

Summary
Aberrant Wnt/β-catenin signaling following loss of the tumor suppressor adenomatous polyposis coli (APC) is thought to initiate colon adenoma formation. Using zebrafish and human cells, we show that homozygous loss of APC causes failed intestinal cell differentiation, but that this occurs in the absence of nuclear β-catenin and increased intestinal cell proliferation. Therefore, loss of APC is insufficient for causing β-catenin nuclear localization. APC mutation-induced intestinal differentiation defects instead depend on the transcriptional corepressor CtBP1 whereas proliferation defects, and nuclear accumulation of β-catenin, require the additional activation of KRAS. These findings suggest that, following APC loss, CtBP1 contributes to adenoma initiation as a first step, while KRAS activation and β-catenin nuclear localization promotes adenoma progression to carcinomas as a second step. Consistent with this model, human FAP adenomas showed robust upregulation of CtBP1 in the absence of detectable nuclear β-catenin, whereas nuclear β-catenin was detected in carcinomas.
Introduction
Mutations in the adenomatous polyposis coli (APC) tumor suppressor gene initiate the process of colon carcinogenesis (Kinzler and Vogelstein, 1996). Current studies indicate that APC functions in normal colon to negatively regulate the Wnt signaling by targeting β-catenin for degradation by the proteosome (Bienz and Clevers, 2000; Clevers, 2006). Wildtype APC plays a central role in a destruction complex that includes Axin, GSK-3β and casein kinase 1 (CK1). This complex directs a series of phosphorylation events on β-catenin that target it for ubiquitination and subsequent proteolysis (Polakis, 2002). In cells harboring mutated APC, β-catenin accumulates and, following its translocation into the nucleus, is thought to co-activate TCF-LEF (Morin, 1999; Polakis, 1999). β-catenin/TCF-LEF-dependent transcriptional activation of cell cycle regulatory genes, like c-myc (He et al., 1998) and cyclin D1 (Tetsu and McCormick, 1999), serves as the mechanistic basis for explaining the APC mutation-dependent initiation of colon adenomas.
The current model of APC function predicts an immediate activation of Wnt signaling upon homozygous loss of APC. This view places aberrant Wnt signaling, along with APC mutation, as the initiating event for intestinal adenoma formation. Manipulation of Apc in murine models offer considerable support for the immediate dysregulation of Wnt signaling following APC loss (Hinoi et al., 2007; Kongkanuntn et al., 1999; Sansom et al., 2004). In these models, dysregulation and nuclear accumulation of β-catenin appear coincident with the development of early adenomas. The presence of stabilizing mutations in β-catenin, rather than APC, in approximately 7% of sporadic human colon carcinomas provides additional genetic evidence directly linking APC and β-catenin (Iwao et al., 1998; Morin et al., 1997; Sparks et al., 1998). Indeed, transgenic mice carrying stabilized, mutant β-catenin develop numerous intestinal adenomas (Romagnolo et al., 1999). Taken together, these studies offer dysregulation of β-catenin as a compelling model that describes the molecular events that accompany APC loss and colon tumor initiation.
Despite accumulating evidence supporting the current model, several studies have failed to confirm the nuclear accumulation of β-catenin upon APC mutation in early human colon adenomas. Anderson et al. examined the subcellular localization of β-catenin in grossly uninvolved and adenoma tissues taken from FAP patients and were unable to detect nuclear β-catenin in greater than 90% of the adenomas examined. However, nuclear β-catenin was readily detected in sporadic adenocarcinomas (Anderson et al., 2002). Similarly, Blaker et al. examined early adenomas with mild dysplasia and late adenomas with significant dysplasia. Although they observed elevated levels of β-catenin in the early adenomas, this β-catenin was confined to the cytoplasm. Again, they did detect nuclear β-catenin in the late adenomas (Blaker et al., 2003). Based on these observations, both groups concluded that homozygous loss of APC alone is insufficient for nuclear accumulation of β-catenin. In agreement with these findings, recent analysis of microadenomas and late adenomas taken from a novel rat model (PIRC) carrying a targeted Apc truncation revealed detectable levels of nuclear β-catenin only in the late adenomas (Amos-Landgraf et al., 2007). Taken together, these findings suggest that Wnt activation, as assessed by nuclear accumulation of β-catenin, may require events in addition to loss of APC. Further, these findings raise the possibility that initiation of adenoma formation following APC loss occurs independently of β-catenin.
KRAS mutations are evident in as many as 40% of colon cancers (Bos et al., 1987; Forrester et al., 1987) and can arise early in adenoma development. Consistent with the multi-hit hypothesis for colon tumor development (Fearon and Vogelstein, 1990), reports in mice indicate that the combination of APC loss and KRAS mutation causes an increase in adenoma size, number and invasiveness (Janssen et al., 2006; Sansom et al., 2006) and promotes the expansion of cells bearing putative stem cell markers within the tumor epithelium (Haigis et al., 2008). Although these studies have demonstrated a critical relationship between APC and KRAS, the models employed limit analysis of the immediate consequences of APC loss on intestinal cells. As such, the precise sequence of events and underlying mechanisms contributing to adenoma formation remain unclear. Herein, we employ comparative studies in zebrafish and human cell lines to show that APC mutation induced intestinal cell fating defects result from dysregulation of C-terminal binding protein-1 (CtBP-1) and precede activation of β-catenin. Rather, the activation of β-catenin controls cell proliferation but requires KRAS, RAF and RAC1 in addition to APC loss.
Results
KRAS promotes intestinal cell proliferation and activation of β-catenin following loss of APC
Homozygous apc mutant zebrafish (apcmcr) embryos display a variety of profound developmental defects and facilitate interrogation of the immediate morphologic and mechanistic consequences of apc loss (Hurlstone et al., 2003; Nadauld et al., 2004). In examining sagital- and cross-sections of the entire intestine of apcmcr zebrafish embryos, we noted an unexpected 34% decrease in the total number of intestinal epithelial cells (Figures 1A, S1 and Table S1) suggesting an absence of Wnt signaling in these cells. Consistent with this possibility, immunofluorescence staining revealed that while β-catenin was robustly upregulated in intestinal cells, its localization was confined to the cytoplasm (Figure 1A). To test whether oncogenic KRAS might constitute an additional trigger needed to stimulate intestinal cell proliferation and accumulation of nuclear β-catenin following loss of apc, we injected apcmcr zebrafish with mRNA encoding oncogenic V5-KRASG12D (hereafter referred to as KRAS). Western analysis confirmed expression of KRAS protein in injected embryos and indicated activation of the KRAS signaling pathway by the presence of increased phospho-ERK (Figure 1B). Injection of KRAS alone failed to induce intestinal cell proliferation or nuclear accumulation of β-catenin (Figure 1A and Table S1). In contrast, injection of KRAS into apcmcr embryos (hereafter referred to as APC-KRAS) caused a robust proliferative response as indicated by a four-fold increase in intestinal cell number relative to apcmcr and robust expression of pcna (Figure 1A and Table S1). Consistent with a requirement for KRAS activity in the nuclear accumulation of β-catenin and a requirement for β-catenin in directing intestinal cell proliferation, nuclear β-catenin was evident only in the proliferative APC-KRAS intestinal cells (Figure 1A).
Figure 1. KRAS promotes intestinal cell proliferation and nuclear localization of β-catenin following loss of APC.
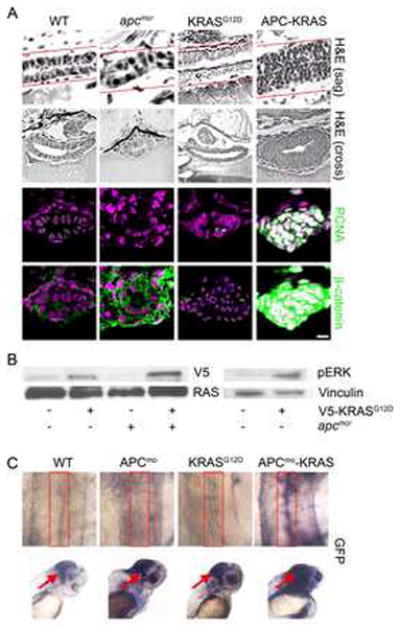
(A) apcmcr zebrafish embryos were injected at the one cell stage with KRAS mRNA. 72 hpf embryos were fixed, sectioned and stained for hematoxylin and eosin (H&E) (Row 1, sagital section; row 2, cross section), DNA (magenta- middle and bottom) and either PCNA (middle-green) or β-catenin (bottom- green). Overlapping expression is shown in white. (B) Protein lysates from 72 hpf embryos were subjected to western blot analysis for either V5-tag (top-left) and total RAS (bottom-left) or phospho-Erk (top-right) and vinculin (bottom-right). (C) Zebrafish harboring an integrated β-catenin TOPGFP reporter were injected with KRAS mRNA, APC morpholino or both. 72 hpf embryos were subjected to whole mount in situ hybridization for GFP. Boxes indicate the intestine (top) and arrows indicate the hindbrain (bottom). All images were captured using the same exposure and represent at least three independent experiments. (Scale Bar: 10μm)
As predicted from the above findings, zebrafish harboring a TOPdGFP reporter construct lacked detectable expression of intestinal GFP upon knock down of apc (Figures 1C and S1) or injection of KRAS only (Figure 1C). However, APCmoKRAS-TOPdGFP embryos displayed robust expression of GFP (Figure 1C) within the intestines, thereby confirming the activation of β-catenin coincident with its nuclear localization and the proliferative state of the intestinal cells. In contrast to the intestines, control injected TOPdGFP embryos showed strong expression of GFP within the hindbrain region and the lens as seen previously (Nadauld et al., 2006a). Consistent with a role for apc and kras in these tissues, knockdown of apc alone or injection of KRAS mRNA each increased GFP expression in comparison to control embryos. This induction was further enhanced by coincident knockdown of apc along with injection of KRAS (Figure 1C).
KRAS and RAF1 mediate nuclear localization of β-catenin
Next, we determined which KRAS effectors mediated targeting of β-catenin to the nucleus. Injection of either constitutively active RAF1 (RAF1) or MEK1 (MEK1) into wildtype embryos failed to induce nuclear accumulation of β-catenin or intestinal cell proliferation. Again increased phospho-erk confirmed the presence of active RAF1 and MEK1 in these embryos (Figure S2). As seen with KRAS, injection of apcmcr embryos with RAF1 caused increased intestinal cell proliferation and accumulation of nuclear β-catenin (Figure 2A and Table S1). In contrast, injection of MEK1 into apcmcr embryos failed to enhance intestinal cell proliferation or accumulation of nuclear β-catenin (Figure 2A and Table S1), thereby suggesting a requirement for RAF1, but not MEK1, in KRAS regulation of β-catenin. A mek1-independent role of kras and raf1 was also evident in that APC-KRAS and APC-RAF1 embryos shared distinct morphologic phenotypes not present in APC-MEK1 embryos (Figure S2). For example, we noted the occurrence of a cyclops phenotype in approximately 40% of the APC-KRAS or APC-RAF1 embryos that was absent in the APC-MEK1 embryos (Figure 5A and data not shown).
Figure 2. KRAS and RAF1 are necessary for nuclear localization of β-catenin and intestinal cell proliferation following loss of APC.
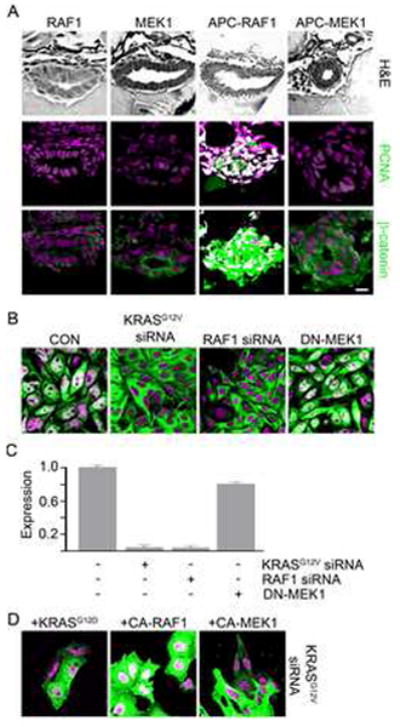
(A) apcmcr zebrafish embryos injected with constitutively active RAF1 or MEK1. 72 hpf embryos were stained for hematoxylin and eosin (H&E) staining (top), DNA (magenta-middle and bottom) and either PCNA (middle-green) or β-catenin (bottom-green). (B) SW-480 cells transfected with control, KRASG12V, RAF1 directed siRNA or DN-MEK1 constructs were stained for DNA (magenta) and β-catenin (green). (C) RNA was harvested and subjected to rt-PCR for Axin2. (D) SW-480 cells transfected with KRASG12V-specific siRNA were cotransfected with constitutively active KRAS, RAF1 or MEK1 and stained for DNA (magenta) and β-catenin (green). Overlapping expression is shown in white. All images were captured using the same exposure and represent three independent experiments. (Scale Bar: 10μm)
Figure 5. KRAS-mediated intestinal cell proliferation following loss of APC requires β-catenin.
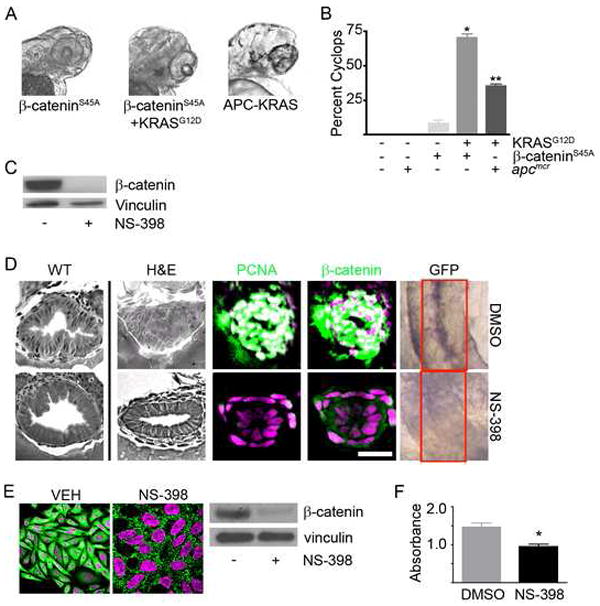
(A) Zebrafish embryos were injected with β-cateninS45A mRNA (left panel) along with KRAS mRNA (middle panel). Also shown is a representative image of the APC-KRAS embryo (right panel). At 72hpf, the embryos were fixed and photographed. (B) The percent cyclops was analyzed (*p<0.01 WT vs β-catenin-KRAS, **p<0.01 WT vs. APC-KRAS). (C) Protein was harvested from apcmcr embryos treated with DMSO or NS-398 and subjected to western blot analysis for β-catenin (top) or vinculin (bottom). (D) Wildtype uninjected or apcmcr embryos injected with KRAS mRNA treated with VEH (top) or NS-398 (bottom) were stained by H&E (WT-left, APC-KRAS-right) and for DNA (magenta), PCNA (green) and β-catenin (green). TOPGFP-APCmo-KRAS embryos were stained for GFP expression (purple). Boxes indicate the intestine. (E) SW-480 cells treated with DMSO or NS-398 were stained for DNA (magenta) and β-catenin (green). Protein lysates were subjected to western blot analysis for β-catenin (top) and vinculin (bottom). (F) Cells were subjected to MTT analysis (*p<0.05 vs DMSO). Overlapping expression is shown in white. All images were captured using the same exposure and represent at least three independent experiments. (Scale Bar: 10μm)
To confirm the above findings in human cell lines, we employed the SW-480 colon cancer cell line as it harbors mutated APC, oncogenic KRAS and shows robust staining for nuclear β-catenin (Figure 2B). Specific targeting of endogenous KRAS or RAF1 using multiple, distinct siRNAs caused a redistribution of nuclear β-catenin to the cytoplasm (Figure 2B, S2 and S3). This was paralleled by the down-regulation of the known β-catenin target gene Axin2 (Figure 2C). Inhibition of MEK1 by transfection of a dominant negative construct or using the pharmacologic inhibitor U0126 failed to alter β-catenin subcellular localization (Figure 2B and S3) despite decreasing phospho-ERK levels (Figure S3). In confirming specificity of the knock down studies, co-transfection of cells with siKRASG12V and constitutively active KRAS or RAF1 (Figure 2D and S3) restored localization of β-catenin to the nucleus. Again, CA-MEK1 was ineffective (Figure 2D and S3).
Next, we sought to manipulate the accumulation and subcellular distribution of β-catenin in zebrafish and human 293 cells containing wildtype APC. Recent studies have implicated PGE2 in stabilizing β-catenin following loss of APC (Castellone et al., 2005; Eisinger et al., 2007; Shao et al., 2005). In agreement with these findings, treatment of wildtype zebrafish with PGE2 mirrored loss of apc and increased cytoplasmic levels of β-catenin without affecting cell proliferation (Figure 3A). Injection of KRAS mRNA coincident with PGE2 treatment increased pcna expression and nuclear localization of β-catenin (Figure 3A) within the intestinal epithelial cells.
Figure 3. KRAS and RAF1 direct stabilized β-catenin to the nucleus.
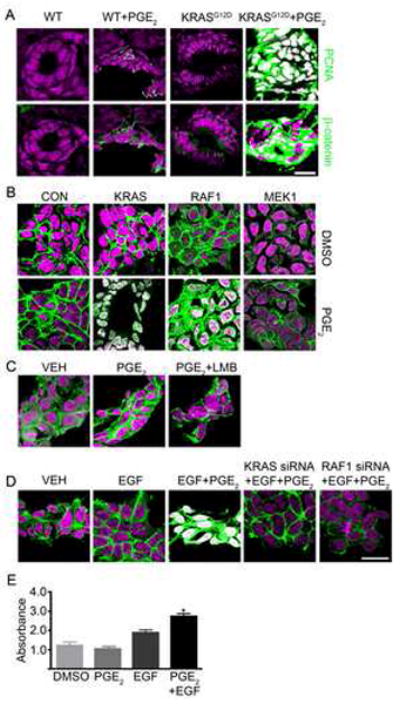
(A) WT and KRAS-injected zebrafish embryos treated with PGE2 were stained for DNA (magenta), PCNA (green) and β-catenin (green). (B) Human 293 cells were transfected with constitutively active KRAS, RAF1 or MEK1 and either DMSO (top) or PGE2 (bottom). Cells were stained for DNA (magenta) and β-catenin (green). (C) 293 cells treated with DMSO, PGE2 or PGE2 and leptomycin B were stained for DNA (magenta) and β-catenin (green). (D) 293 cells were transfected with control or KRAS- or RAF1-directed siRNA and treated with EGF and PGE2 or vehicle then stained for DNA (magenta) and β-catenin (green). (E) 293 cells were subjected to an MTT assay (*p<0.05 vs DMSO). Overlapping expression is shown in white. All images were captured using the same exposure and represent three independent experiments. (Scale Bar: 10μm)
In assessing this observation in human cells, treatment of 293 cells with PGE2 alone or transfection with KRAS showed little change in the subcellular localization of β-catenin (Figure 3B). In contrast, treatment with PGE2 along with transfection with KRAS showed redistribution of β-catenin to the nucleus (Figure 3B and S3), thereby supporting a requirement for KRAS activity in stimulating the translocation of PGE2-stabilized β-catenin into the nucleus of human cells. In support of the results above, expression of RAF1, but not MEK1 (Figure 3B), along with PGE2 exposure also promoted robust redistribution of β-catenin into the nucleus. This redistribution was confirmed by western blotting of nuclear fractions (Figure S3). Exposure of cells to PGE2 and the nuclear export inhibitor leptomycin B failed to stimulate accumulation of nuclear β-catenin (Figure 3C) and eliminated nuclear retention as an underlying mechanism. As a control, we found robust nuclear retention of Mondo A in response to leptomycin B (data not shown). Interestingly, EGF effectively recapitulated the action of KRAS (Figure 3D) and caused nuclear accumulation of β-catenin within 15 minutes of exposure (Figure S3). Consistent with a role for KRAS and RAF1, siRNA targeting of either block the actions of EGF. Further, EGF enhanced cell growth following treatment of cells with PGE2 (Figure 3E). This demonstrates that activation of KRAS and RAF1, either through oncogenic mutation or growth factor stimulation, signals to regulate β-catenin nuclear localization.
KRAS/RAF1 regulation of β-catenin requires the activity of RAC1
Previous studies have implicated RAC1 in controlling the nuclear localization of β-catenin in cells responding to Wnt ligand (Wu et al., 2008). As phosphorylation of serine 191 and 605 on β-catenin depended upon RAC1, we transfected SW-480 cells with β-catenin constructs carrying mutations in either of these sites and noted that β-catenin remained in the cytoplasm (Figure 4A), despite the presence of mutant APC. In contrast, constructs bearing mutations in known PKA-dependent phosphorylation sites (Taurin et al., 2006) (552 and 675) effectively accumulated in the nucleus (Figure 4B). Consistent with these findings, inhibition of RAC1 prevented the nuclear accumulation of β-catenin (Figure 4C and S4) and was accompanied by decreased cell growth (Figure 4D). Inhibition of the closely related Cdc42 failed to affect the localization of β-catenin (Figure 4C and S4). As predicted from the above findings, inhibition of either KRAS or RAF1 reduced the levels of GTP-bound RAC1. In contrast, dominant negative inhibition of MEK1 failed to deactivate RAC1 (Figure 4E). We also noted reduced levels of phospho-cJun (a downstream readout of RAC1 activation) upon inhibition of RAC1, but not Cdc42 (Figure 4F). As expected, inhibition of KRAS or RAF1, but not MEK1, also resulted in decreased phospho-cJun staining (Figure 4F). Finally, expression of constitutively active RAC1 (CA-RAC1) along with stimulation by PGE2 substituted fully for KRAS or RAF1 in promoting the nuclear accumulation of β-catenin in 293 cells (Figure 4G).
Figure 4. KRAS/RAF1 Regulation of β-catenin requires the Activity of RAC1.
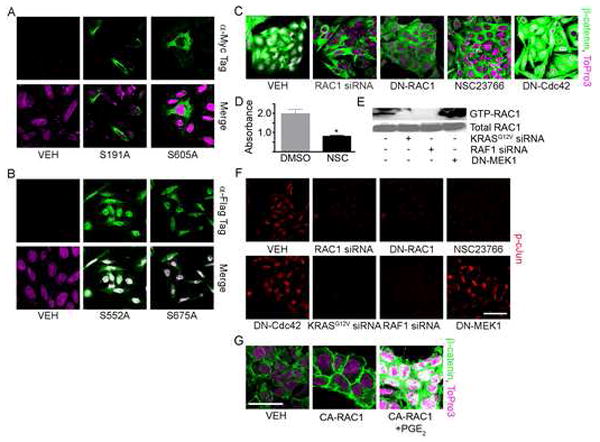
(A, B) SW-480 cells transfected with vehicle or myc-tagged (A, S191A or S605A) or flag-tagged (B, S552A or S675A) β-catenin mutants were stained for DNA (magenta) and α-myc (A, green) or α-flag (B, green). (C) SW-480 cells were transfected with RAC1-directed siRNA, DN-RAC1, DN-Cdc42 or treated with the RAC1-specific inhibitor NSC23766 were stained for DNA (magenta) and β-catenin (green). (D) SW-480 cells were subjected to an MTT assay (*p<0.05 vs DMSO). (E) SW-480 cells were transfected with vehicle, KRASG12V-targeted siRNA, RAF1 siRNA or DN-MEK1 and subjected to a RAC1 activity assay. The western blot was probed for RAC1 (top). Control lysates were probed for total RAC1 (bottom). (F) SW-480 cells treated as above were stained for phospho-cJun. (G) Human 293 cells transfected with constitutively active RAC1 and treated with PGE2 or DMSO were stained for DNA (magenta) and β-catenin (green). All images were captured using the same exposure and are representative of at least three independent experiments. (Scale Bar: 10μm)
KRAS enhances the activity of oncogenic β-catenin
Since APC has been implicated in regulating a number of cellular functions (Bienz and Hamada, 2004; Hanson and Miller, 2005; Nadauld et al., 2006a; Nadauld et al., 2006b; Nadauld et al., 2004; Nadauld et al., 2005; Takacs et al., 2008), it was important to test whether the observed KRAS synergy was mediated through β-catenin. We, therefore, asked whether KRAS enhanced the activity of mutated, stabilized β-catenin injected into wildtype embryos. Upon injection of low levels of stabilized β-catenin alone, we noticed that approximately 5% of the embryos displayed the cyclops phenotype present in the APC-KRAS embryos (Figure 5A). We, therefore, decided to exploit this phenotype to determine whether the effects of KRAS were synergistic with β-catenin. Consistent with a genetic relationship between KRAS and β-catenin, injection of wildtype zebrafish with stabilized, oncogenic β-catenin and KRAS gave similar results to the APC-KRAS embryos (Figure 5B) in that nearly 75% of the embryos displayed the cyclops phenotype (Figure 5B). Injection of KRAS showed no effect. (Note: Examination of the intestines of these embryos showed an apparent increase in cell number. However, the gut tube also duplicated. We were, therefore, unable to distinguish gut duplication from increased cell number due to proliferation). As a measure of in vivo activity, the 191 and 605 mutants failed to synergize with KRAS in promoting a cyclops phenotype in zebrafish embryos (data not shown).
APC mutation induced intestinal cell differentiation defects occur independently of β-catenin
To evaluate the role of β-catenin in intestinal cell proliferation and differentiation defects following apc loss, we used the COX-2 inhibitor (NS-398) (Eisinger et al., 2007) to decrease the level of β-catenin in APC-KRAS zebrafish (Figure 5C). We found that this treatment effectively abrogated the KRAS-dependent increase in intestinal cell number and pcna staining (Figure 5D and Table S1). This change was accompanied by reduced levels of nuclear β-catenin and expression of intestinal TOPdGFP (Figure 5D). Similar to findings seen in zebrafish, inhibition of COX-2 resulted in decreased nuclear accumulation of β-catenin (Figure 5E) and cell growth (Figure 5F). Notably inhibiton of COX-2 failed to restore morphological features typical of differentiated intestinal epithelial cells (Figure 5D).
As a number of studies have demonstrated a relationship between APC and the transcriptional corepressor C-terminal binding protein-1 (CtBP1) (Fang et al., 2006; Hamada and Bienz, 2004; Nadauld et al., 2006b; Sierra et al., 2006), we determined the contribution of ctbp and β-catenin to the observed lack of differentiation present in homozygous apcmcr embryos. As seen previously (Nadauld et al., 2006b), injection of a wildtype fragment of APC (APC955-2075) into apcmcr zebrafish embryos caused coincident down-regulation of β-catenin and ctbp1 (Figure 6A) and restored the normal expression pattern of the intestinal differentiation marker i-fabp (Figure 6B). In contrast, injection of an APC construct containing site-directed mutants in the three SAMP motifs (APC955-2075-AALP) needed for AXIN binding (Spink et al., 2000) failed to regulate either β-catenin or ctbp1 (Figure 6A) and failed to restore i-fabp expression (Figure 6B) and indicated a role for the AXIN binding motifs in regulating both β-catenin and CtBP.
Figure 6. apc control of cellular fate and differentiation are mediated by ctbp1 and are β-catenin-independent.

(A) Protein from 48 hpf apcmcr zebrafish embryos injected with either 6xHis-APC955-2075 or 6xHis-APC955-2075-AALP were subjected to western blot analysis for β-catenin (top), ctbp1 (second), 6xHis (third) or vinculin (bottom). (B) Embryos injected as above were fixed at 72 hpf and subjected to in situ hybridization for i-fabp. (C) apcmcr embryos were treated with DMSO or NS-398 or injected with ctbp1-directed morpholino in the presence or absence of NS-398. At 72 hpf, the embryos were fixed and subjected to in situ hybridization for i-fabp, NaPi, hoxa13a or evx1. All images are representative of at least three independent experiments.
Since APC955-2075 regulated both β-catenin and CtBP, we examined the β-catenin and ctbp1 dependence of several intestinal cell fate (hoxa13a and evx1) and terminal differentiation markers (i-fabp and NaPi) that show aberrant expression patterns within the intestine of apcmcr zebrafish embryos. For example, hoxa13a and evx1 showed expansion of their expression domain from confinement within the proctodium to expression throughout the intestinal tube (Figure 6C). On the other hand, apcmcr zebrafish embryos lacked expression of i-fabp and NaPi (Figure 6C) present in wildtype embryos. To further delineate the roles of ctbp1 and β-catenin in controlling the aberrant expression of these markers, we treated the embryos with the COX-2 inhibitor NS-398 or injected apcmcr embryos with two separate morpholinos targeting ctbp1 (Figure 6C and S6). As shown in Figure 6C and consistent with our above findings, NS-398 mediated down-regulation of β-catenin failed to restore the normal expression of ifabp, NaPi, hoxa13a or evx1 indicating the β-catenin-independence of these defects. In contrast, knockdown of ctbp1 rescued each of the markers. Further, inclusion of NS-398 failed to affect rescue following knock down of ctbp1 and indicated that ctbp1 suppresses intestinal differentiation following loss of apc independently of β-catenin.
JNK activation is coincident with nuclear β-catenin in human carcinoma samples
To extend our findings to human tissues, we examined 20 matched sets of grossly uninvolved and adenoma tissues from FAP patients along with 20 sporadic carcinoma samples for expression and localization of CtBP1, β-catenin, and phospho-cJun (as an indicator of JNK activity). This analysis revealed upregulation of CtBP1 in the nucleus of both human adenoma and carcinoma tissues (Figure 7A). In these same samples, and similar to our finding in apcmcr zebrafish intestines above, there was an apparent increase in the level of β-catenin staining in the adenomatous tissues compared to the grossly uninvolved tissue (Figure 7B). However, as reported previously this staining was confined to cytoplasm (Anderson et al., 2002; Blaker et al., 2003). Adjacent tissue sections showed no evidence of cJun phoshorylation (Figure 7C). In contrast, nuclear β-catenin was detected in carcinomas (Figure 7B) and was paralleled by increased levels of phopsho-cJun (Figure 7C). [Note: Figure 6 shows two representative images. The remaining 18 samples are shown in Figure S7, S8 and S9. These 18 samples showed similar qualitative staining patterns as those utilized as representative images. Demographic information for each sample is provided in Table S2.]
Figure 7. JNK1 activation is coincident with nuclear accumulation of β-catenin in human tumor samples.

FFPE human matched grossly uninvolved and adenoma samples obtained from FAP patients and unmatched sporadic carcinomas were stained to indicate DNA (magenta) and (A) CtBP (green), (B) β-catenin* (green) or (C) phospho-cJun (green) as an indicator of JNK activity. Overall, 20 patient matched grossly uninvolved and adenoma or unmatched carcinoma tissue samples were stained. Shown are two representative samples. All images were captured using the same exposure and overlapping expression is shown in white. *Note: a section from each of the β-catenin-stained adenomas was enlarged to the right. Each antibody was evaluated using serial sections. (Scale Bar: 5μm)
Discussion
Considerable evidence implicates dysregulated Wnt/β-catenin signaling as the initiating event underlying colon adenoma formation following loss of APC (Bienz and Clevers, 2000; Clevers, 2006). Despite this evidence, studies examining human FAP adenomas raise the possibility that APC loss alone is insufficient to promote aberrant Wnt/β-catenin signaling (Anderson et al., 2002; Blaker et al., 2003). Further, although the genetic relationship between APC and KRAS mutation in promoting colon tumor development is well-established, (Fearon and Vogelstein, 1990; Janssen et al., 2006; Sansom et al., 2006) the roles for APC loss and KRAS mutation are largely viewed as independent in the process of tumorigenesis and that KRAS may only exacerbate Wnt signaling following APC mutation. Through juxtaposition of zebrafish with human cell lines, we present evidence that APC and KRAS play distinct but essential roles in controlling the stability and nuclear accumulation of β-catenin. Our findings support a model wherein APC loss alone stabilizes the levels of cytoplasmic β-catenin. This stabilization, however, is insufficient for stimulating nuclear accumulation of β-catenin and intestinal cell proliferation. Rather, the nuclear accumulation of β-catenin, and attendant proliferation, requires the activities of KRAS and RAF1. Signaling between KRAS/RAF1 and β-catenin occurs independently of MEK1 and instead relies on a novel role of RAC1 and JNK2 in promoting nuclear localization of β-catenin. These findings suggest that β-catenin-dependent intestinal cell proliferation may contribute to adenoma progression, rather than initiation following loss of APC.
Our findings offer a mechanistic explanation for data that have failed to confirm the presence of nuclear β-catenin in early adenomas taken from both human and rat tissues harboring mutated APC. For example, Anderson et al. reported that greater than 90% of adenomas taken from FAP patients lacked detectable nuclear β-catenin (Anderson et al., 2002). Further, Blaker et al. examined adenomas from FAP patients and concluded that, although all of the adenomas acquired homozygous loss of APC, none of the specimens displayed detectable nuclear accumulation of β-catenin (Blaker et al., 2003). In each of these studies nuclear β-catenin was readily observed in more advanced lesions. In addition, in a recently developed Apc mutation-dependent rat model of colorectal cancer, the PIRC model, nuclear β-catenin was not evident in microadenomas (Amos-Landgraf et al., 2007) but detectable in later stage adenomas. As with these studies, we were unable to detect nuclear β-catenin in the intestines of homozygous apcmcr zebrafish embryos and in human FAP adenomas. However, we found a mechanistic requirement for KRAS in promoting the nuclear localization of β-catenin in the intestinal cells of zebrafish carrying apc mutation and in human colon cancer cell lines. Furthermore, we observed that the presence of nuclear β-catenin in sporadic human carcinomas was paralleled by evidence of RAC1 activation. A need for KRAS activity in regulating nuclear accumulation of β-catenin provides a mechanistic basis for the differential staining of nuclear β-catenin in early versus late adenomas and carcinomas (Amos-Landgraf et al., 2007; Anderson et al., 2002; Blaker et al., 2003) and suggests a dependent, common regulatory point for APC loss and KRAS activity in the development of colon cancer.
The findings examining β-catenin subcellular localization in human tissues and zebrafish differ from a large body of data regarding the consequence of Apc mutation in mice. These studies have demonstrated the occurrence of nuclear β-catenin in very early lesions following loss of Apc (Hinoi et al., 2007; Kongkanuntn et al., 1999; Sansom et al., 2006; Sansom et al., 2004). Furthermore, a recent study ruled out the presence of mutated Kras in early adenoma development (Hinoi et al., 2007). As an important distinction, these studies have not formally tested a role for Kras or Raf1 in adenoma formation or in directing the nuclear localization of β-catenin following loss of APC. Interestingly, Sansom et al. did observed activation of Tiam1, the major Rac1-GEF at the earliest time point showing evident nuclear β-catenin in mice (Sansom et al., 2004). Our data implicating KRAS, RAF1 and RAC1 in the control of β-catenin subcellular distribution suggested a previously unappreciated molecular link between growth factor signaling and APC loss. Indeed, we found that treatment of human cells with EGF substituted for mutational activation of KRAS or RAF in controlling the nuclear accumulation of β-catenin. Previous work has implicated EGF signaling in neoplastic tissue development in the murine intestine. First, Roberts et al. demonstrated an essential role for EGF in the establishment and maintenance of adenomas in the Apcmin mouse (Roberts et al., 2002). In addition, Moran et al. reported that adenomas from Apcmin mice display increased EGFR activity (Moran et al., 2004). Taken together, the current data suggest that EGF signaling could actively support the nuclear accumulation of β-catenin in the absence of Kras mutation.
Apcmcr zebrafish contain a truncating mutation in apc at position 1318, which is within the mutation cluster region of APC as defined based on the occurrence of mutations underlying human colon cancer (Hurlstone et al., 2003). Studies examining adult apcmcr heterozygous zebrafish revealed infrequent tumor formation that was enhanced by administration of the non-specific chemical carcinogen DMBA (Haramis et al., 2006). Since these studies did not examine the mutational status of apc or kras or a requirement for raf1 or rac1 in tumor formation, it is unclear how the mutagen enhanced tumor formation. Similarly, the APC1638N mutation in mice results in few lesions within the intestines at 3.5 months. Notably, when crossed with the villin-driven KRASG12V mouse, Apc1638N mice develop numerous lesions within the intestine. In agreement with our findings, lesions taken from the APC1638N-KRAS mice showed enhanced nuclear β-catenin (Janssen et al., 2006). The requirement for Kras in the development of adenomas in the Apc1638N mouse appears in contrast to murine models that have utilized more severe truncation or complete loss of Apc. In comparison, these mice develop more numerous lesions following manipulation of Apc alone. However, several of these models also appear to show synergy between Apc and Kras. For example, Sansom et al. demonstrated that activation of Kras along with Apc mutation caused a more aggressive phenotype with more invasive lesions (Sansom et al., 2006). Additonally, Haigis et al. demonstrated that adenocarcinomas from mice carrying an activated Kras and a truncated Apc allele had an expanded stem cell population in intestinal adenomas (Haigis et al., 2008).
A number of examples now invoke post-translational modification of β-catenin as critical to its subcellular localization and activity. Our data implicating RAC1 in controlling β-catenin nuclear localization downstream of KRAS is in direct agreement with recent studies by Wu et al. These investigators demonstrated that Wnt-ligand-mediated β-catenin nuclear accumulation depended on Rac1 activity and that Rac1 control of β-catenin was essential for normal development of the hindlimb in mouse embryos (Wu et al., 2008). In agreement with these studies, we have shown a requirement for RAC1 in regulating nuclear localization of β-catenin in the presence of mutated APC and oncogenic KRAS. This finding elucidates a novel signaling pathway downstream of KRAS that relies on RAF, but not MEK, and may provide a mechanistic basis for several recent observations. First, Haigis et al. found that tumor growth in APC-K-ras mice was refractory to MEK inhibition but that colon cancer cell line growth depends on RAF1. In addition, data from a recent phase II clinical trial showed that MEK inhibition did not significantly affect colorectal cancer progression as measured by tumor growth (Rinehart et al., 2004). It is interesting to note that human colon tumors bear mutations in both KRAS and RAF. However, to date, there are no reports of oncogenic mutation in MEK1 in colorectal cancer (Gripp et al., 2007). In contrast, two groups have reported expression of the constitutively active RAC1 splice variant, RAC1b in colon tumors (Esufali et al., 2007; Singh et al., 2004).
Undetectable nuclear β-catenin in early human adenomas and apcmcr zebrafish intestines is paradoxical in that these tissues show profound morphologic and molecular abnormalities. Indeed defects in cell fating and differentiation have been appreciated in zebrafish (Haramis et al., 2006) and murine (Haigis et al., 2008) models of Apc truncation and attributed to dysregulation of β-catenin. Our findings indicate that defects in intestinal cell fating and differentiation following loss of Apc occur independently of β-catenin. Although it is possible that the levels of nuclear β-catenin are below the limits of detection in these tissues, this level of β-catenin does not appear sufficient for promoting intestinal cell proliferation nor is it essential for suppressing intestinal differentiation in apcmcr zebrafish. Rather, ctbp1 accumulation following apc loss underlies these defects. Consistent with this model, CtBP1 is highly expressed in human adenomas lacking evident nuclear β-catenin. APC control of both CtBP1 and β-catenin could shed light on the distinct clinical phenotypes of colon tumors arising from mutation in APC versus those harboring mutations in β-catenin (Samowitz et al., 1999). Taken together the above findings indicate that other functions of Apc, such as regulation of Ctbp, play important roles in early adenoma development and provide an important new perspective on the ordering of molecular events that may underlie colon tumor initiation and progression.
Experimental Procedures
Zebrafish embryo culture and manipulation
Wildtype, apcmcr and TOPGFP Danio rerio (zebrafish) were maintained on a 14h:10h light:dark cycle. Additionally, morpholino oligonucleotides targeting apc and ctbp1, whole mount in situ hybridization and hematoxylin and eosin staining were carried out as described previously (Nadauld et al., 2004). Inhibition of COX-2 and treatment with PGE2 was accomplished as described previously (Eisinger et al., 2007). mRNA for KRASG12D or β-cateninS45A was synthesized using mMESSAGE mMACHINE (Ambion) according to the manufacturer’s instructions. mRNA was injected into embryos at the one-cell stage. pCEP4-DNA plasmids containing constitutively active RAF1 and MEK1 were gifts from C. Jette (Harvard University) and were injected into apcmcr embryos at the one-cell stage.
Human and zebrafish tissue Immunofluorescence
Immunofluorescence was carried out on formalin-fixed human tissue sections or on 72-hpf zebrafish embryos. Embryos were fixed in 4% paraformaldehyde in PBS then dehydrated to 70% ethanol. 7-μm-thick, paraffin-embedded sections were deparaffinized in xylenes and rehydrated in graded alcohols and then permeabilized for 15 min in 0.1% Triton X-100 in PBS. Antigen retrieval was performed by boiling for 15 min in 10 mM sodium citrate buffer, pH 6.0, followed by cooling to room temperature. Sections were then incubated in blocking buffer (2% goat serum and 1% bovine serum albumin in 1x PBS) for 4 hrs at room temperature and incubated in wash buffer (0.2% goat serum, 0.1% bovine serum albumin, and 5 mM glycine in 1x PBS) for 2 hrs at room temperature. Primary antibody (ms-α-PCNA, AbCam, rb-α-β-catenin, Sigma, ms-α-β-catenin, BD Transduction Laboratories, rb-α-p-cJun, Calbiochem, diluted 1:250 in PBS) was applied overnight at 4°C. Sections were rinsed twice in PBS and incubated for 2 hrs at room temperature in secondary antibody (gt-α-ms Alexa 488 and gt-α-rb Alexa 568, Molecular Probes; dilution 1:250) and the nuclear stain TO-PRO-3 (Molecular Probes; dilution 1:1000). Sections were rinsed in water and coverslips were applied using ProLong Gold antifade reagent (Molecular Probes).
Human cell immunofluorescence
Cells were grown on coverslips and grown to 70% confluency before treatments. Following treatment, cells were washed twice with cold PBS and fixed for 15 min at room temperature in 3.7% paraformaldehyde in PBS. Cells were then washed three times in cold PBS and permeabilized for 30 min in PBS-T. Cells were then incubated overnight at 4°C in primary antibody (ms-α-β-catenin, BD Transduction Laboratories, rb-α-p-cJun, Calbiochem, diluted 1:250 in PBS-T). The following day, cells were washed three times in cold PBS and incubated for 1 hr at room temperature in secondary antibody (gt-α-ms Alexa 488 or gt-α-rb Alexa 568, Molecular Probes, diluted 1:250 in PBS-T). Cells were then washed three times and incubated for 15 min with the nuclear stain TO-PRO-3 (Molecular Probes, diluted 1:1000 in ddH2O). Cells were rinsed with ddH2O and coverslips were applied using ProLong Gold antifade reagent (Molecular Probes).
Imaging
Images were collected using an Olympus FluoView FV300 confocal laser scanning microscope.
Human Cell Lines
Transfections were performed using LipoFectamine2000 (Invitrogen) according to the manufacturer’s recommendations. The siRNA sequences for KRAS, RAF1 and RAC1 are provided in the supplemental procedures. We expressed 1μg pDEST53-KRASG12D, pCEP4-BxB-RAF1, pCEP4-MEK1-ERK2, pCEP4-DN-MEK1, pEF1-RAC1V12, pEF1-RAC1N17, pEF1-Cdc42N17, pCMV-β-cateninS552A, pCMV-β-cateninS675A, CS2-β-cateninS191A, CS2-β-cateninS246A, CS2-β-cateninS605A using LipoFectamine2000. pCMV-β-cateninS552A and pCMV-β-cateninS675A were generous gifts from Dr. Nickolai Dulin and CS2-β-cateninS191A. CS2-β-cateninS246A, and CS2-β-cateninS605A were generous gifts from Dr. Fanxin Long. pEF1-RAC1V12, pEF1-RAC1N17, pEF1-Cdc42N17 were generous gifts from Dr. Matthew Topham. Inhibition of COX-2 and treatment with PGE2 were accomplished as described previously (Eisinger et al., 2007). The MTT assay was performed according to manufacturer’s recommendations (Roche, Switzerland).
Acknowledgments
We thank Dr. Richard Dorsky (University of Utah) for providing the TOPGFP zebrafish. This work is supported by grants from the National Cancer Institute (CA073992 and CA96934) and the Huntsman Cancer Foundation awarded to D.A.J. The work was also supported by access to technical cores supported by a Cancer Center Support Grant (CA042014). We thank Dr. Diana Stafforini for helpful discussions. R.A.P. and T.B. are supported in part by the University of Utah MD/PHD program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amos-Landgraf JM, Kwong LN, Kendziorski CM, Reichelderfer M, Torrealba J, Weichert J, Haag JD, Chen KS, Waller JL, Gould MN, et al. A target-selected Apc-mutant rat kindred enhances the modeling of familial human colon cancer. Proc Natl Acad Sci U S A. 2007;104:4036–4041. doi: 10.1073/pnas.0611690104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CB, Neufeld KL, White RL. Subcellular distribution of Wnt pathway proteins in normal and neoplastic colon. Proc Natl Acad Sci U S A. 2002;99:8683–8688. doi: 10.1073/pnas.122235399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311–320. doi: 10.1016/s0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- Bienz M, Hamada F. Adenomatous polyposis coli proteins and cell adhesion. Curr Opin Cell Biol. 2004;16:528–535. doi: 10.1016/j.ceb.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Blaker H, Scholten M, Sutter C, Otto HF, Penzel R. Somatic mutations in familial adenomatous polyps. Nuclear translocation of β-catenin requires more than biallelic APC inactivation. American journal of clinical pathology. 2003;120:418–423. doi: 10.1309/4E4W-G3AY-GJNC-D11P. [DOI] [PubMed] [Google Scholar]
- Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ, Vogelstein B. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327:293–297. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-β-catenin signaling axis. Science. 2005;310:1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- Clevers H. Wnt/β-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Eisinger AL, Nadauld LD, Shelton DN, Prescott SM, Stafforini DM, Jones DA. Retinoic acid inhibits beta -catenin through suppression of cox-2: A role for truncated adenomatous polyposis coli. J Biol Chem. 2007 doi: 10.1074/jbc.M609768200. [DOI] [PubMed] [Google Scholar]
- Esufali S, Charames GS, Pethe VV, Buongiorno P, Bapat B. Activation of tumor-specific splice variant Rac1b by dishevelled promotes canonical Wnt signaling and decreased adhesion of colorectal cancer cells. Cancer Res. 2007;67:2469–2479. doi: 10.1158/0008-5472.CAN-06-2843. [DOI] [PubMed] [Google Scholar]
- Fang M, Li J, Blauwkamp T, Bhambhani C, Campbell N, Cadigan KM. C-terminal-binding protein directly activates and represses Wnt transcriptional targets in Drosophila. Embo J. 2006;25:2735–2745. doi: 10.1038/sj.emboj.7601153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Forrester K, Almoguera C, Han K, Grizzle WE, Perucho M. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature. 1987;327:298–303. doi: 10.1038/327298a0. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Lin AE, Nicholson L, Allen W, Cramer A, Jones KL, Kutz W, Peck D, Rebolledo MA, Wheeler PG, et al. Further delineation of the phenotype resulting from BRAF or MEK1 germline mutations helps differentiate cardio-facio-cutaneous syndrome from Costello syndrome. American journal of medical genetics. 2007;143:1472–1480. doi: 10.1002/ajmg.a.31815. [DOI] [PubMed] [Google Scholar]
- Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008 doi: 10.1038/ngXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada F, Bienz M. The APC tumor suppressor binds to C-terminal binding protein to divert nuclear β-catenin from TCF. Dev Cell. 2004;7:677–685. doi: 10.1016/j.devcel.2004.08.022. [DOI] [PubMed] [Google Scholar]
- Hanson CA, Miller JR. Non-traditional roles for the Adenomatous Polyposis Coli (APC) tumor suppressor protein. Gene. 2005;361:1–12. doi: 10.1016/j.gene.2005.07.024. [DOI] [PubMed] [Google Scholar]
- Haramis AP, Hurlstone A, van der Velden Y, Begthel H, van den Born M, Offerhaus GJ, Clevers HC. Adenomatous polyposis coli-deficient zebrafish are susceptible to digestive tract neoplasia. EMBO reports. 2006;7:444–449. doi: 10.1038/sj.embor.7400638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, Williams BO, Cho KR, Fearon ER. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 2007;67:9721–9730. doi: 10.1158/0008-5472.CAN-07-2735. [DOI] [PubMed] [Google Scholar]
- Hurlstone AF, Haramis AP, Wienholds E, Begthel H, Korving J, Van Eeden F, Cuppen E, Zivkovic D, Plasterk RH, Clevers H. The Wnt/β-catenin pathway regulates cardiac valve formation. Nature. 2003;425:633–637. doi: 10.1038/nature02028. [DOI] [PubMed] [Google Scholar]
- Iwao K, Nakamori S, Kameyama M, Imaoka S, Kinoshita M, Fukui T, Ishiguro S, Nakamura Y, Miyoshi Y. Activation of the β-catenin gene by interstitial deletions involving exon 3 in primary colorectal carcinomas without adenomatous polyposis coli mutations. Cancer Res. 1998;58:1021–1026. [PubMed] [Google Scholar]
- Janssen KP, Alberici P, Fsihi H, Gaspar C, Breukel C, Franken P, Rosty C, Abal M, El Marjou F, Smits R, et al. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology. 2006;131:1096–1109. doi: 10.1053/j.gastro.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- Kongkanuntn R, Bubb VJ, Sansom OJ, Wyllie AH, Harrison DJ, Clarke AR. Dysregulated expression of β-catenin marks early neoplastic change in Apc mutant mice, but not all lesions arising in Msh2 deficient mice. Oncogene. 1999;18:7219–7225. doi: 10.1038/sj.onc.1203181. [DOI] [PubMed] [Google Scholar]
- Moran AE, Hunt DH, Javid SH, Redston M, Carothers AM, Bertagnolli MM. Apc deficiency is associated with increased Egfr activity in the intestinal enterocytes and adenomas of C57BL/6J-Min/+ mice. J Biol Chem. 2004;279:43261–43272. doi: 10.1074/jbc.M404276200. [DOI] [PubMed] [Google Scholar]
- Morin PJ. β-catenin signaling and cancer. Bioessays. 1999;21:1021–1030. doi: 10.1002/(SICI)1521-1878(199912)22:1<1021::AID-BIES6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Nadauld LD, Chidester S, Shelton DN, Rai K, Broadbent T, Sandoval IT, Peterson PW, Manos EJ, Ireland CM, Yost HJ, et al. Dual roles for adenomatous polyposis coli in regulating retinoic acid biosynthesis and Wnt during ocular development. Proc Natl Acad Sci U S A. 2006a;103:13409–13414. doi: 10.1073/pnas.0601634103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadauld LD, Phelps R, Moore BC, Eisinger A, Sandoval IT, Chidester S, Peterson PW, Manos EJ, Sklow B, Burt RW, et al. Adenomatous polyposis coli control of C-terminal binding protein-1 stability regulates expression of intestinal retinol dehydrogenases. J Biol Chem. 2006b doi: 10.1074/jbc.M602119200. [DOI] [PubMed] [Google Scholar]
- Nadauld LD, Sandoval IT, Chidester S, Yost HJ, Jones DA. Adenomatous polyposis coli control of retinoic acid biosynthesis is critical for zebrafish intestinal development and differentiation. J Biol Chem. 2004;279:51581–51589. doi: 10.1074/jbc.M408830200. [DOI] [PubMed] [Google Scholar]
- Nadauld LD, Shelton DN, Chidester S, Yost HJ, Jones DA. The zebrafish retinol dehydrogenase, rdh1l, is essential for intestinal development and is regulated by the tumor suppressor adenomatous polyposis coli. J Biol Chem. 2005;280:30490–30495. doi: 10.1074/jbc.M504973200. [DOI] [PubMed] [Google Scholar]
- Polakis P. The oncogenic activation of β-catenin. Curr Opin Genet Dev. 1999;9:15–21. doi: 10.1016/s0959-437x(99)80003-3. [DOI] [PubMed] [Google Scholar]
- Polakis P. Casein kinase 1: a Wnt’er of disconnect. Curr Biol. 2002;12:R499–R501. doi: 10.1016/s0960-9822(02)00969-7. [DOI] [PubMed] [Google Scholar]
- Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, Hamid O, Varterasian M, Asbury P, Kaldjian EP, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22:4456–4462. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- Roberts RB, Min L, Washington MK, Olsen SJ, Settle SH, Coffey RJ, Threadgill DW. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc Natl Acad Sci U S A. 2002;99:1521–1526. doi: 10.1073/pnas.032678499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnolo B, Berrebi D, Saadi-Keddoucci S, Porteu A, Pichard AL, Peuchmaur M, Vandewalle A, Kahn A, Perret C. Intestinal dysplasia and adenoma in transgenic mice after overexpression of an activated β-catenin. Cancer Res. 1999;59:3875–3879. [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Munemitsu S, Polakis P. Loss of β-catenin regulation by the APC tumor suppressor protein correlates with loss of structure due to common somatic mutations of the gene. Cancer Res. 1997;57:4624–4630. [PubMed] [Google Scholar]
- Samowitz WS, Powers MD, Spirio LN, Nollet F, van Roy F, Slattery ML. β-catenin mutations are more frequent in small colorectal adenomas than in larger adenomas and invasive carcinomas. Cancer Res. 1999;59:1442–1444. [PubMed] [Google Scholar]
- Sansom OJ, Meniel V, Wilkins JA, Cole AM, Oien KA, Marsh V, Jamieson TJ, Guerra C, Ashton GH, Barbacid M, et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc Natl Acad Sci U S A. 2006;103:14122–14127. doi: 10.1073/pnas.0604130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, Batlle E, Simon-Assmann P, Clevers H, Nathke IS, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18:1385–1390. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao J, Jung C, Liu C, Sheng H. Prostaglandin E2 Stimulates the β-catenin/T cell factor-dependent transcription in colon cancer. J Biol Chem. 2005;280:26565–26572. doi: 10.1074/jbc.M413056200. [DOI] [PubMed] [Google Scholar]
- Sierra J, Yoshida T, Joazeiro CA, Jones KA. The APC tumor suppressor counteracts β-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev. 2006;20:586–600. doi: 10.1101/gad.1385806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Karnoub AE, Palmby TR, Lengyel E, Sondek J, Der CJ. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene. 2004;23:9369–9380. doi: 10.1038/sj.onc.1208182. [DOI] [PubMed] [Google Scholar]
- Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/β-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998;58:1130–1134. [PubMed] [Google Scholar]
- Spink KE, Polakis P, Weis WI. Structural basis of the Axin-adenomatous polyposis coli interaction. Embo J. 2000;19:2270–2279. doi: 10.1093/emboj/19.10.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takacs CM, Baird JR, Hughes EG, Kent SS, Benchabane H, Paik R, Ahmed Y. Dual positive and negative regulation of wingless signaling by adenomatous polyposis coli. Science. 2008;319:333–336. doi: 10.1126/science.1151232. [DOI] [PubMed] [Google Scholar]
- Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. Phosphorylation of β-catenin by cyclic AMP-dependent protein kinase. J Biol Chem. 2006;281:9971–9976. doi: 10.1074/jbc.M508778200. [DOI] [PubMed] [Google Scholar]
- Tetsu O, McCormick F. β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- Wu X, Tu X, Joeng KS, Hilton MJ, Williams DA, Long F. Rac1 activation controls nuclear localization of β-catenin during canonical Wnt signaling. Cell. 2008;133:340–353. doi: 10.1016/j.cell.2008.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]