

Abstract
Rapidly progressive dementias (RPDs) are neurological conditions that develop subacutely over weeks to months, or rarely acutely over days. In contrast to most dementing conditions that take years to progress to death, RPD can be quickly fatal. It is critical to evaluate the RPD patient without delay, usually in a hospital setting, as they may have a treatable condition. In this review, we discuss a differential diagnostic approach to RPD, emphasizing neurodegenerative, toxic/metabolic, infectious, autoimmune, neoplastic, and other conditions to consider.
Introduction
As a major prion disease referral center in the United States, we are referred several rapidly progressive dementias (RPD) cases every week, most of which are referred with a potential diagnosis Jakob-Creutzfeldt disease (CJD). The number of referrals increased dramatically with the identification of quinacrine as a potential therapy for CJD and commencement of the first U.S. CJD treatment trail in 20051.[1, 2] We recognized the need for a broader diagnostic approach to RPD when we observed that 15-20% of these referrals were due to other non-prion conditions, many of which were treatable. Physicians, and even neurologists, generally are not trained formally in the assessment of RPDs. In this review, we hope to provide a more thorough appreciation of the myriad of etiologies for RPDS and to offer a possible diagnostic decision tree or algorithm, based largely on the experience of our center.
Most dementias develop slowly, allowing an unhurried outpatient evaluation. Algorithms for the assessment of these patients have been developed and refined, and most neurologists are well acquainted with these approaches. A careful history will usually detect dementia secondary to medications or depression and routine laboratory assessments help to eliminate metabolic conditions that can cause dementia including anemia, electrolyte imbalance, liver or kidney failure, thyroid disease and vitamin B12 deficiency. The majority of slowly progressive dementias are secondary to Alzheimer's disease (AD), although there is an increasing recognition of non-AD dementias, including frontotemporal dementia (FTD; see Chapter 1), subcortical ischemia vascular disease (SIVD; see Chapter 8), dementia with Lewy bodies (DLB; see Chapter 9), and other parkinsonian dementias such as cortical basal degeneration (CBD) and progressive supranuclear palsy.[3] (see Chapter 11). However, with the possible exceptions of DLB and CBD, the disorders that commonly lead to slowly progressive adult dementia, such as AD and FTD, rarely present as RPDs. [4-6] The patient with an RPD often requires consideration of a different set of disorders.
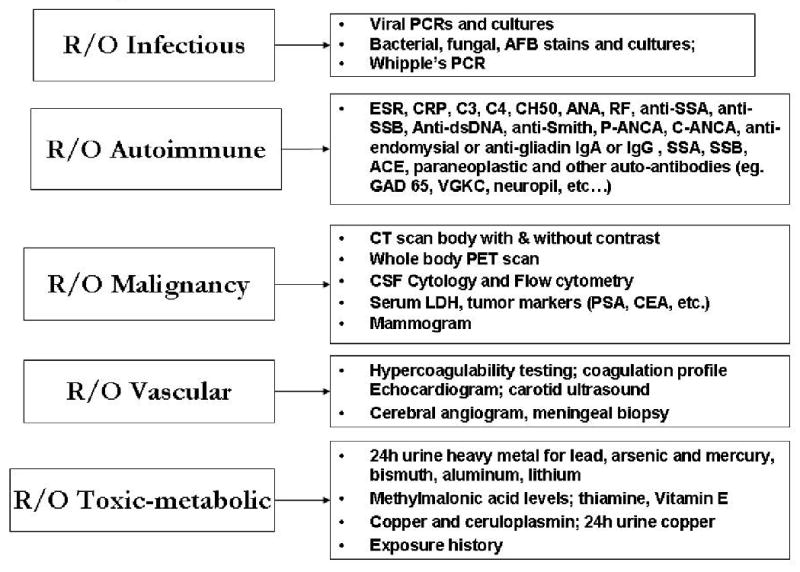
The primary aim of this chapter is to instruct clinicians in an approach to the differential diagnosis of RPD that will broaden their scope of inquiry and, particularly, to identify RPDs that are treatable and potentially reversible. We have organized this chapter around the following categories: neurodegenerative, autoimmune, toxic/metabolic, infectious, neoplastic, vascular, and we will emphasize the RPDs that are most difficult to diagnose or least likely to be recognized. As many types of conditions can cause RPD and they can progress quickly, it is important to have an organized, systematic approach to diagnosis. The mnemonic VITAMINS is often useful for summarizing some of the major categories of etiologies for RPDs (Table 13.1). Table 13.2 lists many etiologies of RPD, many of which there is unfortunately not space to discuss in this review. When considering an RPD patient, it may be helpful to use these tables to ensure a complete differential has been considered. RPDs that present with space occupying brain lesions easily identified by CT or MRI scan, are not discussed in this chapter. Figures 13.1 and 13.2 provide an outline for the diagnostic approach that we use in evaluating a patient with RPD. Figure 13.1 shows the overall approach, while 13.2 details an expanded evaluation when standard testing is inconclusive.
Table 13.1. VITAMINS - mnemonic for categories of conditions causing RPDs.
| Vascular |
| Infectious |
| Toxic-Metabolic |
| Autoimmune |
| Metastases/Neoplasm |
| Iatrogenic |
| Neurodegenerative |
| Systemic |
Table 13.2. Differential Diagnosis of Rapidly Progressive Dementias.
| Neurodegenerative |
| Jakob-Creutzfeldt disease (CJD; sporadic, iatrogenic, familial) |
| Alzheimer's disease (AD) |
| Dementia with Lewy Bodies (DLB) |
| Frontotemporal dementia (FTD) |
| Corticobasal degeneration (CBD) |
| Progressive Supranuclear Palsy (PSP) |
| Infectious |
| Viral encephalitis, including HSV |
| HIV dementia |
| Progressive Multifocal Leukoencephalopathy (PML) |
| Subacute sclerosing panencephalitis (SSPE; young adults) |
| Fungal infections (immunosuppression e.g., CNS aspergillosis,) |
| Syphilis |
| Parasites |
| Lyme disease (rarely encephalopathy) |
| Balamuthia |
| Whipple's Disease |
| Toxic/Metabolic |
| Vitamin B12 (cyanocobalmin) deficiency |
| Vitamin B1 (Thiamine) deficiency |
| Niacin deficiency |
| Folate deficiency (dementia rare) |
| Uremia |
| Wilson's disease |
| Portosystemic encephalopathy |
| Acquired hepatocerebral degeneration |
| Porphyria |
| Bismuth toxicity |
| Lithium toxicity |
| Mercury toxicity |
| Arsenic toxicity |
| Electrolyte Abnormalities |
| Autoimmune |
| Hashimoto's Encephalopathy |
| Paraneoplastic (autoimmune) limbic encephalopathy (PLE) |
| Non-paraneoplastic autoimmune (e.g., anti-VGKC mediated) |
| Lupus cerebritis |
| Other CNS Vasculitides |
| Sarcoid |
| Endocrine Abnormalities |
| Thyroid disturbances |
| Parathyroid abnormalities |
| Adrenal diseases |
| Neoplasm-related |
| Non-autoimmune paraneoplastic conditions |
| Metastases to CNS |
| Primary CNS lymphoma |
| Intravascular lymphoma |
| Lymphomatoid granulomatosis |
| Gliomatosis cerebri |
Figure 13.1. RPD Evaluation.
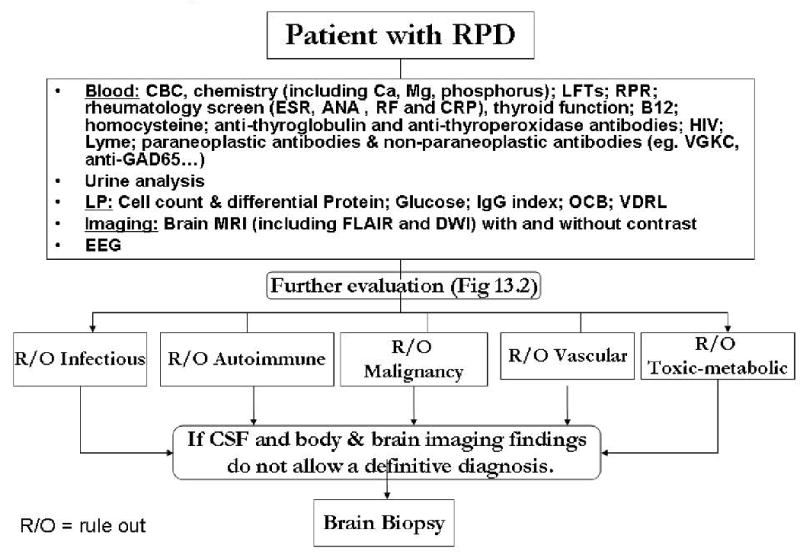
Figure 13.2. Further RPD Evaluation.
Over the past five years, our dementia center has been referred approximately 825 RPD cases, many with a presumptive diagnosis of CJD. After reviewing records and in many cases evaluating the patients, we determined the diagnostic breakdown of this group as 54% prion disease (37% probable or definite sporadic; 15% genetic, and 2% acquired), 28% undetermined (due to insufficient records, although most met criteria for possible CJD; see Chapter 12), and, importantly, 18% with other non-prion conditions, many of which were treatable. The diagnostic breakdown of these non-prion RPDs was: 26% neurodegenerative, 15% autoimmune, 11% infectious, 11% psychiatric, and 9% miscellaneous other, while twenty-eight percent were still undetermined, often leukoencephalopathies or encephalopathies of unknown etiology (unpublished data). Because our efforts in improving diagnosis of RPDs was prompted by the necessity to differentiate prion diseases from other causes of RPD, we begin with a very brief discussion of some of the salient issues in diagnosis of the prototypical RPD, prion disease, which is discussed in detail in the previous chapter, Chapter 11.
Degenerative Dementias
Prion diseases
When considering a rapidly progressive dementia, particularly a patient with prominent motor and/or cerebellar dysfunction, CJD should be high on the differential. CJD is discussed in detail in Chapter 12 in this edition, but some key features to consider with prion disease, based on our experiences, are mentioned here.
The most commonly used clinical criteria for probable sCJD [7, 8] does not allow early diagnosis of CJD and uses ancillary tests (EEG and CSF 14-3-3 protein) that many consider to have poor sensitivity and/or specificity and not to be very useful in clinical practice.[9-11] A major problem with these criteria is that they include signs or symptoms, such as akinetic mutism and the characteristic EEG, that often do not occur until late stages of the illness. These criteria also do not include features that are often early signs of the illness, such as behavioral changes or aphasia.[12] We identified the first symptom in 114 sCJD subjects referred to our center and found the most common were cognitive (39% of patients), followed by cerebellar (21%), behavioral (20%), constitutional (20%), sensory (11%), motor (9%) and visual (7%). Three of these categories - behavioral, constitutional and sensory symptoms (e.g., headache, malaise, vertigo, etc….) - are not included in current diagnostic criteria. Furthermore, we have found that ancillary tests in WHO criteria that are required for a probable sCJD diagnosis, EEG or 14-3-3, are neither sensitive nor specific. In a recent evaluation of an RPD cohort (150 sCJD and 47 non-prion RPD subjects) we found the 14-3-3 to have a sensitivity of 48% and a specificity of 66%. The EEG had a sensitivity of less than 45% by the time patients were had an EEG at our institution, which was generally not their first EEG. This increased to about 50% when patients were then followed prospectively during their entire disease course (M. Geschwind, unpublished data).[13] In our cohort, our preliminary data suggest that two other surrogate biomarkers for sCJD, total tau (t-tau), neuron specific enolase (NSE)have somewhat higher sensitivity and specificity for CJD than 14-3-3 or EEG. We feel these CSF biomarkers are merely signs of rapid neuronal injury, are not specific for prion disease and are therefore of questionable diagnostic utility. A prion-specific test is needed.[14-16]
Typically Chronic Degenerative Dementias
Alzheimer's disease (AD) is rarely rapid, but unusual presentations can be mistaken for CJD.[4] Several cases of AD have been reported in conjunction with angiopathy (CAA) presenting as adult onset RPD. [17-19] Other non-prion neurodegenerative diseases that can also present, albeit rarely, in a more fulminant fashion, include DLB, FTD (particularly the subtype with motor neuron disease, CBD and PSP. Patients with AD typically survive a median of 11.7 (SD ±0.6) years, FTD patients 11 years (SD ±0.9) and PSP/CBD patients 11.8 years (SD ±0.6) [20], and PSP alone 5.6 years [21], from first symptom. More rapid onset and/or progression can occur. [20, 22-25]. In a large German study, out of 413 autopsied suspected cases of CJD 7% had AD and 3% had DLB. Myoclonus and extrapyramidal signs occurred in more than 70% of the DLB and more than 50% of the AD patients. [4] Similarly, in a French pathologic study of 465 suspected CJD patients, the two most frequent non-CJD pathologic diagnoses were AD and DLB.[26]
Parkinsonian dementias, such as DLB and FTD-spectrum disorders including PSP, CBD and FTD are discussed in Chapters 1, 2 and 11 and thus will only will be briefly mentioned here. DLB is a progressive dementia often associated with fluctuations in cognitive function, persistent well-formed visual hallucinations, and/or parkinsonism. [27] Duration of DLB is often shorter than for many other neurodegenerative dementias; one study suggests 3 year survival [28], although rapid decline with death in one year can occur. Periodic sharp waves may be seen on EEG, leading to confusion with CJD. [26, 29]. In several large cohort studies, DLB was the second most common condition mistaken for CJD.[4, 26] FTD is rarely rapidly progressive, although it typically has a faster course than AD. Patients typically present with a frontal syndrome including behavioral, personality and cognitive changes occurring over years, followed by dementia. Fifteen percent or more of FTD patients develop amyotrophic lateral sclerosis (ALS) and these patients typically die within 1.4 years from the time of diagnosis. [30-33] Corticobasal degeneration (CBD) is a clinically and pathologically heterogeneous atypical parkinsonian dementia often confused clinically with AD, PSP, or FTD. [34-38] Many features of CBD, including myoclonus, alien limb and visual, sensory and motor deficits overlap with features of CJD. The converse is also true; CJD can sometimes present as a rapid cortical basal syndrome (CBS) [39] or with a protracted course over two to three years with features indistinguishable from CBD; however, the FLAIR and DWI MRI abnormalities seen in CJD are not found in CBD.[40] As in CJD, PSP patients develop dementia, akinetic-rigid parkinsonism (symmetric bradykinesia and axial rigidity), postural instability, swallowing and speech problems, and often progress to a hypokinetic, mute state. [41-46] Abnormalities of eye movements, particularly slowed of velocity of saccades progressing to supranuclear gaze palsy, are part of the PSP syndrome. CJD mimicking PSP has been reported.[47]
Neurofilament inclusion body disease (NIBD) is a recently described pathologic condition that can clinically present as FTD or CBD. The four index cases were all more rapid than typical degenerative dementias, with duration of only two to four years. Brain MRI and pathology showed frontal, temporal and caudate atrophy. A distinguishing feature of NIBD is the presence of intracytopasmic neuronal inclusions that stain strongly with antibodies to neurofilament proteins and ubiquitin, but not tau or α-synuclein.[48] Once case of NIBD has also presented as an early onset rapidly progressive FTD with features of primary lateral sclerosis.[49]
Fahr's disease is a neurodegenerative disease of unknown etiology with basal ganglia calcification that typically presents with a movement and neuropsychiatric disorder. Although usually very slowly progressive, a 50 year old patient was reported with a rapidly progressive frontal behavioral and cognitive presentation of Fahr's disease (idiopathic basal ganglia calcification) resulting in dementia within six months. Fahr's patients have extensive basal ganglia calcification, a finding which is not present in CJD. [50] Rarely, genetic neurodegenerative diseases may also present as a rapidly progressive dementia. A case was recently reported of a male with the fragile X premutation who presented in his mid-60s with a rapid course of tremor, gait ataxia, parkinsonism, and cognitive deficits [51]
Autoimmune encephalopathies (paraneoplastic and non-paraneoplastic)
Over the past few years, there has been a growing awareness and identification of autoimmune causes of encephalopathy or RPDs. Initially, most of these autoimmune conditions were thought to be paraneoplastic –due antibodies, or other components of the immune system, against the cancer, cross-reacting with antigens of the nervous system. However, in many of these conditions, no cancers have been identified, despite repeated comprehensive searches for a tumor.[mg1] In this section, we will discuss both paraneoplastic and non-paraneoplastic autoimmune encephalopathies.
Paraneoplastic neurologic disorders (PNDs) often present as a rapidly progressive limbic encephalopathy. PNDs that involve the CNS are often divided into two forms: those with isolated involvement of one part of the nervous system (e.g., limbic encephalitis/ encephalopathy, cerebellar syndromes, retinal degeneration) or those with more diffuse, multifocal symptoms, sometimes referred to as paraneoplastic encephalomyelitis (PEM). Paraneoplastic limbic encephalopathy (PLE) can occur as an isolated syndrome or as PEM with involvement of other parts of the nervous system (i.e., brainstem, cerebellum, peripheral nerves). The most common symptoms are a subacute amnestic syndrome, presenting as problems with short-term anterograde memory or more variable retrograde amnesia. Depression, personality changes, anxiety and emotional lability often precede the cognitive dysfunction. Seizures are common.[52, 53] PNDs occur in patients with a known diagnosis of a cancer or may precede the detection of the cancer by weeks, months or rarely a few years. In patients without a known cancer diagnosis, various signs or symptoms may suggest a PEM or PLE, including: subacute development of multifocal neurologic symptoms, CSF evidence of inflammation, elevated tumor markers (e.g., CEA, CA-125, PSA, etc…), a family history of cancer, unexplained anorexia or weight loss or other symptoms suggestive of cancer, and the presence of certain paraneoplastic antibodies in the serum and/or CSF. [52, 53]
The most common tumors associated with PLE are small cell lung cancer (SCLC) (75% of cases), germ-cell tumors (ovarian or testicular), thymoma, Hodgkin's lymphoma and breast cancer. [52, 53], while the most common antibodies associated with PLE are anti-Hu (ANNA-1), Anti-Ma2 (also called Anti-Ta; antigen is Ma2), CV2 (Anti-CMRP-5), Yo (PCA-1), and probably anti-neuropil. [52, 54-56] Anti-Hu antibodies are found in 50% of cases of PLE cases with SCLC. Identification of anti-neural antibodies is highly suggestive of an underlying neoplasm. Furthermore, the type of autoantibody may suggest the tumor type rather than the neurological syndrome. [52, 57] Almost one-third of patients with a neurological syndrome and autoantibodies will have more than one auto-antibody.[57, 58] In PLE associated with anti-Ma2 (Ta) antibodies and testicular cancer, about half of patients have dramatic improvement of their neurologic syndrome after treatment of their cancer. [56, 59]. This may be in part due to the ability to remove all the cancer through orchiectomy.[60] Hypothalamic involvement is common in patients with anti-Ma2 antibodies.[56] Antibodies to CRMP-5 (Anti-CV2 or Anti-CRMP-5), a protein in the collapsin response-mediator protein family are often associated with PNDs, including PLE. Peripheral neuropathy (47%) and autonomic neuropathy (31%) are the most common neurological signs. Subacute dementia and cerebellar ataxia each occur in about ¼ of patients, followed by neuromuscular junction disorders (12%), chorea (11%) and cranial neuropathy (17%, including optic neuropathy and loss of taste. Spinal fluid is often inflammatory. Anti-CV2 is most often seen with small-cell lung cancers, followed by thymomas. [61, 62] FLAIR MRI in Anti-CV2 antibody syndrome often shows caudate, anterior putamen with or without medial temporal lobe hyperintensity,[58] although thalamic T2-weighted hyperintensity can also occur (manuscript submitted). The striatal and thalamic involvement can appear similar to findings in CJD, however, unlike CJD the T2-weighted hyperintensity may extend beyond the deep grey nuclei into adjacent white matter and there are no diffusion-weighted abnormalities. Most patients with limbic encephalopathy and thymoma (often anti-CV2 or anti-VGKC antibodies) have significant neurologic improvement following tumor removal and treatment. [63] Table 13.3 summarizes some of the major antibodies, with their clinical phenotypes, that are associated with limbic encephalopathy.
Table 13.3.
Paraneoplastic and non-paraneoplastic antibodies often associated with limbic encephalopathy
| Antibodies | Tumors | Usual Age of Onset | Gender | Associated Symptoms |
|---|---|---|---|---|
| Anti-Hu | SCLC, neuroblastoma, prostate | 55-65 | F>=M | PEM; SSN |
| Anti- Ma2 (anti-Ta) | Germinoma, testicular; lung Ca | 22-45 | M≫F | PLE; Brainstem; Cerebellar; Hypothalamic |
| Anti-CV2 (Anti-CRMP-5) | Thymoma, lung Ca, renal cell | 50-70 | F=M | Neuropathy, Cerebellar, PLE, Chorea |
| Anti- VGKC | 80% none; 20% tumor thymoma, lung, | variable | ??? | Isaac's and Morvan's Syndromes; neuromyotonia, cramps, hyperhydrosis, sleep disorder, seizures PLE |
| Anti-amphiphysin | Breast, SCLC | PEM, Stiff Person's Syndrome, Opsoclonus-myoclonus | ||
| Anti-Yo | Gynecologic and breast, adenoCa | 26-85 | F≫M | PCD; Limbic encephalopathy |
| Anti-nCMAg (some anti-neuropil) | Teratomas, thymus | 20-50s | F>M | Acute PLE, abnormal movements, decreased consciousness |
| Anti-Ma1 | Lung, other (breast, parotid, colon) | 60 | F | PCD; brainstem |
| Anti-Ri (ANNA-2) | Breast, gynecologic, lung, bladder | Ataxia; opsoclonus/myoclonus |
Recently, there has been increasing awareness of several immune-mediated encephalopathies that are not always associated with cancers.[54, 64] In some of these syndromes, antibodies, and sometimes their antigens, have been identified. Two such syndromes of limbic encephalopathy are due to anti-voltage-gated potassium channels antibodies (VGKC) and to anti-neuropil antibodies. Patients with anti-VGKC antibodies present along a spectrum of nervous system involvement, from the peripheral to the central nervous system. Involvement of the peripheral nervous system alone may manifest as neuromyotonia (Isaac's Syndrome). Isolated CNS involvement may present as a seizure disorder or limbic encephalopathy.[54, 65-69] However, combinations of peripheral and central involvement, such as in Morvan's syndrome also occur. Some of these patients have limbic encephalopathy in isolation, while others have been shown also to exhibit different degrees of Morvan's fibrillary chorea, a syndrome characterized by neuromyotonia, myalgias, hyperhydrosis, and disordered sleep. [70] Anti-GAD antibodies, although commonly associated with stiff person syndrome, can also cause subacute ataxia, sometimes with mild cognitive complaints.[71] Novel antibodies against components of the CNS are continually being identified.[54] If autoimmune syndrome is strongly suspected, due to CSF or serological findings, concurrent or family history of autoimmune disorders, one should have a low threshold for sending serum and CSF to a research laboratory that specializes in identifying such antibodies.
Hashimoto's encephalopathy (HE) is a rare but probably under-diagnosed, treatable autoimmune disorder associated with chronic lymphocytic Hashimoto's thyroiditis [72, 73]. Often, it begins with a prodrome of depression, personality change or psychosis and progresses into a cognitive decline associated with myoclonus, ataxia, pyramidal and extrapyramidal signs, stroke-like episodes, altered levels of consciousness, confusion and/or seizures. Hallucinations or other psychoses are common. [72-74]. It is often confused with CJD because of their overlapping clinical profile. [5, 74]. Compared to CJD, HE is more frequently is associated with seizures and tends to have a more fluctuating course [74]. For unclear reasons, more women (85%) than men have been diagnosed with HE [74]. Patients may be euthyroid, hypothyroid, and even hyperthyroid, although the diagnosis cannot be made until a patient is euthyroid [74]. Elevated levels of either anti-thyroglobulin or anti-thyroperoxidase (anti-TPO) and neurologic and psychiatric symptoms when patients are euthyroid, in the absence of other possible etiologies suggest the diagnosis. The EEG frequently shows nonspecific abnormalities with asynchronous background slowing and intermittent diffuse or focal slow activity; however, as in CJD, triphasic waves or periodic sharp waves may occur [72, 75]. MRI is not specific but commonly shows increased T2-weighted subcortical, mesial-temporal or white matter signal, which may disappear after treatment [72, 76-78]. CSF often has increased protein, a non-specific finding that occurs in many other RPDs, including CJD [13, 72, 73]. The etiology of HE but may be due to the presence of a shared antigen in the brain and thyroid [72, 73, 79]. More than 90% of patients respond favorably to immunosuppression, typically high dose steroids followed by a long, slow taper, although some patients may have persistent symptoms or a fluctuating course [72, 75, 80]. Plasmapharesis may also be helpful [81].
Many other autoimmune disorders present as RPDs, and are important to consider because of potential for reversibility with immunosuppression. A new clinicopathologic entity called “cerebral amyloid inflammatory vasculopathy” recently has been described. These patients show acute or rapid onset of dementia. MRI shows evidence of amyloid-related hemorrhages and sometime large confluent white matter hyperintensities. Brain biopsy revealed Aβ amyloid cerebral angiopathy associated with chronic non-granulomatous vasculitis. With a single dose of dexamethasone, a patient made a rapid and nearly complete recovery over a few months.[82]
Collagen vascular and granulomatous diseases also affect the CNS through mechanisms other than vasculitis. Several of these disorders may cause an encephalopathy or rapidly progressive dementia, including primary angiitis of the CNS (PACNS), polyarteritis nodosa (PAN), sarcoidosis, systemic lupus erythematosus (SLE), Sjögren's syndrome, celiac disease (Sprue), Behçet's disease, and hypereosinophilic syndrome. [83-89] Some authors group these encephalopathies of non-vasculitic origin under the term non-vasculitic autoimmune inflammatory meningoencephalopathies (NAIM); this group includes Hashimoto's and Sjogren's encephalopathies; which almost uniformly have abnormal EEGs and respond to high dose steroids. [90] The heralding features of the disorder may be neurological
Sarcoid, a systemic illness of unknown etiology characterized by the formation of non-necrotizing granulomas, can be successfully treated like other autoimmune conditions with immunosuppresion. Only about 5% of patients with sarcoidosis have involvement of the nervous system, but when it involves the CNS it sometimes presents as an RPD. When there is brainstem involvement, cranial neuropathies may occur. MRI is highly variable and may be normal, show enhancing granulomas (often at the base of the brain), or non-enhancing T2-weighted white matter hyperintensities consistent with a leukoencephalopathy. CT of the chest may reveal hilar lymphadenopathy. CSF may be normal, but often shows elevated protein and from a mild to severe pleiocytosis. CSF angiotensin converting enzyme (ACE) levels are elevated in only 33-58% of cases and this test also lacks specificity. Biopsy of affected tissue is needed for diagnosis, Steroid treatment or other immunosuppression may be helpful, as are plasmapheresis and IVIG. It is important to rule out other granulomatosis diseases, particularly tuberculosis prior to initiating immunosuppression [91](M. Geschwind unpublished data).
Vascular
Depending on the location, strokes can present as rapidly progressive dementia. Large vessel occlusions as well as thalamic, anterior corpus callosal or multiple diffuse infarcts in particular have all presented as RPDs. [92, 93] Thrombotic thrombocytopenic purpura (TTP) can cause microangiopathic thromboses producing global cerebral ischemia, resulting in an encephalopathy. Hyperviscosity syndromes from blood dyscrasias, such as polycythemia or gammopathies, such as Waldenstrom's Macroglobulinemia can present as rapidly progressive dementias by causing global cerebral microvessel ischemia.
Although it is an autoimmune condition CNS vasculitis is discussed in this section because of its direct effect on the vasculature as the cause of RPD. Criteria for classification of certain vasculitides are largely based on a combination of clinical symptoms or signs and laboratory findings.[94, 95] A vasculitis may be limited to the central nervous system without any systemic or peripheral nervous system signs or initially may present as a systemic disorder with accompanying fever, weight loss, rash, neuropathy, and other organ involvement. Urinalysis may contain red cells as a sign of renal involvement. Ophthalmologic exam may identify uveitis, scleritis or signs of ophthalmic artery vasculitis. If a rash is present, a skin biopsy can be diagnostic. There may be signs of a hemolytic anemia. A basic rheumatologic screen may include, ESR, CRP, C3, C4, CH50, ANA, RF, anti-SSA, anti-SSB, P-ANCA, and C-ANCA, with other testing depending on results of this initial screen Serological testing is likely to abnormal in systemic forms of vasculitis, but in primary CNS vasculitis patients typically have normal non-specific tests, such as ESR, ANA and CRP.[96] Vasculitides are often distinguished from other RPDs by brain MRI abnormalities, such as strokes and/or hemorrhage involving both the white or gray matter.[96] Similarly, body imaging for systemic involvement may be helpful [97] When primary CNS vasculitis is suspected, cerebral angiogram or brain and meningeal brain biopsy of the affected area may be required for diagnosis. Intravascular lymphoma sometimes mimics CNS vasculitis on angiogram; if this condition is suspected (based on an elevated serum LDH or MRI findings), then doctors should avoid the angiogram and go directly to biopsy. [98, 99]
Infectious
AIDS-dementia complex, HIV-encephalopathy or HIV-associated dementia is a neurological complication of acquired immunodeficiency syndrome, eventually occurring in 1/4th of patients with AIDS. It typically occurs in the later stages of HIV infection [100] and has diminished since the introduction of highly active anti-retroviral therapy. Some individuals, however, develop RPD during seroconversion or immune reconstitution. In general, more rapid neurological impairment is associated with symptomatic HIV seroconverting illness [101]. Concomitant use of methamphetamine or cocaine may also synergize with HIV infection to cause an accelerated course of HIV dementia. [102]. As dementia can be a presenting feature of AIDS [103], HIV testing should be considered in the evaluation of every RPD.
Subacute and chronic opportunistic infections associated with HIV infection and other immunocompromised states must always be considered in the differential diagnosis of RPD. Cryptococcus and JC virus infections typically present with meningitis or progressive focal neurologic deficits respectively, however, they also can present with rapid progression of dementia. [104] CNS infection with Mycobacteria may present as an inflammatory meningitis. A recent case report identified an atypical acid fast bacillus, Mycobacterium neoaurum, by PCR in autopsy brain tissue from a patient with rapidly progressive dementia who was on low dose steroids. CSF cell count, mycobacterial culture, and Ziehl-Nielsen staining all failed to demonstrate the presence of mycobacterium. It is possible that many undiagnosed RPDs could be caused by infectious organisms that escape detection using standard microbiological techniques. [9, 105, 106]. (For a review on diagnosis and etiology of encephalitis see Glaser et al 2006).
Spirochete infections are unusual causes of cognitive impairment, but important to consider as they are treatable. No workup for dementia, including RPD, is complete without an evaluation for CNS infection with Treponema pallidum, or “neurosyphilis.” Cognitive dysfunction is the most common neurological syndrome, although usually a late complication, of syphilis[107]. It occasionally presents with rapid progression, particularly in immunocompromised patients. [108] Serological testing with RPR and VDRL and CSF VDRL suggest the diagnosis. The CSF in neurosyphilis usually shows a pleiocytosis and an elevated protein. [107] Lyme disease is a systemic infection with the spirochette, Borrelia burgdorferi, which is transmitted to people from a tick bite. Neurological manifestations are rare in Lyme disease, but can include cranial nerve palsy, meningitis, polyradiculopathy, depression, psychosis, and dementia.[109] Although rapidly progressive dementia caused by Lyme disease has been reported, it is rare, [110, 111] but it is important to consider because it responds readily to treatment. [112]
Subacute sclerosing panencephalitis (SSPE) is a chronic CNS infection from the virus that causes measles and is still occurs in individuals from countries where measles infections are common. It typically occurs in children, but can occur in adults. [113]Patients develop progressive dementia, seizures (focal and/or generalized), myoclonus, ataxia, rigidity, and visual disturbances. In the late stage of the illness, patients are unresponsive, with spastic quadriparesis, brisk deep tendon reflexes and positive Babinski signs. EEG often reveals periodic slow-wave complexes with associated sharp waves every 3-10 seconds that are often temporally associated with myoclonus. Definitive diagnosis is made with elevated antibody titers to the measles virus in the blood and CSF in the appropriate clinical setting. [114]
Whipple's disease is a rare bacterial (Tropheryma whippelii) infection, involving numerous organ systems, that can present as a neuropsychiatric syndrome that although typically insidious, can progress rapidly over months. More than 80% of the cases have been diagnosed in men. Although the age range varies from childhood to the elderly, onset is typically in the fifth through seventh decades, with a mean age of onset of about 50. Clinical presentation is varied. It most commonly presents as a malabsorption syndrome with diarrhea, abdominal pain, weight loss, arthralgias, wasting, fever, and lymphadenopathy; but as many as 15% of cases do not exhibit gastrointestinal symptoms. CNS involvement occurs in 5 to 45% of cases, with 5% of cases having neurologic presenting symptoms. [115] Dementia or mental status changes occur in more than 50% of the cases with neurologic involvement. [115, 116] Cognitive impairment occurs in 71% and psychiatric signs in 44% of cases of CNS Whipple's. [117] Eye movement abnormalities, myoclonus or other abnormal involuntary movements, headache, and abnormal hypothalamic function are frequently seen. Seizures, aseptic meningitis, ataxia and focal cerebral signs may occur [46, 77, 115-117]. Ataxia has been reported to occur in 45% of CNS Whipple's cases. [118] About 10% of cases have a triad of dementia, ophthalmoplegia, and myoclonus, which is highly suggestive of this condition,[115] Oculomasticatory myorrythymia is uncommon, but pathognomic.[46, 118] Clinically Whipple's may be mistaken for CBD or PSP.[35] Brain imaging is non-specific. CSF shows elevated protein or pleiocytosis in about half of cases with CNS involvement. Diagnosis is made by identification of PAS-positive inclusions or T. whipellii in foamy macrophages on jejunal biopsy or by T. whipellii PCR of CSF or jejunal biopsy. PCR in serum is probably less sensitive. Diagnosis can be challenging, as many of the symptoms are non-specific and is particularly difficult when Whipple's presents as an isolated neurologic syndrome without gastrointestinal symptoms. [115] Although very rare, Whipple's is important to recognize as it is readily treatable with antibiotics. [115, 117, 119, 120]
Malignancies
Several primary and secondary malignancies can cause an acute or subacute RPD. RPDs that can readily be identified by MRI will not be discussed in detail in this chapter, as once identified the work-up is somewhat routine. Three malignancies that often present as RPDs, and present with varied abnormalities on MRI, are primary CNS lymphoma (PCNSL), intravascular lymphoma (i.e. angiotropic lymphoma), and lymphomatoid granulomatosis (also known as angiocentric immunoproliferative lesions). Only the first two will be discussed in this chapter.
PCNSL is an extranodal form of non-Hodgkin's lymphoma. It typically presents with symptoms of intracranial mass lesions, such as headaches, seizures, and focal neurological deficits, but can present as a rapidly progressive dementia.[121] A diffusely infiltrating PCNSL, sometimes called lymphomatosis cerebri, also occurs.[122] Symptoms of PCNSL include personality changes, irritability, memory loss, lethargy, confusion, disorientation, psychosis, dysphasia, ataxia, gait disorder, and myoclonus. [121-124] CNS lymphoma can mimic CJD. [5, 96, 125] PCNSL accounts for 2-3% of all CNS neoplasms. The vast majority of PCNSL are non-Hodgkin diffuse large B-cell type, but T-cell, Burkitt's and poorly characterized forms also occur.[121, 126] The incidence increased from the mid-1970s to the mid-1980s due to an escalating number of immunocompromised patients from transplants, chemotherapy, and patients with HIV before the era of HAART, but seems to have stabilized over the past decade. [126] We focus here on PCNSL in immunocompetent individuals. PCNSL occurs most commonly in the 6th to 7th decades, but can occur at any age, with a slight male predominance. [123]. Uveitis or vitreitisis present in about 10% of cases, preceding the tumor sometimes by months, in about 75% of cases; identifying the uveitis or vitreitis may allow earlier diagnosis of the cancer.[127]. In immunocompetent individuals, brain MRI may show isointense to mildly hyperintense T2-weighted signal consistent with mass lesions with minimal to moderate edema, often involving the cerebral hemispheres, basal ganglia, periventricular white matter, or corpus callosum. Lesions may be isolated or multiple and generally show contrast enhancement.[128] When presenting as lymphomatosis cerebri, imaging reveals progressive, diffuse white matter signal abnormality without significant (or any) enhancement or mass effect – likely from a diffusely infiltrative process without interruption of the blood-brain barrier. [122, 123] CSF can show a lymphocytosis, increased protein, and low glucose. Serial CSF cytologic evaluations are typically required to identify the lymphoma.[126] EEG may show symmetric or asymmetric nonspecific diffuse slowing [122, 123]Unfortunately, definitive diagnosis often requires brain biopsy. In cases of ocular involvement, diagnosis can sometimes be made by vitrectomy. When possible, it is important to avoid giving corticosteroids prior to biopsy, as steroids can cause tumor cell necrosis, resulting in temporarily shrinkage of the tumor, but preventing tissue diagnosis.[96, 126] Prognosis is poor with patients surviving only a median of 4 months or less without treatment, 12-18 months with whole-brain radiation therapy (WBRT) alone, however upwards of 40 or more months with a combination of aggressive chemo and radiotherapy. Chemotherapy includes high dose systemic methotrexate. The use of chemotherapy alone versus chemotherapy plus WBRT is controversial. Due to the increased risk of neurotoxicity, WBRT in patients over 60 is not recommended. Neurotoxicity presents as an RPD with dementia, ataxia and incontinence, with median onset just over one year post-WBRT [126, 129]
Intravascular lymphoma can occur in almost any organ, but commonly has one of four presentations: CNS, skin, hemophagocytic or fever of unknown origin. It is caused by the proliferation of clonal lymphocytes within blood vessels, with relative sparing of parenchyma.[130] The more acute form of CNS intravascular lymphoma typically presents in middle age as an acute or subacute dementia, often with transient ischemic attacks or strokes. Systemic symptoms (e.g., fever and weight loss) may be present. The tumor cells are thought to be activated or transformed lymphocytes and are typically an angiotropic large B cell lymphoma, although cell-type forms also occur. These clonal lymphocytes preferentially bind endothelium. Imaging in CNS intravascular lymphoma is variable. MRI may show multiple areas of T2-weighted hyperintensity with patchy enhancement on T1-weighting, with or without edema. [131] Unfortunately most cases reported in the literature were diagnosed post-mortem; therefore a high index of suspicion, and a low threshold for brain biopsy, is required for patients with a rapidly progressive dementia and focal T2-weighted abnormalities on MRI. [99, 127, 132] Laboratory findings can include elevated ESR, serum lactate dehydrogenase, CSF pleiocytosis and increased protein. [132, 133] Survival in intravascular lymphoma is usually poor, especially without treatment. Aggressive treatment is needed for both PCNSL and intravascular lymphoma. The combination of chemo- and radiotherapy is better than radiotherapy alone. [127, 130, 132]
At our center, several patients referred as potential CJD were determined to have encephalopathy due to metastatic cancers, including lymphoma. A recently published case of a 79 year old woman with an RPD presenting with early visual hallucinations, followed by severe memory impairment, and extrapyramidal signs was thought to have CJD due to her course and a positive 14-3-3- CSF protein. Unfortunately, the diagnosis of miliary adenocarcinoma was only made at autopsy. [134]
Toxic-metabolic conditions
Metabolic causes of RPD include vitamin deficiences, endocrinologic disturbances and adult-presentations of inborn errors of metabolism. Vitamin deficiencies can result in significant neurologic deficits, including cognitive impairment. Pellagra (“rough skin”) is due to niacin deficiency and is classically described as “the three Ds” - dermatitis, diarrhea and dementia (on a historical note, the original term nicotinic acid was changed to niacin due to its confusion with nicotine). Niacin deficient causes abnormalities of the skin and gastrointestinal tract, as well as peripheral neuropathy, myelopathy and cognitive dysfunction. With a careful history, the dementia of pellagra is typically found to be insidious, not rapid. In industrialized nations, niacin deficiency, should be considered in patients with nutritional deficiency, such as alcoholics and patients with anorexia nervosa, as well as in those taking isoniazid. [135-138] Although diagnosis can be made by finding nicotinic acid metabolites in the urine; given the ease of treatment with 40-250 mg per day of niacin, diagnosis is usually based on clinical suspicion. Treatment often results in rapid resolution of symptoms [135, 139]. Deficiency of thiamine (Vitamin B1), a necessary cofactor in oxidative metabolism, can cause Wernicke's Encephalopathy, which classically presents as a triad of ophthalmoparesis (with vertical and/or horizontal nystagmus), ataxia and memory loss. DWI MRI can show diffusion abnormalities in mammilary bodies and dorsomedial nucleus of the thalamus, areas in which hemorrhagic necrosis is found pathologically.] The thalamic involvement on DWI MRI can appear similar to that seen in CJD. [40, 140-142] All patients with dementia should be screened for Vitamin B12 deficiency, as it is potentially reversible.
Adult presentations of metabolic disorders that typically afflict children can in rare instances also present as dementia in the adult. These dementias are usually associated with a constellation of symptoms such as weakness, spasticity, and ataxia and tend to be more slowly progressive. They can present with rapid cognitive decline. In the proper clinical context, of gastrointestinal disturbance, fluctuating course, an unexplained pain syndrome, and/or worsening after use of new medicines, porphyria should be ruled out. Adult onset metachromatic leukodystrophy (MLD) presented as an RPD in a woman who developed psychiatric symptoms and severe cognitive decline over 18 months with no weakness or ataxia.[143] Other leukodystrophies can also present as RPD. Orthochromatic leukodystrophies (OLD) are a heterogeneous group of metabolic disorders in which the specific enzymatic defects have not been found. Most are sporadic, but dominant inheritance has been reported. One report described 2 family members (57 and 38 at presentation) with a dominantly-inherited OLD who developed rapid dementia progressing to death over 2-3 years.[144] Finally, Kuf's disease, the rare adult form of Neuronal Ceroid Lipofuscinoses (NCLs), can present as RPD. Kuf's is an autosomal recessive lysosomal storage disease in which acid phosphatase staining ceroid and lipofuscins accumulate in neurons, causing a progressive encephalopathy. Kuf's disease typically presents in early adulthood. Type A Kuf's patients present with a progressive myoclonic epilepsy, while type B patients present with dementia which often begins with psychosis. A case reported in 1997 involved a 49 year old woman who initially presented with alternating catatonia and acute psychosis over 5 months before the development of dementia over the next 2 months. Diagnosis was made by acid phosphatase staining of brain and skin biopsies.[145] We recently diagnosed a 50 year old woman with a methylmalonic (and malonic) academia (MMA) who had developed significant cognitive impairment after a several month prodrome of gastrointestinal disturbance and psychiatric disturbance. She had low normal vitamin B12 levels and normal homocysteine. Despite thorough evaluation of her vitamin B12 pathways, the cause of her metabolic disorder is still unknown. (M. Geschwind, unpublished data)
Several toxins can cause an RPD. Exposure to heavy metals, such as arsenic, mercury, aluminum, lithium, lead can lead to cognitive decline, particularly following acute exposure. However, most cases of acute exposures result in florid encephalopathies that progress over hours to days and thus would not be confused with rapidly progressive dementias which progress over weeks to months. Manganese toxicity, found usually in miners, can present with significant parkinsonism. [138]
Bismuth is a metal used to treat gastrointestinal disorders, principally peptic ulcer disease and diarrhea. Bismuth intoxication, typically caused by overdosing on bismuth containing products such as Pepto-bismol®, can cause a disorder mimicking CJD. Patients initially manifest with apathy, mild ataxia, headaches, which progresses to myoclonus, dysarthria, severe confusion, hallucinations (auditory and visual), seizures, and, in severe cases, even death. [146-149]. Blood levels of bismuth greater than 50 mcg/L are considered in the toxic range [147, 149]. The condition is usually reversible, however, extremely prolonged use can result in permanent tremors. [146, 147]. A careful history may be needed to make the diagnosis.
Non-organic (psychiatric) causes of RPD
Psychological processes can sometimes mimic RPD, although it is essential to rule out a neurological etiology in these cases. Pseudodementia, due to depression, occurs in patients with a past history of major depression. There are usually signs that the patient is severely depressed and cognitive dysfunction, particularly on testing, is found to be due to decreased effort. Many of the features of patients with true dementia are seen in atypical psychiatric disorders, including personality disorders, conversion disorders, psychosis, and malingerers [150] and a full assessment is required to rule out potentially treatable or organic disorders. These cases can have many of the features of a true dementia Furthermore, psychiatric features may be an early symptom of many neurodegenerative conditions, including CJD, DLB, CBD and others.[12]
Conclusion
Although the evaluation of a patient with RPD may appear quite daunting, it can be greatly facilitated through a structured approach. Common things being common, most cases of RPD in an elderly person are probably due to urinary infection or pneumonia causing a delirium. However, when the simple causes have been ruled out, we find it most helpful to consider various categories of potential etiologies and then to rule out each category systematically. As numerous tests may be necessary, an inpatient evaluation can expedite the process. We often have found that a body CT with and without contrast to be of great assistance in diagnosing numerous difficult cases, helping to identify such conditions as Sarcoid, malignancies, and paraneoplastic conditions. Unfortunately, in many cases a standard laboratory evaluation is not sufficient and brain biopsy may be necessary. If prion disease is in the differential, prion precautions must be used in the operating room and when handling brain tissue.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Korth C, Peters PJ. Emerging pharmacotherapies for Creutzfeldt-Jakob disease. Arch Neurol. 2006 Apr;63(4):497–501. doi: 10.1001/archneur.63.4.497. [DOI] [PubMed] [Google Scholar]
- 2.Korth C, May BCH, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacoptherapeutics for prion disease. Proc Natl Acad Sci U S A. 2001;98(17):9836–9841. doi: 10.1073/pnas.161274798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greicius MD, Geschwind MD, Miller BL. Presenile dementia syndromes: an update on taxonomy and diagnosis. J Neurol Neurosurg Psychiatry. 2002;72(6):691–700. doi: 10.1136/jnnp.72.6.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tschampa HJ, Neumann M, Zerr I, et al. Patients with Alzheimer's disease and dementia with Lewy bodies mistaken for Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2001 Jul;71(1):33–39. doi: 10.1136/jnnp.71.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poser S, Mollenhauer B, Kraubeta A, et al. How to Improve the Clinical Diagnosis of Creutzfeldt-Jakob Disease. Brain. 1999 Dec;122(Pt 12):2345–2351. doi: 10.1093/brain/122.12.2345. [DOI] [PubMed] [Google Scholar]
- 6.Olichney JM, Galasko D, Salmon DP, et al. Cognitive decline is faster in Lewy body variant than in Alzheimer's disease. Neurology. 1998 Aug;51(2):351–357. doi: 10.1212/wnl.51.2.351. [DOI] [PubMed] [Google Scholar]
- 7.WHO. Global surveillance, diagnosis and therapy of human transmissible spongiform encephalopathies: Report of a WHO consultation. Paper presented at: World Health Organization: Emerging and other communicable diseases, surveillance and control; 9-11 February, 1998; Geneva. [Google Scholar]
- 8.Masters CL, Harris JO, Gajdusek DC, Gibbs CJ, Jr, Bernoulli C, Asher DM. Creutzfeldt-Jakob disease: patterns of worldwide occurrence and the significance of familial and sporadic clustering. Ann Neurol. 1979;5(2):177–188. doi: 10.1002/ana.410050212. [DOI] [PubMed] [Google Scholar]
- 9.Geschwind MD, Martindale J, Miller D, et al. Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease. Arch Neurol. 2003 Jun;60(6):813–816. doi: 10.1001/archneur.60.6.813. [DOI] [PubMed] [Google Scholar]
- 10.Chapman T, McKeel DW, Jr, Morris JC. Misleading results with the 14-3-3 assay for the diagnosis of Creutzfeldt-Jakob disease. Neurology. 2000;55(9) doi: 10.1212/wnl.55.9.1396. [DOI] [PubMed] [Google Scholar]
- 11.Huang N, Marie SK, Livramento JA, Chammas R, Nitrini R. 14-3-3 protein in the CSF of patients with rapidly progressive dementia. Neurology. 2003 Aug 12;61(3):354–357. doi: 10.1212/01.wnl.0000078890.89473.ed. [DOI] [PubMed] [Google Scholar]
- 12.Rabinovici GD, Wang PN, Levin J, et al. First symptom in sporadic Creutzfeldt-Jakob disease. Neurology. 2006 Jan 24;66(2):286–287. doi: 10.1212/01.wnl.0000196440.00297.67. [DOI] [PubMed] [Google Scholar]
- 13.Geschwind MD, Haman A, Torres-Chae C, et al. CSF findings in a United States sporadic CJD cohort. Paper presented at: PRION 2006: Strategies, advances and trends towards protection of society; October 3-6, 2006; Torino, Italy. 2006. [Google Scholar]
- 14.Safar J, Wille H, Itri V, et al. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med. 1998;4(10):1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 15.Safar JG, Geschwind MD, Deering C, et al. Diagnosis of human prion disease. PNAS. 2005 March 1;102(9):3501–3506. doi: 10.1073/pnas.0409651102. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Safar JG, Wille H, Geschwind MD, et al. Human prions and plasma lipoproteins. Proc Natl Acad Sci USA. 2006;103:11312–11317. doi: 10.1073/pnas.0604021103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez O, Claassen D, Boller F. Alzheimer's disease, cerebral amyloid angiopathy, and dementia of acute onset. Aging (Milano) 1991 Jun;3(2):171–175. doi: 10.1007/BF03323998. [DOI] [PubMed] [Google Scholar]
- 18.Barcikowska M, Mirecka B, Papierz W, Bogucki M, Niewodniczy A, Liberski PP. A case of Alzheimer's disease simulating Creutzfeldt-Jakob disease. Neurol Neurochir Pol. 1992 Sep-Oct;26(5):703–710. [PubMed] [Google Scholar]
- 19.Caselli RJ, Couce ME, Osborne D, Deen HG, Parisi JP. From slowly progressive amnesic syndrome to rapidly progressive Alzheimer disease. Alzheimer Dis Assoc Disord. 1998 Sep;12(3):251–253. [PubMed] [Google Scholar]
- 20.Roberson ED, Hesse JH, Rose KD, et al. Frontotemporal dementia progresses to death faster than Alzheimer disease. Neurology. 2005 September 13;65(5):719–725. doi: 10.1212/01.wnl.0000173837.82820.9f. 2005. [DOI] [PubMed] [Google Scholar]
- 21.Litvan I, Mangone CA, McKee A, et al. Natural history of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) and clinical predictors of survival: a clinicopathological study. J Neurol Neurosurg Psychiatry. 1996;60(6):615–620. doi: 10.1136/jnnp.60.6.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caine D, Patterson K, Hodges JR, Heard R, Halliday G. Severe anterograde amnesia with extensive hippocampal degeneration in a case of rapidly progressive frontotemporal dementia. Neurocase. 2001;7(1):57–64. doi: 10.1093/neucas/7.1.57. [DOI] [PubMed] [Google Scholar]
- 23.Kleiner-Fisman G, Lang AE, Bergeron C, Burn DJ, Paviour DC. Rapidly progressive behavioral changes and parkinsonism in a 68-year-old man. Mov Disord. 2004;19(5):534–543. doi: 10.1002/mds.10694. [DOI] [PubMed] [Google Scholar]
- 24.Hodges JR, Davies R, Xuereb J, Kril J, Halliday G. Survival in frontotemporal dementia. Neurology. 2003 Aug 12;61(3):349–354. doi: 10.1212/01.wnl.0000078928.20107.52. [DOI] [PubMed] [Google Scholar]
- 25.Catani M, Piccirilli M, Geloso MC, et al. Rapidly progressive aphasic dementia with motor neuron disease: a distinctive clinical entity. Dement Geriatr Cogn Disord. 2004;17(12):21–28. doi: 10.1159/000074139. [DOI] [PubMed] [Google Scholar]
- 26.Haik S, Brandel JP, Sazdovitch V, et al. Dementia with Lewy bodies in a neuropathologic series of suspected Creutzfeldt-Jakob disease. Neurology. 2000;55(9):1401–1404. doi: 10.1212/wnl.55.9.1401. [DOI] [PubMed] [Google Scholar]
- 27.McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 28.Walker Z, Allen R, Shergill S, Mullan E, Katona C. Three years survival in patients with a clinical diagnosis of dementia with Lewy bodies. Int J Geriatr Psychiatry. 2000 Mar;15(3):267–273. doi: 10.1002/(sici)1099-1166(200003)15:3<267::aid-gps107>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 29.Byrne EJ, Lennox G, Lowe J, Godwin-Austen RB. Diffuse Lewy body disease: clinical features in 15 cases. J Neurol Neurosurg Psychiatry. 1989;52:709–717. doi: 10.1136/jnnp.52.6.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitsuyama Y. Presenile dementia with motor neuron disease. Dementia. 1993;4(34):137–142. doi: 10.1159/000107312. [DOI] [PubMed] [Google Scholar]
- 31.Nasreddine ZS, Loginov M, Clark LN, et al. From genotype to phenotype: a clinical pathological, and biochemical investigation of frontotemporal dementia and parkinsonism (FTDP-17) caused by the P301L tau mutation. Ann Neurol. 1999;45(6):704–715. doi: 10.1002/1531-8249(199906)45:6<704::aid-ana4>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 32.Levy ML, Miller BL, Cummings JL, Fairbanks LA, Craig A. Alzheimer disease and frontotemporal dementias. Behavioral distinctions. Arch Neurol. 1996;53(7):687–690. doi: 10.1001/archneur.1996.00550070129021. [DOI] [PubMed] [Google Scholar]
- 33.Rosen HJ, Lengenfelder J, Miller B. Frontotemporal dementia. Neurol Clin. 2000;18(4):979–992. doi: 10.1016/s0733-8619(05)70235-8. [DOI] [PubMed] [Google Scholar]
- 34.Schneider JA, Watts RL, Gearing M, Brewer RP, Mirra SS. Corticobasal degeneration: neuropathologic and clinical heterogeneity. Neurology. 1997;48(4):959–969. doi: 10.1212/wnl.48.4.959. [DOI] [PubMed] [Google Scholar]
- 35.Litvan I, Agid Y, Goetz C, et al. Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology. 1997;48(1):119–125. doi: 10.1212/wnl.48.1.119. [DOI] [PubMed] [Google Scholar]
- 36.Gimenez-Roldan S, Mateo D, Benito C, Grandas F, Perez-Gilabert Y. Progressive supranuclear palsy and corticobasal ganglionic degeneration: differentiation by clinical features and neuroimaging techniques. J Neural Transm Suppl. 1994;42:79–90. doi: 10.1007/978-3-7091-6641-3_7. [DOI] [PubMed] [Google Scholar]
- 37.Mathuranath PS, Xuereb JH, Bak T, Hodges JR. Corticobasal ganglionic degeneration and/or frontotemporal dementia? A report of two overlap cases and review of literature. J Neurol Neurosurg Psychiatry. 2000;68(3):304–312. doi: 10.1136/jnnp.68.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kertesz A, Martinez-Lage P, Davidson W, Munoz DG. The corticobasal degeneration syndrome overlaps progressive aphasia and frontotemporal dementia. Neurology. 2000;55(9):1368–1375. doi: 10.1212/wnl.55.9.1368. [DOI] [PubMed] [Google Scholar]
- 39.Avanzino L, Marinelli L, Buccolieri A, Trompetto C, Abbruzzese G. Creutzfeldt-Jakob disease presenting as corticobasal degeneration: a neurophysiological study. Neurol Sci. 2006 Jun;27(2):118–121. doi: 10.1007/s10072-006-0611-1. [DOI] [PubMed] [Google Scholar]
- 40.Young GS, Geschwind MD, Fischbein NJ, et al. Diffusion-weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis. AJNR Am J Neuroradiol. 2005 Jun-Jul;26(6):1551–1562. [PMC free article] [PubMed] [Google Scholar]
- 41.Grafman J, Litvan I, Stark M. Neuropsychological features of progressive supranuclear palsy. Brain Cogn. 1995;28(3):311–320. doi: 10.1006/brcg.1995.1260. [DOI] [PubMed] [Google Scholar]
- 42.Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47(1):1–9. doi: 10.1212/wnl.47.1.1. [DOI] [PubMed] [Google Scholar]
- 43.Litvan I, Agid Y, Jankovic J, et al. Accuracy of clinical criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) Neurology. 1996;46(4):922–930. doi: 10.1212/wnl.46.4.922. [DOI] [PubMed] [Google Scholar]
- 44.Litvan I, Mega MS, Cummings JL, Fairbanks L. Neuropsychiatric aspects of progressive supranuclear palsy. Neurology. 1996;47(5):1184–1189. doi: 10.1212/wnl.47.5.1184. [DOI] [PubMed] [Google Scholar]
- 45.Yagishita A, Oda M. Progressive supranuclear palsy: MRI and pathological findings. Neuroradiology. 1996;38 1:S60–66. doi: 10.1007/BF02278121. [DOI] [PubMed] [Google Scholar]
- 46.Leigh RJ, Zee DS. The neurology of eye movements. 3. New York: Oxford University Press; 1999. [Google Scholar]
- 47.Josephs KA, Tsuboi Y, Dickson DW. Creutzfeldt-Jakob disease presenting as progressive supranuclear palsy. Eur J Neurol. 2004 May;11(5):343–346. doi: 10.1111/j.1468-1331.2004.00780.x. [DOI] [PubMed] [Google Scholar]
- 48.Josephs KA, Holton JL, Rossor MN, et al. Neurofilament inclusion body disease: a new proteinopathy? Brain. 2003 Oct;126(Pt 10):2291–2303. doi: 10.1093/brain/awg231. [DOI] [PubMed] [Google Scholar]
- 49.Mackenzie IR, Feldman H. Neurofilament inclusion body disease with early onset frontotemporal dementia and primary lateral sclerosis. Clin Neuropathol. 2004 Jul-Aug;23(4):183–193. [PubMed] [Google Scholar]
- 50.Benke T, Karner E, Seppi K, Delazer M, Marksteiner J, Donnemiller E. Subacute dementia and imaging correlates in a case of Fahr's disease. J Neurol Neurosurg Psychiatry. 2004 August 1;75(8):1163–1165. doi: 10.1136/jnnp.2003.019547. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mothersead P, Conrad K, Hagerman R, Greco C, Hessl D, T F. GRAND ROUNDS: An Atypical Progressive Dementia in a Male Carrier of the Fragile X Premutation: An Example of Fragile X-Associated Tremor/Ataxia Syndrome. Appl Neuropsychol. 2005;12(3):169–178. doi: 10.1207/S15324826AN1203_7. [DOI] [PubMed] [Google Scholar]
- 52.Dropcho EJ. Paraneoplastic Diseases of the Nervous System. Curr Treat Options Neurol. 1999;1(5):417–427. doi: 10.1007/s11940-996-0005-y. [DOI] [PubMed] [Google Scholar]
- 53.Gultekin SH, Rosenfeld MR, Voltz R, Eichen J, Posner JB, Dalmau J. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients. Brain. 2000;123(Pt 7):1481–1494. doi: 10.1093/brain/123.7.1481. [DOI] [PubMed] [Google Scholar]
- 54.Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain. 2005 Aug;128(Pt 8):1764–1777. doi: 10.1093/brain/awh526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosenfeld MR, Eichen JG, Wade DF, Posner JB, Dalmau J. Molecular and clinical diversity in paraneoplastic immunity to Ma proteins. Ann Neurol. 2001 Sep;50(3):339–348. [PubMed] [Google Scholar]
- 56.Dalmau J, Graus F, Villarejo A, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain. 2004 Aug;127(Pt 8):1831–1844. doi: 10.1093/brain/awh203. [DOI] [PubMed] [Google Scholar]
- 57.Pittock SJ, Kryzer TJ, Lennon VA. Paraneoplastic antibodies coexist and predict cancer, not neurological syndrome. Ann Neurol. 2004 Nov;56(5):715–719. doi: 10.1002/ana.20269. [DOI] [PubMed] [Google Scholar]
- 58.Vernino S, Tuite P, Adler CH, et al. Paraneoplastic chorea associated with CRMP-5 neuronal antibody and lung carcinoma. Ann Neurol. 2002 May;51(5):625–630. doi: 10.1002/ana.10178. [DOI] [PubMed] [Google Scholar]
- 59.Burton GV, Bullard DE, Walther PJ, Burger PC. Paraneoplastic limbic encephalopathy with testicular carcinoma. A reversible neurologic syndrome. Cancer. 1988;62(10):2248–2251. doi: 10.1002/1097-0142(19881115)62:10<2248::aid-cncr2820621029>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 60.Mathew RM, Vandenberghe R, Garcia-Merino A, et al. Orchiectomy for suspected microscopic tumor in patients with anti-Ma2-associated encephalitis. Neurology. 2007 Mar 20;68(12):900–905. doi: 10.1212/01.wnl.0000252379.81933.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu Z, Kryzer TJ, Griesmann GE, Kim K, Benarroch EE, Lennon VA. CRMP-5 neuronal autoantibody: marker of lung cancer and thymoma-related autoimmunity. Ann Neurol. 2001 Feb;49(2):146–154. [PubMed] [Google Scholar]
- 62.Honnorat J, Antoine JC, Belin MF. Are the “newly discovered” paraneoplastic anticollapsin response-mediator protein 5 antibodies simply anti-CV2 antibodies? Ann Neurol. 2001 Nov;50(5):688–691. doi: 10.1002/ana.1270. [DOI] [PubMed] [Google Scholar]
- 63.Antoine JC, Honnorat J, Anterion CT, et al. Limbic encephalitis and immunological perturbations in two patients with thymoma. J Neurol Neurosurg Psychiatry. 1995;58(6):706–710. doi: 10.1136/jnnp.58.6.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vernino S, Geschwind MD, Boeve B. Autoimmune encephalopathies. The Neurologist. 2007 May;13(3) doi: 10.1097/01.nrl.0000259483.70041.55. [DOI] [PubMed] [Google Scholar]
- 65.Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol. 2001 Jul;50(1):73–78. doi: 10.1002/ana.1097. [DOI] [PubMed] [Google Scholar]
- 66.Pozo-Rosich P, Clover L, Saiz A, Vincent A, Graus F. Voltage-gated potassium channel antibodies in limbic encephalitis. Ann Neurol. 2003 Oct;54(4):530–533. doi: 10.1002/ana.10713. [DOI] [PubMed] [Google Scholar]
- 67.Thieben MJ, Lennon VA, Boeve BF, Aksamit AJ, Keegan M, Vernino S. Potentially reversible autoimmune limbic encephalitis with neuronal potassium channel antibody. Neurology. 2004 Apr 13;62(7):1177–1182. doi: 10.1212/01.wnl.0000122648.19196.02. [DOI] [PubMed] [Google Scholar]
- 68.Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004 Mar;127(Pt 3):701–712. doi: 10.1093/brain/awh077. [DOI] [PubMed] [Google Scholar]
- 69.Bataller L, Kleopa KA, Wu GF, Rossi JE, Rosenfeld MR, Dalmau J. Autoimmune Limbic Encephalitis in 39 Patients: Immunophenotypes and Outcomes. J Neurol Neurosurg Psychiatry. 2007 April 1;78(4):381–385. doi: 10.1136/jnnp.2006.100644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liguori R, Vincent A, Clover L, et al. Morvan's syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain. 2001 Dec;124(Pt 12):2417–2426. doi: 10.1093/brain/124.12.2417. [DOI] [PubMed] [Google Scholar]
- 71.Chang CC, Eggers SD, Johnson JK, Haman A, Miller BL, Geschwind MD. Anti-GAD antibody cerebellar ataxia mimicking Creutzfeldt-Jakob disease. Clin Neurol Neurosurg. 2007;109:54–57. doi: 10.1016/j.clineuro.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 72.Kothbauer-Margreiter I, Sturzenegger M, Komor J, Baumgartner R, Hess CW. Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment. J Neurol. 1996;243(8):585–593. doi: 10.1007/BF00900946. [DOI] [PubMed] [Google Scholar]
- 73.Ghika-Schmid F, Ghika J, Regli F, et al. Hashimoto's myoclonic encephalopathy: an underdiagnosed treatable condition? Mov Disord. 1996;11(5):555–562. doi: 10.1002/mds.870110511. [DOI] [PubMed] [Google Scholar]
- 74.Seipelt M, Zerr I, Nau R, et al. Hashimoto's encephalitis as a differential diagnosis of Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 1999;66(2):172–176. doi: 10.1136/jnnp.66.2.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Henchey R, Cibula J, Helveston W, Malone J, Gilmore RL. Electroencephalographic findings in Hashimoto's encephalopathy. Neurology. 1995;45(5):977–981. doi: 10.1212/wnl.45.5.977. [DOI] [PubMed] [Google Scholar]
- 76.Bohnen NI, Parnell KJ, Harper CM. Reversible MRI findings in a patient with Hashimoto's encephalopathy. Neurology. 1997;49(1):246–247. doi: 10.1212/wnl.49.1.246. [DOI] [PubMed] [Google Scholar]
- 77.Schwartz MA, Selhorst JB, Ochs AL, et al. Oculomasticatory myorhythmia: a unique movement disorder occurring in Whipple's disease. Ann Neurol. 1986;20(6):677–683. doi: 10.1002/ana.410200605. [DOI] [PubMed] [Google Scholar]
- 78.McCabe DJ, Burke T, Connolly S, Hutchinson M. Amnesic syndrome with bilateral mesial temporal lobe involvement in Hashimoto's encephalopathy. Neurology. 2000;54(3):737–739. doi: 10.1212/wnl.54.3.737. [DOI] [PubMed] [Google Scholar]
- 79.Shein M, Apter A, Dickerman Z, Tyano S, Gadoth N. Encephalopathy in compensated Hashimoto thyroiditis: a clinical expression of autoimmune cerebral vasculitis. Brain Dev. 1986;8(1):60–64. doi: 10.1016/s0387-7604(86)80121-8. [DOI] [PubMed] [Google Scholar]
- 80.Peschen-Rosin R, Schabet M, Dichgans J. Manifestation of Hashimoto's encephalopathy years before onset of thyroid disease. Eur Neurol. 1999;41(2):79–84. doi: 10.1159/000008007. [DOI] [PubMed] [Google Scholar]
- 81.Nieuwenhuis L, Santens P, Vanwalleghem P, Boon P. Subacute Hashimoto's encephalopathy, treated with plasmapheresis. Acta Neurol Belg. 2004 Jun;104(2):80–83. [PubMed] [Google Scholar]
- 82.Harkness KA, Coles A, Pohl U, Xuereb JH, Baron JC, Lennox GG. Rapidly reversible dementia in cerebral amyloid inflammatory vasculopathy. Eur J Neurol. 2004 Jan;11(1):59–62. doi: 10.1046/j.1351-5101.2003.00707.x. [DOI] [PubMed] [Google Scholar]
- 83.Moore PM, Richardson B. Neurology of the vasculitides and connective tissue diseases. J Neurol Neurosurg Psychiatry. 1998;65(1):10–22. doi: 10.1136/jnnp.65.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Younger DS, Hays AP, Brust JC, Rowland LP. Granulomatous angiitis of the brain. An inflammatory reaction of diverse etiology. Arch Neurol. 1988;45(5):514–518. doi: 10.1001/archneur.1988.00520290042012. [DOI] [PubMed] [Google Scholar]
- 85.Younger DS, Kass RM. Vasculitis and the nervous system. Historical perspective and overview. Neurol Clin. 1997;15(4):737–758. doi: 10.1016/s0733-8619(05)70345-5. [DOI] [PubMed] [Google Scholar]
- 86.Ferro JM. Vasculitis of the central nervous system. J Neurol. 1998;245(12):766–776. doi: 10.1007/s004150050285. [DOI] [PubMed] [Google Scholar]
- 87.Yazici H, Yurdakul S, Hamuryudan V. Behcet disease. Curr Opin Rheumatol. 2001;13(1):18–22. doi: 10.1097/00002281-200101000-00004. [DOI] [PubMed] [Google Scholar]
- 88.Briani C, Baracchini C, Zanette G, Zanusso G, Carollo C, Monaco S. Rapidly progressive dementia in hypereosinophilic syndrome. Eur J Neurol. 2001 May;8(3):279–280. doi: 10.1046/j.1468-1331.2001.00197.x. [DOI] [PubMed] [Google Scholar]
- 89.Bruzelius M, Liedholm L, Hellblom M. Celiac disease can be associated with severe neurological symptoms. Analysis of gliadin antibodies should be considered in suspected cases. Lakartidningen. 2001 August 22;98(34):3538–3542. [PubMed] [Google Scholar]
- 90.Caselli RJ, Boeve BF, Scheithauer BW, O'Duffy JD, Hunder GG. Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): a reversible form of encephalopathy. Neurology. 1999 Oct 22;53(7):1579–1581. doi: 10.1212/wnl.53.7.1579. [DOI] [PubMed] [Google Scholar]
- 91.Schielke E, Nolte C, Muller W, Bruck W. Sarcoidosis presenting as rapidly progressive dementia: clinical and neuropathological evaluation. J Neurol. 2001 Jun;248(6):522–524. doi: 10.1007/s004150170164. [DOI] [PubMed] [Google Scholar]
- 92.Rabinstein AA, Romano JG, Forteza AM, Koch S. Rapidly Progressive Dementia Due to Bilateral Internal Carotid Artery Occlusion with Infarction of the Total Length of the Corpus Callosum. J Neuroimaging. 2004 April 1;14(2):176–179. [PubMed] [Google Scholar]
- 93.Auchus AP, Chen CP, Sodagar SN, Thong M, Sng EC. Single stroke dementia: insights from 12 cases in Singapore. J Neurol Sci. 2002 Nov 15;:203–204. 85–89. doi: 10.1016/s0022-510x(02)00272-1. [DOI] [PubMed] [Google Scholar]
- 94.Hunder GG, Arend WP, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis. Introduction. Arthritis Rheum. 1990 Aug;33(8):1065–1067. doi: 10.1002/art.1780330802. [DOI] [PubMed] [Google Scholar]
- 95.American College of Rheumatology. Bibliography of Criteria, Guidelines, and Health Status Assessments Used in Rheumatology. [October 10, 2005];American College of Rheumatology. Available at: http://rheumatology.org/publications/abbreviations/
- 96.Josephson SA, Papanastassiou AM, Berger MS, et al. The diagnostic utility of brain biopsy procedures in patients with rapidly deteriorating neurological conditions or dementia. J Neurosurg. 2007 Jan;106(1):72–75. doi: 10.3171/jns.2007.106.1.72. [DOI] [PubMed] [Google Scholar]
- 97.Wynne PJ, Younger DS, Khandji A, Silver AJ. Radiographic features of central nervous system vasculitis. Neurol Clin. 1997;15(4):779–804. doi: 10.1016/s0733-8619(05)70347-9. [DOI] [PubMed] [Google Scholar]
- 98.Heinrich A, Vogelgesang S, Kirsch M, Khaw AV. Intravascular lymphomatosis presenting as rapidly progressive dementia. Eur Neurol. 2005;54(1):55–58. doi: 10.1159/000087719. [DOI] [PubMed] [Google Scholar]
- 99.Menendez Calderon MJ, Segui Riesco ME, Arguelles M, Nuno Mateo J. Intravascular lymphomatosis. A report of three cases. An Med Interna. 2005 Jan;22(1):31–34. doi: 10.4321/s0212-71992005000100008. [DOI] [PubMed] [Google Scholar]
- 100.Brew BJ. AIDS dementia complex. Neurol Clin. 1999;17(4):861–881. doi: 10.1016/s0733-8619(05)70170-5. [DOI] [PubMed] [Google Scholar]
- 101.Wallace MR, Nelson JA, McCutchan JA, Wolfson T, Grant I. Symptomatic HIV seroconverting illness is associated with more rapid neurological impairment. Sex Transm Infect. 2001;77(3):199–201. doi: 10.1136/sti.77.3.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nath A, Maragos WF, Avison MJ, Schmitt FA, Berger JR. Acceleration of HIV dementia with methamphetamine and cocaine. J Neurovirol. 2001 Feb;7(1):66–71. doi: 10.1080/135502801300069737. [DOI] [PubMed] [Google Scholar]
- 103.Navia BA, Price RW. The acquired immunodeficiency syndrome: dementia as the presenting sole manifestation of human immunodeficiency virus infection. Arch Neurol. 1987;44:65–69. doi: 10.1001/archneur.1987.00520130051017. [DOI] [PubMed] [Google Scholar]
- 104.Ala TA, Doss RC, Sullivan CJ. Reversible dementia: a case of cryptococcal meningitis masquerading as Alzheimer's disease. J Alzheimers Dis. 2004 Oct;6(5):503–508. doi: 10.3233/jad-2004-6507. [DOI] [PubMed] [Google Scholar]
- 105.Heckman GA, Hawkins C, Morris A, Burrows LL, Bergeron C. Rapidly progressive dementia due to Mycobacterium neoaurum meningoencephalitis. Emerg Infect Dis. 2004 May;10(5):924–927. doi: 10.3201/eid1005.030711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Glaser CA, Honarmand S, Anderson LJ, et al. Beyond viruses: clinical profiles and etiologies associated with encephalitis. Clin Infect Dis. 2006 Dec 15;43(12):1565–1577. doi: 10.1086/509330. [DOI] [PubMed] [Google Scholar]
- 107.Timmermans M, Carr J. Neurosyphilis in the modern era. J Neurol Neurosurg Psychiatry. 2004 Dec;75(12):1727–1730. doi: 10.1136/jnnp.2004.031922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fox PA, Hawkins DA, Dawson S. Dementia following an acute presentation of meningovascular neurosyphilis in an HIV-1 positive patient. AIDS. 2000 Sep 8;14(13):2062–2063. doi: 10.1097/00002030-200009080-00031. [DOI] [PubMed] [Google Scholar]
- 109.Kaplan RF, Jones-Woodward L. Lyme encephalopathy: a neuropsychological perspective. Semin Neurol. 1997;17(1):31–37. doi: 10.1055/s-2008-1040910. [DOI] [PubMed] [Google Scholar]
- 110.Waniek C, Prohovnik I, Kaufman MA, Dwork AJ. Rapidly progressive frontal-type dementia associated with Lyme disease. J Neuropsychiatry Clin Neurosci. 1995;7(3):345–347. doi: 10.1176/jnp.7.3.345. [DOI] [PubMed] [Google Scholar]
- 111.Andersson C, Nyberg C, Nyman D. Rapid development of dementia of an elderly person, diagnosis and successful treatment. Duodecim. 2004;120(15):1893–1896. [PubMed] [Google Scholar]
- 112.Wormser G, Dattwyler R, Nowakowski J, Nadelman R. Diagnosis and treatment of Lyme disease in the United States. Resident & Staff Physician. 2001 April;:15–25. [Google Scholar]
- 113.Kouyoumdjian JA. Subacute sclerosing panencephalitis in an adult: report of a case. Arq Neuropsiquiatr. 1985 Sep;43(3):312–315. doi: 10.1590/s0004-282x1985000300011. [DOI] [PubMed] [Google Scholar]
- 114.Davis L. Chapter 41. Nervous System Complications of Systemic Viral Infections. In: Aminoff MJ, editor. Medicine and Neurology. St. Louis, Mo: W.B. Saunders, Harcourt Health Sciences; 2002. [Google Scholar]
- 115.Anderson M. Neurology of Whipple's disease. J Neurol Neurosurg Psychiatry. 2000;68(1):2–5. doi: 10.1136/jnnp.68.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Durand DV, Lecomte C, Cathebras P, Rousset H, Godeau P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple Disease. Societe Nationale Francaise de Medecine Interne. Medicine (Baltimore) 1997;76(3):170–184. doi: 10.1097/00005792-199705000-00003. [DOI] [PubMed] [Google Scholar]
- 117.Louis ED, Lynch T, Kaufmann P, Fahn S, Odel J. Diagnostic guidelines in central nervous system Whipple's disease. Ann Neurol. 1996;40(4):561–568. doi: 10.1002/ana.410400404. [DOI] [PubMed] [Google Scholar]
- 118.Matthews BR, Jones LK, Saad DA, Aksamit AJ, Josephs KA. Cerebellar ataxia and central nervous system whipple disease. Arch Neurol. 2005 Apr;62(4):618–620. doi: 10.1001/archneur.62.4.618. [DOI] [PubMed] [Google Scholar]
- 119.Singer R. Diagnosis and treatment of Whipple's disease. Drugs. 1998;55(5):699–704. doi: 10.2165/00003495-199855050-00007. [DOI] [PubMed] [Google Scholar]
- 120.Ramzan NN, Loftus E, Jr, Burgart LJ, et al. Diagnosis and monitoring of Whipple disease by polymerase chain reaction. Ann Intern Med. 1997;126(7):520–527. doi: 10.7326/0003-4819-126-7-199704010-00004. [DOI] [PubMed] [Google Scholar]
- 121.Bataille B, Delwail V, Menet E, et al. Primary intracerebral malignant lymphoma: report of 248 cases. J Neurosurg. 2000 Feb;92(2):261–266. doi: 10.3171/jns.2000.92.2.0261. [DOI] [PubMed] [Google Scholar]
- 122.Bakshi R, Mazziotta JC, Mischel PS, Jahan R, Seligson DB, Vinters HV. Lymphomatosis cerebri presenting as a rapidly progressive dementia: clinical, neuroimaging and pathologic findings. Dement Geriatr Cogn Disord. 1999;10(2):152–157. doi: 10.1159/000017116. [DOI] [PubMed] [Google Scholar]
- 123.Carlson BA. Rapidly progressive dementia caused by nonenhancing primary lymphoma of the central nervous system. AJNR Am J Neuroradiol. 1996;17(9):1695–1697. [PMC free article] [PubMed] [Google Scholar]
- 124.Rollins KE, Kleinschmidt-DeMasters BK, Corboy JR, Damek DM, Filley CM. Lymphomatosis cerebri as a cause of white matter dementia. Hum Pathol. 2005 March;36(3):282–290. doi: 10.1016/j.humpath.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 125.Haman A, DeArmond S, Johnson JK, Schardein K, Miller BL, Geschwind MD. When CJD is not CJD. Paper presented at: 131st Annual Meeting of the American Neurological Association; October 8-11, 2006; Chicago, IL. [Google Scholar]
- 126.Batchelor T, Loeffler JS. Primary CNS lymphoma. J Clin Oncol. 2006 Mar 10;24(8):1281–1288. doi: 10.1200/JCO.2005.04.8819. [DOI] [PubMed] [Google Scholar]
- 127.Fetell MR. Chapter 52. Lymphomas. In: Rowland L, editor. Merrit's Textbook of Neurology. 9. Baltimore: Williams & Wilkins; 1995. pp. 351–359. [Google Scholar]
- 128.Kuker W, Nagele T, Korfel A, et al. Primary central nervous system lymphomas (PCNSL): MRI features at presentation in 100 patients. J Neurooncol. 2005 Apr;72(2):169–177. doi: 10.1007/s11060-004-3390-7. [DOI] [PubMed] [Google Scholar]
- 129.Abrey LE, DeAngelis LM, Yahalom J. Long-term survival in primary CNS lymphoma. J Clin Oncol. 1998 Mar;16(3):859–863. doi: 10.1200/JCO.1998.16.3.859. [DOI] [PubMed] [Google Scholar]
- 130.Zuckerman D, Seliem R, Hochberg E. Intravascular lymphoma: the oncologist's “great imitator”. Oncologist. 2006 May;11(5):496–502. doi: 10.1634/theoncologist.11-5-496. [DOI] [PubMed] [Google Scholar]
- 131.NICP. Neurology in Clinical Practice - Web Version. New York: Butterworth & Heiman; 2001. Chapter 58. Primary Brain Tumors. [Google Scholar]
- 132.Vieren M, Sciot R, Robberecht W. Intravascular lymphomatosis of the brain: a diagnostic problem. Clin Neurol Neurosurg. 1999 Mar;101(1):33–36. doi: 10.1016/s0303-8467(98)00074-2. [DOI] [PubMed] [Google Scholar]
- 133.Chapin JE, Davis LE, Kornfeld M, Mandler RN. Neurologic manifestations of intravascular lymphomatosis. Acta Neurol Scand. 1995 Jun;91(6):494–499. doi: 10.1111/j.1600-0404.1995.tb00452.x. [DOI] [PubMed] [Google Scholar]
- 134.Rivas E, Sanchez-Herrero J, Alonso M, et al. Miliary brain metastases presenting as rapidly progressive dementia. Neuropathology. 2005;25(2):153–158. doi: 10.1111/j.1440-1789.2005.00595.x. [DOI] [PubMed] [Google Scholar]
- 135.Kinsella LJ, Riley DE. Nutritional deficiency and syndromes associated with alcoholics. In: Goetz C, Pappaert, editors. Clinical Neurology. 1999. [Google Scholar]
- 136.Cummings JL, Benson DF. Dementia: a clinical approach. Boston: Butterworths; 1983. Chapter 5. Dementia in vascular and infectious disorders; pp. 125–167. [Google Scholar]
- 137.Ishii N, Nishihara Y. Pellagra among chronic alcoholics: clinical and pathological study of 20 necropsy cases. J Neurol Neurosurg Psychiatry. 1981;44(3):209–215. doi: 10.1136/jnnp.44.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Geschwind MD, Jay C. Assessment of Rapidly Progressive Dementias Concise Review Related to Chapter 362: Alzheimer's Disease and Other Primary Dementias in Harrison's Textbook of Internal Medicine. McGraw Hill; [6/9/2003]. Available at: http://harrisons.accessmedicine.com/ [Google Scholar]
- 139.Gehring WJ, Knight RT. Prefrontal-cingulate interactions in action monitoring. Nat Neurosci. 2000 May;3(5):516–520. doi: 10.1038/74899. [DOI] [PubMed] [Google Scholar]
- 140.Chu K, Kang DW, Kim HJ, Lee YS, Park SH. Diffusion-weighted imaging abnormalities in wernicke encephalopathy: reversible cytotoxic edema? Arch Neurol. 2002 Jan;59(1):123–127. doi: 10.1001/archneur.59.1.123. [DOI] [PubMed] [Google Scholar]
- 141.Unlu E, Cakir B, Asil T. MRI findings of Wernicke encephalopathy revisited due to hunger strike. Eur J Radiol. 2006 Jan;57(1):43–53. doi: 10.1016/j.ejrad.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 142.Halavaara J, Brander A, Lyytinen J, Setala K, Kallela M. Wernicke's encephalopathy: is diffusion-weighted MRI useful? Neuroradiology. 2003 Aug;45(8):519–523. doi: 10.1007/s00234-003-1043-8. [DOI] [PubMed] [Google Scholar]
- 143.Marcao AM, Wiest R, Schindler K, et al. Adult onset metachromatic leukodystrophy without electroclinical peripheral nervous system involvement: a new mutation in the ARSA gene. Arch Neurol. 2005 Feb;62(2):309–313. doi: 10.1001/archneur.62.2.309. [DOI] [PubMed] [Google Scholar]
- 144.Letournel F, Etcharry-Bouyx F, Verny C, Barthelaix A, Dubas F. Two clinicopathological cases of a dominantly inherited, adult onset orthochromatic leucodystrophy. J Neurol Neurosurg Psychiatry. 2003 May;74(5):671–673. doi: 10.1136/jnnp.74.5.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hinkebein JH, Callahan CD. The neuropsychology of Kuf's Disease: a case of atypical early onset dementia. Arch Clin Neuropsychol. 1997;12(1):81–89. [PubMed] [Google Scholar]
- 146.Gorbach SL. Bismuth therapy in gastrointestinal diseases. Gastroenterology. 1990;99(3):863–875. doi: 10.1016/0016-5085(90)90983-8. [DOI] [PubMed] [Google Scholar]
- 147.Jungreis AC, Schaumburg HH. Encephalopathy from abuse of bismuth subsalicylate (Pepto-Bismol) Neurology. 1993;43(6):1265. doi: 10.1212/wnl.43.6.1265. [DOI] [PubMed] [Google Scholar]
- 148.Gordon MF, Abrams RI, Rubin DB, Barr WB, Correa DD. Bismuth toxicity. Neurology. 1994;44(12):2418. doi: 10.1212/wnl.44.12.2418-a. [DOI] [PubMed] [Google Scholar]
- 149.Benet LZ. Safety and pharmacokinetics: colloidal bismuth subcitrate. Scand J Gastroenterol Suppl. 1991;185:29–35. doi: 10.3109/00365529109093217. [DOI] [PubMed] [Google Scholar]
- 150.Hampel H, Berger C, Muller N. A case of Ganser's state presenting as a dementia syndrome. Psychopathology. 1996;29(4):236–241. doi: 10.1159/000284999. [DOI] [PubMed] [Google Scholar]