

Abstract
The present study tested the hypotheses that vulnerability to ethanol depends upon (1) population-based characteristics of the neuronal progenitors and (2) the maturation of that population by examining the effects of prenatal exposure to ethanol on brainstem nuclei derived from different rhombomeres and from the alar and basal plates. Macaca nemestrina received an ethanol-containing solution one day per week during the first six (Et6) or 24 (Et24) weeks of gestation. Control animals received an equivalent volume of saline. The treatment regime for some animals included early gastrulation (gestational day (G) 19 or G20), whereas others were treated later (on G21 or G24). Brainstems were cryosectioned and stained with cresyl violet. Stereological methods were used to determine the numbers of neurons in six different nuclei: the abducens (VI), vagal (X), and hypoglossal (XII) motor nuclei and sensory components of the trigeminal brainstem nuclear complex (the principal (PSN), oral (SpVo), and interpolar (SpVi) subnuclei). There were no differences in the numbers of neurons in any of the nuclei between controls or Et6- and Et24-treated monkeys. In contrast, the number of trigeminal sensory neurons was significantly (p<0.05) lower in animals treated on G19/G20 than in control. No differences between controls and monkeys treated on G21/G24 were detected. No motor nuclei exhibited an ethanol-induced change. These data together with data on the trigeminal motor nucleus show that vulnerability to ethanol (1) is greater in sensory nuclei than in motor nuclei and (2) is temporally restricted to the time of gastrulation.
Keywords: alar plate, autism, brainstem, fetal alcohol spectrum disorder, fetal alcohol syndrome, gastrulation, rhombomere
INTRODUCTION
Developmental exposure to ethanol can reduce the final number of neurons in a particular structure. This results from a reduction in cell proliferation and an increase in neuronal death (Miller, 1995a; 1999; 2006; Mooney et al., 2006). It has been hypothesized that the vulnerability to teratogen-induced damage is defined by the developmental derivation of the population (Rodier, 1996). Two major contributors to this vulnerability are (1) population-based characteristics including position, or location, of the structure and (2) the timing of the insult. The effect of position is apparent in cerebral cortex wherein populations of cells in different parts of the cortex respond differently to ethanol insult. For example, the pattern of cortical neuronogenesis is differentially disrupted in rostral and caudal cortex by prenatal exposure to ethanol (Miller, 1988). Moreover, the complement of neurons in early developing somatosensory cortex is unaffected by exposure to ethanol during the first two postnatal weeks, whereas the effect in the later developing visual cortex is Dtransient (Mooney and Napper, 2005).
A second critical factor defining ethanol vulnerability is the timing of the exposure. In the cerebral cortex, exposure during early gastrulation (Ashwell and Zhang, 1996; Miller, 2007) or the period of neuronal generation (Miller, 1995; 1996; 1997) affects neuronal number and projections. The effects of timing are also evident in the cerebellum. The number of Purkinje neurons is lower in animals exposed to ethanol on postnatal day (P) 4 or P5 than in control animals or in animals exposed to ethanol before P3 or on P8 or P9 (Goodlett et al., 1990; Pierce et al., 1999; Light et al., 2002). At least part of the response is shaped by the maturational state of the neurons. Earlier developing lobules (e.g., I-V, IX, and X) are more affected by exposure to ethanol on P4 than are later developing lobules (VI and VII) (Goodlett et al., 1990; Light et al., 2002).
The brainstem is uniquely suited to examine issues of regional and temporal vulnerability to ethanol. This is because during early development the brainstem is transiently compartmentalized into segments that are arranged orthogonal to the longitudinal neuraxis. Each segment, called a rhombomere, has a unique pattern of gene expression (Lumsden and Keynes, 1989; Murphy et al., 1989; Wilkinson et al., 1989; Lumsden, 1990). In addition, the developing brainstem can be subdivided into the alar and basal plates that give rise to sensory and motor components, respectively (Nieuwenhuys et al., 1998; Ju et al., 2004). These plates are oriented orthogonal to the rhombomeres and parallel to the neuraxis.
A key time for brainstem development appears to be gastrulation. The present study tested the hypothesis that the timing of the exposure to ethanol relative to gastrulation has different effects on brainstem nuclei derived from (a) different rhombomeres and (b) alar or basal plates. Six cranial nerve nuclei were examined (Fig. 1). Three were sensory nuclei derived from the alar plate across three different segments (rhombomere (r) 2, r4, and r6). Three motor nuclei were also examined. These were basal plate derivatives from r6 and r8.
Figure 1. Schematic diagram of developing brainstem.

During development the brainstem transiently forms at least eight rhomobomeres (r1-r8). Sensory neurons are derived from the alar, or roof, plate (right). Motor neurons are derived from the basal, or floor, plate (left).
MATERIALS AND METHODS
Animals
Fifteen pregnant Macaca nemestrina were housed in the Primate Center at the University of Washington, Seattle WA. All procedures were approved by the local Institutional Animal Care and Use Committee. The husbandry and care of these animals is described in detail elsewhere (Clarren and Astley, 1992). Briefly, animals received a solution by nasogastic intubation one day per week, i.e., every seventh day, for six or 24 weeks of gestation. Nine animals received an ethanol-containing solution (1.8 g/kg; Table 1). Of these, five were given ethanol during the first six (Et6) weeks of gestation and four received ethanol in all 24 (Et24) weeks of gestation. The remaining six animals received an equivalent volume of saline (Ct).
Table 1.
Ethanol exposure
| Timing of Exposure | Duration of Exposure | n | |
|---|---|---|---|
| 6 weeks | 24 weeks | ||
| G19/G20 | 2 | 2 | 4 |
| G21/G24 | 3 | 2 | 5 |
| n | 5 | 4 | |
The timing of the ethanol dosing was critical. The feeding regime was set so that for four animals, the third feeding occurred at the time of gastrulation, on gestational day (G) 19 or G20. These animals selectively exhibited the features of cranial dysmorphia associated with fetal alcohol syndrome (Astley et al., 1999), as well as neocortical changes (Miller, 2007). The remaining five ethanol-treated animals did not get dosed at the time of gastrulation, instead they received ethanol before (e.g., their second or third feedings on G14 or G17, respectively) and after gastrulation (e.g., their third or fourth feedings on G21 or G24, respectively). Venous blood was collected from mothers 2 hr after ethanol administration for determination of blood ethanol concentration (BEC) using gas chromatography.
Animals were allowed to give birth and offspring were reared normally. At four to five years of age, offspring were anesthetized (2.0 mg/kg xylazine and 10 mg/kg ketamine) and perfused intracardially with 200−300 ml saline, followed by 2−3 liter of 4.0% paraformaldehyde in 0.10 M phosphate buffer (PB; pH 7.4), and finally 1 liter of 10% sucrose in PB. Brains were removed and stored in a solution of 30% sucrose in PB for approximately 24 months.
Brainstems were separated by cutting through the rostral superior colliculus in the coronal plane and were hemisected (Mooney and Miller, 2001). The right half of each brainstem was frozen and cut into 40 μm thick sections in the horizontal plane. A series of every fifth section was collected for staining with cresyl violet.
Stereological methods
Neuronal counts were made using stereological methods (Mooney and Miller, 2001). The total numbers of neurons (N) in six brainstem nuclei (Table 2; Fig. 1) were calculated as the product of the volume of the nucleus (V) and the density of constituent neurons (Nv).
Table 2.
Derivation of each brainstem nucleus.
| Name | Abbreviation | Plate Derivation | Rhombomeric Derivation* |
|---|---|---|---|
| principal sensory nucleus of the trigeminal nerve | PSN | Alar | r2 |
| spinal trigeminal nucleus, oral portion | SpVo | Alar | r4 |
| spinal trigeminal nucleus, interpolar portion | SpVi | Alar | r6 |
| motor nucleus of the abducens nerve | MoVI | Basal | r5−6 |
| motor nucleus of the vagal nerve | MoX | Basal | r8 |
| motor nucleus of the hypoglossal nerve | MoXII | Basal | r8 |
Data from Cambronero and Puelles (2000).
The Cavalieri estimator was used to determine V (Gundersen and Jensen, 1987; Miller and Muller, 1989). Identification of each brainstem nucleus relied on cytoarchitectonic features (Paxinos et al., 2000). In every section that contained a profile of the nucleus, the cross-sectional area (a) of that nucleus was measured using the Bioquant Image Analysis System (R&M Biometrics, Nashville TN). Volume of a nucleus (V) was calculated:
wherein Ea was the sum of all cross-sectional areas for each nucleus, t was the section thickness, and f was the frequency of sections used in the analysis (Gundersen and Jensen, 1987; Miller and Muller, 1989; Mooney and Miller, 2001). The section thickness used in this calculation was initial section thickness, i.e., the thickness prior to processing. Thus, V represents the actual volume of each nucleus in the tissue and is independent of any processing-induced shrinkage.
The optical disector method was applied to determine neuronal density (Nv; Gundersen et al., 1988; Mooney and Miller, 2001). Images of the section were projected to a monitor and a sampling frame of known area (a frame) was overlaid. Moving the microscope stage through a known depth (d) in the z-axis turned the two-dimensional frame into a three-dimensional box. The depth of the box used for counting was 20 μm, however, tissue processing resulted in 25% reduction of section thickness (from 40 μm to 30 μm). Thus, d in the calculation was adjusted for this shrinkage and was 26.67 μm. Guard zones of approximately 5 μm above and below the counting box were used for verification of cell identity. Three planes of the box were designated inclusion planes, and all neuronal nuclei completely within the box, or intercepting one of the inclusion planes, were counted (Q). Thus, the neuronal packing density was calculated by the following formula:
Statistical analysis
Data for the ethanol-exposed animals were divided into groups based upon the duration of ethanol exposure (Et6, n = 5 or Et24, n = 4) or by the timing of the exposure (G19/G20, n = 4 or G21/G24, n = 5). A mean (± the standard error of the mean) was determined for each value (e.g., nuclear volume, packing density, and neuronal number) for each group of animals. Multivariate one-way analyses of variance (MANOVA) were used to determine the effect of treatment. This was followed by individual one-way analyses of variance (ANOVAs) for each cranial nerve nucleus. In cases where an ANOVA showed a significant difference (p<0.05), post-hoc Tukey B tests were performed.
RESULTS
Blood ethanol concentrations
Mean BEC (± standard error of the mean) for all nine ethanol-treated macaques was 231 ± 20 mg/dl (Clarren and Astley, 1992). Parsing the data based on the duration or timing of the ethanol exposure did not reveal any differences in BEC (Mooney and Miller, 2001; Miller, 2007).
Brainstem nuclei
Exposure to ethanol did not affect the size or weight of the brainstems (Mooney and Miller, 2001). All six brainstem nuclei were identifiable in both Ct- and Et-treated macaques (Fig. 2). Motor nuclei were identified by their large polygonal cell bodies. This contrasted with the sensory nuclei which tended to be populated by smaller, rounder neuronal somata.
Figure 2. Appearance of the macaque brainstem.
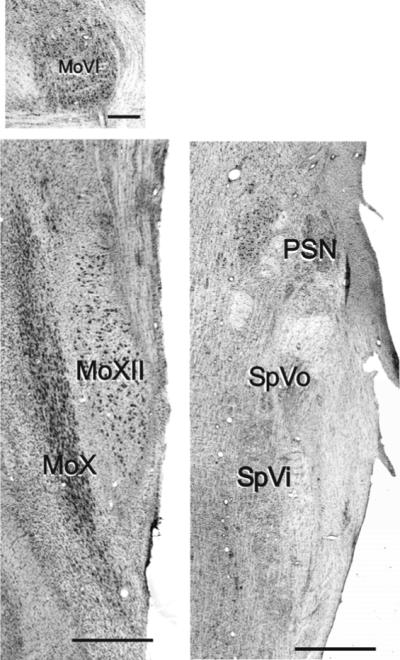
All six nuclei were readily identifiable in horizontal sections stained with cresyl violet. Three sensory nuclei of the trigeminal system are apparent within the same section; the principal sensory nucleus (PSN), and two parts of the spinal trigeminal nucleus - the oralis (SpVo) and the interpolaris (SpVi).
The motor nuclei are shown separately. MoVI motor nucleus of the abducens nerve. MoX motor nucleus of the vagus. MoXII motor nucleus of the hypoglossal. Scale bar is 500μm for all images.
The data were parsed for analysis in two different ways: (1) according to the duration of the exposure to ethanol and (2) based on the timing of the exposure during the third week of gestation. The number of neurons in each nucleus was calculated as the product of the nuclear volume and the cell packing density. None of these parameters were significantly affected by the duration of the ethanol exposure, i.e., whether the treatment was confined to the first six weeks of gestation or occurred during all 24 weeks of gestation.
There was an effect of treatment at different times relative to gastrulation. Based on a MANOVA, both nuclear volume and neuronal number were significantly (p<0.05) affected by the timing of the ethanol exposure. Cell packing density was not significantly different among the groups in this data set.
Sensory nuclei
PSN. In the PSN, the ANOVA showed a significant (F2,11 = 4.800; p = 0.032) effect of treatment when data on the number of neurons was parceled based on timing of exposure (Fig. 3). Post-hoc comparison with the Student-Newman-Keuls test showed that number of neurons was significantly lower in monkeys treated with ethanol during early gastrulation compared with Ct-treated animals (p<0.05). This effect was largely due to a significant (F2,11 = 5.914; p = 0.018) change in the volume of the PSN. Post-hoc analysis showed that it was significantly (p<0.05) smaller in macaques treated on G19/G20 than in controls or in animals treated with ethanol on G21/24. There was no difference between Ct-treated monkeys and those exposed to ethanol on G21/24. Neuronal density was also significantly (F2,11 = 6.844; p = 0.012) lower in animals exposed to ethanol, however, in contrast with N and V, the effect on density was seen in animals treated on G21/24 compared with either controls or macaques that received ethanol on G19/G20.
Figure 3. The effect of ethanol on the number of sensory neurons.

Stereological methods were used to determine volume and cell packing density in each of three motor nuclei. These two parameters were multiplied to generate an estimate of the total number of neurons in each nucleus. Data on the left within each graph are organized according to the duration of exposure; animals were given ethanol on day per week for the first six or all 24 weeks of gestation. On the right within each graph, data are organized based on the day of exposure during the critical time of gastrulation; early gastrulation (gestational day (G)19 or G20) vs. late gastrulation (G21 or G24).
Left. The number of neurons in the PSN was significantly (p<0.05) lower in animals exposed to ethanol on G19 or G20 than in control animals. There were no significant differences among control animals and those exposed to ethanol for six or 24 weeks of gestation.
Middle. The number of neurons in the spinal trigeminal nucleus oralis was significantly lower in animals exposed to ethanol during early gastrulation compared with control-treated animals. This was a time-dependent effect, and was not seen in animals treated on G21 or G24. There was no significant effect of duration of exposure to ethanol.
Right. The time-dependent effect on the number of neurons in the spinal trigeminal nucleus interpolaris was also apparent. Exposure to ethanol during early gastrulation significantly reduced the number of neurons in this nucleus
* significantly different to control, p<0.05; # significantly different to animals exposed to ethanol on G21 or G24.
SpVo. Neither volume nor neuronal packing density was significantly affected by exposure to ethanol (Fig. 3). Despite this, the SpVo showed a significant (F2,11 = 5.647; p = 0.026) effect of exposure to ethanol on neuron number in macaques exposed during early gastrulation. Neuronal number was lower in animals exposed to ethanol on G19/G20 than Ct-treated animals (p = 0.033).
SpVi. There was no significant effect of exposure to ethanol on volume or neuronal packing density in the interpolar nucleus (Fig. 3). As seen in the SpVo, the number of neurons in the SpVi was significantly (F2,11 = 4.529; p = 0.037) affected by treatment. Post-hoc Tukey B tests showed that the number of neurons was significantly (p<0.05) lower in monkeys treated during early gastrulation than in Ct-treated animals.
Motor nuclei
The numbers of neurons in the three motor nuclei, abducens, vagus and hypoglossal, were not affected by exposure to ethanol, regardless of the duration or the timing of the exposure (Fig. 4). There was, similarly, no effect of ethanol on the volume or on the density of neurons within any nucleus.
Figure 4. Quantification of the effect of ethanol on motor nuclei.

Data were generated and organized as per Figure 3. Exposure to ethanol did not significantly affect the number of neurons in MoVI (left), MoX (middle), or MoXII (right).
DISCUSSION
Population-based ethanol vulnerability
Including the nuclei examined in a previous study (Mooney and Miller, 2001), the effects of ethanol on nine cranial nerve nuclei have been examined. Five of the nuclei are derivatives of the basal plate (motor nuclei) and four are alar plate derivatives (sensory nuclei; Fig. 1). The studies quantify the number of constituent neurons. Only one motor structure, the trigeminal motor nucleus, is affected by exposure to ethanol (Mooney and Miller, 2001). In contrast, the numbers of neurons in three of the four sensory nuclei (the PSN, SpVo, and SpVi, but not the medial superior olivary nucleus (MSO)), are affected in a time-dependent manner. A critical determinant of neuronal number in select brainstem nuclei is the timing of the exposure, i.e., the number of neurons is lower when the ethanol exposure includes early gastrulation. Thus, the alar plate is more vulnerable to the effects of ethanol exposure during gastrulation.
Vulnerability of cranial nerve nuclei to teratogenic agents, including ethanol, may be defined by the rhombomeric derivation of the population and timing of the insult (Rodier et al., 1996). This hypothesis is based on data from human children showing that r2 is especially vulnerable to teratogens such as valproic acid and thalidomide, but only when the exposure includes gastrulation. The present data on non-human primates are consistent with the concept that gastrulation is a critical window, but it does not support the rhombomere-specific vulnerability. Though both motor and sensory nuclei in r2 are affected, in more caudal rhombomeres, e.g., r4 and r6, only the sensory nuclei are altered.
It is noteworthy that though the SpVo in the r4 is affected by ethanol exposure on G19/G20, another sensory nucleus in r4 (the MSO) is not. There are two potential explanations for this apparent discrepancy. (1) Ethanol targets nuclei subserving general somatic afferents. The MSO does not transmit such information. (2) Susceptibility is defined by the timing of neuronal generation as well as position. In the rat, the peak production of MSO neurons is about a day earlier than the peak production of neurons in the SpVo (Altman and Bayer, 1980a; 1980b). Thus, it is possible that in the macaque the difference in maturational state of MSO neurons is protective.
The trigeminal-somatosensory system is preferentially targeted by ethanol. Interestingly, this includes motor and sensory components. It is uncertain whether (a) the primary effect is on both basal and alar plate derivatives within this system or (b) the change in the motor nucleus is secondary to a primary insult to the sensory nuclei. That there is no change in the facial motor nucleus (MoVII) and the two motor nuclei are linked such that a change in one can cause a change in the other (Byrd, 1988) argues against the latter possibility.
There is one caveat to this data, and that is that the sample sizes are small. The low “n” may result in a type II error, i.e., reporting a “not significant” finding when there is a difference.
Timing of ethanol vulnerability
A second contributor to ethanol vulnerability is the timing of the ethanol exposure. Damage to the trigeminal system occurs when the ethanol exposure includes early gastrulation. The implications of such targeting are (a) that system specification occurs at the time of gastrulation and (b) that a unique feature of trigeminal progenitors renders them vulnerable to ethanol exposure during gastrulation. It could be argued that the trigeminal specific effects follow the time-dependent effects of ethanol on craniofacial morphology. This argument is countered by the lack of a time-dependent effect on the MoVII which provides motor innervation to most of the face.
Interpretation of data from the present study of macaques is somewhat compromised because the subjects received ethanol doses over the course of weeks. This makes it difficult to ascribe exposure at a particular time to the outcome. Despite this impediment, the same animals as those used in the present study show that when the exposure includes G19 or G20, the offspring exhibit cranial dysmorphia (Astley et al., 1999). This concurs with data from mouse studies showing that exposure only at the time of gastrulation is sufficient to produce the cranial dysmorphia (Sulik et al., 1981; 1988; Dunty et al., 2002). Moreover, rats exposed to ethanol at the time of gastrulation show similar changes to brainstem cranial nerve nuclei (Mooney and Miller, 2007).
The greater sensitivity of the trigeminal system to ethanol-induced insult than the facial nucleus agrees with findings from other toxins, e.g., mercury chloride, and anti-epileptics, e.g., valproic acid (Gofflot et al., 1996; van Maele-Fabry et al.,1995; 1996). Interestingly, gestational exposure to another anti-epileptic, hydantoin, produces a syndrome with craniofacial features that are remarkably similar to those characteristic of fetal alcohol syndrome (cf. Jones and Smith, 1973; Hanson and Smith, 1975).
Underlying developmental events
Two contributors to the final number of neurons in a nucleus are cell proliferation and neuronal death. Ethanol can affect the number of neurons by causing a decrease in proliferation and/or by increasing neuronal death (Miller, 1995; 2006; Mooney et al., 2006; Mooney and Miller, 2007). Neural stem cells in vivo (Miller, 1996) and in vitro (Santillano et al., 2005; Prock and Miranda, 2007) appear to be refractory to ethanol-induced cell death. Thus, the reductions in the number of neurons in cranial nerve nuclei likely result from decreases in cell proliferation.
Cells with more restricted lineage are more susceptible to ethanol neurotoxicity, i.e., cell death is not uncommon among these cells (Miller, 1995; 1999; Jacobs and Miller, 2001; Miller et al., 2003). One alternative mechanism is that exposure to ethanol during gastrulation restricts or shifts cell fate (Kentroti and Vernadakis, 1992; Tateno et al., 2005; Miller and Hu, submitted). Accordingly, proportionally more non-neuronal cells are generated at the expense of neurons or more or one type of neuron is produced at the expense of others. Another possibility is that ethanol accelerates the process of cell definition (Miller and Robertson, 1993). The final consequence of any of these individually, or in combination, is that there are fewer cells in the mature structure.
Acknowledgments
Research support was provided by grants from the National Institutes of Health (AA015413 to SMM; AA06916 and AA07568 to MWM) and the Department of Veterans Affairs (MWM).
ABBREVIATIONS
- ANOVA
analysis of variance
- BEC
blood ethanol concentration
- FAS
fetal alcohol syndrome
- FASD
fetal alcohol spectrum disorder
- G
gestational day
- MANOVA
multivariate analysis of variance
- MoV
motor nucleus of the trigeminal nerve
- MoVI
motor nucleus of the abducens nerve
- MoVII
motor nucleus of the facial nerve
- MoX
motor nucleus of the vagal nerve
- MoXII
motor nucleus of the hypoglossal nerve
- MSO
medial superior Dolivary nucleus
- N
total number
- Nv
cell packing density
- P
Epostnatal Pday
- PSN
principal sensory nucleus of the trigeminal nerve
- r
rhombomere
- SpVi
interpolar portion of the spinal trigeminal nucleus
- SpVo
oral portion of the spinal trigeminal nucleus
- V
volume
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Altman J, Bayer SA. Development of the brain stem in the rat. II. Thymidine-radiographic study of the time of origin of neurons of the upper medulla, excluding the vestibular and auditory nuclei. J. Comp. Neurol. 1980a;194:37–56. doi: 10.1002/cne.901940103. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer SA. Development of the brain stem in the rat. III. Thymidine-radiographic study of the time of origin of neurons of the vestibular Rand auditory nuclei of the upper medulla. J. Comp. Neurol. 1980b;194:877–904. doi: 10.1002/cne.901940410. [DOI] [PubMed] [Google Scholar]
- Ashwell KW, Zhang LL. Forebrain hypoplasia following acute prenatal ethanol exposure: quantitative analysis of effects on specific forebrain nuclei. Pathol. 1996;28:161–166. doi: 10.1080/00313029600169803. [DOI] [PubMed] [Google Scholar]
- Astley SJ, Magnuson SI, Omnell LM, Clarren SK. Fetal alcohol syndrome: changes in craniofacial form with age, cognition, and timing of ethanol exposure in the macaque. Teratology. 1999;59:163–172. doi: 10.1002/(SICI)1096-9926(199903)59:3<163::AID-TERA8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Byrd KE. Craniofacial sequelae of lesions to facial and trigeminal motor nuclei in growing rats. Am. J. Phys. Anthropol. 1988;76:87–103. doi: 10.1002/ajpa.1330760108. [DOI] [PubMed] [Google Scholar]
- Cambronero F, Puelles L. Rostrocaudal nuclear relationships in the avian medulla oblongata: a fate map with quail chick chimeras. J. Comp. Neurol. 2000;427:522–545. doi: 10.1002/1096-9861(20001127)427:4<522::aid-cne3>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Clarren SK, Astley SJ. Pregnancy outcomes after weekly oral administration of ethanol during gestation in the pig-tailed macaque: comparing early gestational exposure to full gestational exposure. Teratology. 1992;45:1–9. doi: 10.1002/tera.1420450102. [DOI] [PubMed] [Google Scholar]
- Dunty WC, Jr, Zucker RM, Sulik KK. Hindbrain and cranial nerve dysmorphogenesis result from acute maternal ethanol administration. Dev. Neurosci. 2002;24:328–342. doi: 10.1159/000066748. [DOI] [PubMed] [Google Scholar]
- Gofflot F, van Maele-Fabry G, Picard JJ. Cranial nerves and ganglia are altered after in vitro treatment of mouse embryos with valproic acid (VPA) and 4-en-VPA. Dev. Brain Res. 1996;93:62–69. doi: 10.1016/0165-3806(96)00031-4. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Marcussen B, West JR. A single day of alcohol exposure during the brain growth spurt induces brain weight restriction and cerebellar Purkinje cell loss. Alcohol. 1990;7:107–114. doi: 10.1016/0741-8329(90)90070-s. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB. The efficiency of systematic sampling in stereology and its prediction. J. Microsc. 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
- Gundersen HJG, Bagger P, Bendtsen TF, Evans SM, Korbo L, Marcussen N, Moller A, Nielsen K, Nyengaard JR, Pakkenberg B, Sorensen FB, Vesterby A, West MJ. The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. A.P.M.I.S. 1988;96:857–881. doi: 10.1111/j.1699-0463.1988.tb00954.x. [DOI] [PubMed] [Google Scholar]
- Hanson JW, Smith DW. The fetal hydantoin syndrome. J. Pediatrics. 1975;87:285–290. doi: 10.1016/s0022-3476(75)80604-4. [DOI] [PubMed] [Google Scholar]
- Jacobs JS, Miller MW. Proliferation and death of cultured fetal neocortical neurons: effects of ethanol on the dyna Pmics of cell growth. J. Neurocytol. 2001;30:391–401. doi: 10.1023/a:1015013609424. [DOI] [PubMed] [Google Scholar]
- Jones KL, Smith DW. Recognition of the fetal alcohol syndrome in early infancy. Lancet. 1973;2:999–1001. doi: 10.1016/s0140-6736(73)91092-1. [DOI] [PubMed] [Google Scholar]
- Ju MJ, Aroca P, Luo J, Puelles L, Redies C. Molecular profiling indicates avian branchiomotor nuclei invade the hindbrain alar plate. Neurosci. 2004;128:785–796. doi: 10.1016/j.neuroscience.2004.06.063. [DOI] [PubMed] [Google Scholar]
- Kentroti S, Vernadakis A. Ethanol administration during early embryogenesis affects neuronal phenotypes at a time when neuroblasts are pluripotential. J. Neurosci. Res. 1992;33:617–625. doi: 10.1002/jnr.490330414. [DOI] [PubMed] [Google Scholar]
- Light KE, Belcher SM, Pierce DR. Time course and manner of Purkinje neuron death following a single ethanol exposure on postnatal day 4 in the developing rat. Neurosci. 2002;114:327–337. doi: 10.1016/s0306-4522(02)00344-5. [DOI] [PubMed] [Google Scholar]
- Lumsden A. The cellular basis of segmentation in the developing hindbrain. Trends Neurosci. 1990;13:329–335. doi: 10.1016/0166-2236(90)90144-y. [DOI] [PubMed] [Google Scholar]
- Lumsden A, Keynes R. Segmental patterns of neuronal development in the chick hindbrain. Nature. 1989;337:424–428. doi: 10.1038/337424a0. [DOI] [PubMed] [Google Scholar]
- Miller MW. Effect of prenatal exposure to ethanol on the development of cerebral cortex: I. Neuronal generation. Alcohol. Clin. Exp. Res. 1988;12:440–449. doi: 10.1111/j.1530-0277.1988.tb00223.x. [DOI] [PubMed] [Google Scholar]
- Miller MW. Effect of pre- or postnatal exposure to ethanol on the total number of neurons in the principal sensory nucleus of the trigeminal nerve: cell proliferation and neuronal death. Alcohol. Clin. Exp. Res. 1995;19:1359–1363. doi: 10.1111/j.1530-0277.1995.tb01625.x. [DOI] [PubMed] [Google Scholar]
- Miller MW. Limited ethanol exposure selectively alters the proliferation of precursor cells in the cerebral cortex. Alcohol. Clin. Exp. Res. 1996;20:139–143. doi: 10.1111/j.1530-0277.1996.tb01056.x. [DOI] [PubMed] [Google Scholar]
- Miller MW. A longitudinal study of the effects of prenatal ethanol exposure on neuronal acquisition and death in the principal sensory nucleus of the trigeminal nerve: interaction with changes induced by transection of the infraorbital nerve. J. Neurocytol. 1999;28:999–1015. doi: 10.1023/a:1007088021115. [DOI] [PubMed] [Google Scholar]
- Miller MW. Exposure to ethanol during gastrulation alters somatosensory-motor cortices and the underlying white matter in the macaque. Cereb Cortex. 2007;17:2961–2971. doi: 10.1093/cercor/bhm024. [DOI] [PubMed] [Google Scholar]
- Miller MW. Normal Processes and the Effects of Alcohol and Nicotine. Oxford University Press; Oxford: 2006. Brain Development. [Google Scholar]
- Miller MW, Hu H. Lability of neuronal lineage decisions is revealed by acute exposures to ethanol. Dev. Neurosci. doi: 10.1159/000207493. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Miller MW, Muller SJ. Structure and histogenesis of the principal sensory nucleus of the trigeminal nerve: effects of prenatal exposure to ethanol. J. Comp. Neurol. 1989;282:570–580. doi: 10.1002/cne.902820408. [DOI] [PubMed] [Google Scholar]
- Miller MW, Robertson S. Prenatal exposure to ethanol alters the postnatal development and transformation of radial glia to astrocytes in the cortex. J. Comp. Neurol. 1993;337:253–266. doi: 10.1002/cne.903370206. [DOI] [PubMed] [Google Scholar]
- Miller MW, Peter A, Wharton SB, Wyllie AH. Proliferation and death of conditionally immortalized neural cells from murine neocortex: p53 alters the ability of neuron-like cells to re-enter the cell cycle. Brain Res. 2003;965:57–66. doi: 10.1016/s0006-8993(02)04119-7. [DOI] [PubMed] [Google Scholar]
- Mooney SM, Miller MW. Episodic exposure to ethanol during development differentially affects brainstem nuclei in the macaque: a model of fetal alcohol syndrome and autism. J. Neurocytol. 2001;30:973–982. doi: 10.1023/a:1021832522701. [DOI] [PubMed] [Google Scholar]
- Mooney SM, Miller MW. Time-specific effects of ethanol exposure on cranial nerve nuclei: gastrulation and neuronogenesis. Exp. Neurol. 2007;205:56–63. doi: 10.1016/j.expneurol.2007.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney SM, Miller MW, Henderson GI. Intracellular events in ethanol-induced cell death. In: Miller MW, editor. Brain Development. Normal Processes and the Effects of Alcohol and Nicotine. Oxford University Press; Oxford: 2006. pp. 267–278. [Google Scholar]
- Mooney SM, Napper RMA. Early postnatal exposure to alcohol reduces the number of neurons in the occipital but not the parietal cortex of the rat. Alcohol. Clin. Exp. Res. 2005;29:683–691. doi: 10.1097/01.alc.0000158936.40150.5a. [DOI] [PubMed] [Google Scholar]
- Murphy P, Davidson PR, Hill RE. Segment-specific expression of a homoeobox-containing gene in the mouse hindbrain. Nature. 1989;341:156–159. doi: 10.1038/341156a0. [DOI] [PubMed] [Google Scholar]
- Nieuwenhuys R, ten Donkelaar HJ, Nicholson C. The central nervous system of vertebrates. Springer; Heidelberg: 1998. [Google Scholar]
- Paxinos G, Huang X-F, Toga AW. The Rhesus Monkey Brain in Stereotaxic Coordinates. Academic Press; San Diego: 2000. [Google Scholar]
- Pierce DR, Williams DK, Light KE. Purkinje cell vulnerability to developmental ethanol exposure in the rat cerebellum. Alcohol. Clin. Exp. Res. 1999;23:1650–1659. [PubMed] [Google Scholar]
- Prock TL, Miranda RC. Embryonic cerebral cortical progenitors are resistant to apoptosis, but increase expression of suicide receptor DISC-complex genes and suppress autophagy following ethanol exposure. Alcohol. Clin. Exp. Res. 2007;31:694–703. doi: 10.1111/j.1530-0277.2007.00354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier PM, Ingram JL, Tisdale B, Nelson A, Romano J. Embryological origin for autism: Developmental anomalies of the cranial nerve motor nuclei. J. Comp. Neurol. 1996;370:247–261. doi: 10.1002/(SICI)1096-9861(19960624)370:2<247::AID-CNE8>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Santillano DR, Kumar LS, Prock TL, Camarillo C, Tingling JD, Miranda RC. Ethanol induces cell-cycle activity and reduces stem cell diversity to alter both regenerative capacity and differentiation potential of cerebral cortical neuroepithelial precursors. B.M.C. Neurosci. 2005;6:59. doi: 10.1186/1471-2202-6-59. doi:10.1186/1471−2202−6−59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulik KK, Cook CS, Webster WS. Teratogens and craniofacial malformations: relationships to cell death. Development. 1988;103(Suppl):213–231. doi: 10.1242/dev.103.Supplement.213. [DOI] [PubMed] [Google Scholar]
- Sulik KK, Johnston MC, Webb MA. Fetal alcohol syndrome: embryogenesis in a mouse model. Science. 1981;214:936–938. doi: 10.1126/science.6795717. [DOI] [PubMed] [Google Scholar]
- Tateno M, Ukai W, Yamamoto M, Hashimoto E, Ikeda H, Saito T. The effect of ethanol on cell fate determination of neural stem cells. Alcohol. Clin. Exp. Res. 1995;29:225S–229S. doi: 10.1097/01.alc.0000190658.56149.d4. [DOI] [PubMed] [Google Scholar]
- van Maele-Fabry G, Gofflot F, Picard JJ. Defects in the development of branchial nerves and ganglia induced by in vitro exposure of mouse embryos to mercuric chloride. Teratology. 1996;53:10–20. doi: 10.1002/(SICI)1096-9926(199601)53:1<10::AID-TERA2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- van Maele-Fabry G, Gofflot F, Clotman F, Picard JJ. Alterations of mouse embryonic branchial nerves and ganglia induced by ethanol. Neurotoxicol Teratol. 1995;17:497–506. doi: 10.1016/0892-0362(95)00009-g. [DOI] [PubMed] [Google Scholar]
- Wilkinson DG, Bhatt S, Cook M, Boncinelli E, Krumlauf R. Segmental expression of Hox-2 homoeobox-containing genes in the developing mouse hindbrain. Nature. 1989;341:405–409. doi: 10.1038/341405a0. [DOI] [PubMed] [Google Scholar]