

Abstract
For more than two decades, Biotechnology and Bioengineering has documented research focused on natural and engineered microbial biofilms within aquatic and subterranean ecosystems, wastewater and waste-gas treatment systems, marine vessels and structures, and industrial bioprocesses. Compared to suspended culture systems, intentionally engineered biofilms are heterogeneous reaction systems that can increase reactor productivity, system stability, and provide inherent cell: product separation. Unwanted biofilms can create enormous increases in fluid frictional resistances, unacceptable reductions in heat transfer efficiency, product contamination, enhanced material deterioration, and accelerated corrosion. Missing from B&B has been an equivalent research dialogue regarding the basic molecular microbiology, immunology, and biotechnological aspects of medical biofilms. Presented here are the current problems related to medical biofilms; current concepts of biofilm formation, persistence, and interactions with the host immune system; and emerging technologies for controlling medical biofilms.
Keywords: medical biofilms, nosocomial infections, host immune response, engineering immunity
Introduction
Biofilms can kill.
“In American hospitals alone, healthcare-associated infections (i.e., nosocomial infections) account for an estimated 1.7 million infections and 99,000 associated deaths each year. Of these infections: 32 % of all healthcare-associated infection are urinary tract infections; 22 percent are surgical site infections; 15 percent are pneumonia (lung infections); and 14 percent are bloodstream infections” (Centers for Disease Control and Prevention Report, 2007).
“The European Centre for Disease Prevention and Control (ECDC) (2007) said in a June 2007 report that every year some 3 million people in European Union countries catch an infectious disease associated with healthcare and that around 50,000 die as a result.” (The European Centre for Disease Prevention and Control, 2007; http://www.ecdc.eu.int/index.html).
As these headlines infer, biofilms cause a significant amount of all human microbial infections. Nosocomial (hospital acquired) infections are the fourth leading cause of death in the U.S. with 2 million cases annually (or ~10% of American hospital patients) leading to more than $5 billion in added medical cost per annum (Wenzel, 2007). About 60–70% of nosocomial infections are associated with some type of implanted medical device. It is estimated that over 5 million medical devices or implants are used per annum in the U.S. alone. Microbial infections have been observed on most, if not all, such devices, including: prosthetic heart valves, orthopedic implants, intravascular catheters, artificial hearts, left ventricular assist devices, cardiac pacemakers, vascular prostheses, cerebrospinal fluid shunts, urinary catheters, ocular prostheses and contact lenses, and intrauterine contraceptive devices. Worldwide production of biomedical devices and tissue engineering-related materials is a $180 billion per year industry and expanding rapidly. Regardless of the sophistication of the biomedical implant (catheters versus a three-dimensional stem cell-containing polymer scaffold), all medical devices or tissue engineering constructs are susceptible to microbial colonization and infection (Bryers and Ratner, 2004; Castelli et al., 2006).
Upon adhesion to a surface (inanimate material or tissue), replicating adherent bacteria can secrete mostly insoluble gelatinous exopolymers, forming a three-dimensional cell: polymer matrix known as a biofilm (Fig. 1). From a medical perspective, both commensal and pathogenic microorganisms form biofilm-like conglomerates that are either associated with the epithelial or endothelial lining; embedded in the lung, intestinal or vaginal mucus layer; attached to the teeth or medical implant surfaces, or formed intracellularly. Biofilm formation and persistence has profound implications for the patient, because microorganisms growing as biofilms are significantly less susceptible to antibiotics and host defenses than are planktonic forms of the same microorganisms. Many biofilm infections are notoriously difficult to resolve and they commonly manifest as chronic or recurrent infections. Biofilm infections constitute a number of clinical challenges, including diseases involving uncultivable species, chronic inflammation, impaired wound healing, rapidly acquired antibiotic resistance, and the spread of infectious emboli.
Figure 1.
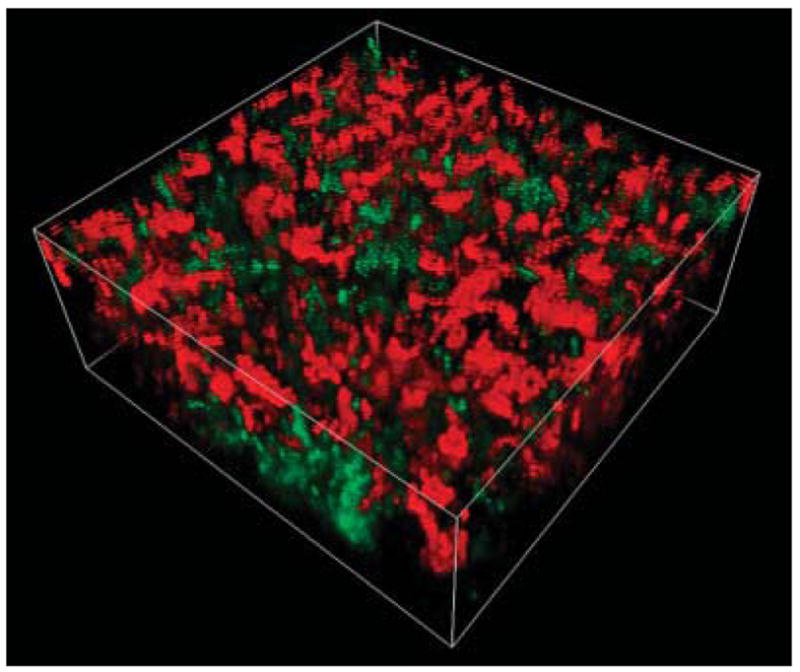
Three-dimensional reconstruction of a binary culture biofilm collected by confocal laser scanning microscopy. Biofilm z-direction “height” is 25 μm. The biofilm comprises the bacterial species, Klebsiella pneumoniae (green) and Pseudomonas aeruginosa (red), which have been visualized using species-specific monoclonal antibody-conjugated quantum dots (QDs). Unlike traditional fluorochrome stains, quantum dot luminescence is photostable and size tunable, allowing for multi-color emitted light from QDs of varying size all with a single excitation wavelength. Here, K. pneumoniae is labeled with monoclonal antibody-conjugated to CdSe-core, ZnS-shell nanocrystals (Green 560 nm emission maxima) and P. aeruginosa (red) monoclonal antibody-conjugated to CdSe/ZnS nanocrystals (Red 600 nm emission maxima).
Antimicrobial resistance in bacteria is also a major worldwide health care issue. The increase in bacteria that are resistant to frequently used anti-infective agents is now well known. Whereas resistant bacteria were previously common only in intensive-care units, more recently, such organisms have been recovered from non-intensive care hospital inpatient areas and are now isolated with increasing frequency in extended-care facilities, ambulatory surgical units, home healthcare sites, and other healthcare settings. The bacterium, Staphylococcus epidermidis, has evolved into one of the leading causes of nosocomial sepsis; this opportunistic pathogen is now the most common organism isolated from nosocomial infections.
Due to space constraints, I urge the reader to explore the many excellent recent literature reviews of medical biofilms that are directed at such topics as overall biofilm formation and infection (Donlan, 2002; Hall-Stoodley and Stoodley, 2005; Hall-Stoodley et al., 2004; Parsek and Singh, 2003; Reisner et al., 2005), biofilm diagnosis and treatment (Fux et al., 2003), general survival strategies of infectious biofilms (Foster, 2005; Fux et al., 2005), biofilm antibiotic/antimicrobial resistance (Döring et al., 2000; Mah and O’Toole, 2001; Stewart, 2002), quorum sensing control of biofilm formation (Bjarnsholt and Givskov, 2007; Harraghy et al., 2007; Smith and Iglewski, 2003), and biofilm issues related to specific infections, such as: cystic fibrosis (Høiby, 2002) osteomyelitis (Brady et al., 2007; Wagner et al., 2005), otitis media (Bakaletz, 2007; Post et al., 2007), and dental caries and oral infections (Bryers and Ratner, 2006; Offenbacher et al., 2007; Selwitz et al., 2007; Slayton et al., 2006).
In this perspective, I will discuss (1) the current concepts of biofilm formation, (2) the interplay between infection, inflammation, and host immune response, and (3) current and emerging approaches for detecting and controlling medical biofilms.
Processes Governing Biofilm Formation
Biofilm formation comprises a number of physical, biological, and chemical processes (Fig. 2), with the relative contribution of each changing throughout biofilm development and depending on prevailing environmental and hydrodynamic conditions (Bryers, 2000). Until non-fouling biomaterials are available and widely used, biomedical devices made with current materials will be subjected to protein fouling. The body typically reacts to biomedical devices by coating them ➀ with a film consisting of proteins and glycoproteins, such as fibronectin, vitronectin, fibrinogen, albumin, and immunoglobulins, many of which serve as binding ligands to receptors on colonizing bacteria or incoming mammalian cells. Regardless of what material (e.g., glass, ceramic, titanium, polyether urethane) is employed, the surface chemistry is instantly altered by macromolecule adsorption (Rosenberg, 2006; Strevett and Chen, 2003; Verran and Whitehead, 2005; Yongsunthon and Lower, 2006). Biofilm formation continues with ➁ the transport of cells to the substratum: liquid interface, which is governed by a combination of transport mechanisms (i.e., diffusion, convection, sedimentation, and motility) (Bryers, 2000). Once at a substratum surface, bacterial cells adhere ➂ by non-specific and/or specific receptor: ligand adhesion mechanisms (An and Friedman, 1998; Busscher et al., 1992; Glantz et al., 1999). Within minutes of attachment, adherent cells up-regulate the secretion of ➃ certain cell signal molecules that orchestrate community-wide phenotypic responses, through a process termed quorum sensing (Bjarnsholt and Givskov, 2007; Camilli and Bassler, 2006; Davies et al., 1998; Harraghy et al., 2007; Hodgkinson et al., 2007; Horswill et al., 2007; Pritchard, 2006; Rumbaugh, 2007). Such population responses include ➄ the up-regulation of virulence factors (Branda et al., 2005; Otto, 2006) and secretion of extracellular polymers (Davies and Geesey, 1995; Davies et al., 1993; Hall-Stoodley and Stoodley, 2005). Biofilms continue to mature by consuming soluble nutrients and recruiting other bacterial species or mammalian cells (e.g., platelets) ➅ (Brunstedt et al., 1995). Depending on other quorum cell signals, fragments of the mixed bacterial: platelet biofilm can detach ➆ (Sauer et al., 2004; Xavier et al., 2005) and be carried downstream (a potentially life threatening event known as a thromboembolism) (Wenzel, 2007).
Figure 2.

Processes governing biofilm formation. Blue plot shows time course of net accumulation of biofilm on an initially clean substratum. Initially, ➀ substratum is biased either by naturally occurring macromolecular adsorption to surface or intentionally pre-treatment with molecules meant to attract mammalian cells. Suspended bacteria are then ➁ deposited at the substratum by a combination of many transport mechanisms. Cells may leave the surface or be permanently attached➂ soon after which they secrete quorum signals ➃ that initiate up-regulation of various genes (many related to virulence) on a community-wide basis. Attached cells secrete ➄ copious polymers (polysaccharides, proteins, oligonucleotides) forming a 3-D extracellular biofilm matrix. Biofilm continues to accumulate; ➅ consuming ambient nutrients, electron donors and acceptors, and attracting other bacterial species or mammalian cells. As a result of increases in shear stress or the onset of other cell: cell signaling events, portions or entire sections of biofilm can ➆ detach or slough off, to move downstream (if in the cardiovasculature, this is a deadly event known as a thromboembolism). Insets from left to right: atomic force microscope of protein pre-treatment of a glass substratum (Jarvis and Bryers, 2005); Live (green) Dead (red) Baclight™ stained adherent P. aeruginosa (Wagner and Bryers, 2004); CLSM image of a developing P. aeruginosa biofilm (TUDelft University); OCT gel-embedded P. aeruginosa biofilm stained with DAPI and CTC (courtesy G. McFeters and ASM.org image library).
Infection, Inflammation, and Host Immune Response
Regrettably the majority, if not all, studies of immune response to bacteria have been carried out using intact freely suspended bacterial cell cultures or bacterial cell components isolated from suspended bacterial cells. Quoting from the National Institutes of Health’s Program on Immunology of Biofilms (PA07-288), “One of the limitations of early approaches used to study infectious organisms in disease is that planktonic (pure freely suspended cultures of) bacteria were employed. Although much information on the immune response came from the study of planktonic bacteria, it is now clear that bacteria in the clinical environment live more often as communities of microorganisms (biofilms) than as single cell suspensions.” While there are not many studies of immune cell interaction with biofilm systems, there are a few that have appeared recently and these works will be discussed at the end of this section.
The host response to biomaterials follows a cascade of events initially similar to, but ultimately diverging from, normal wound healing. The initial injury (a wound or the implantation of a medical device) causes a perturbation of the homeostatic mechanisms, setting off the process of healing (Anderson, 2001). The classic view of the host response to biomaterials divides the response into several overlapping phases: blood–material interactions, acute inflammation, chronic inflammation, foreign body reaction, and fibrous encapsulation. The various different cell phenotypes participating in the immune and inflammatory host response are depicted in Figure 3.
Figure 3.

Phenotypes of cells comprising the immune system.
Inflammatory cell recruitment requires enhanced leukocyte chemotaxis to the implant site, as well as leukocyte transmigration through the endothelium. Various molecular signals at the implant site can act as chemoattractants for the initial recruitment of inflammatory cells. Initial injury from the implantation procedure induces injured cells to release a variety of distress signals such as, ATP, uric acid, and bioactive lipids. Activated platelets release several growth factors and activated mast cells likewise release various degranulation products (e.g., histamine, proteoglycans, proteases) and cytokines (e.g., tumor necrosis factor (TNF)-α, and interleukin IL-16) (Mekori and Metcalfe, 2000). Mast cells can also initiate the production of various other cytokines and chemokines, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-4, IL-6, IL-10, IL-13, macrophage inflammatory protein (MIP)-α, and monocyte chemotactic protein (MCP)-1. As leukocytes arrive at the site of injury, they, too, begin to produce various growth factors and cytokines that attract more inflammatory cells. MIP-1α and MCP-1 have been implicated in phagocyte chemotaxis towards implants (Tang et al., 1998).
Following their chemotaxis toward the medical implant and transmigration through the endothelium, leukocytes adhere to the implant surface and become activated to carry out their functions. Since neutrophils arriving first at the implant site are “spent” within hours to less than a day (Savill, 1996), the focus of most cell–biomaterial interaction studies has been on the next incoming cell line, macrophages. Macrophages are widely considered the central cell type in directing and determining the outcome of the host response against implanted materials, because they are the pre-dominant cell type found at the tissue–material interface in the later stages of the host response (Schwartz et al., 1999). It is commonly hypothesized that host cells interact with surface-adsorbed proteins, rather than with the biomaterial surface itself (Hu et al., 2001; Wilson et al., 2005). Macrophages function is a dynamic process, which is dependent on the modulation of macrophages activation states by either the changing local microenvironment (Stout and Suttles, 2004; Stout et al., 2005) or by the macrophages themselves, as they release a wide variety of cytokines, extracellular matrix (ECM) proteins, and growth factors. Presence of adherent infecting bacteria can lead to a chronic inflammatory situation at the device interface. Chronic inflammation is associated with macrophages, lymphocytes, proliferation of connective tissues, deposition of matrix proteins, and capillary neogenesis (Shankar and Griesler, 1996). Ultimately, macrophages may form foreign body giant cells (FBGCs) in an attempt to “wall off” the implanted device leading to fibrous encapsulation.
Monocyte-derived macrophages are key mediators of the inflammatory response, as evidenced by changes in wound healing exhibited by animals with altered macrophages (Leibovich and Ross, 1975; Martin et al., 2003). In the setting of inflammation, macrophages can be activated in a polarized manner by local factors such as cytokines and microbial organisms (for review, see Mantovani et al., 2002; Mosser, 2003). The classic form of the activated macrophages (termed “M1”) is elicited by cytokines including interferon-gamma (IFN-γ) and TNF as well as by Gram-negative bacterial cell wall lipopolysaccharides (LPS). M1 macrophages generate large amounts of reactive oxygen species and inflammatory cytokines, which all serve to augment killing of phagocytosed intracellular parasites and local cell-mediated immune response (the Th1 response). While M1 macrophages are critical for the early response to injury and infection, prolonged or aberrant activation of M1 macrophages can also result in undesired tissue destruction and chronic inflammation.
Designation of an “M2” macrophages is a broad category that encompasses macrophages activated in a manner different than the classic M1 macrophages, and includes macrophages activated by IL-4 and IL-10, immune complexes, and glucocorticoids (Goldmann et al., 2007; Mantovani et al., 2004; Mosser, 2003). M2 macrophages promote angiogenesis, tissue remodeling, parasite encapsulation, and can act as immunoregulators by suppressing the inflammatory response. While M2 macrophages can be activated via different mechanisms, they are generally characterized by low levels of IL-12 and IL-23 along with high levels of IL-10. M1 macrophages can be distinguished from M2 macrophages by the expression of distinct sets of chemokines and chemokine receptors (Anderson and Mosser, 2002; Gordon, 2003). Polarization of macrophages toward the M2 form can create an environment that is favorable toward wound healing, tissue regeneration and implantable biomaterials. Consequently, the identification of pathways that can be modified to regulate macrophages polarization will be extremely beneficial for harnessing inflammation in pathologic states.
Note that all such reported activation results were based primarily on monocyte interaction with freely suspended cells or their components, which may not reflect true monocyte response to biofilm-bound bacteria (see Biofilm:Immune Cell Interaction Section below).
Immune Cell Response
The immune system has evolved to protect the host from infection in two ways: innate and adaptive immunity. Innate immunity is the ability to produce a response within minutes or hours after infection, through recognition of molecules expressed by pathogens (e.g., bacterial cell wall components, bacterial nucleic acids, formylated peptides, and viral double-stranded RNA). Such molecules are recognized by specialized receptors on cells of the innate immune system, which include: dendritic cells, macrophage, neutrophils, natural killer cells, and gamma-delta T cells. Binding of microbial components to innate immune cell receptors triggers signaling cascades within the cells that induce phagocytosis and the production of antimicrobial products (Fig. 4). Phagocytes also produce growth factors that can regulate adaptive immune responses. While innate immune responses are immediate (and may induce cross-protective immunity; Schmid et al., 2006), they are typically short-lived.
Figure 4.

Triggering dendritic cell response through TOL-like receptors and other adaptor proteins results in distinctive dendritic cell response depending on signal and activated receptor (from Pulendran et al., 2001).
While macrophages can eliminate invading pathogens by phagocytosis and then present various antigens, macrophages are not potent activators of naïve T cells. In contrast, dendritic cells are bone marrow-derived leukocytes, specialized for the uptake and presentation of antigens to naïve T cells (Hart, 1997; Matzinger, 1994; Steinman, 1991). Only one dendritic cell is required to activate 100–3,000 naïve T cells (Banchereau and Steinman, 1998). Dendritic cells rapidly circulate the peripheral tissues in their “immature” form but accumulate very rapidly at sites of injury, device implantation, or infection (Banchereau et al., 2000), suggesting that circulating DC pre-cursors are recruited in response to local inflammatory cues. Immature DCs—characterized by their high phagocytic activity and lack of surface co-stimulatory B.7 molecules (CD80, CD86) (Janeway et al., 2005)—can capture exogenous antigens via several pathways; including: macropinocytosis, receptor-mediated endocytosis, and phagocytosis of particles such as necrotic cell fragments and microbes (Banchereau et al., 2000). Dendritic cells can be activated to mature DCs via several different signals (Fig. 4), such as: interaction with pathogen-related molecules (e.g., LPS), stimulation by cytokines in the local microenvironment (e.g., TNF, IL-1, IL-10, TGF-β), and engagement of other receptors (e.g., mannose receptors, complement receptors) (Banchereau et al., 2000; Janeway et al., 2005). Upon capture of antigen, dendritic cells go through phenotypic changes in which their rate of antigen uptake briefly increases and then almost completely ceases. Dendritic cells then go through morphological changes, down-regulation of chemokine receptors responsible for the migration to the inflammation site, and up-regulation of receptors that mediate translocation to the lymph nodes. Surface expression by dendritic cells of “co-stimulatory” molecules is also up-regulated as well. These co-stimulatory molecules are required for the activation of T-cells, together with presentation of the antigen on major histocompatibility complexes (MHC) Classes I and II, which are also up-regulated upon “maturation.” Once in the lymph nodes, where most T-cells reside, surface co-stimulatory and secreted cytokine signals communicate between the dendritic cell and T-cell structural information about the antigen that was captured. T-cells, specific to the peptide-MHC complex, will then proliferate into clones capable of either destroying a target cell that expresses that same complex on its surface (CD8+ T cell) or, in the case of helper T-cells (CD4+ Th), capable of secreting soluble signals to B and T cells, which direct the immune response required. Cell signaling between lymphocytes can shift the immune response to either a cytotoxic cell-mediated response, called a Th1 response (characterized by secretion of cytokines such as IFN-γ, IL-2, and IL-12 along with IgG2a antibody) or to B-cell secretion of antibody, called a Th2 response (characterized by secretion of IL-4 and IL-10 along with IgG1 antibody).
Immune Evasion by Bacteria
The primary defense against infection is the innate immunity provided by neutrophils, macrophages, and dendritic cells. However, for example, Staphylococcus aureus has the ability to thwart neutrophils and macrophages by (a) inhibiting chemotaxis (blocking formylated peptide recognition, blocking C5a binding, secreting leukotoxins, and blocking LFA1-ICAM1 mediated extravasation), (b) negating opsonization (via protein G reverse IgG binding, plasmin degradation of bound IgG and C3b, and blocking C3b binding), and (c) thwarting phagocytosis (cell wall modifications to resist low endosomal pH, enzymatic degradation of endosomes) (Foster, 2005). S. epidermidis, another Gram-positive and the main species isolated in the majority of nosocomial infections, avoids the immune system also through protein G reverse binding of IgGs and through adhesion and biofilm formation (Sun et al., 2005; Vuong et al., 2004). Otto and co-workers (Li et al., 2007) have documented that Gram-positive bacteria actually sense antimicrobial peptides released by neutrophils and macrophage and can co-ordinate a directed defensive response. They discovered an antimicrobial peptide sensor system that controls major specific resistance mechanisms; the sensor contains a classical two-component signal transducer and an unusual third protein, all of which are indispensable for signal transduction and antimicrobial peptide resistance.
Complement evasion by many Gram-positive bacteria involves incorrect binding of complement recognition factors by bacterial cell wall proteins (Protein A, G, M) (Rooijakkers and van Strijp, 2007). Gram-negative bacteria have evolved secretory proteins that can degrade complement factors or their binding components, or that prove antichemotactic or toxic to immune cells.
Entry of the oral pathogen, Porphyromonas gingivalis, into monocyte-derived dendritic cells in vitro, leading to sub-optimal DC maturation, requires P. gingivalis to express the major fimbriae, FimA (Evans et al., 1992; Jotwani and Cutler, 2004). P. gingivalis also expresses unique immunosuppressive LPS (Cohen et al., 2004; Jotwani et al., 2003; Pulendran et al., 2001) and proteolytic gingipains (Potempa et al., 2003). LPS of P. gingivalis, relative to those of Escherichia coli, stimulate dendritic cells to secrete IL-10, but not IL-12, in vitro (Jotwani et al., 2003) and in vivo (Pulendran et al., 2001). These two factors (sub-optimal DC maturation and truncated cytokine expression) leads to the induction of a Th2-effector response, which suggests that P. gingivalis may target dendritic cell C-type lectin receptors (e.g., dendritic cell-specific ICAM-3-grabbing non-integrins) for entry, and for blunting of dendritic cell maturation. Bacteria that target C-type lectin receptors are unique in their ability to survive in the host DCs and induce chronic life-long infection. C-type lectin receptors function primarily to interact with conserved carbohydrates shared by a large group of microbes, and internalize these pathogens for processing and antigen-presentation. While Toll-like receptors (TLRs) rapidly transmit pathogen-associated molecular patterns through Toll-Interleukin 1 receptor activation domains, C-type lectin receptors appear to lack these activation domains, and thus do not generally lead directly to dendritic cell maturation and cytokine secretion (van Kooyk and Geijtenbeck, 2003; van Kooyk et al., 2004). Thus, some pathogens obviate the immune response by specifically targeting C-type lectin receptors, particularly dendritic cell-specific ICAM-3-grabbing non-integrins and mannose receptors, which benefits the pathogen by down-regulating intracellular signaling, inhibiting maturation, and cytokine secretion. Pathogens, like P. gingivalis, that interact with dendritic cell-specific ICAM-3-grabbing non-integrins cause chronic infections that can last a lifetime by manipulating Th1/Th2 balance (van Kooyk and Geijtenbeck, 2003).
Recent work by Gantner et al. (2003) suggests that there may be recognition signaling through these C-type lectin receptors that may co-operate with TLR signaling in defining inflammatory responses. They examined how dectin-1, a lectin family receptor for β-glucans, co-operates with TLRs in recognizing microbes. Dectin-1 expression enhances TLR-mediated activation of NF-Kb by β-glucan-containing particles. In both macrophages and dendritic cells, dectin-1 and TLRs are synergistic in mediating production IL-12 and TNF-α. Additionally, dectin-1 triggers production of reactive oxygen species, an inflammatory response that is primed by TLR activation. Data demonstrate that collaborative recognition of distinct microbial components by different classes of innate immune receptors is crucial in orchestrating inflammatory responses. Brown (2006) provides an excellent review of the interplay between the more studied TLR activation pathways of inflammation and emerging evidence that non-TLR pattern recognition receptors (C-lectins, mannose receptors) may participate in initiation of adaptive immunity by inducing intracellular signals.
Biofilm: Immune Cell Interactions
As indicated above, freely suspended microorganisms have evolved a multitude of ways to avoid detection by the immune system. One of these mechanisms is to form biofilms. Historically, there have been few studies that focused on biofilm–bound bacterial cell interaction with the immune system but this is changing. As I discuss here, there appears to be a dynamic feed back between immune cells and bacteria that maybe a causative mechanism in chronic inflammation and reduced wound healing. Biofilm bacteria may subtly manipulate immune responses while reactions of immune cells may exacerbate biofilm formation and reinforce virulence.
For years, one explanation behind a biofilm’s resistance to the immune system was that immune cells could not penetrate into the biofilm. Leid et al. (2002) documented such penetration in an in vitro study of freshly isolated human leukocytes contacting 2-day-old (early, maturing) and 7-day-old (fully developed) S. aureus biofilms, under static and flowing fluid conditions that mimic physiological shear. Leukocytes penetrated 7-day-old S. aureus biofilms under laminar-shear conditions but do not penetrate 2-day-old S. aureus biofilms grown under static conditions. Leukocytes produced a Th1-type response to both the 7-day-old shear and 2-day-old static S. aureus biofilms. That leukocytes were able to penetrate biofilms but unable to phagocytose bacteria suggests that other mechanisms may inhibit normal leukocyte function. Leid et al. (2005) continued this investigation to report that the exopolysaccharide alginate matrix protects Pseudomonas aeruginosa biofilm bacteria from IFN-γ-mediated leukocyte killing.
Jesaitis et al. (2003) exposed P. aeruginosa biofilms to neutrophils then measured in vitro oxygen consumption, cell morphology, and host defensive processes. They report that the consumption of oxygen increases in both the bacteria and neutrophils, but with little accumulation of H2O2, when leukocytes settled on established biofilm. Neutrophils did not exhibit significant motility or the characteristic polarized morphology of motile cells, but were still able to engage in phagocytosis while retaining a roughly spherical shape and modest membrane ruffling. Bacteria, on the other hand, exhibited increased oxygen consumption and detachment response that released a cloud of bacteria from the biofilm that eventually enveloped and obscured the settled neutrophils.
Walker et al. (2005) reports that human neutrophils serve to enhance the initial development of P. aeruginosa biofilms. The mechanism of biofilm enhancement by neutrophils was attributed to neutrophil-generated polymers comprised of actin and DNA. The bacteria bind to F-actin and the free DNA promotes biofilm matrix stability. Disruption of the polymers with DNase results in dispersion of the bacteria and a reduction in biofilm development. The presence of these actin-DNA polymers, with co-localization of P. aeruginosa, was confirmed in both neutrophil lysates and CF sputum. Introduction of additional neutrophils after 24 and 48 h further enhanced P. aeruginosa biofilm development, while exposure to fewer neutrophils resulted in a lesser degree of biofilm enhancement.
Similarly, Chandra et al. (2007) showed that peripheral blood mononuclear cells (PBMCs) enhanced the ability of the yeast, Candida albicans, to form biofilms and that the majority of PBMCs were localized to the basal and middle layers of the biofilm. They also found that only viable PBMCs enhanced Candida biofilm formation and that PBMCs cell surface components did not contribute to this biofilm enhancement; the biofilm-enhancing effect is mediated by a soluble factor released into the medium, and that the supernatant collected from co-cultures contained differential levels of pro- and anti-inflammatory cytokines.
Zimmermann et al. (2006) report that the quorum sensing molecule N-(3-oxododecanoyl)-L-homoserine lactone (3OC12-HSL) synthesized by P. aeruginosa induces directed migration (chemotaxis) of neutrophils in an in vitro assay. They proposed that this early attraction of PMN could contribute to prevention of biofilm formation, which may be an inaccurate conclusion. It could be possible that these bacterial signals are actually used by adherent bacteria to stimulate an immune cell response, which in turns enhances biofilm formation and virulence. In a mini-review, Wagner et al. (2006) discusses the topic of communication between adherent bacteria and host immune cells. Wu et al. (2005) demonstrate that IFN-γ binds to an outer membrane protein in P. aeruginosa, OprF, resulting in the expression of a quorum-sensing dependent virulence determinant, the PA-I lectin. To determine whether IFN-γ induced PA-I by activation of the QS signaling system, they measured rhlI gene expression in response to IFN-γ. RhlI is the gene required for the synthesis of C4-HSL (C4-homoserine lactone), a core QS signaling molecule that plays a central role in the expression of PA-I. IFN-γ induced rhlI transcription in PAO1 and C4-HSL synthesis increased significantly after exposure to IFN-γ. Activation of the QS system by IFN-γ also led to the increase of pyocyanin production, another quorum sensing-dependent virulence product. The results by Wu et al. and others (Smith and Iglewski, 2003) suggest that adherent bacteria not only actively sense but can also manipulate host immune response (Tateda et al., 2003; Smith and Iglewski, 2003).
Mittal et al. (2006) report that strains of P. aeruginosa vary in their ability to induce macrophage response, which effects the composition of secreted products (cytokines, chemokines). Concomitantly, these different compositions of macrophage-secreted products influenced overall biofilm formation and up-regulated expression of a litany of virulence factors in a cytokine composition-dependent manner, again indicating the need to study macrophage response to biofilm not planktonic bacteria.
Wagner and Bryers (2004) developed poly(ethylene glycol):poly(acrylate) PEG-g-PA co-polymers (inherently non-fouling) that were covalently modified with either cell adhesion peptides [YRGDS, YRGES (negative control), or YEILDV] or F(ab′) fragments of monoclonal antibodies (Mabs) to monocyte integrin receptors (anti-VLA4, anti-β1, and anti-β2, anti-CD64) known to enhance macrophage adhesion and modulate inflammation activation. Monocyte adhesion, phagocytic response (oxidative burst), and cytokine expression were determined for each material. Monocyte/macrophages cytokine expression (TNF-α, IL-1β, IL-6, and IL-8) in response to control, base, and modified materials, was also determined. Tissue culture poly(styrene) (TCPS) mediated the greatest number of adherent monocytes. Both YRGDS and YEILDV peptides tethered to PEG-g-PA increased macrophages adhesion versus PEG-g-PA controls by a factor of 2×. However, F(ab′) fragments of all four antibodies promoted enhanced monocyte adhesion (7×) versus PEG-g-PA controls. Monocyte adhering to F(ab′) modified surfaces exhibited a lower oxidative burst without the addition of exogenous stimuli (LPS) versus peptide modified surfaces. Cytokine expression and phagocytosis response by surface adherent macrophages to S. epidermidis and P. aeruginosa bacteria were also quantified on a per macrophages cell basis. Upon bacterial challenges, peptide-modified materials pre-disposed adherent macrophages to over-expressed pro-inflammatory cytokines and exhibited a significant immediate phagocytic response that diminished within 1 h. Conversely, materials modified by F(ab′)s, upon bacterial challenge, exhibited “per cell” cytokine expression levels that were significantly reduced compared to peptide-modified PEG-g-PA. Efficiency of adherent macrophages to ingest and kill both species was determined using radiolabeled- and fluorescently labeled bacterial cell ingestion studies as a function of the PEG-g-PA surface modification. Macrophages adhering to F(ab′) modified surfaces exhibited sustained enhanced phagocytic response (>3 h) and 3× higher bacterial killing efficiencies when compared with macrophages on peptide modified materials. The Wagner study indicates that signal molecules, either “surface-decorated” on or “released from” a biomaterial can directly influence MØ inflammatory response and subsequent interactions with invading bacteria. In addition, this work may indicate that biomaterial signals may be employed to direct the differentiation of monocytes into either M1 or M2 type macrophages.
Detecting Medical Biofilms
The “holy grail” of biofilm infections is an “early-warning” diagnostic method that would allow for non-invasive detection of the early stages of tissue or biomedical implant infection (Bauer et al., 2006) and an expedient response. Such diagnostics are only now just emerging.
Currently, only upon the onset of a cyclical fever in an implant recipient, will a patient receive a battery of blood tests meant to detect any infecting microorganisms; such as colony-forming plate count assays that typically take anywhere from 48–72 h and are only capable of detecting planktonic not sessile cells. Emergence of PCR techniques have shorten the time period but they also sample body fluids (blood, saliva, urine), which will not provide an accurate estimate of the actual biofilm flora colonizing an implant.
Xiong et al. (2005) reports a rapid, continuous method for real-time monitoring of biofilm development, both in vitro and in a mouse infection model, through non-invasive imaging of bioluminescent bacteria (Fig. 5). Two important biofilm-forming bacterial pathogens, S. aureus and P. aeruginosa, were made bioluminescent by insertion of a complete lux operon. These bacteria produce significant bioluminescent signals for both in vitro studies and in an in vivo model, allowing effective real-time assessment of the physiological state of the biofilms. In vitro viable counts and light output correlated well for 10 days or longer, provided that the growth medium was replenished every 12 h. Recovery of the bacteria from the catheters of infected animals showed that the bioluminescent signal corresponded to the CFU and that the lux constructs were highly stable even after many days in vivo. The IVIS™ imaging system, commercially available from the Xenogen Corporation, has been used to follow in vivo either infections of soft tissue or medical biomaterial implants or to assess the subsequent efficacy of a single or multiple antibiotic treatment. As a research tool, the IVIS™ camera system and microscope can detect the emitted photons from both live and dead bacteria.
Figure 5.

Real-time monitoring of Staphylococcus aureus Xen29 in an experimental-rat endocarditis model. Two representative animals infected intravenously with either normal saline (control) or 106 CFU of S. aureus strain are shown. The animals were imaged ventrally, with their chest area shaved, to avoid background signal from animal hair. The process of infection was monitored daily by detecting photon emission around the region of interest (heart area) over a 6-day course (Xiong et al., 2005).
An alternative to bioluminescence is the non-invasive detection of fluorescent bacteria using multi-photon laser scanning microscopy. Currently there are no prokaryotic dyes for in vivo use available commercially. As bacterial expression of fluorescent proteins (e.g., green fluorescent protein and its variants) does not require any substrate for endogenous production, it is the method of choice for prokaryotic labeling. Unfortunately, use of GFP is restricted to only those bacterial species that can produce the fluorescent protein, which eliminates the tracking of some key pathogens, such as S. epidermidis or obligates anaerobes. Månsson et al. (2007) reports on the application of multi-photon microscopy for non-invasive in vivo imaging of ongoing infection. E. coli (GFP) uropathogenic strains were injected by micropuncture directly into the proximal tubule of a nephron in an exteriorized kidney. Then, the rat is immediately placed on the stage of a multi-photon microscope, with the injected kidney placed in a saline-filled glass-bottom Petri dish (far from non-invasive!). Using multi-photon microscopy, the progression of infection was visualized in the anaesthetized rat for an extended period of time. In a similar review by Roux et al. (2004), Cryptococcus neoformans (a yeast labeled with FITC) was injected into the tail vein of a mouse. Fluorescence images of the pathogens arriving in the brain of the infected animal were recorded 10 min later, using a multi-photon microscope.
While both of the imaging methods above are capable of detecting either luminescent or fluorescent bacteria in vivo, they are both dependent on the use of bacteria that have been manipulated in vitro then purposely injected to the a subject to mimic an infection. Unfortunately, real infections are not caused by recombinant bacteria that glow. However, multi-photon microscopy does have the potential, especially in the near IR wavelength range, to detect the onset of biofilm formation provided one could label the biofilm cells in situ. Hypothetically, one could imagine periodically injecting an implant patient with immunoconjugated quantum dots (Su and Li, 2004; Yang and Li, 2006) that are specific to potential implant-colonizing bacteria. The circulating immuno-quantum dots would locate targeted biofilm bacteria thus allowing early stage multi-photon detection of an implant infection.
Controlling Medical Biofilms
Good Intentions Gone Bad
Since native immunity can be circumvented or compromised (by drugs or disease), the medical profession has been attempting to eradicate biofilm-based infections by resorting to disinfectants and antibiotics. These are mostly synthetic compounds evaluated for the most part on their ability to inactivate or kill suspended bacteria but exhibit little efficacy when applied to biofilm infections (Stewart, 2002; Stewart and Costerton, 2001). However, biofilm bacteria are significantly less responsive to antibiotics and antimicrobial stressors than planktonic organisms of the same species (Gilbert et al., 2002; Stewart, 2002). Recent studies have shown that sub-lethal doses of antibiotics can actually enhance biofilm formation. Bagge et al. (2004) report that genes coding for alginate biosynthesis were induced by exposure to the β-lactam antibiotic, imipenem. Exposure to subinhibitory concentrations of imipenem caused structural changes in the biofilm, for example, an increased biofilm volume and alginate polymer matrix. Increased levels of alginate matrix production may be an unintended adverse consequence of imipenem treatment in cystic fibrosis patients. Similarly, Hoffman et al. (2005) report that subinhibitory concentrations of aminoglycoside antibiotics (e.g., tobramycin) induced biofilm formation in P. aeruginosa and E. coli. Hoffman’s results along with those of Bagge et al. suggest enhanced biofilm formation in the presence of antibiotics may be one universal defense mechanism of bacteria in avoiding the lethal effects of antibiotics.
The biomaterials community over the past 25 years has attempted to produce anti-infective devices or implants by either (1) mechanical design alternatives (liquid: air breaks; skin cuffs; antibiotic fills; all for indwelling catheters), (2) tethered anti-infective agents, bound directly to the surface of the material (silver coatings, tethered quaternary ammonium, synthetic antibiotics), or (3) the release of soluble toxic agents (chlorhexidine, antibiotics) into the adjacent surroundings. Mechanical design alternatives have had only marginal success and are only applicable for short-term indwelling catheters. Tethered anti-infective agents are only toxic to the initial wave of incoming bacteria and provide little residual effects once layers of dead cells accumulate, which are also inflammatory. Finally, regardless of the type of “drug-release” method used (passive vs. sustained vs. responsive), release of a toxic agent from a biomaterial of a soluble anti-infective agent will inevitably stop once the entrapped agent is depleted. Further, as shown above, delivery of sub-lethal dosages of antibiotics can lead to accelerated biofilm formation and induced virulence factor expression.
New Directions in Medical Biofilm Control
Traditional treatment of microbial infections is based on compounds that kill or inhibit growth of the microbe. One major concern with this approach is the frequent development of resistance to antibiotics (Geddes, 2000). As stated above, biofilm communities tend to be significantly less responsive to antibiotics and antimicrobial stressors than planktonic organisms of the same species. A further complication is that the spread of antibiotic resistance genes borne on plasmid DNA (pDNA), within and between species, is greatly exacerbated in biofilm communities (Beaudoin et al., 1998a,b). As a consequence to this increase in resistance, researchers have turned to a number of alternatives to synthetic antibiotics, including: bacteriophage (Sulakvelidze et al., 2001) and bacteriophage lytic enzymes (Fischetti, 2005), probiotics (Hong et al., 2005; Persson, 2005), and human antimicrobial peptides (defensins, cathelicidins, and histatins) (De Smet and Contreras, 2005). The success of these alternatives awaits much development and optimization. Unfortunately, most of these alternatives are still based upon some mechanism of killing or terminating the target bacteria; an approach some feel pre-ordains the development of resistance in bacteria.
It has been recently proposed to develop substances that specifically inhibit bacterial virulence. Such “antipathogenic” drugs, in contrast to antibacterial drugs, do not kill bacteria or stop their growth and are assumed not to lead to the development of resistant strains. A very elegant approach comprises the inhibition of regulatory systems that govern the expression of a series of bacterial virulence factors: for example, antiadhesion therapy [passive antibody therapy (Casadevall et al., 2004; Martinez and Casadevall, 2005), and synthetic peptide vaccine and antibody therapy (Cachia and Hodges, 2003)], inhibiting or negating cell–cell signaling (Otto, 2004), negating biofilm formation by disrupting iron metabolism (Kaneko et al., 2007), and up-regulation of biofilm detachment promoters (rhamnolipids) (Boles et al., 2005).
Here, I present examples of three such “antibiofilm” strategies that could be deployed from a biomedical device coating or implant (Fig. 6) as a defense aimed at negating biofilm formation; defenses based on (1) disrupting bacterial iron metabolism, (2) enhancing macrophage phagocytosis, and (3) “self-vaccinating” biomaterials.
Figure 6.

Hypothetical biomaterial engineered to enhance short and long-term infection immune response.
Iron Metabolism Interference
Iron is critical for bacterial growth and the function of key metabolic enzymes (Ankenbauer et al., 1985; Barclay and Ratledge, 1986; Finkelstein et al., 1983) and sequestration of iron is an early evolutionary strategy of host defense (Jurado, 1997). Gallium has many features similar to Fe3+, including a nearly identical ionic radius, and biologic systems are often unable to distinguish Ga from Fe3+. Unlike Fe3+, Ga cannot be reduced to the divalent state and sequential reduction/oxidation is critical for Fe to function in many enzymes. Thus, placing Ga, rather than Fe, in such enzymes renders them non-functional (Narasimhan et al., 1992; Singh, 2004). Importantly, Ga binds to the siderophores of Pseudomonas sp. (Chitambar and Narasimhan, 1991; Narasimhan et al., 1992) and is taken up by other bacteria including S. aureus and S. epidermidis, E. coli, Enterococcus faecalis, and S. typhimurium (Atkinson et al., 1998; Hubbard et al., 1986).
Kaneko et al. (2007) recently reported (Fig. 7A) that concentrations 1–2 μM Ga(NO3)3 did not effect the growth rate or extent of P. aeruginosa. Concentrations of Ga >2 μM decreased P. aeruginosa growth rate in a dose-dependent manner. These results led Dr. Singh’s group to investigate the effects of Ga on biofilm formation. For these studies, they used a Ga concentration (1 μM) that did not impair the growth of suspended P. aeruginosa, since they were interested specifically in antibiofilm effects of Ga. (In a therapeutic application, both growth inhibitory and anti-biofilm actions would be desirable.) Gallium effects were studied using a green fluorescent protein (gfp)-expressing P. aeruginosa strain. In sub-inhibitory concentrations of gallium, P. aeruginosa attached to the growth surface, but biofilm formation was completely inhibited (Fig. 7B). Biofilm formation by mucoid and non-mucoid P. aeruginosa isolates from cystic fibrosis patients was also blocked by 1 μM Ga(NO3)3 (Kaneko et al., 2007).
Figure 7.

A: Effect of Ga on P. aeruginosa suspended growth. Ga(NO3)3 inhibits P. aeruginosa growth in a concentration-dependent manner. Experiments were performed in biofilm medium at 37°C, and data are the mean of four experiments; error bars indicate SEM. B: Ga prevents P. aeruginosa biofilm formation. Confocal microscopic images of GFP-expressing P. aeruginosa in flow cells perfused with biofilm medium without (left) and with (right) Ga, 5 days after inoculation. Experiments were performed at 25°C using 0.5 μg/mL Ga(NO3)3. Higher concentrations [1, 5, and 10 μM Ga(NO3)3] also inhibited biofilm formation. Top images are top-down views (x-y plane); bottom images are side views (x-z plane); the dotted line represents the biofilm growth surface; scale bars: 50 μm. Results are representative of six experiments (Kaneko et al., 2007).
To determine if Ga applications can kill P. aeruginosa biofilms, Kaneko reports growing biofilms for 3 days (with no Ga present) and then switching to medium containing Ga for 48 h. Bacterial viability was assayed using a live-dead stain. While most commercial antibiotic agents show markedly less activity against biofilms than against planktonic organisms, P. aeruginosa bacteria within mature biofilms were killed by concentrations of Ga similar to those that killed planktonic cells (Kaneko et al., 2007).
Enhancing Phagocytosis
Another alternative antibiofilm defense is one that seeks to enhance macrophage phagocytosis of bacteria by developing artificial “opsonins.” Opsonization is the process where microorganisms are coated with host-produced molecules (immunoglobulins, complement factors), which in turn facilitates their binding to specific receptor molecules present on phagocytes (neutrophils, macrophage, dendritic cells).
IgG antibodies bind to their antigens on the surface of bacteria through coupling of the variable binding sites in the Fab region of the antibody, leaving the Fc region exposed. Phagocytes possess Fc gamma receptors and therefore can bind to the coated bacteria and internalize them. Complement fragment, C3b, also specifically binds to surface proteins or polysaccharides on microorganisms thus allowing binding to C3b receptors on the phagocytes. As described earlier, bacteria have evolved numerous ways to circumvent these natural opsonins and thus avoid elimination. One strategy bacteria have evolved to avoid phagocytosis is to avoid opsonization by IgG and complement. We have initiated development of “artificial opsonins,” designed to uniquely bind to both target bacteria and macrophage, thus enhancing phagocytosis. There are several reports of enhancing phagocytosis employing either (a) fusion proteins that couple recognition moieties of both bacteria and macrophage or (b) synthetically derived “opsonins.”
Whitesides’ group (Krishnamurthy et al., 2006) describes the application of a bifunctional polyacrylamide containing both vancomycin and fluorescein groups, that recognized the surfaces of four species of Gram-positive bacteria (S. aureus, SE, Streptococcus pneumoniae, and E. faecalis). Vancomycin groups recognize bacterial cell wall component peptides terminated in D–Ala–D–Ala. Fluorescein groups allowed the imaging of bound opsonin plus they are recognized by antifluorescein Mab, which promoted binding to macrophage. Flow cytometry revealed that bi-specific polymer-labeled S. aureus and S. pneumoniae were opsonized by antifluorescein Mabs 20-fold more than were untreated bacteria and promoted subsequent phagocytosis of the S. aureus bacteria by cultured J774 macrophage-like cells approximately twofold more efficiently than in control groups.
The Taylor group, in series of elegant papers, reports the use of several different bi-specific fusion proteins (BiFPs) that enhanced phagocytosis by macrophage of various pathogens, including: E. coli (Kuhn et al., 1998), P. aeruginosa (Lindorfer et al., 2001), and S. aureus (Gyimesi et al., 2004). In all cases, BiFPs consisted of (1) a molecule that recognizes a surface marker on the pathogen that was chemically coupled with (2) a Mab that is specific to the complement receptor 1 (CR1) present on primate erythrocytes. In in vitro and in vivo studies, these works from the Taylor groups have shown that BiFPs promote binding of the target pathogen first to circulating erythrocytes, which then enhances macrophage phagocytosis of the bacteria. BiFP mediated phagocytosis did not apparently harm the erythrocyte, as verified in both in vitro and in vivo experiments (Kuhn et al., 1998).
Kobayashi et al. (2004) reports improved in vivo and in vitro phagocytosis of a periodontal pathogen, P. gingivalis, using a BiFP composed of two monoclonal antibody fragments; one against (a) the hemagglutinin domain of P. gingivalis (anti-r130k-HMGD antibody) and (b) the polymorphonuclear leukocyte (PMN) FcαRI (CD89) receptor (FcR) (Fig. 8). The Kobayashi work selectively targeted Fc receptors that were dominant on PMNs collected from gingival crevicular fluid of chronic periodontitis patients versus Fc receptors dominant on peripheral blood PMNs. Data show that PMNs exhibited a higher capacity to phagocytose and kill P. gingivalis opsonized with the BiFP targeting P. gingivalis r130k-HMGD to leukocyte Fc RI as compared to opsonizing the bacterial with only the anti-r130k-HMGD antibody.
Figure 8.

A: Use of bi-specific fusion proteins to opsonize pathogenic bacteria and enhance phagocytosis. Human polymorphonuclear neutrophils (PMN) (B) and P. gingivalis (C) were incubated with different concentrations of BiFPs. This was followed by FITC-conjugated antimouse (filled circles) or FITC-conjugated antirabbit IgG (open circles) (Kobayashi et al., 2004).
Engineering Infection Immunity
Imagine a biomedical device that would be capable of generating a life-long protection for itself against infection? Our research group is developing a biomaterials platform that will engineer infection immunity by releasing in multiple waves, vaccines that have as antigen targets—those bacterial surface structures used for bacterial specific adhesion (Fig. 6).
The goal of any vaccine is to produce a long-term protective immune response against a pathogen. For most bacteria, initial attachment to a eukaryotic cell surface or ligand-coated biomedical device leads to biofilm formation, then up-regulation of virulence factors leading to infection. Both an innate and an induced antibody response could prevent attachment and abrogate colonization. The ideal antigens to promote both levels of immune response would be the very surface proteins (bacterial “adhesins”) that mediate specific bacterial cell adhesion to ligands present on host tissue or device surfaces (Wizemann et al., 1999). Given that bacterial specific adhesion can trigger expression of many virulence factors leading to acute and chronic inflammation, a vaccine approach that blocks bacterial adhesion may have multiple advantages. Recent research has greatly expanded the molecular details of the specific adhesin: ligand couples employed by Gram-positive bacteria (S. epidermidis, S. aureus, Group A and B Streptococcus, E. faecalis), Gram-negative bacteria (P. aeruginosa, Klebsiella pneumoniae, P. gingivalis, E. coli), and yeast (Candida spp.) to colonize and infect living tissue and implanted biomedical devices (Wizemann et al., 1999). Thus, the concept of biomaterials designed to engineer infection immunity, targeting specific adhesins, could be applied to numerous situations.
There are essentially three ways to activate a dendritic cell to present antigen with the subsequent immune response being dependent upon the form of the antigen (Fig. 9): direct antigen presentation (e.g., whole attenuated pathogen, isolated antigen molecule, recombinant protein), DNA vaccine (i.e., pDNA encoding for the antigen protein), and mRNA vaccine (i.e., RNA encoding for the antigen protein). Exogenously acquired protein antigens are processed by late endosomes into MHC-II complexes that initiate CD+4 T cell activation, and then activate B cell antibody secretion. Endogenously acquired antigen (e.g., viral infection; DNA/mRNA vaccine) is tagged with ubiquitin, then partially degraded in the proteosome, transported next to the endoplasmic recticulum, and presented on the DC’s surface as a MHC-I complex, thus activating an immediate (but short-lived) cytotoxic CD8+ T cell response. In contrast to protein vaccines, DNA- or RNA-based vaccines can provide the ability to potentially generate both a strong cytotoxic T cell response and humoral response.
Figure 9.
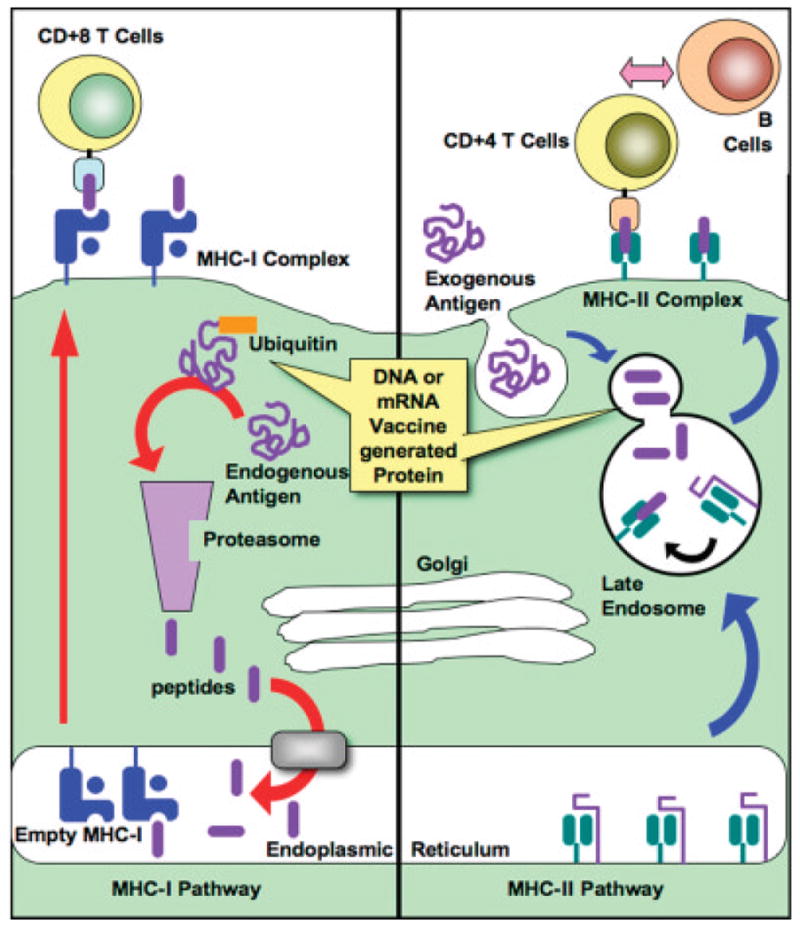
Antigen presentation and pathways of vaccine response. Plasmid DNA or mRNA is taken up by dendritic cells for intracellular expression of antigen. Antigen can be secreted (not shown) and subsequently taken up by another DCs as an exogenous antigen. Antigen expressed intracellularly by a dendritic cell or taken up through cross-priming is presented by MHC-I to CD8+ T-cells (cytotoxic leukocytes; CTLs). Antigen taken in exogenously or directed by DNA or mRNA trafficking signals are processed by the MHC-II pathway and presented to CD4+ TH cells, which can subsequently secrete: soluble cytokine signals (e.g., IL-12) back to the dendritic cell, proliferative signals (e.g., IL-2 and IFN-γ) to Tc cells, or signals directed toward B-cells (e.g., IL-4) to induce B-cell proliferation and antibody secretion.
Upon internalization of a pDNA vaccine carrier by dendritic cells, the carrier must escape endosomal entrapment or be degraded, the carrier must release the pDNA into the cytoplasm, and then the pDNA must be incorporated into the dendritic nucleus for expression. Subsequently expressed antigen can be processed as above into MHC-I complexes to initiate priming of CD8+ T-cell or the antigen can be secreted. The secreted antigen can be taken up exogenously by the same or other DCs and presented by the MHC-II pathway to CD4+ T cells, which can secrete soluble cytokine signals to T- or to B-cells to induce antibody secretion. In dendritic cells, in a process known as “cross-priming,” endogenously expressed antigen could be routed to the MHC-II complex.
DNA vaccines have certain advantages over protein antigens including: (a) DNA can serve as a natural adjuvant by including unmethylated CpG motifs; (b) by coding for multiple gene expression, DNA vaccine can also induce co-stimulatory molecules; (c) pDNA can target expression to certain cellular locations, thus fine tuning the immune response; and (d) pDNAs allow the possibility of multiple antigen expression. Disadvantages of DNA vaccines include: (a) incorporated CpG motifs can lead to overstimulation and toxicity (Heikenwalder et al., 2004; Zhao et al., 2004); (b) the required large amounts of highly characterized pDNA require processing with antibiotics and antibiotic-resistance markers; (c) pDNA contain sequences meant to control gene expression (e.g., promoters, polyA signals, introns) that may deregulate gene expression after integration into the genome; and (d) genome-incorporated pDNA can result in uncontrolled duration and strength of antigen expression.
RNA vaccines offer the same advantages as DNA vaccines versus protein antigens but RNA vaccines have several added benefits versus DNA vaccines (Gilboa and Vieweg, 2004). Due to RNA’s smaller size, a larger amount can be delivered per carrier, thus in general being more efficient. Since RNA does not require nuclear incorporation, expression of antigen in transfected cells occurs much faster than with pDNA. Potentially dangerous side effects are reduced since eukaryotic promoters needed for pDNA are not present in RNA constructs. mRNAs are relatively easy to manufacture in high amounts, to purify to homogeneity, and to characterize. Finally, RNA does not persist in the organism, and RNA is not incorporated into the genome.
One main drawback to both DNA- and RNA-vaccines is that the efficiency of naked oligonucleotide transfection is very low (compared to viral systems) due to lack of protection from systemic nucleases, inability to migrate through cell membranes, or entrapment and degradation within endosomes. Consequently, non-viral gene constructs typically require some type of polymer delivery system. The reader is directed to a number of excellent reviews on the subject of polymer gene delivery published by Dang and Leong (2006), Gao et al. (2007), Keegan and Saltzman (2006), Little and Langer (2005), and Pack et al. (2005).
We are currently developing a platform of polymer constructs that would release condensed RNA vaccines meant to transfect dendritic cells arriving upon implantation of a biomedical device. Our polymer constructs will release nanoparticles of condensed DNA or mRNA vaccine that will target a selected adhesion protein employed by the microorganism to initiate colonization. Such targets could be the fibronectin binding receptors on S. aureus or S. epidermidis used to bind to surface-immobilized fibronectin; cell surface Arg-specific (RgpA) and Lys-specific (Kgp) proteinases (designated the RgpA–Kgp complex) used by the oral pathogen, P. gingivalis, the major cause of chronic periodontitis; Group B Streptococcus to immobilized fibronectin; Steptococcus mutans and S. sangiuns to mucin-coated dental devices; P. aeruginosa binding to a G4 glycolipid on cornea and contact lenses; or E. coli FimH binding to mannose-coated catheters.
One distinct advantage of nucleotide transfection of dendritic cells (vs. direct antigen protein) is that one can modify the DNA or mRNA with targeting signals to better control MHC Classes I and II presentation. For example, incorporation of ubiquitin mRNA with the target antigen will result in an enhanced formation of peptides for MHC Class I presentation (Tobery and Siliciano, 1997, 1999; Varshavsky et al., 2000) whereas targeting sequences from the invariant chain (Ii) or lysosome-associated membrane protein (Lamp1) will lead to presentation of the antigen in the context of both MHC Classes I and II, thus providing antigen-specific help (Bonini et al., 2001; Lin et al., 1996; Thomson et al., 1998; Wu et al., 1995). We are currently developing mRNA vaccines anti- to the S. epidermidis fibronectin binding receptors used to colonize fibronectin-coated cardiovascular devises. S. epidermidis up-regulate these fibronectin binding receptors once exposed to serum at high shear stresses, which distinguishes infecting strains from benign S. epidermidis skin flora.
One might ask: “Why transfect dendritic cells from the medical device rather than simply inoculate the patient?” Transfection rates of DCs cells within the vicinity of a surgically implanted scaffold have been shown by Babensee’s group (Bennewitz and Babensee, 2005) to greatly exceed transfection rates observed by injecting the same amount of antigen within small microparticles. They suggested that “danger signals” associated with the surgical implantation of the larger scaffolds due to tissue injury attracted far greater numbers of localized DCs, leading to an enhanced immune response. Further, Babensee’s group has shown that staged transfections (initial and a series of “boosters”) greatly improved transfections efficiency.
Concluding Remarks
As our population ages, there will be an increase in the number of people experiencing hospitalization and receiving short- or long-term biomedical implants. As engineered biomaterials and tissue regenerative medicine advance, an increasing portion of the population will receive one or multiple biomedical devices, ranging from disposable contact lenses, dental implants, orthopedic implants, and vascular grafts to tissue engineered livers, small diameter vascular grafts that promote stem cells differentiation into endothelial cells, and polymer transfection systems that deliver micro-RNA knockout therapy to control chronic inflammation. The current health care approach to clean and sterilize has done little to prevent an epidemic in nosocomial infections. Biomaterials technologies employing disinfectant rinses, tethered or release antibiotics have also done little to reduce this epidemic and may have contributed to the raise of antibiotic resistant bacteria.
Increasing scientific research over the past 10 years in biofilm formation has provided a wealth of possible targets with which to prevent or eradicate biofilm infections. Advances in the understanding of biofilm formation, coupled with emerging engineered biomaterials, provide many potential platforms and strategies to prevent or significantly reduce biofilm infections in susceptible populations.
References
- An YH, Friedman RJ. Concise review of mechanisms of bacterial adhesion to biomaterial surfaces. J Biomed Mater Res. 1998;43(3):338–348. doi: 10.1002/(sici)1097-4636(199823)43:3<338::aid-jbm16>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Anderson JM. Biological responses to materials. Annu Rev Mater Res. 2001;31:81–110. [Google Scholar]
- Anderson CF, Mosser DM. A novel phenotype for an activated macrophage: The type 2 activated macrophage. J Leukoc Biol. 2002;72:101–106. [PubMed] [Google Scholar]
- Ankenbauer R, Sriyosachati S, Cox CD. Effects of siderophores on the growth of Pseudomonas aeruginosa in human serum and transferrin. Infect Immun. 1985;49:132–140. doi: 10.1128/iai.49.1.132-140.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson RA, Salah El Din AL, Kieffer B, Lefevre JF, Abdallah MA. Bacterial iron transport: 1H NMR determination of the three-dimensional structure of the gallium complex of pyoverdin G4R, the peptidic siderophore of Pseudomonas putida G4R. Biochemistry. 1998;37:15965–15973. doi: 10.1021/bi981194m. [DOI] [PubMed] [Google Scholar]
- Bagge N, Schuster M, Hentzer M, Ciofu O, Givskov M, Greenberg EP, Høiby N. Pseudomonas aeruginosa biofilms exposed to imipenem exhibit changes in global gene expression and β-lactamase and alginate production. Antimicrob Agents Chemother. 2004;48(4):1175–1187. doi: 10.1128/AAC.48.4.1175-1187.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakaletz LO. Bacterial biofilms in otitis media: Evidence and relevance. Pediatr Infect Dis J. 2007;10(Suppl):S17–S19. doi: 10.1097/INF.0b013e318154b273. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Steinman RM. B and T lymphocytes are the mediators of immunity, but their function is under the control of dendritic cells. Dendritic cells in the periphery. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Ann Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- Barclay R, Ratledge C. Participation of iron on the growth inhibition of pathogenic strains of Mycobacterium avium and M. paratuberculosis in serum. Zentralbl Bakteriol Mikrobiol Hyg [A] 1986;262:189–194. doi: 10.1016/s0176-6724(86)80019-0. [DOI] [PubMed] [Google Scholar]
- Bauer TW, Parvizi J, Kobayashi N, Krebs V. Diagnosis of periprosthetic infection. J Bone Joint Surg Am. 2006;88(4):869–882. doi: 10.2106/JBJS.E.01149. [DOI] [PubMed] [Google Scholar]
- Beaudoin D, Bryers JD, Cunningham AB, Peretti SW. Mobilization of broad host range plasmid from Pseudomonas putida to established biofilm of Bacillus azotoformans. I. Experiments. Biotechnol Bioeng. 1998a;57:272–279. [PubMed] [Google Scholar]
- Beaudoin D, Bryers JD, Cunningham AB, Peretti SW. Mobilization of broad host range plasmid from Pseudomonas putida to established biofilm of Bacillus azotoformans. II. Modeling. Biotechnol Bioeng. 1998b;57:280–286. [PubMed] [Google Scholar]
- Bennewitz NL, Babensee JL. The effect of the physical form of poly(lactic-co-glycolic acid) carriers on the humoral immune response to co-delivered antigen. Biomaterials. 2005;26:2991–2999. doi: 10.1016/j.biomaterials.2004.08.023. [DOI] [PubMed] [Google Scholar]
- Bjarnsholt T, Givskov M. Quorum-sensing blockade as a strategy for enhancing host defences against bacterial pathogens. Philos Trans R Soc Lond B Biol Sci. 2007;362(1483):1213–1222. doi: 10.1098/rstb.2007.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boles BR, Thoendel M, Singh PK. Rhamnolipids mediate detachment of Pseudomonas aeruginosa from biofilms. Mol Microbiol. 2005;57:1210–1223. doi: 10.1111/j.1365-2958.2005.04743.x. [DOI] [PubMed] [Google Scholar]
- Bonini C, Lee SP, Riddell SR, Greenberg PD. Targeting antigen in mature dendritic cells for simultaneous stimulation of CD4+ and CD8+ T cells. J Immunol. 2001;166:5250–5257. doi: 10.4049/jimmunol.166.8.5250. [DOI] [PubMed] [Google Scholar]
- Brady RA, Leid JG, Calhoun JH, Costerton JW, Shirtliff ME. Osteomyelitis and the role of biofilms in chronic infection. FEMS Immunol Med Microbiol. 2007 Dec 11; doi: 10.1111/j.1574-695X.2007.00357.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Branda SS, Vik S, Friedman L, Kolter R. Biofilms: The matrix revisited. Trends Microbiol. 2005;13(1):20–26. doi: 10.1016/j.tim.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Brown GD. Dectin-1: A signaling non-TLR pattern-recognition receptor. Nat Rev-Immunol. 2006;6:33–43. doi: 10.1038/nri1745. [DOI] [PubMed] [Google Scholar]
- Brunstedt MR, Sapatnekar S, Rubin KR, Kieswetter KM, Ziats NP, Merritt K, Anderson JM. Bacteria/blood/material interactions. I. Injected and preseeded slime-forming Staphylococcus epidermidis in flowing blood with biomaterials. J Biomed Mater Res. 1995;29(4):455–466. doi: 10.1002/jbm.820290405. [DOI] [PubMed] [Google Scholar]
- Bryers JD, editor. Biofilms. 2. New York, NY: J. Wiley Interscience; 2000. [Google Scholar]
- Bryers JD, Ratner BD. Bioinspired implant materials befuddle bacteria. ASM News. 2004;70:232–237. [Google Scholar]
- Bryers JD, Ratner BD. Biomaterials approaches to combating oral biofilms and dental disease. BMC Oral Health. 2006;6 (Suppl 1):S15. doi: 10.1186/1472-6831-6-S1-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busscher HJ, Cowan MM, van der Mei HC. On the relative importance of specific and non-specific approaches to oral microbial adhesion. FEMS Microbiol Rev. 1992;8(3–4):199–209. doi: 10.1111/j.1574-6968.1992.tb04988.x. [DOI] [PubMed] [Google Scholar]
- Cachia PJ, Hodges RS. Synthetic peptide vaccine and antibody therapeutic development: Prevention and treatment of Pseudomonas aeruginosa. Biopolymers. 2003;71(2):141–168. doi: 10.1002/bip.10395. [DOI] [PubMed] [Google Scholar]
- Camilli A, Bassler BL. Bacterial small-molecule signaling pathways. Science. 2006;311(5764):1113–1116. doi: 10.1126/science.1121357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall A, Dadachova E, Pirofski LA. Passive antibody therapy for infectious diseases. Nat Rev Microbiol. 2004;2(9):695–703. doi: 10.1038/nrmicro974. [DOI] [PubMed] [Google Scholar]
- Castelli P, Caronno R, Ferrarese S, Mantovani V, Piffaretti G, Tozzi M, Lomazzi C, Rivolta N, Sala A. New trends in prosthesis infection in cardiovascular surgery. Surg Infect. 2006;7 (Suppl 2):s45–s47. doi: 10.1089/sur.2006.7.s2-45. [DOI] [PubMed] [Google Scholar]
- Monina Klevens R, editor. Centers for Disease Control and Prevention Public Health Reports. Healthcare-associated infections and deaths in U.S. Hospitals. Vol. 1 2007. March–April, [Google Scholar]
- Chandra J, McCormick TS, Imamura Y, Mukherjee PK, Ghannoum MA. Interaction of Candida albicans with adherent human peripheral blood mononuclear cells increases C. albicans biofilm formation and results in differential expression of pro- and anti-inflammatory cytokines. Infect Immun. 2007;75(5):2612–2620. doi: 10.1128/IAI.01841-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitambar CR, Narasimhan J. Targeting iron-dependent DNA synthesis with gallium and transferrin-gallium. Pathobiology. 1991;59(1):3–10. doi: 10.1159/000163609. [DOI] [PubMed] [Google Scholar]
- Cohen N, Morisset J, Emilie D. Induction of tolerance by Porphyromonas gingivalis on APCS: A mechanism implicated in periodontal infection. J Dent Res. 2004;83:429–433. doi: 10.1177/154405910408300515. [DOI] [PubMed] [Google Scholar]
- Dang JM, Leong KW. Natural polymers for gene delivery and tissue engineering. Adv Drug Deliv Rev. 2006;58(4):487–499. doi: 10.1016/j.addr.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Davies DG, Geesey GG. Regulation of the alginate biosynthesis gene algC in Pseudomonas aeruginosa during biofilm development in continuous culture. Appl Environ Microbiol. 1995;61(3):860–867. doi: 10.1128/aem.61.3.860-867.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies DG, Chakrabarty AM, Geesey GG. Exopolysaccharide production in biofilms: Substratum activation of alginate gene expression by Pseudomonas aeruginosa. Appl Environ Microbiol. 1993;59(4):1181–1186. doi: 10.1128/aem.59.4.1181-1186.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science. 1998;280(5361):295–298. doi: 10.1126/science.280.5361.295. [DOI] [PubMed] [Google Scholar]
- De Smet K, Contreras R. Human antimicrobial peptides: Defensins, cathelicidins and histatins. Biotechnol Lett. 2005;27(18):1337–1347. doi: 10.1007/s10529-005-0936-5. [DOI] [PubMed] [Google Scholar]
- Donlan RM. Biofilms: Microbial life on surfaces. Emerg Infect Dis. 2002;8(9):881–890. doi: 10.3201/eid0809.020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döring G, Conway SP, Heijerman HG, Hodson ME, Høiby N, Smyth A, Touw DJ. Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis: A European consensus. Eur Respir J. 2000;16(4):749–767. doi: 10.1034/j.1399-3003.2000.16d30.x. [DOI] [PubMed] [Google Scholar]
- European Centre for Disease Prevention and Control. Annual epidemiological report on communicable diseases in Europe. Stockholm; Sweden: 2007. ( http://www.ecdc.eu.int/index.html) [PubMed] [Google Scholar]
- Evans RT, Klausen B, Sojar HT, Bedi GS, Sfintescu C, Ramamurthy NS, Golub LM, Genco RJ. Immunization with Porphyromonas (Bacleroides) gingivalis fimbriae protects against periodontal destruction. Infect Immun. 1992;60:2926–2935. doi: 10.1128/iai.60.7.2926-2935.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein RA, Sciortino CV, McIntosh MA. Role of iron in microbe-host interactions. Rev Infect Dis. 1983;5 (Suppl 4):S759–S777. doi: 10.1093/clinids/5.supplement_4.s759. [DOI] [PubMed] [Google Scholar]
- Fischetti VA. Bacteriophage lytic enzymes: Novel anti-infectives. Trends Microbiol. 2005;13(10):491–496. doi: 10.1016/j.tim.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Foster TJ. Immune evasion by Staphylococci. Nat Rev Microbiol. 2005;3:948–958. doi: 10.1038/nrmicro1289. [DOI] [PubMed] [Google Scholar]
- Fux CA, Stoodley P, Hall-Stoodley L, Costerton JW. Bacterial biofilms: A diagnostic and therapeutic challenge. Expert Rev Anti Infect Ther. 2003;1(4):667–683. doi: 10.1586/14787210.1.4.667. [DOI] [PubMed] [Google Scholar]
- Fux CA, Costerton JW, Stewart PS, Stoodley P. Survival strategies of infectious biofilms. Trends Microbiol. 2005;13(1):34–40. doi: 10.1016/j.tim.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med. 2003;197(9):1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Gu W, Chen L, Xu Z, Li Y. A multifunctional nano device as non-viral vector for gene delivery: In vitro characteristics and transfection. J Control Release. 2007;118(3):381–388. doi: 10.1016/j.jconrel.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Geddes A. Infection in the twenty-first century: Predictions and postulates. J Antimicrob Chemother. 2000;46:873–878. doi: 10.1093/jac/46.6.873. [DOI] [PubMed] [Google Scholar]
- Gilbert P, Allison DG, McBain AJ. Biofilms in vitro and in vivo: Do singular mechanisms imply cross-resistance? J Appl Microbiol. 2002;92 (Suppl):98S–110S. [PubMed] [Google Scholar]
- Gilboa E, Vieweg J. Cancer immunotherapy with mRNA transfected dendritic cells. Immunol Rev. 2004;199:251–263. doi: 10.1111/j.0105-2896.2004.00139.x. [DOI] [PubMed] [Google Scholar]
- Glantz PO, Arnebrant T, Nylander T, Baier RE. Bioadhesion—A phenomenon with multiple dimensions. Acta Odontol Scand. 1999;57(5):238–241. doi: 10.1080/000163599428634. [DOI] [PubMed] [Google Scholar]
- Goldmann O, von Köckritz-Blickwede M, Höltje C, Chhatwal GS, Geffers R, Medina E. Transcriptome analysis of murine macrophages in response to infection with Streptococcus pyogenes reveals an unusual activation program. Infect Immun. 2007;75(8):4148–4157. doi: 10.1128/IAI.00181-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Gyimesi E, Bankovich AJ, Schuman TA, Goldberg JB, Lindorfer MA, Taylor RP. Staphylococcus aureus bound to complement receptor 1 on human erythrocytes by bispecific monoclonal antibodies is phagocytosed by acceptor macrophages. Immunol Lett. 2004;95:185–192. doi: 10.1016/j.imlet.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Hall-Stoodley L, Stoodley P. Biofilm formation and dispersal and the transmission of human pathogens. Trends Microbiol. 2005;13(7):7–10. doi: 10.1016/j.tim.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: From the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2(2):95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- Harraghy N, Kerdudou S, Herrmann M. Quorum-sensing systems in staphylococci as therapeutic targets. Anal Bioanal Chem. 2007;387(2):437–444. doi: 10.1007/s00216-006-0860-0. [DOI] [PubMed] [Google Scholar]
- Hart D. Dendritic cells: Unique leukocyte populations that control the primary immune response. Blood. 1997;90:3245–3287. [PubMed] [Google Scholar]
- Heikenwalder M, Polymenidou M, Junt T, Sigurdson C, Wagner H, Akira S, Zinkernagel R, Aguzzi A. Lymphoid follicle destruction and immunosuppression after repeated CpG oligodeoxynucleotide administration. Nat Med. 2004;10:187–192. doi: 10.1038/nm987. [DOI] [PubMed] [Google Scholar]
- Hodgkinson JT, Welch M, Spring DR. Learning the language of bacteria. ACS Chem Biol. 2007;2(11):715–717. doi: 10.1021/cb700227k. [DOI] [PubMed] [Google Scholar]
- Hoffman LR, D’Argenio DA, MacCoss MJ, Zhang Z, Jones RA, Miller SI. Aminoglycoside antibiotics induce bacterial biofilm formation. Nature. 2005;436(25):1171–1175. doi: 10.1038/nature03912. [DOI] [PubMed] [Google Scholar]
- Høiby N. Understanding bacterial biofilms in patients with cystic fibrosis: Current and innovative approaches to potential therapies. J Cyst Fibros. 2002;1(4):249–254. doi: 10.1016/s1569-1993(02)00104-2. [DOI] [PubMed] [Google Scholar]
- Hong HA, Ducle H, Cutting SM. The use of bacterial spore formers as probiotics. FEMS Microbiol Rev. 2005;29(4):813–835. doi: 10.1016/j.femsre.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Horswill AR, Stoodley P, Stewart PS, Parsek MR. The effect of the chemical, biological, and physical environment on quorum sensing in structured microbial communities. Anal Bioanal Chem. 2007;387(2):371–380. doi: 10.1007/s00216-006-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Eaton J, Tang L. Molecular basis of biomaterial-mediated foreign body reactions. Blood. 2001;98:1231–1238. doi: 10.1182/blood.v98.4.1231. [DOI] [PubMed] [Google Scholar]
- Hubbard JA, Lewandowska KB, Hughes MN, Poole RK. Effects of iron-limitation of Escherichia coli on growth, the respiratory chains and gallium uptake. Arch Microbiol. 1986;146(1):80–86. doi: 10.1007/BF00690163. [DOI] [PubMed] [Google Scholar]
- Janeway CJ, Travers P, Walport M, Shlomchik M. Immunobiology: The immune system in health and disease. 6. New York: Garland Science Publishing; 2005. [Google Scholar]
- Jarvis R, Bryers JD. Effects of controlled fibronectin surface orientation on subsequent Staphylococcus epidermidis adhesion. J Biomed Mater Res-A. 2005;75:41–55. doi: 10.1002/jbm.a.30404. [DOI] [PubMed] [Google Scholar]
- Jesaitis AJ, Franklin MJ, Berglund D, Sasaki M, Lord CI, Bleazard JB, Duffy JE, Beyenal H, Lewandowski Z. Compromised host defense on Pseudomonas aeruginosa biofilms: Characterization of neutrophil and biofilm interactions. J Immunol. 2003;171(8):4329–4339. doi: 10.4049/jimmunol.171.8.4329. [DOI] [PubMed] [Google Scholar]
- Jotwani R, Cutler CW. Fimbriated Porphyromonas gingivalis is more efficient than fimbria-deficient P. gingivalis in entering human dendritic cells in vitro and induces an inflammatory Th1 effector response. Infect Immun. 2004;72:1725–1732. doi: 10.1128/IAI.72.3.1725-1732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jotwani R, Pulendran B, Agrawal S, Cutler CW. Human dendritic cells respond to Porphyromonas gingivaux LPS by promoting Th2 effector response in vitro. Eur J Immunol. 2003;33:2980–2986. doi: 10.1002/eji.200324392. [DOI] [PubMed] [Google Scholar]
- Jurado RL. Iron, infections, and anemia of inflammation. Clin Infect Dis. 1997;25(4):888–895. doi: 10.1086/515549. [DOI] [PubMed] [Google Scholar]
- Kaneko Y, Thoendel M, Olakanmi O, Britigan BE, Singh PK. The transition metal gallium disrupts Pseudomonas aeruginosa iron metabolism and has antimicrobial and antibiofilm activity. J Clin Invest. 2007;117(4):877–888. doi: 10.1172/JCI30783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan ME, Saltzman WM. Surface-modified biodegradable microspheres for DNA vaccine delivery. Methods Mol Med. 2006;127:107–113. doi: 10.1385/1-59745-168-1:107. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Takauchi A, van Spriel AB, Vilé HA, Hayakawaf M, Shibata Y, Abiko Y, van de Winkel JGJ, Yoshiea H. Targeting of Porphyromonas gingivalis with a bispecific antibody directed to FcαRI (CD89) improves in vitro clearance by gingival crevicular neutrophils. Vaccine. 2004;23:585–594. doi: 10.1016/j.vaccine.2004.07.015. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy VM, Quinton LJ, Estroffa LA, Metalloa SJ, Isaacs JM, Mizgerd JP, Whitesides GM. Promotion of opsonization by antibodies and phagocytosis of Gram-positive bacteria by a bifunctional polyacrylamide. Biomaterials. 2006;27:3663–3674. doi: 10.1016/j.biomaterials.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Kuhn SE, Nardin A, Klebba PE, Taylor RP. Escherichia coli bound to the primate erythrocyte complement receptor via bispecific monoclonal antibodies are transferred to and phagocytosed by human monocytes in an in vitro model. J Immunol. 1998;160:5088–5097. [PubMed] [Google Scholar]
- Leibovich SJ, Ross R. The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am J Pathol. 1975;78:71. [PMC free article] [PubMed] [Google Scholar]
- Leid JG, Shirtliff ME, Costerton JW, Stoodley AP. Human leukocytes adhere to, penetrate, and respond to Staphylococcus aureus biofilms. Infect Immun. 2002;70(11):6339–6345. doi: 10.1128/IAI.70.11.6339-6345.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leid JG, Willson CJ, Shirtliff ME, Hassett DJ, Parsek MR, Jeffers AK. The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-gamma-mediated macrophage killing. J Immunol. 2005;175(11):7512–7518. doi: 10.4049/jimmunol.175.11.7512. [DOI] [PubMed] [Google Scholar]
- Li M, Lai Y, Villaruz AE, Cha DJ, Sturdevant DE, Otto M. Gram-positive three-component antimicrobial peptide-sensing system. Proc Natl Acad Sci USA. 2007;104(22):9469–9474. doi: 10.1073/pnas.0702159104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KY, Guarnieri FG, Staveley-O’Carroll KF, Levitsky HI, August JT, Pardoll DM, Wu TC. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–26. [PubMed] [Google Scholar]
- Lindorfer MA, Nardin A, Foley PL, Solga MD, Bankovich AJ, Martin EN, Henderson AL, Price CW, Gyimesi E, Wozencraft CP, Goldberg JB, Sutherland WM, Taylor RP. Targeting of Pseudomonas aeruginosa in the bloodstream with bispecific monoclonal antibodies. J Immunol. 2001;167:2240–2249. doi: 10.4049/jimmunol.167.4.2240. [DOI] [PubMed] [Google Scholar]
- Little SR, Langer R. Nonviral delivery of cancer genetic vaccines. Adv Biochem Eng Biotechnol. 2005;99:93–118. doi: 10.1007/10_004. [DOI] [PubMed] [Google Scholar]
- Mah TF, O’Toole GA. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001;9(1):34–39. doi: 10.1016/s0966-842x(00)01913-2. [DOI] [PubMed] [Google Scholar]
- Månsson LE, Melican K, Molitoris BA, Richter-Dahlfors A. Progression of bacterial infections studied in real time; novel perspectives provided by multiphoton microscopy. Cell Microbiol. 2007 doi: 10.1111/j.1462-5822.2007.01019.x. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Martin P, D’Souza D, Martin J, Grose R, Cooper L, Maki R, McKercher SR. Wound healing in the PU.1 null mouse—tissue repair is not dependent on inflammatory cells. Curr Biol. 2003;13:1122. doi: 10.1016/s0960-9822(03)00396-8. [DOI] [PubMed] [Google Scholar]
- Martinez LR, Casadevall A. Specific antibody can prevent fungal biofilm formation and this effect correlates with protective efficacy. Infect Immun. 2005;73(10):6350–6362. doi: 10.1128/IAI.73.10.6350-6362.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- Mekori Y, Metcalfe D. Mast cells in innate immunity. Immunol Rev. 2000;173:131–140. doi: 10.1034/j.1600-065x.2000.917305.x. [DOI] [PubMed] [Google Scholar]
- Mittal R, Sharma S, Chhibber S, Harjai K. Effect of macrophage secretory products on elaboration of virulence factors by planktonic and biofilm cells of Pseudomonas aeruginosa, Comparative Immunology. Microbiol Infect Dis. 2006;29:12–26. doi: 10.1016/j.cimid.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- Narasimhan J, Antholine WE, Chitambar CR. Effect of gallium on the tyrosyl radical of the iron-dependent M2 subunit of ribonucleotide reductase. Biochem Pharmacol. 1992;44:2403–2408. doi: 10.1016/0006-2952(92)90686-d. [DOI] [PubMed] [Google Scholar]
- Offenbacher S, Barros SP, Singer RE, Moss K, Williams RC, Beck JD. Periodontal disease at the biofilm-gingival interface. J Periodontol. 2007;78(10):1911–1925. doi: 10.1902/jop.2007.060465. [DOI] [PubMed] [Google Scholar]
- Otto M. Quorum-sensing control in Staphylococci—A target for antimicrobial drug therapy? FEMS Microbiol Lett. 2004;241:135–141. doi: 10.1016/j.femsle.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Otto M. Bacterial evasion of antimicrobial peptides by biofilm formation. Curr Top Microbiol Immunol. 2006;306:251–258. doi: 10.1007/3-540-29916-5_10. [DOI] [PubMed] [Google Scholar]
- Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov. 2005;4(7):581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- Parsek MR, Singh PK. Bacterial biofilms: An emerging link to disease pathogenesis. Annu Rev Microbiol. 2003;57:677–701. doi: 10.1146/annurev.micro.57.030502.090720. [DOI] [PubMed] [Google Scholar]
- Persson GR. Immune responses and vaccination against periodontal infections. J Clin Periodontol. 2005;32 (Suppl 6):39–53. doi: 10.1111/j.1600-051X.2005.00800.x. [DOI] [PubMed] [Google Scholar]
- Post JC, Hiller NL, Nistico L, Stoodley P, Ehrlich GD. The role of biofilms in otolaryngologic infections: Update 2007. Curr Opin Otolaryngol Head Neck Surg. 2007;15(5):347–351. doi: 10.1097/MOO.0b013e3282b97327. [DOI] [PubMed] [Google Scholar]
- Potempa J, Sroka A, Imamura T, Travis J. Gingipains, the major cysteine proteinases and virulence factors of Porphyromonas gingivalis: Structure, function and assembly of multidomain protein complexes. Curr Protein Pept Sci. 2003;4:397–407. doi: 10.2174/1389203033487036. [DOI] [PubMed] [Google Scholar]
- Pritchard DI. Immune modulation by Pseudomonas aeruginosa quorum-sensing signal molecules. Int J Med Microbiol. 2006;296(2–3):111–116. doi: 10.1016/j.ijmm.2006.01.037. [DOI] [PubMed] [Google Scholar]
- Pulendran B, Kumar P, Cutler CW, Mohamadzadeh M, Van Dyke T, Banchereau J. Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J Immunol. 2001;167:5067–5076. doi: 10.4049/jimmunol.167.9.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisner A, Høiby N, Tolker-Nielsen T, Molin S. Microbial pathogenesis and biofilm development. Contrib Microbiol. 2005;12:114–131. doi: 10.1159/000081692. [DOI] [PubMed] [Google Scholar]
- Rooijakkers SH, van Strijp JA. Bacterial complement evasion. Mol Immmunol. 2007;44(1–3):23–32. doi: 10.1016/j.molimm.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Rosenberg M. Microbial adhesion to hydrocarbons: Twenty-five years of doing MATH. FEMS Microbiol Lett. 2006;262(2):129–134. doi: 10.1111/j.1574-6968.2006.00291.x. [DOI] [PubMed] [Google Scholar]
- Roux P, Münter S, Frischknecht F, Herbomel P, Shorte SL. Focusing light on infection in four dimensions. Cell Microbiol. 2004;6(4):333–343. doi: 10.1111/j.1462-5822.2004.00374.x. [DOI] [PubMed] [Google Scholar]
- Rumbaugh KP. Convergence of hormones and autoinducers at the host/pathogen interface. Anal Bioanal Chem. 2007;387(2):425–435. doi: 10.1007/s00216-006-0694-9. [DOI] [PubMed] [Google Scholar]
- Sauer K, Cullen MC, Rickard AH, Zeef LA, Davies DG, Gilbert P. Characterization of nutrient-induced dispersion in Pseudomonas aeruginosa PAO1 biofilm. J Bacteriology. 2004;186(21):7312–7326. doi: 10.1128/JB.186.21.7312-7326.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savill J. Phagocyte recognition of apoptotic cells. Biochem Soc Trans. 1996;24:1065–1069. doi: 10.1042/bst0241065. [DOI] [PubMed] [Google Scholar]
- Schmid D, Dengjel J, Schoor O, Stevanovic S, Münz C. Autophagy in innate and adaptive immunity against intracellular pathogens. J Mol Med. 2006;84:194–202. doi: 10.1007/s00109-005-0014-4. [DOI] [PubMed] [Google Scholar]
- Schwartz B, Karsan A, Bombeli T, Harlan J. A novel β-1 integrin-dependent mechanism of leukocyte adherence to apoptotic cells. J Immunol. 1999;162:4842–4848. [PubMed] [Google Scholar]
- Selwitz RH, Ismail AI, Pitts NB. Dental caries. Lancet. 2007;369(9555):51–59. doi: 10.1016/S0140-6736(07)60031-2. [DOI] [PubMed] [Google Scholar]
- Shankar R, Griesler H. In: Biomaterials science: An introduction to materials in medicine. Ratner BD, Hoffman AS, Schoen FJ, Lemons JE, editors. New York: Academic Press; 1996. [Google Scholar]
- Singh PK. Iron sequestration by human lactoferrin stimulates P. aeruginosa surface motility and blocks biofilm formation. Biometals. 2004;17:267–270. doi: 10.1023/b:biom.0000027703.77456.27. [DOI] [PubMed] [Google Scholar]
- Slayton RL, Bryers JD, Milgrom P. Biotech and biomaterials research to reduce the caries epidemic. BMC Oral Health. 2006;6 (Suppl 1):S1. doi: 10.1186/1472-6831-6-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RS, Iglewski BH. P. aeruginosa quorum-sensing systems and virulence. Curr Opin Microbiol. 2003;6(1):56–60. doi: 10.1016/s1369-5274(03)00008-0. [DOI] [PubMed] [Google Scholar]
- Steinman R. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- Stewart PS. Mechanisms of antibiotic resistance in bacterial biofilms. Int J Med Microbiol. 2002;292:107–113. doi: 10.1078/1438-4221-00196. [DOI] [PubMed] [Google Scholar]
- Stewart PS, Costerton JW. Antibiotic resistance of bacteria in biofilms. Lancet. 2001;358:135–138. doi: 10.1016/s0140-6736(01)05321-1. [DOI] [PubMed] [Google Scholar]
- Stout R, Suttles J. Functional plasticity of macrophages: Reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76:509–513. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. 2005;175:342–349. doi: 10.4049/jimmunol.175.1.342. [DOI] [PubMed] [Google Scholar]
- Strevett KA, Chen G. Microbial surface thermodynamics and applications. Res Microbiol. 2003;154(5):329–335. doi: 10.1016/S0923-2508(03)00038-X. [DOI] [PubMed] [Google Scholar]
- Su XL, Li Y. Quantum dot biolabeling coupled with immunomagnetic separation for detection of Escherichia coli O157: H. Anal Chem. 2004;76(16):4806–4810. doi: 10.1021/ac049442+. [DOI] [PubMed] [Google Scholar]
- Sulakvelidze A, Alavidze Z, Morris JG., Jr Bacteriophage therapy. Antimicrob Agents Chemother. 2001;45(3):649–659. doi: 10.1128/AAC.45.3.649-659.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Accavitti MA, Bryers JD. Inhibition of biofilm formation by monoclonal antibodies against Staphylococcus epidermidis RP62A accumulation-associated protein. Clin Diagn Lab Immunol. 2005;12:93–100. doi: 10.1128/CDLI.12.1.93-100.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Jennings T, Eaton J. Mast cells mediate acute inflammatory responses to implanted biomaterials. Proc Natl Acad Sci USA. 1998;95:8841–8846. doi: 10.1073/pnas.95.15.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateda K, Ishii Y, Horikawa M, Matsumoto T, Miyairi S, Pechere JC, Standiford TJ, Ishiguro M, Yamaguchi K. The Pseudomonas aeruginosa autoinducer N-3-oxododecanoyl homoserine lactone accelerates apoptosis in macrophages and neutrophils. Infect Immun. 2003;71(10):5785–5793. doi: 10.1128/IAI.71.10.5785-5793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson SA, Burrows SR, Misko IS, Moss DJ, Coupar BE, Khanna R. Targeting a polyepitope protein incorporating multiple class II-restricted viral epitopes to the secretory/endocytic pathway facilitates immune recognition by CD4+ cytotoxic T lymphocytes: A novel approach to vaccine design. J Virol. 1998;72:2246–2252. doi: 10.1128/jvi.72.3.2246-2252.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobery TW, Siliciano RF. Targeting of HIV-1 antigens for rapid intracellular degradation enhances cytotoxic T lymphocyte (CTL) recognition and the induction of de novo CTL responses in vivo after immunization. J Exp Med. 1997;185:909–920. doi: 10.1084/jem.185.5.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobery T, Siliciano RF. Cutting edge: Induction of enhanced CTL-dependent protective immunity in vivo by N-end rule targeting of a model tumor antigen. J Immunol. 1999;162:639–642. [PubMed] [Google Scholar]
- van Kooyk Y, Geijtenbeck TB. DC-SIGN: Escape mechanism for pathogens. Nat Rev Immunol. 2003;3:697–709. doi: 10.1038/nri1182. [DOI] [PubMed] [Google Scholar]
- van Kooyk Y, Engering A, Lekkerkerker AN, Ludwig IS, Geijtenbeck TB. Pathogens use carbohydrates to escape immunity induced by dendritic cells. Curr Opin Immunol. 2004;16:488–493. doi: 10.1016/j.coi.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Varshavsky A, Turner G, Du F, Xie Y. Felix Hoppe-Seyler Lecture 2000. The ubiquitin system and the N-end rule pathway. Biol Chem. 2000;381:779–789. doi: 10.1515/BC.2000.101. [DOI] [PubMed] [Google Scholar]
- Verran J, Whitehead K. Factors affecting microbial adhesion to stainless steel and other materials used in medical devices. Int J Artif Organs. 2005;28(11):1138–1145. doi: 10.1177/039139880502801111. [DOI] [PubMed] [Google Scholar]
- Vuong C, Kocianova S, Voyich JM, Yao Y, Fischer ER, DeLeo FR, Otto M. A crucial role for exopolysaccharide modification in bacterial biofilm formation, immune evasion, and virulence. J Biol Chem. 2004;279:54881–54886. doi: 10.1074/jbc.M411374200. [DOI] [PubMed] [Google Scholar]
- Wagner VE, Bryers JD. Poly(ethylene glycol)-polyacrylate copolymers modified to control adherent monocyte-macrophage physiology: Interactions with attaching Staphylococcus epidermidis or Pseudomonas aeruginosa bacteria. J Biomed Mater Res A. 2004;69(1):79–90. doi: 10.1002/jbm.a.20115. [DOI] [PubMed] [Google Scholar]
- Wagner C, Obst U, Hänsch GM. Implant-associated posttraumatic osteomyelitis: Collateral damage by local host defense? Int J Artif Organs. 2005;28(11):1172–1180. doi: 10.1177/039139880502801115. [DOI] [PubMed] [Google Scholar]
- Wagner VE, Frelinger JG, Barth RK, Iglewski BH. Quorum sensing: Dynamic response of Pseudomonas aeruginosa to external signals. Trends Microbiol. 2006;14(2):55–58. doi: 10.1016/j.tim.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Walker TS, Tomlin KL, Worthen GS, Poch KR, Lieber JG, Saavedra MT, Fessler MB, Malcolm KC, Vasil ML, Nick JA. Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infect Immun. 2005;73(6):3693–3701. doi: 10.1128/IAI.73.6.3693-3701.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel RP. Health care-associated infections: Major issues in the early years of the 21st century. Clin Infect Dis. 2007;45 (Suppl 1):S85–S88. doi: 10.1086/518136. [DOI] [PubMed] [Google Scholar]
- Wilson C, Clegg R, Leavesley D, Pearcy M. Mediation of biomaterial-cell interactions by adsorbed proteins: A review. Tissue Eng. 2005;11:1–18. doi: 10.1089/ten.2005.11.1. [DOI] [PubMed] [Google Scholar]
- Wizemann TM, Adamou JE, Langermann S. Adhesins as targets for vaccine development. Emerg Infect Dis. 1999;5:395–403. doi: 10.3201/eid0503.990310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TC, Guarnieri FG, Staveley-O’Carroll KF, Viscidi RP, Levitsky HI, Hedrick L, Cho KR, August JT, Pardoll DM. Engineering an intracellular pathway for major histocompatibility complex class II presentation of antigens. Proc Natl Acad Sci USA. 1995;92:11671–11675. doi: 10.1073/pnas.92.25.11671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Estrada O, Zaborina O, Bains M, Shen L, Kohler JE, Patel N, Musch MW, Chang EB, Fu YX, Jacobs MA, Nishimura MI, Hancock RE, Turner JR, Alverdy JC. Recognition of host immune activation by Pseudomonas aeruginosa. Science. 2005;309(5735):774–777. doi: 10.1126/science.1112422. [DOI] [PubMed] [Google Scholar]
- Xavier JB, Picioreanu C, van Loosdrecht MCM. A general description of detachment for multidimensional modelling of biofilms. Biotechnol Bioeng. 2005;91(6):651–669. doi: 10.1002/bit.20544. [DOI] [PubMed] [Google Scholar]
- Xiong YQ, Willard J, Kadurugamuwa JL, Yu J, Francis KP, Bayer AS. Real-time in vivo bioluminescent imaging for evaluating the efficacy of antibiotics in a rat Staphylococcus aureus endocarditis model. Anti-microb Agents Chemother. 2005;49(1):380–387. doi: 10.1128/AAC.49.1.380-387.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li Y. Simultaneous detection of Escherichia coli O157:H7 and Salmonella typhimurium using quantum dots as fluorescence labels. Analyst. 2006;131(3):394–401. doi: 10.1039/b510888h. [DOI] [PubMed] [Google Scholar]
- Yongsunthon R, Lower SK. Force measurements between a bacterium and another surface in situ. Adv Appl Microbiol. 2006;58:97–124. [PubMed] [Google Scholar]
- Zhao H, Hemmi H, Akira S, Cheng SH, Scheule RK, Yew NS. Contribution of Toll-like receptor 9 signaling to the acute inflammatory response to nonviral vectors. Mol Ther. 2004;9:241–248. doi: 10.1016/j.ymthe.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Zimmermann S, Wagner C, Müller W, Brenner-Weiss G, Hug F, Prior B, Obst U, Hänsch GM. Induction of neutrophil chemotaxis by the quorum-sensing molecule N-(3-oxododecanoyl)-L-homoserine lactone. Infect Immun. 2006;74(10):5687–5692. doi: 10.1128/IAI.01940-05. [DOI] [PMC free article] [PubMed] [Google Scholar]