

Abstract
Objective
Delayed cerebral vasospasm has long been recognized as an important cause of poor outcome after an otherwise successful treatment of a ruptured intracranial aneurysm, but it remains a pathophysiological enigma despite intensive research for more than half a century.
Method
Summarized in this review are highlights of research from North America, Europe and Asia reflecting recent advances in the understanding of delayed ischemic deficit.
Result
It will focus on current accepted mechanisms and on new frontiers in vasospasm research.
Conclusion
A key issue is the recognition of events other than arterial narrowing such as early brain injury and cortical spreading depression and of their contribution to overall mortality and morbidity.
Keywords: Early brain injury, subarachnoid hemorrhage, vasospasm
INTRODUCTION
When persons in good health are suddenly seized with pains in the head, and straightway are laid down speechless, and breathe with stertor, they die in seven days.
Hippocrates 460–370 BC, Aphorisms on Apoplexy1
Hippocrates’ 2400-year-old description of delayed death probably caused by a ruptured intracranial aneurysm with subsequent vasospasm is still valid today. Aneurysmal subarachnoid hemorrhage (SAH) affects about 10 out of 100,000 adults annually, and up to half of those affected die soon after2; most of the rest are successfully treated surgically and/or endovascularly. Despite obliterating the offending aneurysm and removing the risk of rebleeding, up to half of the treated patients develop a syndrome of focal and/or cognitive deficits due to cerebral vasospasm (delayed ischemic neurological deficit, symptomatic vasospasm) between the fourth and ninth day after the SAH3. As a result, many die or suffer permanent morbidity2, and it has been described as the single most important cause of morbidity and mortality in patients whose ruptured aneurysm is successfully treated4.
Patients require vigilant monitoring and treatment for up to 2 weeks, including invasive monitoring of blood pressure, cerebral blood flow and metabolism and often complex treatment with calcium antagonists, hypertensive drugs, hemodilution and hypervolemia (triple H therapy), plus risky and often only temporarily effective intra-arterial administration of vasodilator drugs or balloon angioplasty5. These treatments have been documented in nine international conferences on cerebral vasospasm (Table 1).
Table 1.
Summary of international conferences on cerebral vasospasm, 1972–2006
| Year | Location | Chairman | Honored guest | Honorary president | Other officers |
|---|---|---|---|---|---|
| 1972 | Jackson, MI, USA | RR Smith, JT Robertson | FA Echlin | ||
| 1979 | Amsterdam, The Netherlands | RH Wilkins | C Miller Fisher | AJM van der Werf, S Ishii, SJ Peerless, L Symon | |
| 1987 | Charlottesville, VA, USA | NF Kassell | K Sano | CG Drake | D Vollmer |
| 1990 | Tokyo, Japan | K Sano | BK Weir | L Symon | K Takakura, NF Kassell, I Saito, T Sasaki |
| 1993 | Alberta, Canada | BK Weir | NF Kassell | JM Findlay | |
| 1996 | Sydney, Australia | NWC Dorsch | RR Smith | ||
| 2000 | Zurich, Switzerland | R Seiler | H Nornes | ||
| 2003 | Chicago, IL, USA | RL Macdonald | T Ohta, S Suzuki | J Zhang | |
| 2006 | Istanbul, Turkey | T Kiris | R Seiler | Young Investigator: T Ogawa |
Since the demonstration of arterial narrowing in the syndrome of cerebral vasospasm in 19516 and the further emphasis in 1978 by Weir et al.3, it has been proven that SAH gives rise to arterial narrowing and in turn ischemia, causing infarction and poor outcome. Most research into delayed deterioration after SAH has been conducted in concordance with this axiom, with the goal of interrupting this perceived chain of events. There have been many clinical trials, but until the arrival of clazosentan, a selective endothelin 1A receptor antagonist, it has not been possible to reproducibly break this chain. Clazosentan did, however, effectively prevent and reverse arterial narrowing in one work7, providing what was thought may at last be an effective treatment. However, the subsequent multi-center CONSCIOUS trial, despite significant reductions in angiographic vasospasm, failed to show any effect on long-term outcome.
The axiom has thus been challenged in such a fashion that it amounts to a paradigm shift.
Accumulated evidence suggests that (1) arterial narrowing is not the only cause of delayed clinical deterioration, (2) arterial narrowing is not necessarily multifactorial but (3) may actually be an effect of a single factor and finally (4) the entire picture of delayed clinical deterioration may be multifactorial. These facts should lead to a search for a more comprehensive and adequate theory that not only can explain observed discrepancies but also will lead to development of a specific and effective treatment strategy.
In recent years, two major concepts in pre-vasospasm research have developed: early brain injury and cortical spreading depression. Basic animal works and some clinical observations have long pointed to the importance of the pre-vasospasm period, with recognition of the importance of transitory ischemia at the onset of SAH 8, the opening of the blood–brain barrier9,10, the existence of early arterial narrowing in clinical settings11 and the detection of cortical spreading ischemia after SAH12. One or more of these events may replace arterial narrowing as important causes of poor outcomes after SAH13.
PATHOPHYSIOLOGY OF ARTERIAL NARROWING: NEW DEVELOPMENTS
The idea of arterial narrowing has previously been central to understanding the syndrome of cerebral vasospasm, but as outlined above, a paradigm shift is underway. Even so, the association of arterial narrowing with delayed ischemic deficits and the fact that reversal of narrowing by angioplasty can reverse deficits make consideration of pathophysiological events in cerebral arteries still very relevant, as shown in several reviews. Highlights of recent developments are presented below.
Hemoglobin
Ferrous hemoglobin released from subarachnoid clot undoubtedly leads to delayed arterial narrowing by mechanisms, which are multiple and poorly understood. Possibilities include neuronal apoptosis14, scavenging or decreased production of nitric oxide (NO)15, increased endothelin 1 levels16, direct oxidative stress on smooth muscle cells13, free radical production and lipid peroxidation of cell membranes17, modification of potassium and calcium channels18 and differential up-regulation of genes19,20.
Recent research has focused on oxidative stress as causing or contributing to vasospasm, possibly via direct activation of calcium channels in smooth muscle cells as well as on vasoactive proteins21 or through covalent modification by reactive oxygen species producing vasoactive molecules. For example, reactive oxygen species may act on arachidonic acid to produce vasoactive lipids, which in turn may contribute to vessel contraction. The lack of success of antioxidants such as tirilazad makes non-specific oxidation unlikely22. Other possible vasoactive compounds are bilirubin oxidation products (Figure 1), synthesized by the oxidation associated with oxidative stress. However, the location of the oxidations leading to the production of bilirubin oxidation products is unclear. It could be speculated that lysed blood cells are phagocytosed by lymphocytes, with resultant heme or bilirubin release after lysosomatic breakdown. In such a scenario, antioxidants that primarily target membranes would have relative little effect22.
Figure 1.

A putative pathway for the production of bilirubin-oxidized fragments (BOXes) from blood present post-SAH. Key steps are the liberation of heme from blood and oxidation from free radicals. The dotted line between the heme and oxidation of bilirubin is a pathway for the production of BOXes that has not yet been demonstrated
Oxidative stress in the subarachnoid space has also been reported to activate protein kinase C and Rho kinase, leading to smooth muscle cell constriction. Rho kinase initiates vascular contraction through protein kinase Cδ activity (Figure 2), which also induces proliferation and growth of vascular smooth muscle cells, a possible mechanism for the phenotypic change and remodeling of vascular smooth muscle seen in vasospasm23. Further support for a reorganization hypothesis comes from observations of increased β-actin messenger RNA (mRNA), of structural change in the 3′ untranslated region of β-actin mRNA and of induction of the embryonal isoform of myosin heavy chain accompanied by a decrease in the expression of smooth muscle myosin heavy chain in arteries in spasm23. Histological morphometric analysis also showed an increase in the area of the arterial wall without changes in the number of nuclei of smooth muscle cells24. Therapy for cerebral vasospasm may thus also need to address cerebral vascular remodeling.
Figure 2.

Relations among Rho kinase, Rho A, PKCI′ and PKCI± for the regulation of myosin and actin in the contraction and relaxation of vascular smooth muscle cells. Rho kinase and Rho A activate myosin light chain (MLC) phosphorylation directly. Rho kinase and Rho A inhibit MLC phosphatase, resulting in long lasting MLC phosphorylation. They also activate PKCI′, which enhances the contraction of vascular smooth muscle cells. PKCI± independently activates the contraction of vascular smooth muscle cells. Caldesmon is an actin-side regulatory protein acting on the detaching between actin and MLC and relaxation of the vascular smooth muscle cells. PKCI′ inhibits the activity of caldesmon through phosphorylates of caldesmon, which causes long lasting interaction between actin and MLC. MLC, myosin light chain; MLCK, myosin light chain kinase; P, phosphorylation; PKC, protein kinase C
Endothelin 1
The potent vasoconstricting peptide endothelin 1 was isolated from pig endothelial cells in 198825, and elevated endothelin 1 levels in the cerebrospinal fluid of patients and animals with vasospasm were soon reported16,26,27. This basic observation was supported by a number of experimental results. Oxyhemoglobin causes an increase in endothelin 1 synthesis from endothelial cells28, astrocytes may synthesize endothelin 1 in response to ischemia26 and leucocytes that have infiltrated the subarachnoid space after SAH may produce endothelin 129. Furthermore, cerebral arteries may become more sensitive to endothelin 1, leading to increased cerebrovascular tone even in the absence of an increase in endothelin 130. These findings provided the impetus for trials of endothelin receptor antagonists7. A phase II study of the endothelin receptor antagonist clazosentan in aneurysmal SAH randomized 413 patients, well balanced for prognostic factors. Moderate to severe angiographic vasospasm was significantly reduced in a dose-dependent manner from 66% in the placebo group to 36% in the high-dose clazosentan group. Despite this reduction, there was no significant effect on outcome based on the modified Rankin scale at 3 months31. Why this efficacy against angiographic vasospasm did not translate into improved outcome is a key question and a fundamental reason for the paradigm shift necessary to produce a more complete understanding of cerebral vasospasm and delayed deterioration.
Nitric oxide
Inhibition of relaxation can also cause or contribute to arterial narrowing because of the basic myogenic tone of cerebral arteries. Hence, the fate of NO (a powerful endogenous vasodilator) has attracted interest. The disappearance of neuronal NO synthase immunoreactivity from arteries in spasm, endothelial NO synthase dysfunction in cerebral vessels after SAH and the affinity for NO of the heme moiety in hemoglobin (‘NO sink effect’) suggest a role for NO depletion in the pathophysiology of arterial narrowing (Figure 3)15. New strategies for NO-based therapy against vasospasm include gene therapy32,33 preclinical and early clinical trials of NO donors administered intra-arterially34, intrathecally35, locally36,37 or intravenously38.
Figure 3.

Signal transduction for nitric oxide (NO) and endothelin 1 (ET-1). These are antagonistic regulators of cerebral blood flow, released from endothelial cells in response to changes in the shear stress, transmural pressure, concentration of CO2 and O2, ischemia or presence of hemoglobin. NO (vasodilator) and ET-1 (vasoconstrictor) regulate blood vessel tension via smooth muscle cells. NO, due to its high affinity to the heme moiety (1000 times higher than oxygen), stimulates guany(ly)l cyclase, leading to an increase of 3,5′ cyclic guanosine monophosphate and dephosphorylation of MLCs, smooth muscle cell hyperpolarization and closure of calcium channels resulting in vasodilation and an increase of blood flow. ET-1 is a product of several post-translational modifications of pre-pro-ET-1 and big ET-1. It acts on smooth muscles via two types of receptors: ETA, present mostly on smooth muscle cells, whose stimulation leads to smooth muscle constriction (paracrine action), and ETB, present mostly on endothelial cells, stimulation of which leads to an increased NO release and to smooth muscle relaxation (endocrine action). ET-1 stimulation of the ETA receptor leads to the formation of diacylglycerol and inositol 1,4,5-triphosphate, which in turn increases the concentration of intracellular calcium directly or via protein kinase C, resulting in vasoconstriction and decrease of blood flow
In terms of genetic predisposition in the NO synthase system, Khurana et al.39 reported that the endothelial NO synthase T-786C single-nucleotide polymorphism, associated with a significant reduction in endothelial NO synthase gene promoter activity, was significantly more common in patients with cerebral vasospasm. Another development in relation to NO synthase is the statins, which improve endothelial function by up-regulating endothelial NO synthase expression40. A threefold increase in endothelial NO synthase mRNA, protein and enzymatic activity has been demonstrated following statin treatment, resulting in an increase in cerebral blood flow41,42. Statin treatment has attenuated cerebral vasospasm and prevented delayed ischemic deficits in a murine SAH model40. In a retrospective series, patients who received statin therapy for at least 1 month before SAH demonstrated an 11-fold decrease in the risk of developing symptomatic vasospasm after SAH43. In recent prospective, double-blind, randomized placebo-controlled clinical trials of statins given for 14 days after SAH, the incidence of symptomatic cerebral vasospasm was significantly reduced in treated patients44,45.
Membrane pathology
Small diameter cerebral arteries (<200 μm) play important roles in the autoregulation of cerebral blood flow, matching local blood supply in the brain to neuronal activity (Figure 4)46. Although angiography, which can assess arteries >1 mm in diameter, has long been the standard to diagnose vasospasm47, constriction of small cerebral arteries may also contribute to ischemia after SAH21,48–51.
Figure 4.

Potential mechanisms of increased vascular smooth muscle intracellular Ca2+ and enhanced contraction of cerebral artery myocytes following SAH. Enhanced Ca2+ influx through voltage-dependent Ca2+ channels (VDCCs) may result from a combination of increased VDCC expression (L- and R-type) and increased VDCC activity due to membrane depolarization. Mechanisms contributing to depolarization include oxyhemoglobin-induced internalization of voltage-dependent (KV) K+ channels and decreased activity of large-conductance Ca2+ -activated (BK) K+ channels due to inhibition of Ca2+ sparks and/or increased levels of the cytochrome P450 metabolite 20-hydroxyeicosatetraenoic acid
The concentration of free intracellular Ca2+ ([Ca2+]i), and thus the contractile state of smooth muscle cells in cerebral resistance arteries, is determined primarily by Ca2+ influx through voltage-dependent Ca2+ channels52, the open-state probability of which is dictated by the membrane potential of the smooth muscle cells53. Following SAH, changes have been reported in the electrical properties of smooth muscle cells of small diameter cerebral arteries leading to enhanced Ca2+ influx, with vasoconstriction and decreased cerebral blood flow54. Cerebral arteries from healthy animals express only L-type voltage-dependent Ca2+ channels encoded by the gene CaV 1.2. Expression of an additional type of voltage-dependent Ca2+ channels (R-type, CaV 2.3) occurs after SAH, leading to increased Ca2+ channel density, increased Ca2+ influx and vasoconstriction55.
After SAH, calcium entry is further enhanced by membrane depolarization54,56. Oxyhemoglobin causes suppression of the voltage-dependent K+ channel (KV) current in cerebral artery smooth muscle cells through a mechanism involving tyrosine kinase-mediated channel endocytosis55. Decreased activity of large conductance calcium-activated K+ channels may also contribute to membrane depolarization via potential mechanisms, such as a decrease in Ca2+ spark frequency57 and increased production of the cytochrome P450 metabolite 20-hydroxyarachidonic acid58. This combination of increased voltage-dependent Ca2+ channel density and membrane depolarization will increase [Ca2+]i and lead to vasoconstriction. At the same time, decreased sensitivity of voltage-dependent Ca2+ channels s to L-type Ca2+ channel antagonists55 may limit the utility of agents, such as nimodipine, in the treatment of arterial narrowing. Targeting voltage-dependent Ca2+ channels and K+ channels in small diameter cerebral arteries may lead to safer and more effective treatments for SAH.
NEW FRONTIERS OF CEREBRAL VASOSPASM RESEARCH
Acute and early changes after aneurysmal SAH
One of the most important advances in recent years is the recognition of early brain injury after SAH, from the impact of the initial bleed and its detrimental effect on patient outcome. The term early brain injury has only recently been coined and refers to the injury to the brain as a whole within the first 72 hours after the ictus59, i.e. before the development of vasospasm. This early brain injury includes an elevation of intracranial pressure, a global reduction of cerebral blood flow, blood–brain barrier disruption, brain edema and neuronal cell death.
Decreased perfusion pressure after SAH was reported in patients by Nornes8, but its influence on outcome has only recently been recognized. The initial event of intracranial aneurysm rupture is the stop flow phenomenon14, with an acute transient global cerebral ischemia, which may itself be lethal2. When the patient survives, there may be a secondary ischemic insult due to blood–brain barrier disruption60 progressing to global cerebral edema61 or to delayed neuronal apoptosis14. Brain edema contributes to a further rise in intracranial pressure13 and thus a further reduction of cerebral blood flow. The mechanism of blood–brain abrrier disruption is unclear, but apoptosis affecting the brain and arteries may be involved13,60.
Cortical spreading depression
In experimental works by Dreier et al.62, fluid with a composition similar to the cerebrospinal fluid after SAH was applied in the subarachnoid space. This induced spreading depolarization waves over the cortex, which in turn triggered spreading microvascular spasm and spreading ischemia62, leading to widespread cortical necrosis12. This pathomorphological finding corresponds with autopsy works of non-operated SAH patients, in which 80% of fatal cases showed widely scattered triangular, round or laminar ischemic cortical lesions, 13 times more common than infarcts in the territories of large arteries63,64. In a clinical work of 18 patients undergoing craniotomy for hematoma evacuation or aneurysm treatment following aneurysmal SAH65, 13 showed waves of spreading depolarization. In several patients, clusters of spreading depolarizations occurred at the onset of neurological deterioration, and in some, prolonged periods of depressed electrocorticographic activity were followed by radiographic evidence of ischemia. There is thus clinical evidence for the first part of the ‘spreading ischemia theory’ of cortical infarcts after SAH, i.e. association with a cluster of spreading depolarizations (Figure 5). The decisive second part of the hypothesis, that a marked propagating decrease of cortical blood flow occurs in conjunction with spreading depolarizations and leads to delayed infarcts, remains open.
Figure 5.
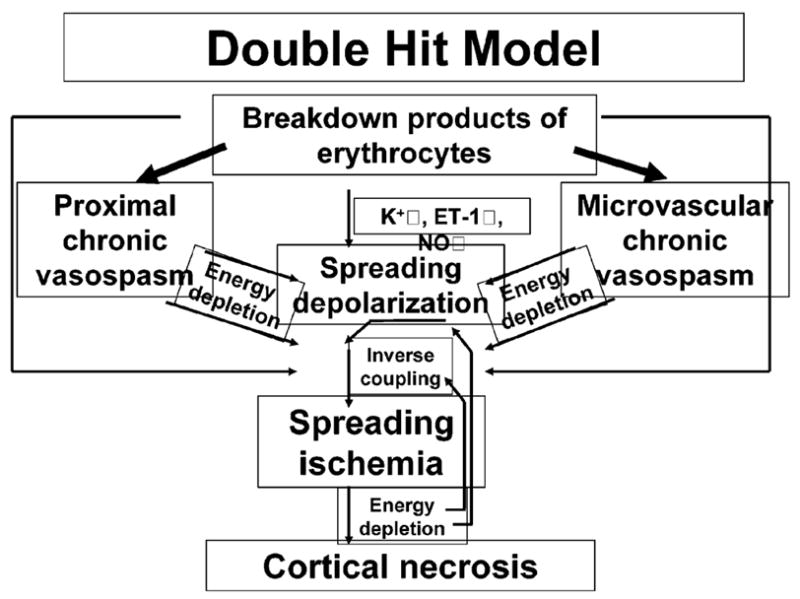
Double-hit model of delayed ischemic neurological deficits after SAH based on Dreier et al.12. The two hits on the brain parenchyma consist of acutely triggered microvascular spasm in response to spreading depolarizations, superimposed on chronic vasospasm
It is assumed that breakdown products of erythrocytes in the subarachnoid space induce delayed neurological deficits after SAH, based on observations that the risk of developing delayed neurological deficits correlates with the amount of blood in the initial computed axial tomography scan and that its onset coincides with the time of peak subarachnoid hemolysis in the primate model of SAH. According to the double hit model (Figure 5), breakdown products of erythrocytes have four major synergistic pathological effects: They induce chronic vasospasm of (1) proximal cerebral arteries and (2) the microcirculation; (3) they promote spreading depolarizations via chronic vasospasm/energy depletion, increase of the baseline extracellular K+ concentration and endothelin 1 and a decrease of NO; (4) they invert the coupling between spreading depolarization of the cortex and cerebral blood flow by direct effects (K+ ↑, NO↓) and indirectly via chronic vasospasm/energy depletion. Consistently with the double hit model, it was recently shown using subdural electrodes that delayed ischemic infarcts in SAH patients are preceded and accompanied by clusters of spreading depolarizations with prolonged depression periods65. There is still no direct evidence of spreading ischemia in the human brain. Spreading depolarization is an energy-demanding perturbation of brain cortical ion homeostasis and energy metabolism; in response to this demand, cerebral blood flow rises during spreading depolarization under physiological conditions. However, in the presence of breakdown products of erythrocytes in rats12,62, as well as in the ischemic penumbra after middle cerebral artery occlusion in mice66, spreading depolarization induces severe acute microarterial spasm and spreading ischemia, rather than microarterial dilation and spreading hyperemia. This process is called ‘inverse coupling’12,62. Spreading ischemia is associated with virtual disappearance of the pial circulation for periods of minutes or hours and leads to widespread cortical infarcts in these rat experiments12.
TREATMENT OF VASOSPASM
The importance of the therapeutic approach of multimodality treatment is stressed
Delayed neurological deficits after aneurysmal SAH are not caused by one single factor and therefore cannot be expected to be prevented or reversed by a single treatment such as clazosentan. Clinical trials have shown promising results for the cisternal placement of controlled-release nicardipine or papaverine pellets, for intravenous magnesium sulfate, for oral pravastatin or simvastatin, for lumbar cerebrospinal fluid drainage and for lamina terminalis fenestration67–69. There have been reports on variations of the head shaking technique with cisternal lavage, enoxaparin, cervical sympathetic block, aortic balloon counterpulsation or partial blockage and nitroglycerine patches5. Many of these works showed significantly reduced delayed neurological deficits and/or lower mortality.
Systemic: fasudil and multimodal therapy
Intra-arterial fasudil hydrochloride (a Rho-kinase inhibitor) is part of the multimodal therapy after SAH in Japan70, along with cisternal urokinase injection71, drainage of subarachnoid clots69 and strict maintenance of general conditions. The result of multimodal treatment with fasudil when compared with patients without multimodal therapy was a decrease in incidence of vasospasm from 57 to 37% with subsequent reduced mortality and improved outcome.
Topical: nicardipine
Clinical trials have tested the efficacy against vasospasm of local prolonged-release nicardipine-loaded polymers72,73 implanted at the time of aneurysm clipping into the basal cisterns close to the proximal arteries, with the drug being released over 14 days. In the first work, arterial narrowing was completely prevented in arteries surrounded by thick clot. The second work confirmed reductions in vasospasm (73% control versus 7% nicardipine-loaded polymers) and delayed ischemic lesions (47% control versus 14% nicardipine), and the outcome was significantly better. This polymer-based drug-delivery system offers a new and promising treatment approach. However, the lack of drug penetration to areas covered by blood clot is a potential limitation, needing further elucidation for the routine clinical setting.
Intra-arterial: nicardipine/verapamil
Safer endovascular treatments for severe refractory arterial narrowing are now available74. In one series of 350 SAH patients, 47 developed severe clinical vasospasm requiring endovascular therapy, including 175 intra-arterial injections of nicardipine (average dose, 6.0 mg; maximum, 22 mg per patient) or verapamil (8.0 mg per vessel, maximum 16 mg per patient) and 49 balloon angioplasties. There was significant improvement after the intra-arterial drug treatment, lasting for 24–48 hours, while balloon angioplasty, mostly (49%) for middle cerebral arteries, was more effective for proximal artery spasm. There were no complications from the angioplasty itself (A Zauner, unpublished data).
CONCLUSION
While our understanding of the pathophysiology of delayed vasospasm has progressed significantly, this knowledge has not been translated into clinically effective treatment. Possible sources of this mismatch include the multifactorial nature of the disease, the use of inadequate animal SAH models (e.g. models that create SAH but do not alter intracranial pressure), the lack of randomization or blinded assessment in many preclinical and some clinical works, underpowered experimental and clinical works, the lack of a priori identified inclusion/exclusion criteria and bias toward publication of positive but not negative findings75. The results of the phase II international clazosentan study support the concept that linking outcome solely to delayed vasospasm is an oversimplification, focusing attention on vasospasm rather than on patients’ well-being (RL Macdonald, unpublished data). This work opens up the question of why the prevention of vasospasm did not translate into improved outcome. It seems clear now that research must focus more on the acute subacute, delayed and late events after aneurysmal SAH and their influence on outcome.
These new developments in the understanding of the pathophysiology of SAH and vasospasm and advances in the treatment, as well as controversies around both pathophysiology and treatment, make it mandatory to ‘spread the word’ among researchers and clinicians that a widening of interest from delayed arterial narrowing to other SAH-evoked events is now essential. Research on the relationship between all post-hemorrhage events and their contribution to outcome should hasten the development of effective treatment for vasospasm and other events. A crucial element will be the use of improved animal models to help elucidate the contributions of the various mechanisms discussed above, as well as others not specifically addressed in this work such as thromboembolism76 platelet activation or inflammation, to delayed vasospasm or deterioration after aneurysmal SAH.
Acknowledgments
This research was support in part by the Intramural Research Program of the NIH, NINDS.
References
- 1.Clarke E. Apoplexy in the Hippocratic writings. Bull Hist Med. 1963;37:301. [PubMed] [Google Scholar]
- 2.Burnett M, Danish S, McKhann G, II, et al. Pathology and pathophysiology of aneurysmal subarachnoid hemorrhage. In: Leroux P, Winn W, Newell D, editors. Management of Cerebral Aneurysms. Philadelphia, PA: Elsevier; 2004. pp. 127–137. [Google Scholar]
- 3.Weir B, Grace M, Hansen J, et al. Time course of vasospasm in man. J Neurosurg. 1978;48:173–178. doi: 10.3171/jns.1978.48.2.0173. [DOI] [PubMed] [Google Scholar]
- 4.Kassell N, Torner J, Haley E., Jr The International Cooperative Study on the Timing of Aneurysm Surgery. Part 1: Overall management results. J Neurosurg. 1990;73:18–36. doi: 10.3171/jns.1990.73.1.0018. [DOI] [PubMed] [Google Scholar]
- 5.Dorsch N. Therapeutic approaches to vasospasm in subarachnoid hemorrhage. Curr Opin Crit Care. 2002;8:128–133. doi: 10.1097/00075198-200204000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Ecker A, Rimenschneider P. Arteriographic demonstration of spasm of the intracranial arteries with special reference to saccular arterial aneurysms. J Neurosurg. 1951;8:660–667. doi: 10.3171/jns.1951.8.6.0660. [DOI] [PubMed] [Google Scholar]
- 7.Vajkoczy P, Meyer B, Weidauer S, et al. Clazosentan (AXV-034343), a selective endothelin A receptor antagonist, in the prevention of cerebral vasospasm following severe aneurysmal subarachnoid hemorrhage: Results of a randomized, double-blind, placebo-controlled, multicenter phase IIa study. J Neurosurg. 2005;103:9–17. doi: 10.3171/jns.2005.103.1.0009. [DOI] [PubMed] [Google Scholar]
- 8.Nornes H. The role of intracranial pressure in the arrest of hemorrhage in patients with ruptured intracranial aneurysms. J Neurosurg. 1973;39:226–234. doi: 10.3171/jns.1973.39.2.0226. [DOI] [PubMed] [Google Scholar]
- 9.Grant G, Janigro D, Winn H. Subarachnoid hemorrhage and the blood–brain barrier. In: Leroux P, Winn W, Newell D, editors. Management of Cerebral Aneurysms. Philadelphia, PA: Elsevier; 2004. p. 121c137. [Google Scholar]
- 10.Trojanowski T. Blood Elsevier brain barrier changes after experimental subarachnoid hemorrhage. Acta Neurochir (Wien) 1982;60:45–54. doi: 10.1007/BF01401749. [DOI] [PubMed] [Google Scholar]
- 11.Baldwin ME, Macdonald R, Huo D, et al. Early vasospasm on admission angiography in patients with aneurysmal subarachnoid hemorrhage is a predictor for in-hospital complications and poor outcome. Stroke. 2004;35:2506–2511. doi: 10.1161/01.STR.0000144654.79393.cf. [DOI] [PubMed] [Google Scholar]
- 12.Dreier J, Ebert N, Priller J, et al. Products of hemolysis in the subarachnoid space inducing spreading ischemia in the cortex and focal necrosis in rats: A model for delayed ischemic neurological deficits after subarachnoid hemorrhage? J Neurosurg. 2000;93:658–666. doi: 10.3171/jns.2000.93.4.0658. [DOI] [PubMed] [Google Scholar]
- 13.Ostrowski RP, Colohan AR, Zhang JH. Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28 (4):399–414. doi: 10.1179/016164106X115008. [DOI] [PubMed] [Google Scholar]
- 14.Cahill WJ, Calvert JH, Zhang JH. Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2006;26:1341–1353. doi: 10.1038/sj.jcbfm.9600283. [DOI] [PubMed] [Google Scholar]
- 15.Pluta R. Delayed cerebral vasospasm and nitric oxide: Review, new hypothesis, and proposed treatment. Pharmacol Ther. 2005;105:23–56. doi: 10.1016/j.pharmthera.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Seifert V, Loffler B, Zimmermann M, et al. Endothelin concentrations in patients with aneurysmal subarachnoid hemorrhage: Correlation with cerebral vasospasm, delayed ischemic neurological deficits and volume of hematoma. J Neurosurg. 1995;82:55–62. doi: 10.3171/jns.1995.82.1.0055. [DOI] [PubMed] [Google Scholar]
- 17.Sehba F, Bederson J. Mechanisms of acute brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28:381–398. doi: 10.1179/016164106X114991. [DOI] [PubMed] [Google Scholar]
- 18.Zuccarello M, Bonasso C, Lewis A, et al. Relaxation of subarchnoid hemorrhage-induced spasm of rabbit basilar artery by the K+ channel activator cromakalim. Stroke. 1996;27:311–316. doi: 10.1161/01.str.27.2.311. [DOI] [PubMed] [Google Scholar]
- 19.Turner C, Bergeron M, Matz P, et al. Heme oxygenase-1 is induced in glia throughout brain by subarachnoid hemoglobin. J Cereb Blood Flow Metab. 1998;18:257–273. doi: 10.1097/00004647-199803000-00004. [DOI] [PubMed] [Google Scholar]
- 20.Vikman P, Beg S, Khurana T, et al. Gene expression and molecular changes in cerebral arteries following subarachnoid hemorrhage in the rat. J Neurosurg. 2006;105:438–444. doi: 10.3171/jns.2006.105.3.438. [DOI] [PubMed] [Google Scholar]
- 21.Dietrich H, Dacey R. Molecular keys to the problems of cerebral vasospasm. Neurosurgery. 2000;46:517–530. doi: 10.1097/00006123-200003000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Clark JF, Sharp FR. Bilirubin oxidation products (BOXes) and their role in cerebral vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2006;26 (10):1223–1233. doi: 10.1038/sj.jcbfm.9600280. [DOI] [PubMed] [Google Scholar]
- 23.Nishizawa S, Laher I. Signaling mechanisms in cerebral vasospasm. Trends Cardiovasc Med. 2005;15 (1):24–34. doi: 10.1016/j.tcm.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 24.Ohkuma H, Tsurutani H, Suzuki S. Changes of beta-actin mRNA expression in canine vasospastic basilar artery after experimental subarachnoid hemorrhage. Neurosci Lett. 2001;311 (1):9–12. doi: 10.1016/s0304-3940(01)02101-2. [DOI] [PubMed] [Google Scholar]
- 25.Yanagisawa M, Kurihara H, Kimura S, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- 26.Pluta R, Boock R, Afshar J, et al. The source and cause of endothelin 1 release to CSF after SAH. J Neurosurg. 1997;87:287–293. doi: 10.3171/jns.1997.87.2.0287. [DOI] [PubMed] [Google Scholar]
- 27.Zimmermann M, Seifert V. Endothelin and subarachnoid hemorrhage: An overview. Neurosurgery. 1998;43:863–875. doi: 10.1097/00006123-199810000-00083. [DOI] [PubMed] [Google Scholar]
- 28.Kasuya H, Weir B, White D, et al. Mechanism of oxyhemoglobin-induced release of endothelin-1 from cultured vascular endothelial cells and smooth-muscle cells. J Neurosurg. 1993;79:892–898. doi: 10.3171/jns.1993.79.6.0892. [DOI] [PubMed] [Google Scholar]
- 29.Fassbender K, Hodapp B, Rossol S, et al. Endothelin-1 in subarachnoid hemorrhage: An acute-phase reactant produced by cerebrospinal fluid leukocytes. Stroke. 2000;31:2971–2975. doi: 10.1161/01.str.31.12.2971. [DOI] [PubMed] [Google Scholar]
- 30.Alabadi JA, Salom J, Torregrosa G, et al. Changes in the cerebrovascular effects of endothelin-1 and nicardipine after experimental subarachnoid hemorrhage. Neurosurgery. 1993;33:707–714. doi: 10.1227/00006123-199310000-00022. [DOI] [PubMed] [Google Scholar]
- 31.Macdonald RL, Pluta RM, Zhang JH. Cerebral vasospasm after subarachnoid hemorrhage: The emerging revolution. Nat Clin Pract Neurol. 2007;3 (5):256–263. doi: 10.1038/ncpneuro0490. [DOI] [PubMed] [Google Scholar]
- 32.Luders J, Weihl C, Lin G, et al. Adenoviral gene transfer of nitric oxide synthase increases cerebral blood flow in rats. Neurosurgery. 2000;47:1206–1214. doi: 10.1097/00006123-200011000-00039. [DOI] [PubMed] [Google Scholar]
- 33.Smith RS, Jr, Agata J, Xia CF, et al. Human endothelial nitric oxide synthase gene delivery protects against cardiac remodeling and reduces oxidative stress after myocardial infarction. Life Sci. 2005;76 (21):2457–2471. doi: 10.1016/j.lfs.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 34.Pluta R, Oldfield E, Boock R. Reversal and prevention of cerebral vasospasm by intracarotid infusions of nitric oxide donors in a primate model of subarachnoid hemorrhage. J Neurosurg. 1997;87:746–751. doi: 10.3171/jns.1997.87.5.0746. [DOI] [PubMed] [Google Scholar]
- 35.Thomas J, Rosenwasser R. Reversal of severe cerebral vasospasm in three patients after aneurysmal subarachnoid hemorrhage: Initial observations regarding the use of intraventricular sodium nitroprusside in humans. Neurosurgery. 1999;44 (1):48–57. doi: 10.1097/00006123-199901000-00026. [DOI] [PubMed] [Google Scholar]
- 36.Clatterbuck RE, Gailloud P, Tierney T, et al. Controlled release of a nitric oxide donor for the prevention of delayed cerebral vasospasm following experimental subarachnoid hemorrhage in nonhuman primates. J Neurosurg. 2005;103:745–751. doi: 10.3171/jns.2005.103.4.0745. [DOI] [PubMed] [Google Scholar]
- 37.Tierney T, Pradilla G, Wang P, et al. Intracranial delivery of the nitric oxide donor diethylenetriamine/nitric oxide from a controlled-release polymer: Toxicity in cynomolgus monkeys. Neurosurgery. 2006;58:952–960. doi: 10.1227/01.NEU.0000210182.48546.8F. [DOI] [PubMed] [Google Scholar]
- 38.Pluta RM, Dejam A, Grimes G, et al. Nitrite infusions prevent cerebral artery vasospasm in a primate model of subarachnoid aneurysmal hemorrhage. JAMA. 2005;293:1477–1484. doi: 10.1001/jama.293.12.1477. [DOI] [PubMed] [Google Scholar]
- 39.Khurana V, Sohni Y, Mangrum W, et al. Endothelial nitric oxide synthase gene polymorphism predict susceptibility to aneurysmal subarachnoid hemorrhage and cerebral vasospasm. J Cereb Blood Flow Metab. 2004;24:291–297. doi: 10.1097/01.WCB.0000110540.96047.C7. [DOI] [PubMed] [Google Scholar]
- 40.McGirt MJ, Lynch J, Parra A, et al. Simvastatin increases endothelial nitric oxide synthase and ameliorates cerebral vasospasm resulting from subarachnoid hemorrhage. Stroke. 2002;33:2950–2956. doi: 10.1161/01.str.0000038986.68044.39. [DOI] [PubMed] [Google Scholar]
- 41.Endres M, Laufs U. [HMG-CoA reductase inhibitor and risk of stroke] Nervenarzt. 1998;69 (8):717–721. doi: 10.1007/s001150050335. [in German] [DOI] [PubMed] [Google Scholar]
- 42.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273 (37):24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 43.McGirt M, Pradilla G, Legnani F, et al. Systemic administration of simvastatin after the onset of experimental subarachnoid hemorrhage attenuates cerebral vasospasm. Neurosurgery. 2006;58:945–951. doi: 10.1227/01.NEU.0000210262.67628.7E. [DOI] [PubMed] [Google Scholar]
- 44.Lynch J, Wang H, McGirt M, et al. Simvastatin reduces vasospasm after aneurysmal subarachnoid hemorrhage: Results of a pilot randomized clinical trial. Stroke. 2005;36:2024–2026. doi: 10.1161/01.STR.0000177879.11607.10. [DOI] [PubMed] [Google Scholar]
- 45.Tseng M-Y, Czosnyka M, Richards H, et al. Effects of acute treatment with pravastatin on cerebral vasospasm, autoregulation, and delayed ischemic deficits after aneurysmal subarachnoid hemorrhage: A phase II randomized placebo-controlled trial. Stroke. 2005;36:1627–1632. doi: 10.1161/01.STR.0000176743.67564.5d. [DOI] [PubMed] [Google Scholar]
- 46.Paulson OB, Newman EA. Does the release of potassium from astrocyte endfeet regulate cerebral blood flow? Science. 1987;237 (4817):896–898. doi: 10.1126/science.3616619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kassell N, Sasaki T, Colohan A, et al. Cerebral vasospasm following aneurysmal subarachnoid hemorrhage. Stroke. 1985;16:562–572. doi: 10.1161/01.str.16.4.562. [DOI] [PubMed] [Google Scholar]
- 48.Jakobsen M, Enevoldsen E, Dalager T. Spasm index in subarachnoid hemorrhage: Consequences of vasospasm upon cerebral blood flow and oxygen extraction. Acta Neurol Scand. 1990;82 (5):311–320. doi: 10.1111/j.1600-0404.1990.tb03309.x. [DOI] [PubMed] [Google Scholar]
- 49.Ohkuma H, Manabe H, Tanaka M, et al. Impact of cerebral microcirculatory changes on cerebral blood flow during cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Stroke. 2000;31 (7):1621–1627. doi: 10.1161/01.str.31.7.1621. [DOI] [PubMed] [Google Scholar]
- 50.Shimoda M, Takeuchi M, Tominago J, et al. Asymptomatic versus symptomatic infarct from vasospasm in patients with subarachnoid hemorrhage: Serial magnetic resonans imaging. Neurosurgery. 2001;49:1341–1348. doi: 10.1097/00006123-200112000-00010. [DOI] [PubMed] [Google Scholar]
- 51.Treggiari-Venzi M, Suter P, Romand J-A. Review of medical prevention of vasospasm after aneurysmal subarachnoid hemorrhage: A problem of neurointensive care. Neurosurgery. 2001;48:249–262. doi: 10.1097/00006123-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 52.Wellman GC, Nathan DJ, Saundry CM, et al. Ca2+ sparks and their function in human cerebral arteries. Stroke. 2002;33 (3):802–808. doi: 10.1161/hs0302.104089. [DOI] [PubMed] [Google Scholar]
- 53.Nelson M. Ca2+-activated potassium channels and ATP-sensitive potassium channels as modulators of vascular tone. Trends Cardiovasc. 1993;3:54–60. doi: 10.1016/1050-1738(93)90037-7. [DOI] [PubMed] [Google Scholar]
- 54.Wellman T, Jenkins J, Penar P, et al. Nitric oxide and reactive oxygen species exert opposing effects on the stability of hypoxia-inducible factor-1alpha (HIF-1alpha) in explants of human pial arteries. FASEB J. 2004;18:379–381. doi: 10.1096/fj.03-0143fje. [DOI] [PubMed] [Google Scholar]
- 55.Ishiguro M, Wellman TL, Honda A, et al. Emergence of a R-type Ca2+ channel (CaV 2.3) contributes to cerebral artery constriction after subarachnoid hemorrhage. Circ Res. 2005;96 (4):419–426. doi: 10.1161/01.RES.0000157670.49936.da. [DOI] [PubMed] [Google Scholar]
- 56.Harder D, Dernbach P, Waters A. Possible cellular mechanism for cerebral vasospasm after subarachnoid hemorrhage in the dog. J Clin Invest. 1987;80:875–880. doi: 10.1172/JCI113146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jewell RP, Saundry CM, Bonev AD, et al. Inhibition of Ca++ sparks by oxyhemoglobin in rabbit cerebral arteries. J Neurosurg. 2004;100 (2):295–302. doi: 10.3171/jns.2004.100.2.0295. [DOI] [PubMed] [Google Scholar]
- 58.Roman RJ, Renic M, Dunn KM, et al. Evidence that 20-HETE contributes to the development of acute and delayed cerebral vasospasm. Neurol Res. 2006;28 (7):738–749. doi: 10.1179/016164106X152016. [DOI] [PubMed] [Google Scholar]
- 59.Kusaka G, Ishikawa M, Nanda A, et al. Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2004;24:916–925. doi: 10.1097/01.WCB.0000125886.48838.7E. [DOI] [PubMed] [Google Scholar]
- 60.Park S, Yamaguchi M, Zhou C, et al. Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke. 2004;35:2412–2417. doi: 10.1161/01.STR.0000141162.29864.e9. [DOI] [PubMed] [Google Scholar]
- 61.Claassen J, Carhuapoma J, Kreiter K, et al. Global cerebral edema after subarachnoid hemorrhage: Frequency, predictors, and impact on outcome. Stroke. 2002;33:1225–1232. doi: 10.1161/01.str.0000015624.29071.1f. [DOI] [PubMed] [Google Scholar]
- 62.Dreier JP, Korner K, Ebert N, et al. Nitric oxide scavenging by hemoglobin or nitric oxide synthase inhibition by N-nitro-L-arginine induces cortical spreading ischemia when K+ is increased in the subarachnoid space. J Cereb Blood Flow Metab. 1998;18 (9):978–990. doi: 10.1097/00004647-199809000-00007. [DOI] [PubMed] [Google Scholar]
- 63.Birse SH, Tom MI. Incidence of cerebral infarction associated with ruptured intracranial aneurysms. A study of 8 unoperated cases of anterior cerebral aneurysm. Neurology. 1960;10:101–106. doi: 10.1212/wnl.10.2.101. [DOI] [PubMed] [Google Scholar]
- 64.Neil-Dwyer G, Lang D, Doshi B, et al. Delayed cerebral ischaemia: The pathological substrate. Acta Neurochir (Wien) 1994;131:137–145. doi: 10.1007/BF01401464. [DOI] [PubMed] [Google Scholar]
- 65.Dreier JP, Woitzik J, Fabricius M, et al. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain. 2006;129 (Pt 12):3224–32237. doi: 10.1093/brain/awl297. [DOI] [PubMed] [Google Scholar]
- 66.Shin H, Dunn A, Jones P, et al. Vasoconstrictive neurovascular coupling during focal ischemic depolarizations. J Cereb Blood Flow Metab. 2006;26:1018–1030. doi: 10.1038/sj.jcbfm.9600252. [DOI] [PubMed] [Google Scholar]
- 67.Andaluz N, Zuccarello M. Fenestration of the lamina terminalis as a valuable adjunct in aneurysm surgery. Neurosurgery. 2004;55 (5):1050–1059. doi: 10.1227/01.neu.0000140837.63105.78. [DOI] [PubMed] [Google Scholar]
- 68.Kiriş T, Erden T, Sahinbas M, et al. CSF drainage for prevention and reversal of cerebral vasospasm after surgical treatment of intracranial aneurysms. In: Macdonald RL, editor. Cerebral Vasospasm. New York: Thieme Verlag; 2005. pp. 255–258. [Google Scholar]
- 69.Klimo P, Kestle J, MacDonald J, et al. Marked reduction of cerebral vasospasm with lumbar drainage of cerebrospinal fluid after subarachnoid hemorrhage. J Neurosurg. 2004;100:215–224. doi: 10.3171/jns.2004.100.2.0215. [DOI] [PubMed] [Google Scholar]
- 70.Yamamoto Y, Ikegaki I, Sasaki Y, et al. The protein kinase inhibitor fasudil protects against ischemic myocardial injury induced by endothelin-1 in the rabbit. J Cardiovasc Pharmacol. 2000;35 (2):203–211. doi: 10.1097/00005344-200002000-00005. [DOI] [PubMed] [Google Scholar]
- 71.Sayama CM, Liu JK, Couldwell WT. Update on endovascular therapies for cerebral vasospasm induced by aneurysmal subarachnoid hemorrhage. Neurosurg Focus. 2006;21 (3):E12. doi: 10.3171/foc.2006.21.3.12. [DOI] [PubMed] [Google Scholar]
- 72.Kasuya H, Onda H, Takeshita M, et al. Efficacy and safety of nicardipine prolonged-release implants for preventing vasospasm in humans. Stroke. 2002;33:1011–1015. doi: 10.1161/01.str.0000014563.75483.22. [DOI] [PubMed] [Google Scholar]
- 73.Kasuya H, Onda H, Sasahara A, et al. Application of nicardipine prolonged-release implants: Analysis of 97 consecutive patients with acute subarachnoid hemorrhage. Neurosurgery. 2005;56 (5):895–902. [PubMed] [Google Scholar]
- 74.Hoh BL, Ogilvy CS. Endovascular treatment of cerebral vasospasm: Transluminal balloon angioplasty, intra-arterial papaverine, and intra-arterial nicardipine. Neurosurg Clin N Am. 2005;16 (3):501–516. doi: 10.1016/j.nec.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 75.Dirnagl U. Bench to bedside: The quest for quality in experimental stroke research. J Cereb Blood Flow Metab. 2006;26:1465–1478. doi: 10.1038/sj.jcbfm.9600298. [DOI] [PubMed] [Google Scholar]
- 76.Ohta T. Cerebral vasospasm revisited: SAH syndrome. In: Macdonald RL, editor. Cerebral Vasospasm: Advances in Research and Treatment. New York: Thieme; 2004. pp. 106–111. [Google Scholar]