

Abstract
Sepsis causes over 200,000 deaths yearly in the US; better treatments are urgently needed. Administering bone marrow stromal cells (BMSCs—also known as mesenchymal stem cells) to mice before or shortly after inducing sepsis by cecal ligation and puncture reduced mortality and improved organ function. The beneficial effect of BMSCs was eliminated by macrophage depletion or pretreatment with antibodies specific for interleukin-10 (IL-10) or IL-10 receptor. Monocytes and/or macrophages from septic lungs made more IL-10 when prepared from mice treated with BMSCs versus untreated mice. Lipopolysaccharide (LPS)-stimulated macrophages produced more IL-10 when cultured with BMSCs, but this effect was eliminated if the BMSCs lacked the genes encoding Toll-like receptor 4, myeloid differentiation primary response gene-88, tumor necrosis factor (TNF) receptor-1a or cyclooxygenase-2. Our results suggest that BMSCs (activated by LPS or TNF-α) reprogram macrophages by releasing prostaglandin E2 that acts on the macrophages through the prostaglandin EP2 and EP4 receptors. Because BMSCs have been successfully given to humans and can easily be cultured and might be used without human leukocyte antigen matching, we suggest that cultured, banked human BMSCs may be effective in treating sepsis in high-risk patient groups.
Sepsis, a serious medical condition that affects 18 million people per year worldwide, is characterized by a generalized inflammatory state caused by infection. Widespread activation of inflammation and coagulation pathways progresses to multiple organ dysfunction, collapse of the circulatory system (septic shock) and death. Because as many people die of sepsis annually as from acute myocardial infarction1, a new treatment regimen is desperately needed. In the last few years, it has been discovered that BMSCs are potent modulators of immune responses2-5. We wondered whether such cells could bring the immune response back into balance, thus attenuating the underlying pathophysiology that eventually leads to severe sepsis, septic shock and death6,7.
As a model of sepsis, we chose cecal ligation and puncture (CLP), a procedure that has been used for more than two decades8. This mouse model closely resembles the human disease: it has a focal origin (cecum), is caused by multiple intestinal organisms, and results in septicemia with release of bacterial toxins into the circulation. With no treatment, the majority of the mice die 24−48 h postoperatively.
RESULTS
BMSC treatment improves survival and organ function after CLP
First, we looked at survival rates after CLP in untreated and BMSC-treated mice. There was a statistically significant (P < 0.01) improvement in the survival of mice given 1 million BMSCs intravenously at the time of surgery; 50% of the mice survived until the end of day 4, when all were killed (Fig. 1a). The beneficial effect on survival was seen when the cells were injected 24 h before or 1 h after CLP (Fig. 1a). In contrast, intravenous injection of isolated skin fibroblasts, whole bone marrow or heat-killed BMSCs did not alter survival (Fig. 1a). BMSCs isolated from different strains of mice (C57/BL6, BALB/c or FVB/NJ) all rescued the C57/BL6 mice that we used in our studies (Fig. 1a).
Figure 1.

Effect of intravenous injection of BMSCs on the course of sepsis after CLP. (a) Survival curves of mice after CLP and a variety of treatments using BMSCs from C57/BL6, FVB/NJ and BALB/c mice, as well as C57/BL6-derived fibroblasts. (b) BMSC treatment effects on kidney function, as reflected by serum concentration of creatinine (SCr). The number of mice in all measurements is as follows: sham, n = 5; CLP, n = 13; CLP + BMSC, n = 14. Tubular injury scores are shown at right. (c) Intense PAS staining of hepatocytes is shown after sham operation and BMSC treatment. No staining can be seen in CLP. After treatment (CLP + BMSC), the red staining by PAS in hepatocytes indicates partial glycogen storage capacity. Scale bar, 20 μm. (d) Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) concentrations in the liver after sham and BMSC, CLP or CLP and BMSC treatment. (e) Serum amylase concentrations after sham and BMSC, CLP or CLP and BMSC treatment. (f) DAB staining of caspase-3 cells in untreated spleen sections and BMSC-treated spleen sections. A quantitative comparison between the numbers of apoptotic splenic cells in treated versus untreated mice (right) shows a significant decrease with BMSC treatment. Scale bar, 100 μm. (g) Serum TNF-α and IL-6 concentrations after sham and BMSC, CLP or CLP and BMSC treatment. (h) Serum IL-10 concentrations at 3, 6 and 12 h after CLP. n = 8−11 at each time point. Error bars represent means ± s.e.m.; *P < 0.05; **P < 0.01.
Because the lethality in sepsis is associated with organ failure, we examined the pathology and function of major organs often injured in human subjects. Kidney function, as measured by serum creatinine and renal tubular injury scores, was markedly improved in the treated mice (Fig. 1b). BMSCs reduced organ damage when they were administered up to 24 h before CLP surgery (Supplementary Fig. 1 online). In the livers, improved glycogen storage was observed in treated mice versus control mice (Fig. 1c). Concentrations of liver enzymes (alanine aminotransferase and aspartate aminotransferase) that are released into the circulation upon injury and death of liver cells were significantly decreased in the serum after treatment (Fig. 1d), as were serum amylase values, which mirror pancreatic damage (Fig. 1e). Similarly, there was a significant decrease in the number of apoptotic (activated caspase-3–positive) and necrotic cells in the spleen (Fig. 1f). These results suggest that BMSCs protect infected mice from death and organ damage.
Effect of BMSCs on plasma cytokine concentrations
We hypothesized that BMSCs might alter the immune response to infection; therefore, we studied TNF-α and IL-6, proinflammatory cytokines that have a central role in sepsis1. Twenty-four hours after injection of BMSCs, there was a significant reduction in serum TNF-α and IL-6 concentrations in treated versus untreated mice (Fig. 1g). Serum concentrations of interferon-γ were unaltered by BMSCs (Supplementary Fig. 2 online), but IL-10 abundance started to rise 3 h after the cells were given, almost doubled by the sixth hour and was still elevated 12 h afterward. (Fig. 1h).
To learn where injected BMSCs go and how long they remain detectable, we prelabeled the cells with carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) fluorescent tracking dye and visualized them 1−24 h later. We found BMSCs in the blood up to 1 h after intravenous injections and saw many cells in the lung parenchyma, with some in the spleen and kidney. The labeled cells in the lung seemed to be surrounded by macrophages (Fig. 2a–c). What attracts these cells to each other remains to be determined. The number of cells in the lung gradually decreased over time; few were visible 24 h after they were administered (data not shown).
Figure 2.

Fate of injected BMSCs and effect of BMSC treatment on survival of normal and immune cell–depleted mice. (a–c) Immunohistochemical staining showing that BMSCs prelabeled with Q-dot (red punctate staining; a) travel to the lung (b) and take up residence in close proximity to macrophages (c). The latter cells were immunostained with an antibody to Iba1 (ionized calcium-binding adaptor molecule-1, a specific marker of the macrophage lineage47) and visualized with Alexa-Fluor-488 conjugated to a secondary antibody (green). Scale bar, 10 μm. (d–f) Summary of the effectiveness of BMSC treatment of mice genetically lacking or depleted of certain subsets of immune cells or soluble mediators. Survival curves show survival percentage of macrophage-depleted mice with or without BMSC treatment (d), survival percentage of BMSC-treated CLP mice and untreated mice after neutralizing IL-10 or blocking the IL-10 receptor (e) and survival percentage of after treatment with BMSCs derived from Il10−/− septic mice (f). *P < 0.05.
Alterations in vascular permeability are central to the pathogenesis of sepsis-induced organ injury, and the benefits of BMSC treatment in reducing vascular permeability have already been reported9-12. We studied this in the peritoneum, lung, liver and kidney by measuring Evans blue dye leakage 24 h after CLP. CLP surgery significantly increased peritoneal, liver and renal vascular permeability (P < 0.01, P < 0.01 and P < 0.001, respectively); all three were significantly (P < 0.05, P < 0.05 and P < 0.05, respectively) decreased in mice treated with BMSCs (Supplementary Fig. 3 online).
Involvement of immune cell subtypes in the effect of BMSCs
The observations summarized above suggested that BMSCs might quickly act to reprogram a specific population of cells involved in mediating the immune response. To test this hypothesis, we examined the effects of BMSCs in mice that genetically lack mature T and B cells (Rag2−/−)13 or are depleted of natural killer (NK) cells with a rabbit ganglio-N-tetraosylceramide (asialo GM1)-specific antibody, resulting in nearly complete elimination of NK cell activity14,15. The effect of BMSC injections on the survival of the mice was still present in both of these models, suggesting that lymphocyte populations of T, B and NK cells do not mediate the effect of BMSCs in the CLP model (Supplementary Fig. 4 online). Because BMSCs were found in close proximity to lung macrophages, we asked whether monocytes and/or macrophages are needed for the beneficial effects of the BMSCs. To deplete monocytes and macrophages, we administered clodronate-filled liposomes to the mice16 before we performed the CLP procedure and then treated them with BMSCs. BMSCs were no longer effective in mice lacking monocytes and macrophages (Fig. 2d).
Macrophage-derived IL-10 is key for the effect of BMSCs
Because IL-10 serum levels were increased in CLP mice that were treated with BMSCs as compared to sham controls, we asked whether IL-10 might be important for the actions of BMSCs. To test this hypothesis, we treated mice with an antibody to IL-10 or an antibody to the IL-10 receptor before CLP. In both of these groups, the BMSC injections were ineffective (Fig. 2e), suggesting that IL-10 is a major mediator of the effect. To see whether the injected BMSCs might be producing an essential pool of IL-10, we used BMSCs isolated from Il10−/− mice. These BMSCs were still effective in improving the survival of mice after CLP (Fig. 2f), suggesting that the source of the IL-10 is an endogenous population of cells. Large amounts of IL-10 are known to be produced by subsets of T cells and monocytes and macrophages17. As mentioned earlier (Fig. 2d), monocyte and consequent macrophage depletion eliminated the beneficial effect of BMSCs; thus, we focused on monocytes and macrophages as the probable source of IL-10 required for survival after CLP. BMSC treatment resulted in a significant decrease in the number of circulating monocytes and an increase in the number of circulating neutrophils (Fig. 3a,b). Because tissue macrophages are derived from circulating monocytes, we speculated that the decrease in monocyte numbers might be due to their tissue invasion. We found a significant increase in the number of lung monocytes and macrophages in CLP versus unoperated mice by immunocytochemistry (data not shown). This increase was completely eliminated when circulating monocytes were depleted with clodronate-filled liposomes (Fig. 3c). To confirm the histological findings, we used FACS to determine the relative percentages of immune cells isolated from the lungs of four treated versus four untreated septic mice. There was an increase in monocytes but not lymphoid cells in the lung (Fig. 4a,b). Monocytes and macrophages (CD11b+) were isolated from the lungs of BMSC-treated and untreated mice18, placed in culture and restimulated with LPS. Three and five hours after LPS stimulation ex vivo, monocytes and macrophages from BMSC-treated septic mice produced and released significantly more IL-10 than did untreated mice (Fig. 4c). To determine whether the change in IL-10 production was due to direct interaction between BMSCs and monocytes and macrophages, we cultured the two cell populations together or placed them in a transwell system in which two cell populations were separated from one another by a permeable membrane. In addition, we put monocytes and macrophages in BMSC-conditioned medium. When the macrophages were in direct contact with BMSCs (Fig. 4d), they produced significantly (P < 0.001) more IL-10 in response to LPS stimulation than when they were cultured in transwell plates without direct contact with the BMSCs or exposed to BMSC-conditioned medium. We also used intracellular cytokine staining by FACS to compare the number of IL-10–producing monocytes and macrophages isolated from treated versus untreated septic mice and found a significantly higher number of IL-10–producing monocytes and macrophages in the treated mice (Fig. 4e).
Figure 3.
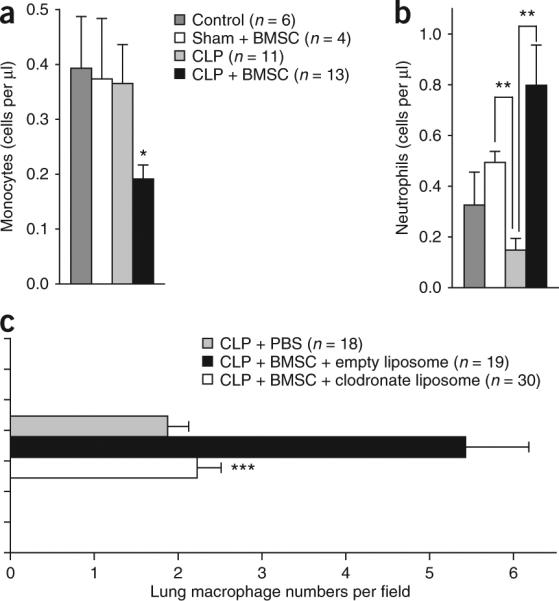
Effect of BMSC treatment on leukocyte trafficking. (a,b) The average number of circulating monocytes (a) and the number of circulating neutrophils (b) after BMSC treatment. The cell counts are the average of data from five mice per group, 24 h after the induction of CLP. (c) The average number of macrophages isolated from lungs of CLP mice with no treatment, with BMSC treatment and BMSC treatment after depletion of circulating monocytes using clodronate. The numbers of macrophages are per random microscope visual field. Five mice were studied in each group. The numbers in parentheses are the total number of fields in which cells were counted. Error bars represent means ± s.e.m. **P < 0.01; ***P < 0.001.
Figure 4.

Characterization of control and stimulated mononuclear cells in CLP mice in vivo and in vitro. (a) The distribution of monocytes (Mon), macrophages (Mac) and polymorphonuclear cells (PMN) among cells isolated from septic lungs. Error bars represent means ± s.e.m. (b) FACS plots of lung mononuclear cells. The cells were first gated on forward scatter (FSC) and CD11b. The R1 group (high FSC, dim CD11b) represents resident macrophages. R2 (high FSC and bright CD11b) represents a group of myeloid cells. R3 (medium FSC and bright CD11b) is a mixed population of monocytes and PMN cells when it is further gated on a monocyte-lineage marker (F4/80) and a PMN marker (GR1). The FACS is a representative example of the four mice shown on the bar graph in a. Blue color indicates monocytes and red indicates polymorphonuclear cells in both a and b expressed as percentage of total number of cells analyzed. (c) Quantification of IL-10 secretion after ex vivo LPS treatment of lung macrophages. Six hours after the induction of CLP, lung macrophages were isolated from mice with or without BMSC treatment (four mice per group), cultured and treated ex vivo with LPS. IL-10 production of these isolated macrophages is shown 3 and 5 h after LPS stimulation. (d) To test whether the increased IL-10 production could be due to a BMSC-macrophage interaction, macrophages from bone marrow were cocultured in vitro with BMSCs and stimulated with LPS. IL-10 production of macrophages cocultured with BMSCs is shown compared to macrophages that had no contact with BMSCs at 1, 3, 5 and 7 h after LPS stimulation. (e) Lungs of four BMSC-treated and four untreated mice were used for a FACS experiment with CD11b and an intracellular marker for IL-10. The number of IL-10 producing monocytes and macrophages after the treatment is shown. (f) Bacterial counts in the peritoneal space and in the circulation in 12 untreated (red) and 10 BMSC treated (blue) mice. (g) Myeloperoxidase (MPO) abundance in the kidney and the liver after BMSC treatment. Error bars represent means ± s.e.m. *P < 0.05, **P < 0.01 and ***P < 0.001.
Because IL-10 has been reported to inhibit the rolling, adhesion19 and transepithelial migration20-22 of neutrophils, we examined the white cell counts in the circulation of treated and untreated CLP mice and found a significant increase in the number of circulating neutrophils in the treated mice (Fig. 3a). It is possible that the high circulating neutrophil counts in the BMSC-treated mice could help lower blood bacterial counts (Fig. 4f), but further studies are necessary to explore this hypothesis. Neutrophils are also known to migrate into the tissues in septic states, where they can cause oxidative organ damage23-25, an unwanted side effect of myeloperoxidase, an enzyme that neutrophils use to eliminate bacteria26. To see whether a change in myeloperoxidase abundance could contribute to BMSC-related organ protection, we measured the amount of myeloperoxidase in the kidney and liver of septic mice with and without BMSC treatment. We found that the amount of myeloperoxidase was significantly decreased (Fig. 4g) in the mice that received BMSC injections, which would be consistent with the lack of neutrophil invasion and the lesser extent of organ damage. The above data suggest that the intravenously injected BMSCs are able to optimally balance circulating and tissue-bound immune cells to maximize bacterial killing in the circulation while minimizing organ damage due to leukocyte invasion.
Molecular basis of the BMSC-macrophage interaction
To understand the molecular basis of the interaction between BMSCs and macrophages, we performed a series of experiments in vitro and in vivo. Wild-type BMSCs showed NF-κB activation 30 min after LPS stimulation (Fig. 5a). In a number of cell types, NF-κB has been shown to induce prostaglandin production and release through a pathway involving cyclooxygenase-2 (COX2; refs. 26-29). BMSCs have been shown to produce prostaglandin E2 and possibly affect other immune cells via EP1–EP4 receptors30,31. We found a significant increase in the expression and activity of COX2 (which produces the substrate for prostaglandin synthase enzymes) in BMSCs 3 h and 5 h after LPS stimulation (Fig. 5b,c), but when the BMSCs were treated with an antibody to TNF-α or collected from Tlr4−/− mice, the COX2 expression did not change (Fig. 5d). This suggested that prostaglandin E2 might indeed be responsible for reprogramming the macrophages. For this reason, we measured the amount of prostaglandin E2 in the coculture medium and found a significant increase in its concentration after LPS stimulation (Fig. 5e). This increase was eliminated if the BMSCs lacked TLR4 or if they were incubated with antibody to TNF-α (Fig. 5e). Inducible nitric oxide synthase (iNOS) inhibition resulted in a significant reduction of COX2 enzyme activity 1 h, 3 h and 5 h after LPS stimulation (Fig. 5f). Although IL-10 concentrations in the medium were increased by LPS when BMSCs from Ptgs1−/− mice (lacking COX1) were used in the assay or when the cells were treated with a COX1 inhibitor (SC-560), the increased IL-10 release was not seen when Ptgs2−/− cells (lacking COX2) were used or a COX2 inhibitor (NS-398) was added (Fig. 6a).
Figure 5.

Studies of molecular alterations underlying the effect of BMSCs on macrophages. (a) NF-κB abundance in nuclei isolated from triplicate samples of BMSCs 30, 60 and 90 min after addition of LPS to the culture medium. (b) Western blot (WB) analysis of COX2 abundance in LPS-stimulated cocultures. Density measurements of three western blots are quantified in the bar graph. (c) The COX2 activity after LPS treatment of cocultures (triplicate samples). (d) Western blot analysis of COX2 abundance in Tlr4−/− (KO) BMSCs or in cultures treated with antibody to TNF-α (anti–TNFα). (e) Prostaglandin E2 (PGE2) abundance in macrophage cultures or coculture supernatants in a variety of conditions 3 and 5 h after LPS stimulation. Four samples were run for each condition. (f) COX2 enzyme activity after iNOS inhibition and 1, 3 and 5 h after LPS stimulation in triplicate samples. Error bars represent means ± s.e.m. *P < 0.05, ***P < .001.
Figure 6.

Summary of studies of the molecular pathways involved in the interaction between BMSC and macrophages. (a) IL-10 concentration changes in supernatants of cocultures in a variety of treatment conditions after LPS stimulation. Colored graphs (except for the blue color that labels the bone marrow macrophages as the source of all consecutive experiments) show treatments that eliminate the effect of BMSCs on macrophages. Black graphs show IL-10 levels after LPS stimulation, whereas open graphs show the control (nonstimulated) values. The experiments where the conditions eliminated the effect are colored. Purple color shows the effect of septic environment, green color shows agents and cellular compartments related to the PGE2 pathway and pink color shows agents related to the nitric oxide pathway. Three separate kinds of macrophages (bone marrow macrophages; peritoneal macrophages and the RAW264.7 cell line) were examined initially. Because they behaved identically in the assay, we used bone marrow derived macrophages (BM MF) for the rest of the experiments. In the box labeled 1, the effect of septic environment on the BMSCs is studied in BMSCs from TLR4-, MyD88-, TNFR1- and TNFR2- deficient mice, or antibody to TNF-α was used to neutralize the effect of TNF. The box labeled with 2 shows the cytokines and agents that have been implicated in the literature in immunomodulation of T cells by BMSCs, including COX1/2 and iNOS inhibitors. The box labeled 3 shows studies of the COX2 pathway, including the prostaglandin receptors EP1–EP4. Finally, in the box labeled with 4, we show studies related to nitric oxide. (b) A summary of our current hypothesis about the mechanisms that underlie the interactions between BMSCs and macrophages in the CLP sepsis model. Bacterial toxins (for example, LPS) and circulating TNF-α act on the TLR4 and TNFR-1 of the BMSCs, respectively. This results in the translocation of NF-κB into the nucleus. This activation process seems to be nitric oxide dependent. Activated NF-κB induces the production of COX2, resulting in increased production and release of PGE2. PGE2 binds to EP2 and EP4 receptors on the macrophage, increasing its IL-10 secretion and reducing inflammation.
To further examine the effect of BMSCs on macrophages, we cultured macrophages with BMSCs, added LPS and measured the IL-10 concentration in the culture medium. Because Toll-like receptor-4 (TLR4) is the receptor to which LPS binds and through which it acts32, we were not surprised to see that BMSCs from Tlr4−/− mice could not increase IL-10 production and secretion in our assay (Fig. 6a). Myeloid differentiation primary response gene-88 (MyD88) is required for activation of nuclear factor-κB (NF-κB) by TLR4, and cells from Myd88−/− mice did not stimulate IL-10 secretion either (Fig. 6a). Because TNF-α has also been implicated in upregulating NF-κB in BMSCs, and both TNF receptor-1 (TNFR-1) and TNFR-2 have been reported to be present in BMSCs33, we next tested whether these receptors are involved in the function. We used antibody to TNF-α and BMSCs from Tnfrsf1a−/− or Tnfrsf1b−/− mice and found that TNF-α and TNFR-1 are also necessary for the activation of BMSCs (Fig. 6a). In the absence of TNFR-2, BMSCs still increased IL-10 concentrations in the cocultures (Fig. 6a).
Because it seemed that prostaglandin E2 might mediate the effect of BMSCs on macrophage cytokine production, we asked whether specific EP receptors could be involved in transducing the prostaglandin signal. To answer this question, we used prostaglandin receptor antagonists and macrophages from EP receptor–knockout mice. EP1 and EP3 receptor antagonists had no effect on LPS-induced IL-10 secretion, and neither did substituting Ptger1−/− macrophages (lacking the gene encoding prostaglandin E receptor-1) or Ptger3−/− macrophages (lacking the gene encoding prostaglandin E receptor-3) for wild-type cells in the assay (Fig. 6a). However, both EP2 and EP4 receptor antagonists prevented the increase in IL-10 secretion, as did using Ptger2−/− or Ptger4−/− macrophages instead of wild-type ones (Fig. 6a). TNFR-1 and TLR4 do not seem to be the only elements in the BMSCs required for COX2 induction. Nitric oxide produced in the BMSCs, the macrophages or both may be involved. Using BMSCs from Nos2−/− mice with WT macrophages or Nos2−/− mice macrophages with WT BMSCs did not eliminate the effect on IL-10 production (Fig. 6a). However, when BMSCs and macrophages were both derived from Nos2−/− mice (lacking iNOS-2), IL-10 was not induced (Fig. 6a). To show that this effect is specifically related to loss of iNOS activity and reduced production of nitric oxide, we added S-nitroso-N-acetylpenicillamine (SNAP), an external nitric oxide donor, to the system34. SNAP fully restored the ability of Nos2−/− cells to increase IL-10 production in response to LPS (Fig. 6a). To determine whether nitric oxide might act through guanylyl cyclase in the BMSCs, we pretreated these cells with 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), an irreversible inhibitor of soluble guanylyl cyclase35 (Fig. 6a). The IL-10 increase was eliminated when the BMSCs, but not the macrophages, were treated with ODQ. This suggests that the BMSCs need nitric oxide to achieve the effect on IL-10. Apparently, the nitric oxide can either be produced by the BMSCs themselves or by the macrophages. Thus, on the basis of our in vitro studies, we concluded that BMSCs respond to the presence of infectious agents by increasing prostaglandin E2 synthesis and secretion, and that subsequent activation of prostaglandin E2 and E4 receptors on the macrophages results in IL-10 induction. We tested this hypothesis in vivo, and, as predicted, BMSCs lacking TNFR1, MyD88 or COX2 all failed to rescue mice subjected to CLP (Supplementary Fig. 5 online). Cells from Tlr4−/− mice had partial efficacy, reflecting the fact that their detection of pathogens is impaired but not absent (Supplementary Fig. 5), raising the possibility of the involvement of other TLRs in vivo.
DISCUSSION
Our results suggest that an intravenous injection of bone marrow stromal cells can beneficially modulate the response of the host immune system to sepsis and improve survival. BMSCs were first reported to have a potent immunosuppressive effect in vivo in humans in 2004, when a young individual with resistant graft-versus-host disease was successfully treated with transplanted haploidentical mesenchymal stem cells2. Subsequently, cases of hemorrhagic cystitis and peritonitis were treated with allogeneic mesenchymal stem cells5. Thus, in humans and in animals3,36-39, bone marrow stromal cells seem to be potent immunomodulators, but their mechanism of action has been unclear. BMSCs are known to inhibit T cell proliferation4 and to modulate B cell function40. In the present study, we show that BMSCs can be used to fight sepsis in mice. We suggest that the injected BMSCs interact with circulating and tissue (mostly lung) monocytes and macrophages and reprogram them. Treated monocytes and macrophages produce large amounts of IL-10, and the treatment decreases the amounts of circulating TNF-α and IL-6. This reduces harm caused by unbridled immune responses to the host tissues. The observation that the macrophages isolated from treated septic mice produce significantly higher amounts of IL-10 than those from nontreated mice suggest a temporary reprogramming of monocyte and macrophage function that persists for at least 12 h. The monocyte and macrophage–derived IL-10 seems to prevent neutrophils from migrating into tissues and causing oxidative damage, thus mitigating multiorgan damage. This inhibition of neutrophil migration into tissues also results in a buildup of neutrophils in the blood, allowing for more efficient bacterial clearance. The combination of our in vitro and in vivo experiments shows that the beneficial effect is likely to be due to an increased release of prostaglandin E2 from the BMSCs acting on the EP2 and EP4 receptors of the macrophages and stimulating the production and release of IL-10, an anti-inflammatory cytokine (Fig. 6b). Simultaneous stimulation of EP2 and EP4 receptor subtypes by prostaglandin E2 has been shown to promote monocyte-derived dendritic cell maturation41. In peritoneal macrophages, prostaglandin E2 was shown to modulate inflammatory reactions via the EP2 and EP4 receptors42, and EP2 and EP4 agonists were shown to cause upregulation of zymosan-induced IL-10 production42. Because both of these receptors work through cyclic AMP, one wonders why both are necessary for the effect to take place. Why eliminating either the EP2 or the EP4 receptor affects IL-10 induction remains to be determined. The two receptors could increase cyclic AMP levels more when they act in concert than they do alone, or they could have partially nonoverlapping actions that are essential for induction to occur43. It is also possible that the receptors form functional heterodimers in macrophages.
Our model is somewhat similar to peritonitis in humans. The mechanism of how BMSCs are beneficial might be universal in infectious diseases, or it might be unique to this model. It is possible that the mechanism described could be harmful in other models or that there are other beneficial mechanisms related to BMSCs in other infectious or noninfectious diseases. Owing to the effectiveness of BMSCs in the CLP model, and because BMSCs have been successfully given to humans2,44 and can easily be cultured and used without human leukocyte antigen matching, we suggest that cultured, banked human BMSCs should be tested for their ability to prevent sepsis in very high-risk groups.
METHODS
Mice
Mouse care was in full compliance with the US NIH criteria for the care and use of laboratory animals in research and all studies were approved by the Animal Care and Use Committee of the NIDDK and NIDCR, NIH. Aged (42−44 weeks old) male C57BL/6 mice (NIH) had free access to water and chow before and after surgery. In addition, 8−12-week-old males with a variety of genetic manipulations were used as listed in the Supplementary Methods online. All reagents used are summarized in Supplementary Table 1 online.
Polymicrobial cecal ligation and puncture sepsis
We performed CLP as previously described, with some modifications, in C57BL/6 mice45,46. We ligated the cecum by silk 4−0 and punctured it twice with a 21-gauge needle, gently squeezed it to express a small amount of fecal material and then returned it to the central abdominal cavity. In sham-operated mice, we located the cecum but neither ligated nor punctured it. We closed the abdominal incision in two layers with 6−0 nylon sutures. After surgery, we gave 1 ml per 30 g body weight of pre-warmed normal saline. All mice received antibiotic and fluid therapy subcutaneously (a combination of imipenem and cilastatin; 14 mg per kg in 1.5 ml of normal saline at 6 h and 7 mg per kg in 1.5 ml of normal saline at 18 h after surgery). The details of the tissue harvest are described in the Supplementary Methods.
Treatment of cecal ligation and puncture mice with bone marrow stromal cells
We injected one million BMSCs in 0.3 ml sterile PBS via the tail vein. We gave untreated control mice 0.3 ml sterile PBS with no cells. There was no difference in organ injury, cytokine abundance, Evans blue dye level or pathology in the sham-operated mice with or without BMSC injection. Details of cell isolation and culture are provided in the Supplementary Methods.
Survival studies
We assessed survival after surgery every 6 h within the first 48 h and then every 8 h for 4 d. We began antibiotic injection and fluid resuscitation 6 h after CLP by subcutaneous injection and repeated it every 12 h for 4 d. We killed all mice at the end of the fourth day.
Ex vivo studies on isolated lung monocytes and macrophages
We killed septic mice 6 h after CLP induction with or without intravenous injection of 1 million BMSCs. We removed the lungs, minced them into small pieces and incubated the pieces in RPMI 1640 medium with 1% penicillin-streptomycin and 1% glutamine for 30 min at 37 °C 5% CO2 in the presence of collagenase type 1 (300 U ml−1) and DNase I (50 U ml−1) (Worthington Biochemicals). After the incubation, we filtered the cell suspension through a 70-μm cell strainer and then washed it with complete RPMI medium. We incubated the resulting cells for 15 min at 4 °C with CD11b magnetic beads and subsequently applied them to MS columns (Miltenyi) for the positive selection of CD11b cells. The cell purity of the CD11b+ cells, as assessed by FACS analysis, was greater than 97%. After washing, we plated the isolated cells in 96-well plates at a concentration of 50,000 cells per well per 200 μl RPMI medium (with 10% FBS, 1% glutamine and 1% penicillin-streptomycin) containing 10 μg ml−1 LPS (from Escherichia coli O111:B4, Sigma). We incubated cells for 1, 3, 5 and 7 h after stimulation and collected supernatants sequentially from wells dedicated to each time point. For each time point, we used triplicate wells. We collected supernatants in microtubes, spun the tubes down at maximum speed for 10 min to get rid of cell debris, aliquoted the supernatants and froze them at −20 °C until use. We performed ELISA for IL-10 (R&D Systems) on the samples. We made measurements in duplicates from three independent experiments. To determine intracellular amounts of IL-10 produced in lung cells, we prepared single-cell suspensions as described above and plated cells in six-well plates containing 10 μg ml−1 LPS and 3 μg ml−1 brefeldin A, a Golgi-blocking agent frequently used to prepare samples for intracellular FACS analysis. After 5 h of LPS restimulation, we collected the cells, washed them and performed intracellular IL-10 staining combined with cell surface staining.
Macrophage–bone marrow stromal cells coculture experiments
First, we plated macrophages in 96-well plates at a concentration of 40,000 cells per 200 μl. After 1 h, we removed the supernatants and added either 40,000 BMSCs in fresh medium or fresh medium alone to the wells. As a control, we plated BMSCs without macrophages. After an overnight incubation, we added LPS to the cocultures to reach a final concentration of 1 μg ml−1. We collected supernatants 1, 3, 5 and 7 h after the stimulation and performed ELISA (R&D Systems) on the samples to detect the released IL-10. For the transwell or conditioned medium experiments, we either added BMSCs on the insert membrane of the transwell system (HTS Transwell 0.4-μm pore size poly-carbonate membrane, from Corning Incorporated) or added conditioned medium diluted 1:1 with fresh medium to the macrophages. For generation of BMSC-conditioned medium, we cultured BMSCs in complete medium. After 3 d, we collected supernatants, spun them down to remove possible cell contamination (500g for 10 min) and stored the resulting supernatants at −20 °C until further use. Further details on ELISA and western blotting are in the Supplementary Methods.
Blood chemistry, natural killer cell and macrophage depletion and IL-10 neutralization, microvascular permeability, bacterial counts, cell tracking, histology and flow cytometry
Detailed methodology is described in the Supplementary Methods.
Statistical analyses
We examined the differences between the groups for statistical significance by Student's t-test or analysis of variance with an appropriate correction. We compared survival curves with a log-rank test (Prism 4.0; Graphpad Software). A P value of <0.05 was accepted as statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank M.J. Brownstein for continuous advice and discussions; J. M. Weiss (NCI, NIH) for supplying the Ifng−/− mice; A. Keane-Myers (NIAID) for supplying the Il10−/− mice; Christophe Cataisson (NCI) for supplying the Tnfrsf1a−/− and Tnfrsf1b−/− mice; T. Merkel (US Food and Drug Administration) for supplying the Tlr4−/− and Myd88−/− mice; K. Holmbeck and L. Szabova (NIDCR) for the FVB/NJ mouse cells; and I. Szalayova and S. Key (NIDCR) for their superb technical help. The research was supported by the intramural programs of the NIDCR and the NIDDK, NIH.
Footnotes
Note: Supplementary information is available on the Nature Medicine website.
References
- 1.Ulloa L, Tracey KJ. The “cytokine profile”: a code for sepsis. Trends Mol. Med. 2005;11:56–63. doi: 10.1016/j.molmed.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 2.Le Blanc K, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;363:1439–1441. doi: 10.1016/S0140-6736(04)16104-7. [DOI] [PubMed] [Google Scholar]
- 3.Noel D, Djouad F, Bouffi C, Mrugala D, Jorgensen C. Multipotent mesenchymal stromal cells and immune tolerance. Leuk. Lymphoma. 2007;48:1283–1289. doi: 10.1080/10428190701361869. [DOI] [PubMed] [Google Scholar]
- 4.Rasmusson I. Immune modulation by mesenchymal stem cells. Exp. Cell Res. 2006;312:2169–2179. doi: 10.1016/j.yexcr.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 5.Ringden O, et al. Tissue repair using allogeneic mesenchymal stem cells for hemorrhagic cystitis, pneumomediastinum and perforated colon. Leukemia. 2007;21:2271–2276. doi: 10.1038/sj.leu.2404833. [DOI] [PubMed] [Google Scholar]
- 6.Bochud PY, Calandra T. Pathogenesis of sepsis: new concepts and implications for future treatment. BMJ. 2003;326:262–266. doi: 10.1136/bmj.326.7383.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calandra T. Pathogenesis of septic shock: implications for prevention and treatment. J. Chemother. 2001;13(Spec No 1):173–180. doi: 10.1179/joc.2001.13.Supplement-2.173. [DOI] [PubMed] [Google Scholar]
- 8.Hubbard WJ, et al. Cecal ligation and puncture. Shock. 2005;24(Suppl 1):52–57. doi: 10.1097/01.shk.0000191414.94461.7e. [DOI] [PubMed] [Google Scholar]
- 9.Togel F, et al. Vasculotropic, paracrine actions of infused mesenchymal stem cells are important to the recovery from acute kidney injury. Am. J. Physiol. Renal. Physiol. 2007;292:F1626–F1635. doi: 10.1152/ajprenal.00339.2006. [DOI] [PubMed] [Google Scholar]
- 10.Togel F, et al. Administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation-independent mechanisms. Am. J. Physiol. Renal Physiol. 2005;289:F31–F42. doi: 10.1152/ajprenal.00007.2005. [DOI] [PubMed] [Google Scholar]
- 11.Semedo P, et al. Mesenchymal stem cells ameliorate tissue damages triggered by renal ischemia and reperfusion injury. Transplant. Proc. 2007;39:421–423. doi: 10.1016/j.transproceed.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 12.Lange C, et al. Administered mesenchymal stem cells enhance recovery from ischemia/reperfusion-induced acute renal failure in rats. Kidney Int. 2005;68:1613–1617. doi: 10.1111/j.1523-1755.2005.00573.x. [DOI] [PubMed] [Google Scholar]
- 13.Shinkai Y, et al. RAG-2–deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- 14.Habu S, et al. In vivo effects of anti-asialo GM1. I. Reduction of NK activity and enhancement of transplanted tumor growth in nude mice. J. Immunol. 1981;127:34–38. [PubMed] [Google Scholar]
- 15.Kasai M, et al. In vivo effect of anti-asialo GM1 antibody on natural killer activity. Nature. 1981;291:334–335. doi: 10.1038/291334a0. [DOI] [PubMed] [Google Scholar]
- 16.van Rooijen N, Sanders A, van den Berg TK. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J. Immunol. Methods. 1996;193:93–99. doi: 10.1016/0022-1759(96)00056-7. [DOI] [PubMed] [Google Scholar]
- 17.Moore KW, O'Garra A, de Waal Malefyt R, Vieira P, Mosmann TR. Interleukin-10. Annu. Rev. Immunol. 1993;11:165–190. doi: 10.1146/annurev.iy.11.040193.001121. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Wakeham J, Harkness R, Xing Z. Macrophages are a significant source of type 1 cytokines during mycobacterial infection. J. Clin. Invest. 1999;103:1023–1029. doi: 10.1172/JCI6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonder CS, et al. P-selectin can support both TH1and TH2 lymphocyte rolling in the intestinal microvasculature. Am. J. Pathol. 2005;167:1647–1660. doi: 10.1016/S0002-9440(10)61248-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perretti M, Szabo C, Thiemermann C. Effect of interleukin-4 and interleukin-10 on leucocyte migration and nitric oxide production in the mouse. Br. J. Pharmacol. 1995;116:2251–2257. doi: 10.1111/j.1476-5381.1995.tb15061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cassatella MA. The neutrophil: one of the cellular targets of interleukin-10. Int. J. Clin. Lab. Res. 1998;28:148–161. doi: 10.1007/s005990050036. [DOI] [PubMed] [Google Scholar]
- 22.Ajuebor MN, et al. Role of resident peritoneal macrophages and mast cells in chemokine production and neutrophil migration in acute inflammation: evidence for an inhibitory loop involving endogenous IL-10. J. Immunol. 1999;162:1685–1691. [PubMed] [Google Scholar]
- 23.Hernandez LA, et al. Role of neutrophils in ischemia-reperfusion—induced micro-vascular injury. Am. J. Physiol. 1987;253:H699–H703. doi: 10.1152/ajpheart.1987.253.3.H699. [DOI] [PubMed] [Google Scholar]
- 24.Jaeschke H, Smith CW. Mechanisms of neutrophil-induced parenchymal cell injury. J. Leukoc. Biol. 1997;61:647–653. doi: 10.1002/jlb.61.6.647. [DOI] [PubMed] [Google Scholar]
- 25.Nussler AK, Wittel UA, Nussler NC, Beger HG. Leukocytes, the Janus cells in inflammatory disease. Langenbecks Arch. Surg. 1999;384:222–232. doi: 10.1007/s004230050196. [DOI] [PubMed] [Google Scholar]
- 26.Matthijsen RA, et al. Myeloperoxidase is critically involved in the induction of organ damage after renal ischemia reperfusion. Am. J. Pathol. 2007;171:1743–1752. doi: 10.2353/ajpath.2007.070184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. IL-1beta-mediated up-regulation of HIF-1α via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003;17:2115–2117. doi: 10.1096/fj.03-0329fje. [DOI] [PubMed] [Google Scholar]
- 28.Nakao S, et al. Tumor necrosis factor α (TNF-α)-induced prostaglandin E2 release is mediated by the activation of cyclooxygenase-2 (COX-2) transcription via NFκB in human gingival fibroblasts. Mol. Cell. Biochem. 2002;238:11–18. doi: 10.1023/a:1019927616000. [DOI] [PubMed] [Google Scholar]
- 29.Ramsay RG, Ciznadija D, Vanevski M, Mantamadiotis T. Transcriptional regulation of cyclo-oxygenase expression: three pillars of control. Int. J. Immunopathol. Pharmacol. 2003;16:59–67. [PubMed] [Google Scholar]
- 30.Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815–1822. doi: 10.1182/blood-2004-04-1559. [DOI] [PubMed] [Google Scholar]
- 31.Sotiropoulou PA, Perez SA, Gritzapis AD, Baxevanis CN, Papamichail M. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells. 2006;24:74–85. doi: 10.1634/stemcells.2004-0359. [DOI] [PubMed] [Google Scholar]
- 32.Pevsner-Fischer M, et al. Toll-like receptors and their ligands control mesenchymal stem cell functions. Blood. 2007;109:1422–1432. doi: 10.1182/blood-2006-06-028704. [DOI] [PubMed] [Google Scholar]
- 33.Crisostomo PR, et al. Gender differences in injury induced mesenchymal stem cell apoptosis and VEGF, TNF, IL-6 expression: role of the 55 kDa TNF receptor (TNFR1). J. Mol. Cell. Cardiol. 2007;42:142–149. doi: 10.1016/j.yjmcc.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park YK, et al. Nitric oxide donor, (+/−)-S-nitroso-N-acetylpenicillamine, stabilizes transactive hypoxia-inducible factor-1α by inhibiting von Hippel-Lindau recruitment and asparagine hydroxylation. Mol. Pharmacol. 2008;74:236–245. doi: 10.1124/mol.108.045278. [DOI] [PubMed] [Google Scholar]
- 35.Bal-Price A, Gartlon J, Brown GC. Nitric oxide stimulates PC12 cell proliferation via cGMP and inhibits at higher concentrations mainly via energy depletion. Nitric Oxide. 2006;14:238–246. doi: 10.1016/j.niox.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 36.Bianco P, Riminucci M, Gronthos S, Robey PG. Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells. 2001;19:180–192. doi: 10.1634/stemcells.19-3-180. [DOI] [PubMed] [Google Scholar]
- 37.Chamberlain G, Fox J, Ashton B, Middleton J. Concise review: mesenchymal stem cells: their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells. 2007;25:2739–2749. doi: 10.1634/stemcells.2007-0197. [DOI] [PubMed] [Google Scholar]
- 38.Mansilla E, et al. Human mesenchymal stem cells are tolerized by mice and improve skin and spinal cord injuries. Transplant. Proc. 2005;37:292–294. doi: 10.1016/j.transproceed.2005.01.070. [DOI] [PubMed] [Google Scholar]
- 39.van Laar JM, Tyndall A. Adult stem cells in the treatment of autoimmune diseases. Rheumatology (Oxford) 2006;45:1187–1193. doi: 10.1093/rheumatology/kel158. [DOI] [PubMed] [Google Scholar]
- 40.Corcione A, et al. Human mesenchymal stem cells modulate B-cell functions. Blood. 2006;107:367–372. doi: 10.1182/blood-2005-07-2657. [DOI] [PubMed] [Google Scholar]
- 41.Kubo S, et al. E-prostanoid (EP)2/EP4 receptor-dependent maturation of human monocyte-derived dendritic cells and induction of helper T2 polarization. J. Pharmacol. Exp. Ther. 2004;309:1213–1220. doi: 10.1124/jpet.103.062646. [DOI] [PubMed] [Google Scholar]
- 42.Shinomiya S, et al. Regulation of TNFα and interleukin-10 production by prostaglandins I2 and E2: studies with prostaglandin receptor—deficient mice and prostaglandin E-receptor subtype-selective synthetic agonists. Biochem. Pharmacol. 2001;61:1153–1160. doi: 10.1016/s0006-2952(01)00586-x. [DOI] [PubMed] [Google Scholar]
- 43.Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol. Ther. 2004;103:147–166. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 44.Le Blanc K, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008;371:1579–1586. doi: 10.1016/S0140-6736(08)60690-X. [DOI] [PubMed] [Google Scholar]
- 45.Miyaji T, et al. Ethyl pyruvate decreases sepsis-induced acute renal failure and multiple organ damage in aged mice. Kidney Int. 2003;64:1620–1631. doi: 10.1046/j.1523-1755.2003.00268.x. [DOI] [PubMed] [Google Scholar]
- 46.Yasuda H, Yuen PS, Hu X, Zhou H, Star RA. Simvastatin improves sepsis-induced mortality and acute kidney injury via renal vascular effects. Kidney Int. 2006;69:1535–1542. doi: 10.1038/sj.ki.5000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem. Biophys. Res. Commun. 1996;224:855–862. doi: 10.1006/bbrc.1996.1112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.