

Abstract
A specific pentasaccharide sequence of heparin binds with high affinity to native antithrombin and induces a conformational change in the inhibitor by a previously described two-step interaction mechanism. In this work, the interactions of heparin with the antiangiogenic latent and cleaved antithrombin forms were studied. Binding of heparin to these antithrombin forms was specific for the same pentasaccharide sequence as native antithrombin. Rapid kinetics studies demonstrated that this pentasaccharide induced a conformational change also in latent and cleaved antithrombin. The binding affinities of these antithrombin forms for the pentasaccharide, as compared to native antithrombin, were ∼30-fold lower due to 2-3 fewer ionic interactions, resulting in less stable conformationally altered states. Affinities of latent and cleaved antithrombin for longer heparin chains, containing the pentasaccharide sequence, were two-fold lower than for the pentasaccharide itself. This contrasts the interaction with native antithrombin and demonstrates that residues flanking the pentasaccharide sequence of heparin are repelled by the latent and cleaved forms. These findings contribute to delineating the mechanism by which heparin or heparan sulfate mediate antiangiogenic activity of antithrombin.
The serpin, antithrombin, has been found to possess potent antiangiogenic properties in addition to being an important physiological anticoagulant. The anticoagulant native form of antithrombin has an intact surface-exposed loop, the reactive center loop (RCL)1, containing a reactive site that is recognized by the target proteases. In contrast, the antiangiogenic activities are exerted by conformationally changed forms of the protein, i.e. cleaved, latent and prelatent antithrombin (1-5). Cleaved antithrombin is formed by proteolytic cleavage in the RCL, which leads to the insertion of the N-terminal part of this loop as a new strand in the center of a large central β-sheet, called β-sheet A, of the inhibitor. Latent antithrombin is formed by mild heat treatment, which gives a conformation similar to cleaved antithrombin, although the RCL is intact. The latent and cleaved forms have lost their ability to inhibit proteases (6, 7). Prelatent antithrombin is formed as an intermediate in the conversion from native to latent antithrombin in the presence of stabilizing ions and possesses both anticoagulant and antiangiogenic properties (3, 8). The antiangiogenic forms of antithrombin have been found to inhibit angiogenesis in several in vivo models and to inhibit tumor growth in several mouse models (1-3, 9). Mechanisms of the antiangiogenic actions include inhibition of FGF-2 and VEGF-induced proliferation of endothelial cells (2, 9). Moreover, latent and cleaved antithrombin down-regulate several proangiogenic genes and upregulate several antiangiogenic genes (10), suggesting that these antithrombin forms have direct signaling functions.
The interaction of heparin or heparan sulfate with native antithrombin plays an essential role in activating the inhibitory potential of antithrombin, by increasing the rate of attack of the inhibitor on its target proteases, partially due to a bridging mechanism and partially due to an allosteric mechanism (11). In the bridging mechanism, protease-inhibitor complex formation is enhanced through the binding to the same heparin chain. The allosteric mechanism is achieved by a global conformational change, induced by the binding of a specific pentasaccharide sequence (12), and is accompanied by a 40% enhancement in tryptophan fluorescence (13). Heparin fractions that comprise this pentasaccharide sequence are denoted high-affinity heparin (HAH) and those that lack it are called low-affinity heparin (LAH).
It has recently been shown that the heparin-binding site is required for the antiangiogenic activities of latent and cleaved antithrombin, suggesting that heparin/heparan sulfate is involved in mediating these activities (9). An essential question is therefore whether the antiangiogenic antithrombin forms have different heparin sequence specificities than native antithrombin. In this work, the interactions of HAH, LAH and two synthetic pentasaccharides with cleaved, latent and native antithrombin were studied by fluorescence measurements and by an affinity matrix method. The heparin preference of latent and cleaved antithrombin and the mechanism of heparin interactions with these antithrombin forms were characterized and compared with those of native antithrombin. Latent and cleaved antithrombin were found to have specificity for the same pentasaccharide sequence as native antithrombin, although with approximately 30-fold lower affinities. Furthermore, the interaction with longer heparin chains differed, because latent and cleaved antithrombin repelled heparin in what is in native antithrombin denoted the extended heparin binding site. This information may be useful in defining the type of heparin or heparan sulfate that is used by the antiangiogenic antithrombin forms to mediate antiangiogenic activity. Moreover, since treatments with various heparin forms have been found to reduce the mortality rate of cancer (14), information on the sequences that interact with the different antithrombin forms may be of importance for achieving the optimal desired effects on angiogenesis as well as coagulation during such treatments.
EXPERIMENTAL PROCEDURES
Antithrombin Preparations
The α-form of antithrombin, i.e. the form that is glycosylated on all four potential N-glycosylation sites and constitutes ∼90% of antithrombin in human blood (15, 16), was purified from human plasma as described previously (17). Latent antithrombin was prepared by incubating native antithrombin at 60°C for 24 h in 10 mM Tris/HCl, 0.5 M sodium citrate and pH 7.4 (2). Human neutrophil elastase (Athens Research and Technology, Athens, GA, USA) was used to cleave the RCL in native antithrombin to produce cleaved antithrombin, as described previously (4, 18, 19). SDS- and native PAGE, according to Laemmli, were used to assess the purity of the antithrombin variants (17, 20). The cleaved antithrombin variant was differentiated from native antithrombin by 10% SDS-PAGE under non-reducing conditions (21). The samples were boiled for 10 min before they were applied to the gel. Concentrations of the antithrombin variants were determined spectrophotometrically from the absorbance of 280 nm with the use of the molar absorption coefficient of 37 700 M-1 cm-1 (22). The purified antithrombin forms were snap-frozen and stored at -70°C. Native, latent and cleaved antithrombin were >95 % homogenous in reducing SDS-PAGE, native PAGE and nonreducing SDS-PAGE, respectively.
Heparins
The α-methyl glycoside form of the heparin pentasaccharide mimicking the antithrombin binding site of heparin (Fondaparinux clinically used under the trade name Arixtra ®) (23) was a gift from Glaxo Smith Kline. This pentasaccharide is referred to as the normal pentasaccharide in this work. The monosaccharide units that it is composed of are denoted DEFGH from the nonreducing end. The synthetic O-methylated, O-sulfated pentasaccharide Idraparinux (23), here referred to as the high-affinity pentasaccharide, was supplied by Sanofi-Aventis. Heparins with low and high affinity for native antithrombin, i.e. lacking or containing the pentasaccharide and denoted LAH and HAH, respectively, with molecular weights of ∼7900, were isolated as described previously (17).
Experimental conditions
Equilibrium binding and kinetics of antithrombin-heparin interactions and thrombin inhibition stoichiometries were analyzed at 25 ± 0.2°C or 10 ± 0.2°C in 20 mM sodium phosphate buffer containing 0.1 mM EDTA and 0.1% polyethyleneglycol at pH 7.4. Sodium chloride was added to give the final ionic strength, which in most cases was 0.15, corresponding to physiological ionic strength.
Stoichiometries of thrombin inhibition
Stoichiometries of thrombin inhibition were determined as detailed previously (17). Briefly, an increasing amount of antithrombin was added to a fixed thrombin concentration of 0.5 μM in a final volume of 20 μl. Molar ratios of antithrombin/thrombin varied between 0 and 2. The mixture was incubated at 25°C for at least 2 h, until complex formation was complete. Residual thrombin activity was determined by diluting the incubation mixture 1:200 into 110 μM of the thrombin substrate S-2238 (Haemochrom Diagnostica AB, Mölndal, Sweden). Substrate hydrolysis was recorded in a dual beam Hitachi U-2000 spectrophotometer at 405 nm. The residual thrombin activity was plotted against the molar ratio of antithrombin/thrombin, and the stoichiometry of inhibition was determined by linear regression from the abscissa intercept. Native antithrombin had a stoichiometry of inhibition indistinguishable from one and latent and cleaved antithrombin were inactive.
Fluorescence titrations
Equilibrium binding was studied by fluorescence titrations, monitored either by the tryptophan fluorescence change accompanying the interaction, or the fluorescence change obtained in the presence of a fluorescent probe, TNS (Invitrogen). TNS binds weakly to antithrombin and the fluorescence of the TNS-bound antithrombin complex decreases upon heparin binding (24), which is useful for assessing certain antithrombin-heparin interactions that are not accompanied by changes in tryptophan fluorescence. The change in TNS-bound antithrombin fluorescence induced by heparin binding was experimentally more difficult to determine than the tryptophan fluorescence, because we found that i) the TNS-antithrombin fluorescence decreased exponentially with time during the first hour after mixing, presumably due to a slow isomerization of the bound probe, and ii) high heparin concentrations caused a fluorescence change with TNS even in the absence of antithrombin. TNS fluorescence was thus the optimal choice mainly for studies of the interactions of pentasaccharide and HAH with cleaved antithrombin, as the binding of the two saccharides to latent antithrombin caused a larger tryptophan but smaller TNS fluorescence change than the binding to cleaved antithrombin. In other cases tryptophan fluorescence was the preferred method. In TNS titrations, antithrombin and TNS were mixed 30-45 minutes before the titration, and the titrations were subtracted by blank titrations of buffer into TNS-bound antithrombin. Heparin concentrations used in these titrations minimally affected the reported changes in TNS fluorescence. The titrations were done in an SLM 4800S spectrofluorimeter (SLM instruments, Rochester, NY). Excitation and emission wavelengths of 280 and 336-340 nm, respectively, and excitation and emission bandwidths of 2 and 16 nm, respectively, were used for tryptophan fluorescence measurements. For TNS titrations, excitation and emission wavelengths were 326 and 455 nm, and excitation and emission bandwidths were 4 and 16 nm, respectively.
Stoichiometric titrations for the binding of pentasaccharide to latent antithrombin were done at 20 μM antithrombin and were based on the tryptophan fluorescence change. Affinity titrations, based on either tryptophan or TNS fluorescence change, were done at 100-500 nM native antithrombin or 0.5-5 μM latent and cleaved antithrombin. The titrations in the presence of TNS were usually conducted in 10-20 μM and, in some cases, 40 μM TNS. Binding stoichiometries and dissociation equilibrium constants, KD, were determined by fitting the data to the equilibrium binding equation by nonlinear least-squares analysis, as described before (17). The ionic strength dependence for the interactions between the saccharides and the antithrombin forms was determined by measuring KD values at different sodium ion concentrations, as described previously (25).
TNS fluorescence spectra
The effects of the interactions of the normal pentasaccharide, LAH and HAH with the antithrombin forms on the fluorescence of AT-bound TNS were determined from emission spectra measured from λem 380 to 530 nm at λex 326 nm. The excitation and emission bandwidths were 5 and 3 nm, respectively. Spectra were run for free TNS, TNS + the heparin forms, TNS + the antithrombin variants and TNS + the antithrombin variants + the heparin forms. The fluorescence change at 455 nm induced by heparin was expressed relative to that of the antithrombin-bound TNS after subtraction of the effect of heparin on the fluorescence of free TNS.
Rapid kinetics of pentasaccharide binding to the antithrombin forms
Rapid kinetics of normal pentasaccharide binding to native and latent antithrombin were measured by monitoring the change in tryptophan fluorescence accompanying the interaction in an SX-17MV stopped-flow instrument (Applied Photophysics, Leatherhead, UK), essentially as described previously (26), at λex 280 nm and with the use of a 330 nm emission bandpass filter. The experiments were conducted at ionic strength 0.15, pH 7.4 and 10°C. The low temperature was introduced to enhance the small fluorescence signal for the interaction of the pentasaccharide with latent antithrombin. Kinetics for normal pentasaccharide binding to cleaved antithrombin was monitored under the same conditions as the binding to native and latent antithrombin, except that the change in TNS fluorescence, monitored at λex 326 nm and with an emission 420 nm cut-off filter, was analysed. No background TNS fluorescence change was observed under these conditions during the time-frame of the fluorescence traces. Pseudo-first order conditions were arranged, with the pentasaccharide concentration in at least a 5-fold and, in most cases a 10-fold, ratio to the protein concentration. Observed pseudo-first order rate constants, kobs, were obtained by least-squares fitting of the fluorescence traces to a single-exponential function by nonlinear regression. Each kobs value reported represents the average ± SEM of 16-37 traces.
Affinity matrix interaction studies
Interactions of the normal pentasaccharide, LAH and HAH with the antithrombin forms were further studied by an affinity matrix method detailed previously (27). This method is based on the binding of the antithrombin forms to a heparin matrix (heparin Sepharose) and the displacement of antithrombin from the matrix by heparin competitors. Control binding experiments were first done by measuring the amount of antithrombin bound to the matrix as a function of the total antithrombin concentration. To this end, increasing concentrations of each antithrombin form were incubated during shaking in the presence (or absence for unbound control) of 50 μl heparin Sepharose in 500 μl final volume for 2 hours at room temperature. The samples were then centrifuged and antithrombin concentration in the supernatant determined. Competitive binding experiments were conducted similarly but with a fixed antithrombin concentration of 200 nM and increasing concentrations of heparin forms. The unbound antithrombin concentration was determined by fluorescence measurements at λex 280 nm and λem 340 nm after addition of 3M NaCl and polybrene to the supernatant to avoid heparin effects on the antithrombin fluorescence. Fluorescence values were corrected for the dilution. The following equation was used for fitting the displacement data (27):
| (Eq. 1) |
where
In this equation A is antithrombin, B is heparin competitor, [A]T and [B]T represent total concentrations of A and B, n is the stoichiometry of the competitor interaction, KAB is the KD for antithrombin binding to competitor heparin in solution and C3 is the slope of the control curve for binding of antithrombin to the matrix. [A]b,X is the concentration of antithrombin bound to the matrix and [A]b,B, i.e. the concentration of antithrombin bound to the heparin competitor, is given above. C3 was used as a fitted parameter to avoid the effects of different binding efficiencies between different experiments. A stoichiometry, n, of 1 was assumed for all interactions.
RESULTS
Changes in tryptophan and TNS fluorescence induced by the interactions of heparins with the antithrombin forms
The different heparin saccharides were titrated into a solution of protein or protein-TNS and the resulting change in tryptophan- or TNS fluorescence was monitored. The titrations were conducted up to saccharide concentrations at which saturation of the proteins was approached. The data were fit well by the equilibrium binding equation, as shown for the interactions of HAH with the antithrombin forms in Fig.1. The changes in tryptophan fluorescence for the binding of the saccharides to latent antithrombin at saturation were considerably smaller than for the binding to native antithrombin (Table 1). Even smaller changes in tryptophan fluorescence were observed for the binding of the saccharides to cleaved antithrombin. Of all the saccharides, LAH caused the smallest tryptophan fluorescence change for the binding to all antithrombin forms.
Fig. 1. Equilibrium binding, studied by fluorescence titrations, for the interactions of high-affinity heparin with native, latent and cleaved antithrombin.

The changes in tryptophan fluorescence are shown for the interactions of high-affinity heparin with native, latent and cleaved antithrombin (A) and the changes in TNS fluorescence for the interactions of high-affinity heparin with native, latent and cleaved antithrombin (B). The protein concentrations used for the titrations were 0.5 μM native antithrombin, 3.5 μM latent antithrombin and 2 μM cleaved antithrombin in (A) and 0.2 μM native antithrombin, 1 μM latent antithrombin and 1.8 μM cleaved antithrombin in (B).●, native antithrombin; ○, latent antithrombin; □, cleaved antithrombin.
TABLE 1. Changes in tryptophan and TNS fluorescence obtained by LAH, HAH and normal pentasaccharide binding to native, latent and cleaved antithrombin (AT).
The changes in tryptophan fluorescence were obtained from the ΔFmax values derived from computer-fits of stoichiometric titration data to the equilibrium binding equation. The changes in TNS fluorescence were obtained from emission spectra recorded at λex 326 nm, as described in Experimental Procedures. The negative changes in TNS fluorescence values at λem 455 nm are reported
| Fluorescence change (%) | ||||
|---|---|---|---|---|
| AT form | Fluorescence form | Normal pentasaccharide | HAH | LAH |
| Native | Tryptophan | 40 | 43 | 8 |
| TNS | 28 | 51 | 0 | |
| Latent | Tryptophan | 6 | 6 | 2 |
| TNS | 24 | 32 | 0 | |
| Cleaved | Tryptophan | 3 | 3 | <1 |
| TNS | 66 | 51 | 0 | |
TNS has previously been used to study certain protein-heparin interactions when the tryptophan fluorescence change is limited (9, 24, 28). Titrating heparin into TNS-bound native antithrombin has previously been shown to result in a decrease of TNS fluorescence (24). Such a decrease was also observed in this work on HAH or pentasaccharide binding to all antithrombin forms studied. In contrast, LAH caused no measurable change in TNS fluorescence, either with native, latent or cleaved antithrombin (Table 1). This low or absent fluorescence change indicates that LAH binds in a different mode than HAH or the pentasaccharides to all these antithrombin forms, as described previously for native antithrombin (29). A different binding mode may result in LAH inducing more limited conformational changes in latent and cleaved antithrombins than HAH from LAH inducing minimal or no conformational change in latent and cleaved antithrombins.
Binding affinities determined by fluorescence
Dissociation equilibrium constants, KD, for the interactions between the saccharides and the antithrombin forms were determined by fluorescence titrations (Table 2). Titration data were computer-fit to the spectroscopic equilibrium binding equation (17). Equimolar binding stoichiometries have previously been reported for the binding of the normal pentasaccharide, the high-affinity pentasaccharide, HAH and LAH to native antithrombin and for the binding of HAH and the high-affinity pentasaccharide to cleaved antithrombin (7, 9, 30, 31). We similarly determined a 1:1 stoichiometry for the binding of the normal pentasaccharide to latent antithrombin. For the remaining saccharide-antithrombin interactions, 1:1 stoichiometries were assumed. Native and latent antithrombin were titrated with the normal pentasaccharide in experiments monitored by both tryptophan and TNS fluorescence, which gave similar KD values (Table 2). The KD value for the binding of the normal pentasaccharide to native antithrombin, 50 nM, is in agreement with previous studies (32). Affinities of the normal pentasaccharide for latent and cleaved antithrombin were 30-fold lower than those for native antithrombin (Table 2), whereas the affinities of the high-affinity pentasaccharide for latent antithrombin was over 100-fold lower and for cleaved antithrombin about 40-fold lower than for the native protein. The affinities of the normal pentasaccharide were up to two-fold higher than those of HAH for latent and cleaved antithrombin, even though high-affinity heparin contains the pentasaccharide sequence. No KD values could be obtained from the low or absent fluorescence changes observed on binding of LAH to latent and cleaved antithrombin, either in the presence or in the absence of TNS. In two previous studies up to 10-fold higher KD values have been reported for the interactions of pentasacharide or HAH with latent and cleaved antithrombin (7, 33). Our values were, however, verified by three different techniques including the more sensitive TNS binding assay and a more recent study reported KD values similar to those presented here for pentasaccharide interactions with latent and cleaved antithrombin (9).
TABLE 2. Dissociation equilibrium constants, determined by fluorescence titrations, for the interactions of pentasaccharides and HAH with the antithrombin forms at 25°C, pH 7.4 and ionic strength 0.15.
Dissociation equilibrium constants were determined by fluorescence titrations, monitored by tryptophan or TNS fluorescence, as described in Experimental Procedures. The values are the means ± SE of at least three titrations
Ionic and nonionic contributions to the affinity
The ionic and nonionic contributions to the interactions between the pentasaccharides and the antithrombin forms were determined from the dependence of lg KD on lg [Na+], as described before (25, 31, 34) (Fig. 2). The normal pentasaccharide binds to native antithrombin with ∼4 ionic interactions and a nonionic KD value of 63 μM (35). Whereas the high-affinity pentasaccharide also binds to native antithrombin with ∼4 ionic interactions, the nonionic KD value is considerably lower, 0.45 μM (36). Binding of the normal pentasaccharide to latent and cleaved antithrombin was shown to involve two to three less ionic interactions than the binding to native antithrombin, whereas the nonionic affinity was similar (Table 3). The binding of the high-affinity pentasaccharide to latent antithrombin similarly involved two less ionic interactions than the binding to native antithrombin. However, the interaction of the high-affinity pentasaccharide with cleaved antithrombin involved only one less ionic interaction compared with native antithrombin, demonstrating a somewhat different binding mode of this pentasaccharide to cleaved than to latent antithrombin. In contrast to normal pentasaccharide binding, the nonionic affinity was lower (5-10-fold) for the binding of the high-affinity pentasaccharide to latent and cleaved antithrombin than for the binding to the native protein.
Fig. 2. Double-logarithmic plots of the dependence of the KD on [NaCl] for the binding of different heparins to native, latent and cleaved antithrombin.

KD values were determined by fluorescence titrations at different sodium ion concentrations for the binding of normal pentasaccharide (A) or high-affinity pentasaccharide (B) to native antithrombin (●), latent antithrombin (○) and cleaved antithrombin (□). The ionic and nonionic contributions to the interactions were obtained as described in Experimental Procedures. The titrations in Figure A and those of cleaved antithrombin in Figure B were based on TNS fluorescence, whereas all other titrations were based on tryptophan fluorescence. The values are the means ± SE of 2-4 titrations. Error bars that are not shown are hidden by the symbols.
TABLE 3. Ionic and nonionic contributions to the interactions of pentasaccharides with native and latent antithrombin at 25°C and pH 7.4.
The number of ionic interactions, Z, and the nonionic affinities, KD’, were determined from the slopes and y-intercepts, respectively, of double-logarithmic plots of KD versus [Na+] (Fig. 1). The slopes and intercepts ± SE were obtained by linear regression
Rapid kinetics of pentasaccharide binding
The kinetics for the interactions of the normal pentasaccharide with native, latent and cleaved antithrombin were determined from the change in tryptophan or, in the case of the cleaved form, TNS fluorescence, under pseudo-first order conditions at 10°C. This low temperature was used because it was experimentally found to give fluorescence traces with improved signal to noise ratio than at 25°C for the interaction of the pentasaccharide with latent antithrombin, for which the fluorescence change at saturation is only ∼6% (Table 1). This improvement is presumably, at least partially, due to the ∼10-fold increase in affinity at this temperature (Table 4), resulting in a larger proportion of antithrombin-pentasaccharide complex at equilibrium. Observed pseudo-first order rate constants, kobs, were determined at different pentasaccharide concentrations. In the low concentration range, 0-0.3 μM, the dependences of kobs on the pentasaccharide concentration were essentially linear for native and latent antithrombin (Fig. 3a), giving the bimolecular association rate constants, kon, from the slope and the dissociation rate constants, koff, from the y-intercept (Table 4). In the case of cleaved antithrombin, the analyses by TNS fluorescence did not give sufficiently reproducible kobs values in the low pentasaccharide concentration range to allow determination of kon and koff. For native antithrombin, kon at 10°C was similar to the values obtained previously at 25°C whereas koff at 10°C was approximately 15-fold lower (31, 35) (Table 4). There was no observable difference in kon for the interactions of the pentasaccharide with latent and native antithrombin at 10°C (30±4 and 33±1 M-1s-1, respectively), whereas koff was substantially higher for the interaction with latent than for that with native antithrombin (1.5±0.9 and 0.1±0.2 s-1 respectively) (Table 4).
TABLE 4. Kinetic and dissociation equilibrium constants for the interactions of the normal pentasaccharide with native, latent and cleaved antithrombin at 10°C, ionic strength 0.15 and pH 7.4.
Kinetic constants were determined by stopped-flow fluorimetry, monitoring the change in tryptophan or TNS fluorescence induced by pentasaccharide binding, as described in Experimental Procedures. kon and koff were determined form the slopes and ordinate intercepts of linear plots of observed pseudo-first order rate constants, kobs, versus pentasaccharide concentration (Fig. 3A). kon and koff values ± SE were calculated by linear regression. K1, k+2 and k-2 were determined by fitting the data from the plots of Fig. 3B to the rectangular hyperbolic equation (37). These values ± SE were calculated by nonlinear regression. KD values are the means ± SE of three fluorescence titrations conducted at 10°C as described in Experimental Procedures
| Antithrombin form | kon (106 M-1s-1) | koff (s-1) | k-2 (s-1) | K1 (μM) | k+2 (s-1) | Kd (nM) |
|---|---|---|---|---|---|---|
| Native | 33 ± 1 | 0.1 ± 0.2 | 0.3 ± 0.9 | 6 ± 0.4 | 200 ± 5 | 4 ± 2 |
| Latent | 30 ± 4 | 1.5 ± 0.9 | 3 ± 1 | 9 ± 1 | 200 ± 15 | 150 ± 50 |
| Cleaved | 14 ± 6a | 2 ± 2 b | 9 ± 2 | 14 ± 4 | 200 ± 30 | 160 ± 90 |
Calculated from K1 and k+2
Calculated from K1, k+2 and KD
Fig. 3. Rapid kinetics for the interactions of the normal pentasaccharide with native, latent and cleaved antithrombin.

Observed rate constants, kobs, were determined under pseudo-first-order conditions at 10°C, ionic strength 0.15 and pH 7.4 in the low (A) and high (B) concentration ranges, as described in Experimental Procedures. Each kobs value represents the average of 16-37 fluorescence traces. ●, Native antithrombin; ○, Latent antithrombin; □, Cleaved antithrombin.
The dependence of kobs for the interactions with native, latent and cleaved antithrombin on the pentasaccharide concentration was extended to higher such concentrations. The resulting curves were hyperbolic and could be well fit to the rectangular hyperbolic equation described previously (31, 37) (Fig. 3B). This behavior indicates that the interactions of the pentasaccharide with latent and cleaved antithrombin are accompanied by a conformational change, similarly to native antithrombin (31), according to the two-step mechanism described in scheme 1.
Scheme 1.
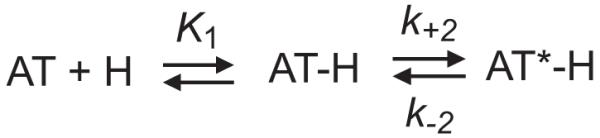
In this scheme, H is HAH or pentasaccharide, K1 is the dissociation equilibrium constant for the first binding step, k+2 is the forward rate constant for the second, conformational change step, and k-2 is the reverse rate constant for the second binding step. The absence of any detectable lag phase for the interaction of the pentasaccharide with latent and cleaved antithrombin over the pentasaccharide concentration range studied is in agreement with a rapid equilibrium being established in the first binding step, as described previously for the interactions of HAH and pentasaccharide with native antithrombin at 25°C (31, 37). The kinetic constants characterizing the two-step mechanism for pentasaccharide binding to native antithrombin at 10°C differed from those determined previously at 25°C. The K1 and k+2 values were 6±0.5 μM and 200±5 s-1 at 10°C, compared with 22±2 μM and 750±50 s-1 at 25°C. The first binding step is thus 3-4-fold tighter, whereas the forward rate constant of the second binding step is 3-fold lower at the lower temperature. K1 was somewhat higher for the interaction of the pentasaccharide with latent antithrombin and twice as high for the interaction with cleaved antithrombin than for that with native antithrombin, whereas k+2 was indistinguishable for the interactions of the pentasaccharide with the three antithrombin forms (Table 4). For cleaved antithrombin, the experimentally determined values of K1 and k+2 were used to calculate kon and subsequently koff. Although there was a small decrease in kon, the values show that the major factor causing the decreased affinity of the normal pentasaccharide for cleaved than for native antithrombin was a substantial increase in koff, as in the case of latent antithrombin.
Affinity matrix results
Due to the inadequacy of fluorescence techniques for quantifying the interactions of LAH with latent and cleaved antithrombin, an affinity matrix method was additionally used (27). Control experiments verified that the amount of antithrombin bound to the heparin-Sepharose gel increased linearly with the total antithrombin concentration up to 500 nM for the three antithrombin forms (Fig. 4A). Under such linear conditions, the equation used for fitting the binding data in the presence of heparin competitor reduces to a quadratic equation (Eq. 1) (27). In the absence of heparin competitor, more native than latent or cleaved antithrombin bound to the matrix, as expected from the affinity difference (Fig. 4A). Furthermore, less latent than cleaved antithrombin was bound, in agreement with affinities determined by fluorescence titrations (Table 2).
Fig. 4. Binding of native, latent and cleaved antithrombin to a heparin matrix in the absence or presence of heparin competitors.

Binding was first studied as a function of increasing total antithrombin concentrations in the absence of heparin competitors (A). The displacement of antithrombin by increasing amounts of heparin competitors was then analysed for native (B), latent (C) and cleaved (D) antithrombin forms. The solid line in (A) represents linear regression fitting. The solid lines in (B-C) represent nonlinear regression fitting to Eq. 1, as described in Experimental Procedures. ○, native antithrombin; □, cleaved antithrombin; △, latent antithrombin; ◆, LAH; ■, HAH; ●, normal pentasaccharide.
Heparin competitors caused the displacement of bound antithrombin from the matrix in a concentration-dependent manner (Fig. 4B-C). The data for the binding of all heparin forms to all three antithrombin variants were readily fit by Eq. 1 (solid lines of Fig. 4B-C), confirming that all heparin forms bind to the same or overlapping sites of all antithrombin forms, which has previously been shown only for native antithrombin (29, 38). Affinities obtained by analysis of the binding of the three types of heparin to the three antithrombin forms are shown in Table 5. KD values were difficult to determine for competitor heparin interactions in cases where antithrombin was tightly bound to the matrix, as for native antithrombin, due to the large excess of antithrombin-binding sites on the matrix and, thus, low amount of unbound antithrombin. This resulted in large errors in the KD values and deviation from the values obtained by fluorescence titrations. In contrast, the KD values for the interactions of the normal pentasaccharide and HAH with latent and cleaved antithrombin were well determined and similar to the values obtained by fluorescence titrations (Table 2), verifying the validity of this method. These values also confirmed the finding obtained by fluorescence titrations that the pentasaccharide binds with approximately two-fold higher affinities than HAH to latent and cleaved antithrombin. With this method the affinities of LAH for latent and cleaved antithrombin could also be determined and were clearly found to be weaker than the affinities of the normal pentasaccharide and HAH for these antithrombin forms. However, the difference in affinities of low- and high-affinity heparins for latent and cleaved antithrombin was considerably smaller than the 1000-fold difference in affinity of the two heparin forms for native antithrombin (29, 30).
TABLE 5. Dissociation equilibrium constants determined by the affinity matrix method for the interactions of the normal pentasaccharide and high- and low-affinity heparins with native, latent and cleaved antithrombin.
The antithrombin forms were bound to a heparin-Sepharose matrix and the displacement from this matrix by increasing pentasaccharide or heparin concentrations was analysed, as described in Experimental Procedures. 26-saccharide heparins were used. KD values ± SE were determined by nonlinear regression of the binding data of Fig. 3B-C to Eq. 1
| KD (nM) | |||
|---|---|---|---|
| Heparin form | Native AT | Latent AT | Cleaved AT |
| Normal pentasaccharide | 450±160 | 1100±350 | 1200±120 |
| HAH | 59±10 | 2700±730 | 2100±400 |
| LAH | 4400±1500 | 8400±2900 | 17000±2700 |
DISCUSSION
In this study we have characterized the interactions of various heparin and pentasaccharide forms with latent and cleaved antithrombin. Rapid kinetics experiments provided evidence for a similar two-step mechanism for the interaction of the normal pentasaccharide with latent and cleaved as with native antithrombin, i.e. an initial weak interaction followed by a conformational change. The considerably lower tryptophan fluorescence changes observed in this study for normal pentasaccharide binding to latent and cleaved than to native antithrombin, reflects that the latent and cleaved forms were unable to undergo the same pentasaccharide-induced conformational change as native antithrombin. This finding is strengthened by the crystal structure of pentasaccharide-bound native and latent antithrombins (39) (Fig. 5), which reveals for instance that pentasaccharide binding induces the formation of a 1.5 turn α-helix, called the P-helix, in both antithrombin forms. In contrast, the pentasaccharide-induced 2-turn elongation of the D-helix and the expulsion of the RCL from sheet A occur in native but not in latent antithrombin.
Fig. 5. Close-up of the pentasaccharide-binding site of native and latent antithrombin forms.

The image shows a close-up of the D- and P-helices and parts of the N-terminal region and the A-helix of the heparin-binding site of native antithrombin (A) and latent antithrombin (B) in complex with a high-affinity pentasaccharide (large pictures; PDB code 1e03) and in the unbound state (inset pictures; PDB code 1e05). A ribbon presentation of the selected parts of the protein backbone is shown in grey. The amino acid side chains of the residues of the N-terminus and the A- D- and P-helices that are known to participate in the interaction of pentasaccharide with native antithrombin are shown. Additionally, the amino acid side chains of Arg132 and Lys133, forming part of the extended heparin binding site of native antithrombin, and Tyr131, are shown. Carbon atoms of amino acid side-chains are drawn in black, nitrogen atoms in blue and oxygen atoms in red. The pentasaccharide is drawn in green. The images were produced in swiss PDB viewer.
The 20-30-fold lower affinities of the normal pentasaccharide for latent and cleaved than for native antithrombin were fully accounted for by 2 and 3 less ionic interactions, respectively, whereas the nonionic contributions to the binding were essentially the same. The decreased affinity was almost completely due to a decrease in koff, whereas K1 was only slightly decreased and k+2 was unchanged. As k+2 values for heparin-antithrombin interactions are independent on ionic strength (31, 36, 37), these findings imply that all the ionic interactions for the binding of the normal pentasaccharide to latent and cleaved antithrombin are made in the first binding step. In fact, the number of ionic interactions made in the first step of the normal pentasaccharide binding to native antithrombin is similar to what was observed here for the overall interaction of this pentasaccharide with latent and cleaved antithrombin. The higher koff for the interaction of the pentasaccharide with latent and cleaved antithrombin thus appears to result from only nonionic interactions, without any ionic contributions, being established in the conformational activation step. N-O binding distances between Arg129, Lys125 and Arg46 and the interacting functional groups of the pentasacharide in PDB structure 1e03 were longer in latent than in native antithrombin, which may explain the reduced number of ionic interactions in the pentasaccharide-bound latent form. The formation of salt bridges between these residues of active antithrombin and the normal pentasaccharide has been verified in the antithrombin-S195A factor Xa - pentasaccharide complex structure (40). The lower extent of pentasaccharide-induced changes in the positions of the pentasaccharide-binding basic residues in latent than in native antithrombin (Fig. 5) thus appears to be due to a reduced number of ionic interactions stabilizing the activated state. Previous studies have suggested that D-helix elongation is important for the allosteric activation of native antithrombin (41, 42). Interestingly, a Lys133Pro antithrombin variant, whose D-helix cannot be elongated, binds the normal pentasaccharide with similar affinity and kinetics as latent and cleaved antithrombin (41). The compaction of the structure that forces the expulsion of the RCL in native antithrombin is likely driven by exposure of Tyr131 when helix D extends (42). In contrast to native antithrombin, Tyr131 is identically positioned in the free and pentasaccharide-bound forms of latent antithrombin (Fig. 5). Together, these findings suggest that activated antithrombin has higher affinity for the pentasaccharide than antithrombin forms incapable of extending their D helix, due to the greater number of ionic interactions being made in the activated state (31, 35).
The limited fluorescence changes observed when pentasaccharide bound to latent and cleaved relative to native antithrombin can be explained by the locations of the four tryptophans, Trp49, Tr189, Trp225 and Trp307 (43). Trp225 at the end of the third strand of β-sheet A and Trp307 on helix H, contribute most to the pentasaccharide-induced fluorescence change of native antithrombin (43), followed by Trp189 on helix F and Trp49 on the A-helix, close to the heparin-binding site. Superimposing the crystal structures of pentasaccharide-bound (1e05L) and free (1e03L) latent antithrombin showed that the position of Trp49 was most affected by pentasaccharide binding, followed by Trp189, whereas Trp225 and Trp307 were similarly positioned in the bound and free states. The lower florescence change observed for pentasaccharide binding to latent than to native antithrombin thus supports the conclusion that the conformational change in latent antithrombin occurs only at or around the pentasaccharide binding site. The similar affinity, kinetics and changes in tryptophan fluorescence observed for the binding of the normal pentasaccharide to cleaved as to latent antithrombin, indicate that the pentasaccharide-induced conformational changes are similar for these antithrombin forms. Differences in changes in TNS fluorescence induced by pentasaccharide binding to latent as compared to cleaved antithrombin are presumably due to TNS binding differently to these antithrombin forms. The finding that the normal pentasaccharide and high-affinity heparin, but not low-affinity heparin, caused a change in TNS fluorescence with latent and cleaved antithrombin suggests that the change in TNS fluorescence, like the tryptophan fluorescence change, reports the conformational change.
The similar fluorescence changes and affinities observed for HAH as for the normal pentasaccharide interacting with latent and cleaved antithrombin indicate that HAH binds to these antithrombin forms through the same pentasaccharide sequence that binds to native antithrombin. Moreover, the lack of fluorescence changes and the low affinities for the interactions of LAH with latent and cleaved antithrombin also support a specificity of latent and cleaved antithrombin for the pentasaccharide sequence, found only in HAH. LAH preparations are reported to have highly similar structures as HAH with the important difference that LAH lacks the 3-O-S group in unit F of the anticoagulant pentasaccharide sequence (44, 45). This 3-O-S group thus appears to enhance the affinity not only for native but also for latent and cleaved antithrombin. The 20- and 100-fold tighter binding of the high-affinity- than of the normal pentasaccharide to latent and cleaved antithrombin, respectively, indicates that the addition of a 3-O-S group in unit H further enhances the affinity for latent and cleaved antithrombin. This finding is supported by a previous study using a structurally different high-affinity pentasaccharide that also contains a 3-O-S group in unit H (9), although it should be noted that both these high-affinity pentasaccharides contain additional structural differences from the normal pentasaccharide that are not believed to be important for antithrombin binding. The higher affinities of the normal pentasaccharide than HAH for latent and cleaved antithrombin, although HAH contains this pentasaccharide sequence, demonstrate that the residues outside the pentasaccharide sequence of HAH that are attracted by native antithrombin (31) are repelled by latent and cleaved antithrombin. The glycan at Asn135 may contribute to this repulsive effect, like it also may do in the first step of binding of HAH to native antithrombin (31, 35). Alternatively, the repulsive effect could be due to acidic residues outside the pentasaccharide-binding site of latent and cleaved antithrombin. In either case, it is likely to occur towards the upper pole of the serpin (in a classical serpin orientation).
The findings presented in this work suggest that the pentasaccharide sequence may be a common site in heparin for mediating both anticoagulant activities of native antithrombin and antiangiogenic activities of latent and cleaved antithrombin forms. The restriction of antiangiogenic properties to latent, cleaved and prelatent antithrombin forms is thus presumably caused by these variants expressing an antiangiogenic epitope which is hidden in native antithrombin but becomes exposed in loop inserted forms, as proposed previously for prelatent antithrombin (5). This epitope is required for antithrombin to bind to a specific endothelial cell receptor and heparan sulfate coreceptor and thereby mediate antithrombin’s antiangiogenic function (9). The suggestion that anticoagulant heparan sulfates also mediate FGF-FGFR interactions (46) suggests that the specific binding of antiangiogenic antithrombin to anticoagulant heparan sulfate may serve not only to promote binding to a specific receptor but also to antagonize growth factor-receptor interactions. Future studies will be required to verify such a mechanism.
ACKNOWLEDGEMENTS
The authors thank Professor Ingemar Björk for helpful comments and critical reading of the manuscript.
Footnotes
This work was supported by the Swedish Research Council grants 2003-6107 and 2005-6412 and by the Magnus Bergvall foundation (to S.S.W.) and by NIH grant HL-39888 (to S.T.O.).
- AT
- antithrombin
- FGF
- fibroblast growth factor
- FGFR
- FGF receptor
- HAH
- high affinity heparin
- LAH
- low affinity heparin
- RCL
- reactive center loop
- SDS
- sodium dodecyl sulphate
- TNS
- 2-(p-toluidinyl)naphtalene-6-sulfonic acid
- VEGF
- vascular endothelial growth factor
- PAGE
- polyacrylamide gel electrophoresis
REFERENCES
- 1.O’Reilly MS, Pirie-Shepherd S, Lane WS, Folkman J. Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science. 1999;285:1926–1928. doi: 10.1126/science.285.5435.1926. [DOI] [PubMed] [Google Scholar]
- 2.Larsson H, Sjoblom T, Dixelius J, Ostman A, Ylinenjarvi K, Björk I, Claesson-Welsh L. Antiangiogenic effects of latent antithrombin through perturbed cell-matrix interactions and apoptosis of endothelial cells. Cancer Res. 2000;60:6723–6729. [PubMed] [Google Scholar]
- 3.Larsson H, Akerud P, Nordling K, Raub-Segall E, Claesson-Welsh L, Björk I. A novel anti-angiogenic form of antithrombin with retained proteinase binding ability and heparin affinity. J. Biol. Chem. 2001;276:11996–12002. doi: 10.1074/jbc.M010170200. [DOI] [PubMed] [Google Scholar]
- 4.Zhang W, Chuang YJ, Swanson R, Li J, Seo K, Leung L, Lau LF, Olson ST. Antiangiogenic antithrombin down-regulates the expression of the proangiogenic heparan sulfate proteoglycan, perlecan, in endothelial cells. Blood. 2004;103:1185–1191. doi: 10.1182/blood-2003-08-2920. [DOI] [PubMed] [Google Scholar]
- 5.Richard B, Swanson R, Schedin-Weiss S, Ramirez B, Izaguirre G, Gettins PG, Olson ST. Characterization of the conformational alterations, reduced anticoagulant activity, and enhanced antiangiogenic activity of prelatent antithrombin. J. Biol. Chem. 2008;283:14417–14429. doi: 10.1074/jbc.M710327200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wardell MR, Chang WS, Bruce D, Skinner R, Lesk AM, Carrell RW. Preparative induction and characterization of L-antithrombin: a structural homologue of latent plasminogen activator inhibitor-1. Biochemistry. 1997;36:13133–13142. doi: 10.1021/bi970664u. [DOI] [PubMed] [Google Scholar]
- 7.Björk I, Fish WW. Production in vitro and properties of a modified form of bovine antithrombin, cleaved at the active site by thrombin. J. Biol. Chem. 1982;257:9487–9493. [PubMed] [Google Scholar]
- 8.Richard B, Swanson R, Schedin-Weiss S, Ramirez B, Izaguirre G, Gettins PG, Olson ST. Characterization of the conformational alterations, reduced anticoagulant activity and enhanced antiangiogenic activity of prelatent antithrombin. J. Biol. Chem. 2008 doi: 10.1074/jbc.M710327200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang W, Swanson R, Izaguirre G, Xiong Y, Lau LF, Olson ST. The heparin-binding site of antithrombin is crucial for antiangiogenic activity. Blood. 2005;106:1621–1628. doi: 10.1182/blood-2005-02-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang W, Chuang YJ, Jin T, Swanson R, Xiong Y, Leung L, Olson ST. Antiangiogenic antithrombin induces global changes in the gene expression profile of endothelial cells. Cancer Res. 2006;66:5047–5055. doi: 10.1158/0008-5472.CAN-05-4449. [DOI] [PubMed] [Google Scholar]
- 11.Olson ST, Chuang YJ. Heparin activates antithrombin anticoagulant function by generating new interaction sites (exosites) for blood clotting proteinases. Trends Cardiovasc. Med. 2002;12:331–338. doi: 10.1016/s1050-1738(02)00183-4. [DOI] [PubMed] [Google Scholar]
- 12.Petitou M, Casu B, Lindahl U. 1976-1983, a critical period in the history of heparin: the discovery of the antithrombin binding site. Biochimie. 2003;85:83–89. doi: 10.1016/s0300-9084(03)00078-6. [DOI] [PubMed] [Google Scholar]
- 13.Nordenman B, Danielsson Å, Björk I. The binding of low-affinity and high-affinity heparin to antithrombin. Fluorescence studies. Eur. J. Biochem. 1978;90:1–6. doi: 10.1111/j.1432-1033.1978.tb12567.x. [DOI] [PubMed] [Google Scholar]
- 14.Rickles FR. Mechanisms of cancer-induced thrombosis in cancer. Pathophysiol. Haemost. Thromb. 2006;35:103–110. doi: 10.1159/000093551. [DOI] [PubMed] [Google Scholar]
- 15.Peterson CB, Blackburn MN. Isolation and characterization of an antithrombin III variant with reduced carbohydrate content and enhanced heparin binding. J. Biol. Chem. 1985;260:610–615. [PubMed] [Google Scholar]
- 16.Picard V, Ersdal-Badju E, Bock SC. Partial glycosylation of antithrombin III asparagine-135 is caused by the serine in the third position of its N-glycosylation consensus sequence and is responsible for production of the beta-antithrombin III isoform with enhanced heparin affinity. Biochemistry. 1995;34:8433–8440. doi: 10.1021/bi00026a026. [DOI] [PubMed] [Google Scholar]
- 17.Olson ST, Björk I, Shore JD. Kinetic characterization of heparin-catalyzed and uncatalyzed inhibition of blood coagulation proteinases by antithrombin. Methods Enzymol. 1993;222:525–559. doi: 10.1016/0076-6879(93)22033-c. [DOI] [PubMed] [Google Scholar]
- 18.Chang WS, Wardell MR, Lomas DA, Carrell RW. Probing serpin reactive-loop conformations by proteolytic cleavage. Biochem. J. 1996;314(Pt 2):647–653. doi: 10.1042/bj3140647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arocas V, Bock SC, Raja S, Olson ST, Björk I. Lysine 114 of antithrombin is of crucial importance for the affinity and kinetics of heparin pentasaccharide binding. J. Biol. Chem. 2001;276:43809–43817. doi: 10.1074/jbc.M105294200. [DOI] [PubMed] [Google Scholar]
- 20.Zhou A, Huntington JA, Carrell RW. Formation of the antithrombin heterodimer in vivo and the onset of thrombosis. Blood. 1999;94:3388–3396. [PubMed] [Google Scholar]
- 21.Olson ST. Heparin and ionic strength-dependent conversion of antithrombin III from an inhibitor to a substrate of alpha-thrombin. J. Biol. Chem. 1985;260:10153–10160. [PubMed] [Google Scholar]
- 22.Nordenman B, Nystrom C, Björk I. The size and shape of human and bovine antithrombin III. Eur. J. Biochem. 1977;78:195–203. doi: 10.1111/j.1432-1033.1977.tb11730.x. [DOI] [PubMed] [Google Scholar]
- 23.Petitou M, van Boeckel CA. A synthetic antithrombin III binding pentasaccharide is now a drug! What comes next? Angew. Chem. Int. Ed. Engl. 2004;43:3118–3133. doi: 10.1002/anie.200300640. [DOI] [PubMed] [Google Scholar]
- 24.Meagher JL, Olson ST, Gettins PG. Critical role of the linker region between helix D and strand 2A in heparin activation of antithrombin. J. Biol. Chem. 2000;275:2698–2704. doi: 10.1074/jbc.275.4.2698. [DOI] [PubMed] [Google Scholar]
- 25.Olson ST, Björk I. Predominant contribution of surface approximation to the mechanism of heparin acceleration of the antithrombin-thrombin reaction. Elucidation from salt concentration effects. J. Biol. Chem. 1991;266:6353–6364. [PubMed] [Google Scholar]
- 26.Schedin-Weiss S, Desai UR, Bock SC, Gettins PG, Olson ST, Björk I. Importance of lysine 125 for heparin binding and activation of antithrombin. Biochemistry. 2002;41:4779–4788. doi: 10.1021/bi012163l. [DOI] [PubMed] [Google Scholar]
- 27.Olson ST, Bock PE, Sheffer R. Quantitative evaluation of solution equilibrium binding interactions by affinity partitioning: application to specific and nonspecific protein-heparin interactions. Arch. Biochem. Biophys. 1991;286:533–545. doi: 10.1016/0003-9861(91)90076-u. [DOI] [PubMed] [Google Scholar]
- 28.O’Keeffe D, Olson ST, Gasiunas N, Gallagher J, Baglin TP, Huntington JA. The heparin binding properties of heparin cofactor II suggest an antithrombin-like activation mechanism. J. Biol. Chem. 2004;279:50267–50273. doi: 10.1074/jbc.M408774200. [DOI] [PubMed] [Google Scholar]
- 29.Streusand VJ, Björk I, Gettins PG, Petitou M, Olson ST. Mechanism of acceleration of antithrombin-proteinase reactions by low affinity heparin. Role of the antithrombin binding pentasaccharide in heparin rate enhancement. J. Biol. Chem. 1995;270:9043–9051. doi: 10.1074/jbc.270.16.9043. [DOI] [PubMed] [Google Scholar]
- 30.Nordenman B, Björk I. Binding of low-affinity and high-affinity heparin to antithrombin. Ultraviolet difference spectroscopy and circular dichroism studies. Biochemistry. 1978;17:3339–3344. doi: 10.1021/bi00609a026. [DOI] [PubMed] [Google Scholar]
- 31.Olson ST, Björk I, Sheffer R, Craig PA, Shore JD, Choay J. Role of the antithrombin-binding pentasaccharide in heparin acceleration of antithrombin-proteinase reactions. Resolution of the antithrombin conformational change contribution to heparin rate enhancement. J. Biol. Chem. 1992;267:12528–12538. [PubMed] [Google Scholar]
- 32.Olson ST, Swanson R, Raub-Segall E, Bedsted T, Sadri M, Petitou M, Herault JP, Herbert JM, Björk I. Accelerating ability of synthetic oligosaccharides on antithrombin inhibition of proteinases of the clotting and fibrinolytic systems. Comparison with heparin and low-molecular-weight heparin. Thromb. Haemost. 2004;92:929–939. doi: 10.1160/TH04-06-0384. [DOI] [PubMed] [Google Scholar]
- 33.Skinner R, Chang WS, Jin L, Pei X, Huntington JA, Abrahams JP, Carrell RW, Lomas DA. Implications for function and therapy of a 2.9 A structure of binary-complexed antithrombin. J. Mol. Biol. 1998;283:9–14. doi: 10.1006/jmbi.1998.2083. [DOI] [PubMed] [Google Scholar]
- 34.Record MT, Jr., Lohman ML, De Haseth P. Ion effects on ligand-nucleic acid interactions. J. Mol. Biol. 1976;107:145–158. doi: 10.1016/s0022-2836(76)80023-x. [DOI] [PubMed] [Google Scholar]
- 35.Turk B, Brieditis I, Bock SC, Olson ST, Björk I. The oligosaccharide side chain on Asn-135 of alpha-antithrombin, absent in beta-antithrombin, decreases the heparin affinity of the inhibitor by affecting the heparin-induced conformational change. Biochemistry. 1997;36:6682–6691. doi: 10.1021/bi9702492. [DOI] [PubMed] [Google Scholar]
- 36.Hjelm R, Schedin-Weiss S. High Affinity Interaction between a Synthetic, Highly Negatively Charged Pentasaccharide and alpha- or beta-Antithrombin Is Predominantly Due to Nonionic Interactions. Biochemistry. 2007;46:3378–3384. doi: 10.1021/bi6024929. [DOI] [PubMed] [Google Scholar]
- 37.Olson ST, Srinivasan KR, Björk I, Shore JD. Binding of high affinity heparin to antithrombin III. Stopped flow kinetic studies of the binding interaction. J. Biol. Chem. 1981;256:11073–11079. [PubMed] [Google Scholar]
- 38.Danielsson Å, Björk I. The binding of low-affinity and high-affinity heparin to antithrombin. Competition for the same binding site on the protein. Eur. J. Biochem. 1978;90:7–12. doi: 10.1111/j.1432-1033.1978.tb12568.x. [DOI] [PubMed] [Google Scholar]
- 39.Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW. The anticoagulant activation of antithrombin by heparin. Proc. Natl. Acad. Sci. U. S. A. 1997;94:14683–14688. doi: 10.1073/pnas.94.26.14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson DJ, Li W, Adams TE, Huntington JA. Antithrombin-S195A factor Xa-heparin structure reveals the allosteric mechanism of antithrombin activation. Embo J. 2006;25:2029–2037. doi: 10.1038/sj.emboj.7601089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Belzar KJ, Zhou A, Carrell RW, Gettins PG, Huntington JA. Helix D elongation and allosteric activation of antithrombin. J. Biol. Chem. 2002;277:8551–8558. doi: 10.1074/jbc.M110807200. [DOI] [PubMed] [Google Scholar]
- 42.dela Cruz RG, Jairajpuri MA, Bock SC. Disruption of a tight cluster surrounding tyrosine 131 in the native conformation of antithrombin III activates it for factor Xa inhibition. J. Biol. Chem. 2006;281:31668–31676. doi: 10.1074/jbc.M604826200. [DOI] [PubMed] [Google Scholar]
- 43.Meagher JL, Beechem JM, Olson ST, Gettins PG. Deconvolution of the fluorescence emission spectrum of human antithrombin and identification of the tryptophan residues that are responsive to heparin binding. J. Biol. Chem. 1998;273:23283–23289. doi: 10.1074/jbc.273.36.23283. [DOI] [PubMed] [Google Scholar]
- 44.Kusche M, Backstrom G, Riesenfeld J, Petitou M, Choay J, Lindahl U. Biosynthesis of heparin. O-sulfation of the antithrombin-binding region. J. Biol. Chem. 1988;263:15474–15484. [PubMed] [Google Scholar]
- 45.Razi N, Lindahl U. Biosynthesis of heparin/heparan sulfate. The D-glucosaminyl 3-O-sulfotransferase reaction: target and inhibitor saccharides. J. Biol. Chem. 1995;270:11267–11275. doi: 10.1074/jbc.270.19.11267. [DOI] [PubMed] [Google Scholar]
- 46.McKeehan WL, Wu X, Kan M. Requirement for anticoagulant heparan sulfate in the fibroblast growth factor receptor complex. J. Biol. Chem. 1999;274:21511–21514. doi: 10.1074/jbc.274.31.21511. [DOI] [PubMed] [Google Scholar]