

Abstract
Twelve analogs of makaluvamines have been synthesized. These compounds were evaluated for their ability to inhibit the enzyme topoisomerase II. Five compounds were shown to inhibit topoisomerase catalytic activity comparable to two known topoisomerase II targeting control drugs, etoposide and m-AMSA. Their cytotoxicity against human colon cancer cell line HCT-116 and human breast cancer cell lines MCF-7 and MDA-MB-468 have been evaluated. Four makaluvamine analogs exhibited better IC50 values against HCT-116 as compared to control drug etoposide. One analog exhibited better IC50 value against HCT-116 as compared to m-AMSA. All twelve of the makaluvamine analogs exhibited better IC50 values against MCF-7 and MDA-MB-468 as compared to etoposide as well as m-AMSA.
For the past quarter of a century, global marine sources have proven to be a rich source of a vast array of new medicinally valuable compounds.1 These natural products exist as secondary metabolites in marine invertebrates such as sponges, bryazoa, tunicates and ascidians. As a result of the potential for new drug discovery, marine natural products have attracted scientists from different disciplines, such as organic chemistry, bioorganic chemistry, pharmacology and biology. About a dozen of marine alkaloids are currently in various phases of human clinical trials for treatment of different cancers2. The largest number of bioactive marine alkaloids with novel structures have been isolated from marine sponges.3,4 Sponges produce a plethora of chemical compounds with widely varying carbon skeletons. Most bioactive compounds from sponges have exhibited a variety of activities such as anti-inflammatory, antitumor, immunosuppressive, neurosuppressive, antiviral, antimalarial and antibiotic activities.4 While a number of these alkaloids have been isolated in quantities sufficient to ascertain their biological profile, many with unique structures are available only in minute quantities, precluding their thorough biological evaluations. Laboratory synthesis of these alkaloids is the only practical solution to this problem.
Marine sponges of the genera Latrunculia, Batzella, Prianos and Zyzzya are a rich source of alkaloids bearing a pyrrolo[4,3,2-de]quinoline skeleton.5,6 This series of alkaloids comprise of about 60 metabolites including discorhabdins,7 epinardins,8 batzellines,9 isobatzellines,9 makaluvamines10–15 and veiutamine16. Pyrrolo[4,3,2-de]quinoline alkaloids have shown a variety of biological activities such as inhibition of topoisomerase I14 and II,10 cytotoxicity against different tumor cell lines,10,17 antifungal14 and antimicrobial activities18. Pyrrolo[4,3,2-de]quinoline alkaloids have recently received increasing attention as a source of new anticancer drugs.19–24 Their unique fused ring skeletons carrying interesting biological properties have made them targets for several synthetic and biological studies. There has been a rapid growth of interest in the synthesis and biological evaluation of this class of compounds and their analogs. Several reviews have been published on the chemistry and bioactivity of this class of compounds.6, 25–26 Our interest is focused on makaluvamines which belong to this family of alkaloids. Makaluvamines A-P is a group of 16 marine alkaloids isolated mainly from four species of marine sponges, namely the Fijian sponge Zyzzya cf. marsailis,10 Indonesian sponge Histodermella sp,14 Pohnpeian sponge Zyzzya fuliginosa11 and Jamaican sponge Smenospongia aurea.15
Makaluvamines have exhibited in vitro cytotoxicity against human colon tumor cell line, HCT-116. The cytotoxicities exhibited by makaluvamines against a Chinese hamster ovary (CHO) cell line Xrs-6 have paralleled the data obtained with HCT-11610. Makaluvamines produce their anticancer activity by targeting the enzyme topoisomerase II as shown by a decatenation inhibition assay10. According to a recent publication it is possible that makaluvamines produce their anticancer activity by other mechanisms as well27.
As a part of our research on marine natural product analogs with potential pharmacological value, we have been interested in studying the cytotoxicity of makaluvamine analogs. We propose to make these analogs by introducing different substituents at the 7-position of the pyrroloiminoquinone ring present in makaluvamines. Many pyrroloiminoquinone alkaloids with proven cytotoxicities were found to have substitutions at the 7-position of the ring. We decided to explore some simple substitutions with increased steric bulk, hydrophobicity and hydrophilicity at the 7-position of the pyrroloiminoquinone ring and study their effects on topoisomerase II inhibition and cytotoxicity. Proposed target structures are given in Figure 2.
Figure 2.

Makaluvamine analogs were synthesized from the pyrroloiminoquinone derivative 5. Synthesis of compound 5 has been reported in the literature by several groups.28 We prepared this compound following the 4,6,7-trimethoxyindole approach described previously.29 This compound was converted to the makaluvamine analogs in two steps as outlined in Scheme 1.
Scheme 1.

Treatment of the pyrroloiminoquinone derivative 5 with different amine derivatives (6a–g) in anhydrous methanol at room temperature provided the aminated compounds 7a–g in 51–68 % yield. Removal of tosyl protecting group from the compound 7a–g was accomplished by treatment with NaOMe in MeOH to obtain the final products 4a–g in 48–62 % yield. In the synthesis of compounds 4a–b, intermediate products 7a–b were not isolated as the amination and detosylation took place in a single step leading to the formation of final products. This type of detosylation in the presence of amines was not unusual and has been previously reported in literature.29 All these compounds were individually synthesized and fully characterized.
Compounds 4a–g and 7c–g were evaluated for their cytotoxicities against human breast cancer cell lines MCF-7 and MDA-MB-468 and human colon cancer cell line HCT-116. The dose of the compound that inhibits 50% cell proliferation (IC50) was calculated using the data generated from 2–4 independent tetrazolium-based (XTT) cytotoxicity assays (R & D Systems Inc., Minneapolis, MN). Two known topoisomerase II targeting drugs, m-AMSA and etoposide, were included for comparisons. Results of cytotoxic assays are summarized in Table 1. HCT-116 cells were shown to be the most sensitive to etoposide and m-AMSA with IC50 doses of 1.7 μM and 0.7 μM, respectively. MDA-MB-468 cells showed IC50 doses of 13.6 μM and 8.5 μM for etoposide and m-AMSA, respectively. MCF-7 cells were shown to be the least sensitive with IC50 values of 35.6 μM and 21.7 μM for etoposide and m-AMSA, respectively. Several of makaluvamine analogs have shown significantly better inhibition than the control drugs in these assays. Four makaluvamine analogs (4c, 7d, 7f and 7g) exhibited better IC50 values against HCT-116 as compared to control drug etoposide. One analog (7d) exhibited better IC50 value against HCT-116 as compared to m-AMSA. All twelve of the makaluvamine analogs exhibited better IC50 values against MCF-7 and MDA-MB-468 as compared to etoposide as well as m-AMSA. Compound that showed best activity against HCT-116 is the N-tosyl-6-phenethylamino derivative (7d) with an IC50 value of 0.5 μM. The compound that showed best activity against MCF-7 is the benzyl amino derivative (4c) with an IC50 value of 1.0 μM. Benzyl amino derivative, 4c and phenethyl amino derivative, 4d showed best activity against MDA-MB-468 with IC50 value of 0.3 μM for each.
Table 1.
In vitro cytotoxicity and inhibition of topoisomerase II catalytic activities of makaluvamine analogs
| Structure | Cpd No | R = | Inhibition of Cancer Cell Proliferation (IC50,μM)a | Topoisom erase II Inhibitionb | ||
|---|---|---|---|---|---|---|
| HCT-116 | MCF-7 | MDA-MB-468 | ||||
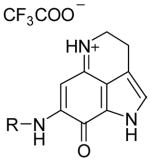
|
4a | −CH3 | 2.5 ± 1.3 | 1.3 ± 0.1 | 0.4 ± 0.05 | - |
| 4b | −CH2CH3 | 3.6 ± 0.7 | 1.3 ± 0.3 | 0.4 ± 0.03 | - | |
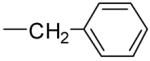
| 4c |
|
1.3 ± 0.2 | 1.0 ± 0.4 | 0.3 ± 0.18 | ++ | |
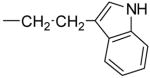
| 4d |
|
3.9 ± 1.2 | 1.7 ± 0.5 | 0.3 ±0.27 | + | |
| 2 |
|
13.2 ± 0.9 | 3.2 ± 0.6 | 0.6 ± 0.05 | - | |
| 4f |

|
14.4 ± 1.2 | 13.2 ± 3.1 | 4.5 ± 0.8 | +++ | |
| 4g |
|
5.2 ± 0.8 | 1.6 ± 0.2 | 0.4 ± 0.01 | - | |
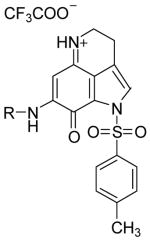
|
7c |
|
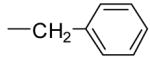
2.7 ± 1.5 | 1.8 ± 0.2 | 1.0 ± 0.01 | +++ |
| 7d |
|
0.5± 0.03 | 1.5 ±0.1 | 0.8 ±0.1 | -d | |
| 7e |
|
5.3 ± 0.6 | 5.1 ± 0.4 | 1.5 ± 0.14 | +++ | |
| 7f |
|
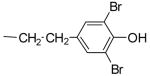
1.0± 0.2 | 2.3 ±1.1 | 1.5 ± 0.9 | -d | |
| 7g |
|
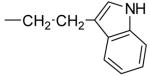
0.8 ± 0.3 | 1.2 ± 0.03 | 1.4 ± 0.9 | -d | |
| Etoposide | -c | 1.7 ± 0.2 | 35.6 ± 3.4 | 13.6 ± 0.6 | + | |
| m-AMSA | -c | 0.7 ± 0.3 | 21.7 ± 2.5 | 8.5 ± 1.2 | ++ | |
The dose that inhibits 50% cell proliferation (IC50) was determined in human colon cancer cell line, HCT-116, and the human breast cancer cell lines, MCF-7 and MDA-MB-468 (ATCC, Manassas, VA). The IC50 dose from 2–4 independent XTT assays performed in triplicate were combined for an average ± standard deviation.
Topoisomerase II (4U) was incubated with 500 ng plasmid DNA containing vehicle (DMSO) or 100 μM drug compound as described in the Topo II Drug Screening Kit protocol (TopoGEN, Inc). m-AMSA and etoposide were used as positive controls. Relaxed DNA was separated using non-ethidium bromide (EtBr) agarose gels, then stained with EtBr and quantified using Kodak Gel Logic Imaging System and Molecular Imaging software (Eastman Kodak Co., Rochester, NY). Inhibition of relaxation of plasmid DNA or catalytic activity was reported as; – none, + low, ++ moderate, or +++ strong.
not applicable;
not determined.
The final products (4a–g) and the intermediate products (7c–g) were also screened for their ability to inhibit topoisomerase II enzymatic activity. Topoisomerase II functions by generating a double-stranded DNA break followed by resealing of the break. Topoisomerase II inhibitors will interfere with the breakage-rejoining reaction thereby trapping the enzyme in a cleavage complex. In order to find out the ability of our compounds to inhibit topoisomerase II enzymatic activity, we used a topoisomerase-II drug screening kit (TopoGEN, Inc., Port Orange, FL) that uses a supercoiled plasmid DNA substrate (pRYG) which contains one topoisomerase II cleavage/recognition site. Two topoisomerase II inhibiting cancer drugs m–AMSA and etoposide were used as controls in these assays. The results of our assays are summarized in table 1. Five out of nine tested makaluvamine analogs exhibited inhibition of topoisomerase II catalytic activity comparable to etoposide and m-AMSA. Three of these compounds (4f, 7c and 7e) showed the strongest inhibition of catalytic activity of topoisomerase II.
In conclusion, several analogs of makaluvamines have been synthesized and characterized. Anticancer activity of these analogs against human colon tumor cell line, HCT-116 and human breast cancer cell lines, MCF-7 and MDA-MB-468 were evaluated. Four makaluvamine analogs exhibited better IC50 values against HCT-116 as compared to control drug etoposide. One analog exhibited better IC50 value against HCT-116 as compared to m-AMSA. All twelve of the makaluvamine analogs exhibited better IC50 values against MCF-7 and MDA-MB-468 as compared to etoposide as well as m-AMSA. Makaluvamine analogs were also evaluated for their ability to inhibit topoisomerase II enzymatic activity and found that five makaluvamine analogs exhibited inhibition of topoisomerase II comparable to etoposide and m-AMSA.
Supplementary Material
Figure 1.

Acknowledgments
Authors wish to acknowledge the financial support by Breast Spore pilot grant from the University of Alabama at Birmingham (UAB). SV also wish to acknowledge the financial support by a faculty development grant from UAB (FGDP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Blunt JW, Copp BR, Munro MHG, Northcote PT, Prinsep MR. Nat Prod Rep. 2005;22:15. doi: 10.1039/b415080p. [DOI] [PubMed] [Google Scholar]
- 2.Newman DJ, Cragg GM. J Nat Prod. 2004;67:1216. doi: 10.1021/np040031y. [DOI] [PubMed] [Google Scholar]
- 3.Sipkema D, Franssen MCR, Osinga R, Tramper J, Wijffels RH. Mar Biotechnol. 2005;7:142. doi: 10.1007/s10126-004-0405-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Higa T, Tanaka J, Kitamura A, Koyama T, Takahashi M, Uchida T. Pure Appl Chem. 1994;66:2227. [Google Scholar]
- 5.Faulkner DJ. Nat Prod Rep. 2002;19:1–48. doi: 10.1039/b009029h. and previous reviews in the series. [DOI] [PubMed] [Google Scholar]
- 6.Antunes EM, Copp BR, Davies-Coleman MT, Samaai T. Nat Prod Rep. 2005;22:62. doi: 10.1039/b407299p. [DOI] [PubMed] [Google Scholar]
- 7.Dijoux MG, Gamble WR, Hallock YF, Cardellina JH, Van Soest R, Boyd MR. J Nat Prod. 1999;62:636. doi: 10.1021/np980465r. [DOI] [PubMed] [Google Scholar]
- 8.D’Ambrosio M, Guerriero A, Chiasera G, Pietra F. Tetrahedron. 1996;52:8899. [Google Scholar]
- 9.Chang LC, Otero-Quintero S, Hooper JNA, Bewley CA. J Nat Prod. 2002;65:776. doi: 10.1021/np010581l. [DOI] [PubMed] [Google Scholar]
- 10.Radisky DC, Radisky ES, Barrows LR, Copp BR, Kramer RA, Ireland CM. J Am Chem Soc. 1993;115:1632. [Google Scholar]
- 11.Schmidt EW, Harper MK, Faulkner DJ. J Nat Prod. 1995;58:1861. doi: 10.1021/np50126a008. [DOI] [PubMed] [Google Scholar]
- 12.Venables DA, Concepcion GP, Matsumota SS, Barrows LR, Ireland CM. J Nat Prod. 1997;60:408. doi: 10.1021/np9607262. [DOI] [PubMed] [Google Scholar]
- 13.Casapullo A, Cutignano A, Bruno I, Bifulco G, Debitus C, Gomez-Paloma L, Riccio R. J Nat Prod. 2001;64:1354. doi: 10.1021/np010053+. [DOI] [PubMed] [Google Scholar]
- 14.Carney JR, Scheuer PJ, Kelly-Borges M. Tetrahedron. 1993;49:8483. doi: 10.1021/np50091a025. [DOI] [PubMed] [Google Scholar]
- 15.Hu JF, Schetz JA, Kelly M, Peng JN, Ang KKH, Flotow H, Leong CY, Ng SB, Buss AD, Wilkins SP, Hamann MT. J Nat Prod. 2002;65:476. doi: 10.1021/np010471e. [DOI] [PubMed] [Google Scholar]
- 16.Venables DA, Barrows LR, Lassota P, Ireland CM. Tetrahedron Lett. 1997;38:721. [Google Scholar]
- 17.Gunasekera SP, Zuleta IA, Longley RE, Wright AE, Pomponi SA. J Nat Prod. 2003;66:1615. doi: 10.1021/np030292s. [DOI] [PubMed] [Google Scholar]
- 18.Perry NB, Blunt JW, Munro MHG. Tetrahedron. 1988;44:1727. [Google Scholar]
- 19.Kokoshka JM, Capson TL, Holden JA, Ireland CM, Barrows LR. Anti-Cancer Drugs. 1996;7:758. doi: 10.1097/00001813-199609000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Bénéteau V, Pierré A, Pfeiffer B, Renard P, Besson T. Bioorg Med Chem Lett. 2000;10:2231. doi: 10.1016/s0960-894x(00)00450-9. [DOI] [PubMed] [Google Scholar]
- 21.Bénéteau V, Besson T. Tetrahedron Lett. 2001;42:2673. [Google Scholar]
- 22.Zhao R, Oreski B, Lown W. Bioorg Med Chem Lett. 1996;6:2169. [Google Scholar]
- 23.Legentil L, Lesur B, Delfourne E. Bioorg Med Chem Lett. 2006;16:427. doi: 10.1016/j.bmcl.2005.09.063. [DOI] [PubMed] [Google Scholar]
- 24.Legentil L, Benel L, Bertrand V, Lesur B, Delfourne E. J Med Chem. 2006;49:2979. doi: 10.1021/jm051247f. [DOI] [PubMed] [Google Scholar]
- 25.Urban S, Hickford SJH, Blunt JW, Munro MHG. Curr Org Chem. 2000;4:778. [Google Scholar]
- 26.Ding Q, Chichak K, Lown JW. Curr Med Chem. 1999;6:1. [PubMed] [Google Scholar]
- 27.Dijoux M-G, Schnabel PC, Hallock YF, Boswell JL, Johnson TR, Wilson JA, Ireland CM, Soest R, Boyd MR, Barrows LR, Cardellina JH., II Bioorg Med Chem. 2005;13:6035. doi: 10.1016/j.bmc.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 28.Harayama Y, Kita Y. Curr Org Chem. 2005;9:1567. [Google Scholar]
- 29.Sadanandan EV, Pillai SK, Lakshmikantham MV, Billimoria AD, Culpepper JS, Cava MP. J Org Chem. 1995;60:1800. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.