer, pH 5.5 at 37°C. The initial rate of substrate hydrolysis (V0) at final concentrations of 0.1-30µM was determined by measuring the appearance of product under initial rate conditions. V0 values were plotted as a function of substrate concentration ([S]) and fit to the Michaelis-Menten equation using GraphPad Prism software (version 4.0b). The sensitivity of wild type and mutated ECE-2s to inhibition by phosphoramidon and inhibitor compounds were determined by establishing dose-dependent inhibition curves and data analysis was done using GraphPad Prism software (version 4.0b).
Supplementary Material
Acknowledgments
We would like to thank Dr. Sherwin Wilk for the help with some of the studies and Dr. Ivone Gomes for reading the manuscript. These studies are supported by NIH grants (NS26880 and DA019521 to LAD and CA87658 to MMZ).
Footnotes
Abbreviations used: ECE-2, endothelin-converting enzyme; ET, endothelin; NEP, neprilysin; PC, prohormone convertase; CPE, carboxypeptidase; PAM, peptidylglycine α-amidating monooxygenase; TLN, thermolysin; PDB, Protein data Bank; McaBk2, (7-methoxycoumarin-4-yl) acetyl-RPPGFSAFL-(2,4-dinitrophenyl); wt, wild type; rmsd, root mean square deviation.
Supporting information available: Ramachandran plot of ECE-2 model, characterization of inhibition and table of purification of ECE-2 activity. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Docherty K, Steiner DF. Post-translational proteolysis in polypeptide hormone biosynthesis. Annu Rev Physiol. 1982;44:625–638. doi: 10.1146/annurev.ph.44.030182.003205. [DOI] [PubMed] [Google Scholar]
- 2.Steiner DF. The proprotein convertases. Curr Opin Chem Biol. 1998;2(1):31–39. doi: 10.1016/s1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]
- 3.Mizuno K, Minamino N, Kangawa K, Matsuo H. A new family of endogenous "big" Met-enkephalins from bovine adrenal medulla: purification and structure of docosa- (BAM-22P) and eicosapeptide (BAM-20P) with very potent opiate activity. Biochem Biophys Res Commun. 1980b;97(4):1283–1290. doi: 10.1016/s0006-291x(80)80005-2. [DOI] [PubMed] [Google Scholar]
- 4.Yang HY, Fratta W, Majane EA, Costa E. Isolation, sequencing, synthesis, and pharmacological characterization of two brain neuropeptides that modulate the action of morphine. Proc Natl Acad Sci U S A. 1985;82(22):7757–7761. doi: 10.1073/pnas.82.22.7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sigafoos J, Chestnut WG, Merrill BM, Taylor LC, Diliberto EJ, Jr., Viveros OH. Novel peptides from adrenomedullary chromaffin vesicles. J Anat. 1993;183( Pt 2):253–264. [PMC free article] [PubMed] [Google Scholar]
- 6.Che FY, Yan L, Li H, Mzhavia N, Devi LA, Fricker LD. Identification of peptides from brain and pituitary of Cpe(fat)/Cpe(fat) mice. Proc Natl Acad Sci U S A. 2001;98(17):9971–6. doi: 10.1073/pnas.161542198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perry SJ, Yi-Kung Huang E, Cronk D, Bagust J, Sharma R, Walker RJ, Wilson S, Burke JF. A human gene encoding morphine modulating peptides related to NPFF and FMRFamide. FEBS Lett. 1997;409(3):426–430. doi: 10.1016/s0014-5793(97)00557-7. [DOI] [PubMed] [Google Scholar]
- 8.Vilim FS, Aarnisalo AA, Nieminen ML, Lintunen M, Karlstedt K, Kontinen VK, Kalso E, States B, Panula P, Ziff E. Gene for pain modulatory neuropeptide NPFF: induction in spinal cord by noxious stimuli. Mol Pharmacol. 1999;55(5):804–811. [PubMed] [Google Scholar]
- 9.Ahn K. Handbook of Proteolytic Enzymes. Academic Press; San Diego: 1998b. pp. 1085–1089. [Google Scholar]
- 10.Molineaux C, Wilk S. Extracellular processing of peptides. Vol. Chapter 10. CRC press; Boca Raton: 1991. pp. 251–282. [Google Scholar]
- 11.Oefner C, D'Arcy A, Hennig M, Winkler FK, Dale GE. Structure of human neutral endopeptidase (Neprilysin) complexed with phosphoramidon. J Mol Biol. 2000;296(2):341–349. doi: 10.1006/jmbi.1999.3492. [DOI] [PubMed] [Google Scholar]
- 12.Emoto N, Yanagisawa M. Endothelin-converting enzyme-2 is a membrane-bound, phosphoramidon-sensitive metalloprotease with acidic pH optimum. J Biol Chem. 1995;270(25):15262–15268. doi: 10.1074/jbc.270.25.15262. [DOI] [PubMed] [Google Scholar]
- 13.Turner AJ, Murphy LJ. Molecular pharmacology of endothelin converting enzymes. Biochem Pharmacol. 1996;51(2):91–102. doi: 10.1016/0006-2952(95)02036-5. [DOI] [PubMed] [Google Scholar]
- 14.Turner AJ, Isaac RE, Coates D. The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. Bioessays. 2001;23(3):261–269. doi: 10.1002/1521-1878(200103)23:3<261::AID-BIES1036>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 15.Xu D, Emoto N, Giaid A, Slaughter C, Kaw S, deWit D, Yanagisawa M. ECE-1: a membrane-bound metalloprotease that catalyzes the proteolytic activation of big endothelin-1. Cell. 1994;78(3):473–485. doi: 10.1016/0092-8674(94)90425-1. [DOI] [PubMed] [Google Scholar]
- 16.Eckman EA, Reed DK, Eckman CB. Degradation of the Alzheimer's amyloid beta peptide by endothelin-converting enzyme. Journal of Biological chemistry. 2001;276(27):24540–24548. doi: 10.1074/jbc.M007579200. [DOI] [PubMed] [Google Scholar]
- 17.Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB. Alzheimer's disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J Biol Chem. 2003;278(4):2081–2084. doi: 10.1074/jbc.C200642200. [DOI] [PubMed] [Google Scholar]
- 18.Mzhavia N, Pan H, Che FY, Fricker LD, Devi LA. Characterization of endothelin-converting enzyme-2. Implication for a role in the nonclassical processing of regulatory peptides. J Biol Chem. 2003;278(17):14704–14711. doi: 10.1074/jbc.M211242200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malfroy B, Schwartz JC. Enkephalinase from rat kidney. Purification, characterization, and study of substrate specificity. J Biol Chem. 1984;259(23):14365–14370. [PubMed] [Google Scholar]
- 20.Devault A, Nault C, Zollinger M, Fournie-Zaluski MC, Roques BP, Crine P, Boileau G. Expression of neutral endopeptidase (enkephalinase) in heterologous COS-1 cells. Characterization of the recombinant enzyme and evidence for a glutamic acid residue at the active site. J Biol Chem. 1988a;263(8):4033–4040. [PubMed] [Google Scholar]
- 21.Devault A, Sales V, Nault C, Beaumont A, Roques B, Crine P, Boileau G. Exploration of the catalytic site of endopeptidase 24.11 by site-directed mutagenesis. Histidine residues 583 and 587 are essential for catalysis. FEBS Lett. 1988b;231(1):54–58. doi: 10.1016/0014-5793(88)80701-4. [DOI] [PubMed] [Google Scholar]
- 22.Vijayaraghavan J, Kim YA, Jackson D, Orlowski M, Hersh LB. Use of site-directed mutagenesis to identify valine-573 in the S'1 binding site of rat neutral endopeptidase 24.11 (enkephalinase) Biochemistry. 1990;29(35):8052–8056. doi: 10.1021/bi00487a009. [DOI] [PubMed] [Google Scholar]
- 23.Beaumont A, Le Moual H, Boileau G, Crine P, Roques BP. Evidence that both arginine 102 and arginine 747 are involved in substrate binding to neutral endopeptidase (EC 3.4.24.11) J Biol Chem. 1991;266(1):214–20. [PubMed] [Google Scholar]
- 24.Kim YA, Shriver B, Quay T, Hersh LB. Analysis of the importance of arginine 102 in neutral endopeptidase (enkephalinase) catalysis. J Biol Chem. 1992;267(17):12330–12335. [PubMed] [Google Scholar]
- 25.Dion N, Le Moual H, Crine P, Boileau G. Kinetic evidence that His-711 of neutral endopeptidase 24.11 is involved in stabilization of the transition state. FEBS Lett. 1993;318(3):301–304. doi: 10.1016/0014-5793(93)80533-z. [DOI] [PubMed] [Google Scholar]
- 26.Dion N, Le Moual H, Fournie-Zaluski MC, Roques BP, Crine P, Boileau G. Evidence that Asn542 of neprilysin (EC 3.4.24.11) is involved in binding of the P2' residue of substrates and inhibitors. Biochem J. 1995;311( Pt 2):623–627. doi: 10.1042/bj3110623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sansom CE, Hoang VM, Turner AJ. Molecular modeling of the active site of endothelin-converting enzyme. J Cardiovasc Pharmacol. 1995;26(Suppl 3):S75–77. [PubMed] [Google Scholar]
- 28.Tiraboschi G, Jullian N, Thery V, Antonczak S, Fournie-Zaluski MC, Roques BP. A three-dimensional construction of the active site (region 507-749) of human neutral endopeptidase (EC.3.4.24.11) Protein Eng. 1999;12(2):141–149. doi: 10.1093/protein/12.2.141. [DOI] [PubMed] [Google Scholar]
- 29.Johnson GD, Ahn K. Development of an internally quenched fluorescent substrate selective for endothelin-converting enzyme-1. Anal Biochem. 2000;286(1):112–118. doi: 10.1006/abio.2000.4772. [DOI] [PubMed] [Google Scholar]
- 30.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. LJournal of Chemical Information and Modeling. 2005;45(1):177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matthews BW. Structural bases of the action of thermolysin and related zinc peptidases. Acc Chem Res. 1988;21(9):333–340. [Google Scholar]
- 32.Bur D, Dale GE, Oefner C. A three-dimensional model of endothelin-converting enzyme (ECE) based on the X-ray structure of neutral endopeptidase 24.11 (NEP) Protein Eng. 2001;14(5):337–41. doi: 10.1093/protein/14.5.337. [DOI] [PubMed] [Google Scholar]
- 33.Voisin S, Rognan D, Gros C, Ouimet T. A three-dimensional model of the neprilysin 2 active site based on the X-ray structure of neprilysin. Identification of residues involved in substrate hydrolysis and inhibitor binding of neprilysin 2. J Biol Chem. 2004;279(44):46172–46181. doi: 10.1074/jbc.M407333200. [DOI] [PubMed] [Google Scholar]
- 34.Lee S, Debnath AK, Redman CM. Active amino acids of the Kell blood group protein and model of the ectodomain based on the structure of neutral endopeptidase 24.11. Blood. 2003;102(8):3028–3034. doi: 10.1182/blood-2003-05-1564. [DOI] [PubMed] [Google Scholar]
- 35.Ferro CJ, Webb DJ. The clinical potential of endothelin receptor antagonists in cardiovascular medicine. Drugs. 1996;51(1):12–27. doi: 10.2165/00003495-199651010-00003. [DOI] [PubMed] [Google Scholar]
- 36.Kohan DE. Endothelins in the normal and diseased kidney. Am J Kidney Dis. 1997;29(1):2–26. doi: 10.1016/s0272-6386(97)90004-4. [DOI] [PubMed] [Google Scholar]
- 37.Humbert M, Simonneau G. Drug Insight: endothelin-receptor antagonists for pulmonary arterial hypertension in systemic rheumatic diseases. Nat Clin Pract Rheumatol. 2005;1(2):93–101. doi: 10.1038/ncprheum0048. [DOI] [PubMed] [Google Scholar]
- 38.Jeng AY, Mulder P, Kwan AL, Battistini B. Nonpeptidic endothelin-converting enzyme inhibitors and their potential therapeutic applications. Can J Physiol Pharmacol. 2002;80(5):440–449. doi: 10.1139/y02-025. [DOI] [PubMed] [Google Scholar]
- 39.Ahn K, Sisneros AM, Herman SB, Pan SM, Hupe D, Lee C, Nikam S, Cheng XM, Doherty AM, Schroeder RL, Haleen SJ, Kaw S, Emoto N, Yanagisawa M. Novel selective quinazoline inhibitors of endothelin converting enzyme-1. Biochem Biophys Res Commun. 1998a;243(1):184–190. doi: 10.1006/bbrc.1998.8081. [DOI] [PubMed] [Google Scholar]
- 40.Sansom CE, Hoang MV, Turner AJ. Molecular modelling and site-directed mutagenesis of the active site of endothelin-converting enzyme. Protein Eng. 1998;11(12):1235–1241. doi: 10.1093/protein/11.12.1235. [DOI] [PubMed] [Google Scholar]
- 41.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen M-Y, Pieper U, A. S. Comparative Protein Structure Modeling with MODELLER. Vol. UNIT 5.6. John Wiley and Sons, Inc.; 2006. [Google Scholar]
- 42.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 43.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;26:283–291. [Google Scholar]
- 44.Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Stereochemical quality of protein structure coordinates. Proteins. 1992;12(4):345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 45.Sipple MJ. Recognition of errors in three-dimensional structures of proteins. Proteins, structure, function, and bioinformatics. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- 46.Ginalski K. Comparative modeling for protein structure prediction. Curr Opin Struct Biol. 2006;16(2):172–177. doi: 10.1016/j.sbi.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 47.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19(14):1639–1662. [Google Scholar]
- 48.Goodsell DS, Morris GM, Olson AJ. Automated Docking of Flexible Ligands: Applications of AutoDock. Journal of molecular recognition. 1996;9(1):1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 49.Solis FJ, Wets RJ. Minimization by Random Search Techniques. Mathematics of Operations Research. 1981;6(1):19–30. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.