

Abstract
ICP0 is a multi-functional herpes simplex virus type 1 (HSV-1) immediate-early (IE) gene product that contributes to efficient virus growth and reactivation from latency. Here we show that HSV-1-induced cell-cycle arrest at the G2/M border requires ICP0 and Chk2 kinase and that ICP0 expression by transfection or infection induces ATM-dependent phosphorylation of Chk2 and Cdc25C. Infection of cells with a replication-defective mutant virus deleted for all the regulatory IE genes except ICP0 (TOZ22R) induced G2/M arrest whereas a mutant virus deleted in addition for ICP0 (QOZ22R) failed to do so. Chk2-deficient cells and cells expressing a kinase-deficient Chk2 did not undergo cell-cycle arrest in response to TOZ22R infection. Chk2 deficiency diminished the growth of wild-type HSV-1, but not the growth of an ICP0-deleted recombinant virus. Together, these results are consistent with the interpretation that ICP0 activates a DNA damage response pathway to arrest cells in G2/M phase and promote virus growth.
Keywords: HSV-1, cell cycle, Chk2, DNA damage response, ICP0
Introduction
Herpes simplex virus type 1 (HSV-1) is a neurotropic human pathogen that establishes life-long latency in sensory neurons (Steiner et al., 2007; Stevens, 1989). During latency, the viral DNA exists in a quiescent episomal state wherein expression is restricted to the latency-associated transcripts (LATs) (Steiner et al., 2007). In response to a variety of stress-associated stimuli, the virus is derepressed resulting in resumption of viral gene expression and synthesis of new virus. In the replicative stage, three classes of genes, referred to as immediate-early (IE), early (E) and late (L), are expressed in a tightly-regulated, well-ordered cascade (Honess and Roizman, 1974; Roizman and Knipe, 2001). One of the IE gene products, ICP0 (also known as Vmw110), is a multifunctional protein that is essential for viral reactivation as well as efficient virus growth following infection at low multiplicity (Cai and Schaffer, 1989; Everett, 2000). Consequently, HSV-1 mutants defective for ICP0 are severely impaired for replication in normal fibroblast cultures and fail to reactivate from latency in rodent models (Sacks and Schaffer, 1987).
The activities of ICP0 are mainly linked to two properties, its transactivation potential and its ability to degrade intracellular proteins (Cai and Schaffer, 1989; Everett, 2000). ICP0 has been reported to bind the Co-REST/REST/HDAC-1 repressor complex and induce the dissociation of HDAC-1, thus promoting the formation of euchromatin within the virus genome (Gu et al., 2005; Zhang and Jones, 2001). Its protein destabilizing property is linked to two functionally independent E3-ubiquitin (Ub) ligase domains. ICP0 associates with nuclear domain 10 (ND10) structures (also known as PML bodies), causing the degradation of ND10 structural components (Boutell et al., 2005; Gu and Roizman, 2003). ICP0 also triggers the degradation of p53, imparting resistance to UV-induced apoptosis in infected cells (Boutell and Everett, 2004), and DNA-PKcs, the catalytic subunit of DNA-dependent protein kinase (DNA-PK) (Lees-Miller et al., 1996; Parkinson et al., 1999).
Recent investigations have suggested a role for ICP0 in HSV-1-induced cell-cycle arrest at both the G1/S and G2/M boundaries (Hobbs and DeLuca, 1999; Lomonte and Everett, 1999). ICP27 plays a key role in the establishment of G1/S arrest while ICP0, along with other viral products, contributes to the inhibition of G1-specific processes such as the induction and maintenance of G1 cyclin kinase activities and consequent hyperphosphorylation of the retinoblastoma protein (Ehmann et al., 2000; Song et al., 2000; Song et al., 2001). Unlike the G1/S block, the mitotic block requires ICP0 as viruses that are deficient for this protein prevent progression into S phase, but not through the G2/M boundary (Lomonte and Everett, 1999). The mechanism by which ICP0 affects the cell cycle is not known, but it has been shown that ICP0-induced degradation of CENP-A and CENP-C, centromeric protein components of the inner plate of human kinetochores, correlates well with cell-cycle stalling during mitosis in HSV-1-infected cells (Everett et al., 1999; Lomonte et al., 2001).
The gene mutated in the human disorder ataxia-telangiectasia (A-T) encodes a protein kinase, termed ATM, that shares homology with PI-3 kinases (McKinnon, 2004). ATM plays a critical role in double-stranded DNA break (DSB)-induced cellular responses, including those induced by ionizing radiation (IR) (Kastan and Lim, 2000; McKinnon, 2004). Following DNA damage, ATM is catalytically activated through dimerization and auto-phosphorylation of S1981 (Bakkenist and Kastan, 2003). Activated ATM, in turn, phosphorylates a number of downstream targets including Brca1, Nbs1, p53, and the cell cycle checkpoint kinase Chk2 (Ahn et al., 2003; Bakkenist and Kastan, 2003; Kastan and Lim, 2000; McKinnon, 2004). ATM activates the kinase activity of Chk2 by phosphorylation of residue T68 (Matsuoka et al., 2000), although ATM-independent Chk2 phosphorylation and activation have also been reported (Ahn et al., 2000; Chen et al., 2002; Matsuoka et al., 1998; Wang et al., 2006). In a parallel signaling pathway, the structurally and functionally related kinase ATR stimulates Chk1 kinase through phosphorylation on S345 and S317 in response to UV light-induced DNA damage and agents that block replication (Liu et al., 2000; Zhao and Piwnica-Worms, 2001). Both Chk1 and Chk2 kinases phosphorylate the cell cycle regulatory phosphatase Cdc25C on residue S216 (Blasina et al., 1999; Matsuoka et al., 1998; Sanchez et al., 1997), facilitating its binding to the molecular chaperone 14-3-3 which inactivates Cdc25C through cytoplasmic sequestration (Peng et al., 1997). In addition, inactivation of Cdc25C in response to certain genotoxins by direct degradation in the ubiquitin-proteosome pathway has been reported (Chen et al., 2002). Since Cdc25C is required for activation of Cdk1/Cdc2 and cell entry into mitosis, Cdc25C inactivation is widely viewed as a key event in establishment of the G2/M checkpoint (Peng et al., 1997; Sanchez et al., 1997; Strausfeld et al., 1991).
In this report, we show that HSV-1 ICP0 expression is associated with activation of Chk2 kinase through an ATM-dependent mechanism and that Chk2 is required for ICP0-induced G2/M arrest. We found that Cdc25C was phosphorylated when Chk2 and ATM were activated by ICP0, suggesting that ICP0-induced G2/M arrest takes effect through nuclear exclusion of Cdc25C. Cellular Chk2 deficiency reduced the growth of wild-type HSV-1, but did not further diminish the already impaired growth of an ICP0-deficient mutant virus. Together, these results support a role for ICP0 activation of the ATM-Chk2-Cdc25C signaling cascade in both the establishment of HSV-1-induced G2/M arrest and HSV-1 growth.
Results
HSV-1 ICP0 activates Chk2 kinase
HSV-1 infection is associated with inhibition of host cell DNA synthesis and cell cycle progression (Hobbs and DeLuca, 1999; Lomonte and Everett, 1999). Although the HSV-1 IE gene product ICP0 has been implicated in halting cell cycle progression, the precise mechanism by which ICP0 triggers this response is not known. Given that the DNA damage checkpoint kinases, Chk1 and Chk2, regulate cell cycle arrest responses to genotoxin-induced stress, we asked whether these kinases are targeted for activation by HSV-1 ICP0. To test this possibility, 293T cells were infected with a replication-defective mutant HSV-1 virus, TOZ22R, in which all of the IE genes are deleted except ICP0 and ICP47 (Fig. 1), and Chk1 and Chk2 activation were assessed by immunoblotting with phospho-specific anti-Chk1 (S317) and anti-Chk2 (T68) antibodies. As a control virus, we used mutant QOZ22R, a derivative of TOZ22R additionally deleted for ICP0 (Fig. 1). As shown in Fig. 2A, infection of 293T cells with TOZ22R at an MOI of 2 resulted in Chk2 activation, as demonstrated by increased Chk2 T68 phosphorylation as early as 4 h post-infection. The control virus QOZ22R, however, failed to activate Chk2. Moreover, neither TOZ22R nor QOZ22R activated Chk1 kinase. Anti-ICP0 immunoblotting confirmed the expression of ICP0 in TOZ22R, but not in QOZ22R virus-infected cells. Thus, ICP0 expression was specifically associated with activation of Chk2, but not Chk1 kinase. Activation of Chk2 by ICP0 is not cell-type specific since similar results were obtained with HCT116 cells (see below) and HeLa cells (data not shown).
Fig. 1.

Virus constructs. (A) Structure of the ICP0+ (TOZ22R) and ICP0− (QOZ22R) HSV-1 strain KOS mutant viruses compared to wild type HSV-1 KOS and parental mutant virus TOZ.1. The HSV-1 genome is a linear, double-stranded DNA of ~152 kb characterized by unique long (UL) and unique short (US) segments, each flanked by inverted repeats (IRL, internal repeat of the long segment; IRS, internal repeat of the short segment; TRL, terminal repeat of the long segment; TRS, terminal repeat of the short segment). TOZ.1 contains a lacZ expression construct in the UL41 gene, has complete deletions of the essential IE genes ICP4 and ICP27, and has an internal portion of the ICP22 gene replaced with the HCMV IE promoter fused to the bovine growth hormone polyadenylation region (Krisky et al., 1998). TOZ22R was derived from TOZ.1 by substitution of the interrupted ICP22 coding sequence with the DsRed2 gene. QOZ22R was derived from TOZ22R by replacement of both copies of the ICP0 coding region with an eGFP expression construct. Black bars, gene deletions; colored bars, gene substitutions. Blue, lacZ; red, DsRed2; green, eGFP. IE genes are identified by their numbers. (B) Southern blot demonstrating deletion of the ICP0 gene in QOZ22R. Viral DNAs were digested with DraI and BamHI and the blot reacted with a probe for the ICP0 gene (top). Fragment sizes (in kbp) are indicated at the left of the autoradiograph. The target locus is illustrated underneath with the location of the probe and relevant restriction sites indicated. Plain numbers refer to HSV-1 strain 17 map positions (GenBank accession no. X14112) while italicized numbers preceded by a + sign indicate the distance in bp to the left boundary (SspI site) of the eGFP insertion in QOZ22R. D, DraI; B, BamHI; H, HpaI; St, StuI; S, SspI; A, AseI. Restriction sites that were lost in constructing the eGFP substitution of ICP0 in QOZ22R are shown between parentheses. Restriction sites derived from the eGFP donor plasmid are shown in italics. The two copies of the ICP0 locus are identical in the region represented here.
Fig. 2.

HSV-1 ICP0 activation of Chk2 kinase. (A) 293T cells were infected at an MOI of 2 with TOZ22R or QOZ22R and harvested at 0–24 hours post-infection (hpi). Cell lysates were electrophoresed and examined by immunoblotting with the indicated general or phospho-specific antibodies. The lanes are labeled by time point (hpi). (B) 293T cells were transfected with plasmid E110 expressing ICP0 or with empty plasmid. Cells were harvested 24 h later and processed for immunoblotting with the indicated antibodies.
We next investigated whether ICP0 expression is sufficient for Chk2 activation. To accomplish this, 293T cells were transfected with an ICP0 expression plasmid and 24 h post-transfection cell extracts were prepared and subjected to immunoblotting with phospho-specific (T68) Chk2 antibody. Consistent with the results obtained for TOZ22R-infected cells, lysates prepared from ICP0 transfectants displayed robust Chk2 T68 phosphorylation indicating activation, but no Chk1 S317 phosphorylation (Fig. 2B). Anti-Chk1 and anti-Chk2 immunoblotting confirmed that the steady-state levels of these proteins were not affected by ICP0 expression. Since the control cells were transfected with empty vector, Chk2 phosphorylation in the ICP0-transfected cells was not due to the introduction of foreign DNA. Thus, ICP0 alone is capable of activating the cell-cycle checkpoint kinase Chk2.
ICP0-induced G2/M arrest is Chk2-dependent and correlates with Cdc25C S216 phosphorylation
As an element of DNA damage checkpoints, the Chk2 kinase regulates cell-cycle arrest at the G2/M border through phosphorylation and inactivation of Cdc25C (Masrouha et al., 2003; McKinnon, 2004). To determine whether the ICP0-dependent G2/M arrest response to HSV infection requires Chk2, we infected HCT116 and HCT116(Chk2−/−) cells with TOZ22R or QOZ22R and monitored cell cycle inhibition by flow cytometry. At 24 h post-infection, the population of HCT116 cells in G2/M phase was increased in the TOZ22R-infected culture compared to the mock-infected control (~39% vs. ~25%; Fig. 3A, left panels), indicating virus-induced G2/M arrest. As expected, ICP0-deleted QOZ22R virus did not inhibit cell cycle progression. Notably, TOZ22R infection failed to induce the arrest response in HCT116(Chk2−/−) cells as infected and uninfected cultures displayed similar percentages of cells at the G2/M border (mock, ~20%; TOZ22R, ~23%; Fig. 3A, right panels).
Fig. 3.


Chk2 kinase requirement for ICP0-induced G2/M arrest. (A) HCT116 and HCT116(Chk2−/−) cells were mock infected or infected with TOZ22R or QOZ22R at an MOI of 2. At 24 hpi, the cells were fixed, stained with propidium iodide and subjected to flow cytometric analysis. The numbers inside the panels show the percentage of cells in G1, S, and G2/M phase. Since the total number of viable cells differs between the panels, absolute peak surfaces can not be compared between the panels. (B) Chk2-deficient HCT15 cells and HCT15 cells complemented with either wild-type Chk2 [HCT15(Chk2+)] or kinase-deficient Chk2 [HCT15(Chk2-T68A)] were mock infected or infected with TOZ22R and cell cycle progression analyzed at 24 hpi by flow cytometry. The percentage of cells in each phase is indicated inside the panels. (C) HCT116 and HCT116(Chk2−/−) cells were mock infected or infected with TOZ22R and harvested at 24 hpi for immunoblotting with the indicated antibodies.
To examine whether the kinase function of Chk2 was necessary for the Chk2-dependent G2/M block imposed by ICP0, we compared HCT15 cells, a colorectal cancer cell line deficient in Chk2 function, and HCT15 cells reconstituted with either wild-type Chk2 or a kinase-defective mutant, Chk2-T68A (Wu and Chen, 2003). Similar to our observations with HCT116 versus HCT116(Chk2−/−) cells, TOZ22R infection of HCT15 cells expressing wild-type Chk2 resulted in G2/M arrest (~39% G2/M compared to ~29% in mock-infected cells), while no increase was observed in the G2/M population of the Chk2-deficient parental HCT15 cell line after TOZ22R infection (~22% G2/M) (Fig. 3B). HCT15 cells expressing the kinase-inactive Chk2-T68A mutant showed little accumulation of cells at the G2/M border in response to TOZ22R infection (~26% G2/M, Fig. 3B), demonstrating that the kinase function of Chk2 is indispensable for ICP0-induced G2/M arrest.
To further support the conclusion from these results that ICP0-induced G2/M arrest occurs through Chk2 activation, we determined whether Cdc25C was altered in TOZ22R-infected cells. Phosphorylation of Cdc25C on residue S216 promotes binding of the molecule to 14-3-3 proteins, which results in cytoplasmic sequestration of Cdc25C and consequent failure to activate nuclear Cdk1/Cdc2, a key player in cell-cycle progression to mitosis (Graves et al., 2001). Compared to uninfected HCT116 cells, we observed a major increase in the amount of S216-phosphorylated Cdc25C in TOZ22R-infected cells (Fig. 3C). In contrast, Cdc25C phosphorylation on S216 was almost undetectable in TOZ22R-infected HCT116(Chk2−/−) cells. The total amount of Cdc25C was constant throughout and no significant decrease was apparent in the level of Cdc25A, a regulator of G1-S transition and S phase progression whose abundance is controlled mainly by Chk1 (Xiao et al., 2003). Thus, the dependence of G2/M arrest on ICP0 and Chk2 correlated with ICP0-dependent Chk2 phosphorylation and Chk2-dependent inactivation of Cdc25C in cells infected with ICP0+ virus.
ATM is required for ICP0-dependent Chk2 activation
As an upstream effector kinase, ATM stimulates the kinase activity of Chk2 by phosphorylating Chk2 on T68 following DNA damage (Ahn et al., 2000). To determine if Chk2 activation by ICP0 is dependent on ATM function, we infected HCT116 cells and three different ATM-deficient cell lines with TOZ22R and examined Chk2 phosphorylation 24 h later. Infected HCT116 cells displayed Chk2 activation along with S1981 phosphorylation of ATM (Fig. 4A). In contrast, no virus-dependent Chk2 activation was observed in the A-T lines, in agreement with previous reports using wild-type HSV-1 (Lilley et al., 2005) or HSV-2 (Shirata et al., 2005). To determine whether these results signified ATM activation by ICP0, we examined the phosphorylation status of ATM in ICP0-transfected 293T cells. The results showed increased S1981 phosphorylation of ATM in ICP0-transfected cells compared to mock-transfected cells (Fig. 4B); γ-irradiated and untreated cells included as positive and negative controls showed the expected activation of ATM by IR. ICP0-transfected and IR-treated cells also displayed increased levels of S139-phosphorylated histone H2AX (γ-H2AX), in addition to increased Chk2 phosphorylation as observed earlier. In further experiments, we found that the Chk2-dependent Cdc25C phosphorylation observed earlier in TOZ22R-infected cells (Fig. 3C) requires ATM (Fig. 4C), and that the ATM-specific inhibitor KU-55933 (Hickson et al., 2004) blocked the ICP0-induced phosphorylation of ATM, Chk2 and Cdc25C (Fig. 4D). Together, these results demonstrated that ICP0 initiates ATM-dependent signaling to cell-cycle control proteins similar to the damage-response pathway activated by IR.
Fig. 4.

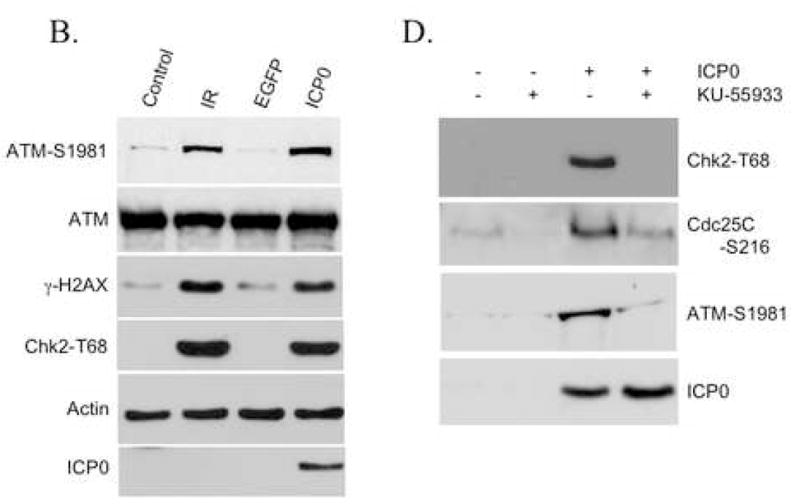
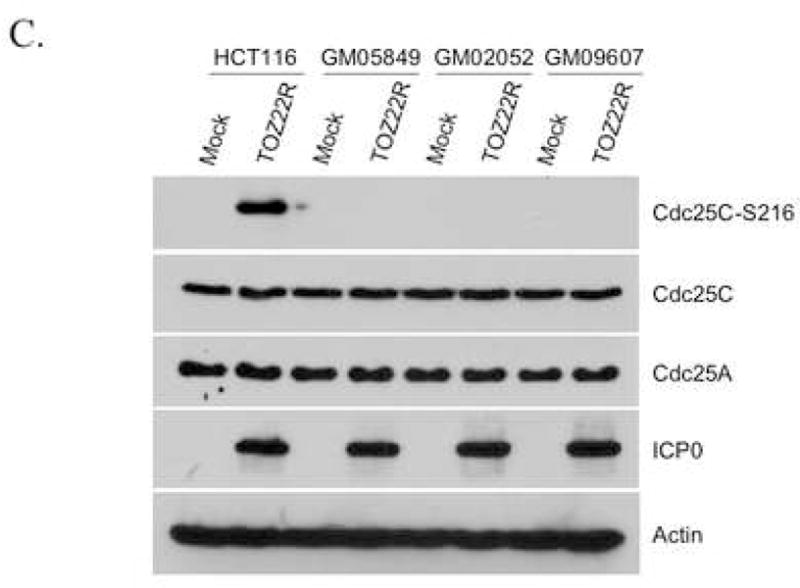
ATM requirement for ICP0-induced Chk2 activation. (A, C) HCT116 cells and A-T fibroblast lines GM05849, GM02052 and GM09607 were mock-infected or infected with TOZ22R at an MOI of 2. At 24 hpi, cells were collected and lysates analyzed by immunoblotting with general and phospho-specific antibodies, as indicated. (B) 293T cells were transfected with ICP0 expression plasmid E110 or eGFP-expressing control plasmid pEGFP-C1. As additional controls, cells were untreated (Control) or treated with ionizing radiation (IR; 5 Gy, 137Cs source). Lysates were prepared 24 h later and analyzed by immunoblotting. (D) 293T cells were incubated for 1 h with or without 10 μM KU-55933 and transfected with plasmid E110 in the presence or absence of the drug. The cells were washed and replenished with fresh medium with or without the drug. Lysates were prepared 24 h later and analyzed by immunoblotting with the indicated antibodies.
ICP0 promotes virus replication through Chk2 activation
The results to this point demonstrated that ICP0 and Chk2 are required for G2/M arrest in the absence of viral replication and that these requirements correlate with the induction of a DNA repair signaling cascade previously identified as a component of the cellular response to HSV infection (Lilley et al., 2005; Shirata et al., 2005). To relate these observations to the lytic cycle of the virus, we compared the growth of wild-type HSV-1 KOS and an ICP0-deficient, replication-competent mutant virus, ICP0:lacZ, on HCT116 and HCT116(Chk2−/−) cells infected at an MOI of 0.2. Virus yields were determined by plaque assay on Vero or ICP0-complementing 7B0-104 cells. ICP0:lacZ infection of HCT116 cells yielded approximately 100-fold less virus at 48 and 60 hpi than infection of the same cells with wild-type virus, confirming that ICP0 contributes to optimal virus growth at low MOI (Fig. 5A, B). Host-cell Chk2 deficiency reduced the growth rate and yields of the wild-type virus (Fig. 5A) to an extent similar to that observed when ICP0 was deleted from the virus (Fig. 5B). In contrast, Chk2 deficiency in the host cells did not further impair the growth of the ICP0-deficient virus (Fig. 5B). These results demonstrated that the growth-promoting effect of ICP0 requires the presence of Chk2.
Fig. 5.

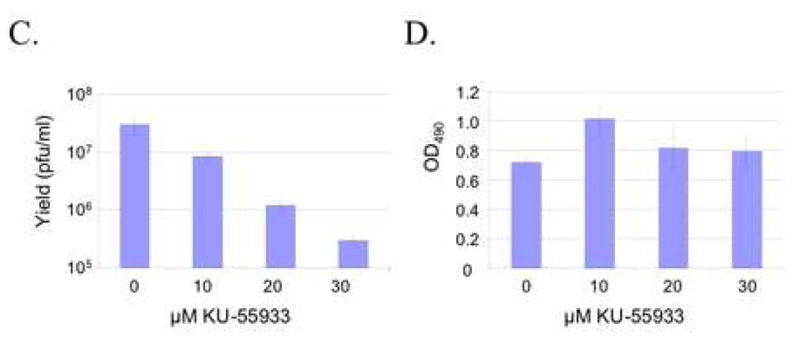
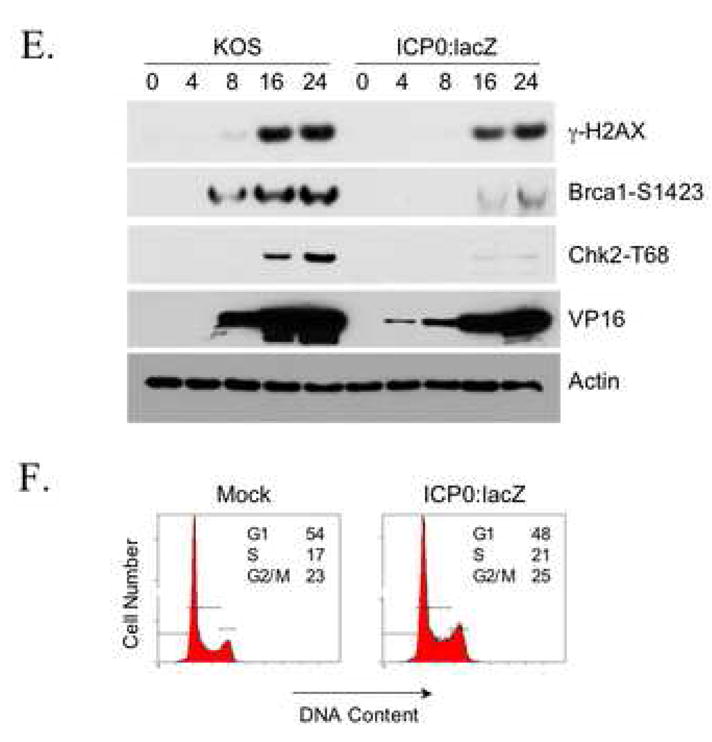
Chk2 requirement for efficient virus growth. (A, B) HCT116 and HCT116(Chk2−/−) cells were infected at an MOI of 0.2 with either wild-type HSV-1 KOS (A) or ICP0-deficient mutant virus ICP0:lacZ (B). Cells plus media were harvested at various times post-infection and viral yields assessed by plaque assays on Vero cells (A) or 7B0-104 cells (B). All infections were carried out at least in duplicate. Error bars in B and for certain data points in A are smaller than the data-point symbols and thus are not visible here. (C) AD-293 cells were incubated for 1 h with 0–30 μM KU-55933 and infected in duplicate with HSV-1 KOS (MOI=0.2) for 4 h in the continued presence or absence of the drug. Cells were washed and fed with fresh medium with or without the drug. Cells and media were harvested at 18 hpi for determination of virus yields by plaque assay on Vero cells. (D) Mock-infected AD-293 cells were incubated with KU-55933 as in (C) and cell viability measured by MTS assay at 18 hpi for 10 wells per drug concentration. (E) 293T cells were infected with either HSV-1 KOS (MOI=0.5) or ICP0:lacZ (MOI=5) and harvested at the times indicated in hpi above the lanes. Immunoblotting was performed with the antibodies shown. (D) 293T cells were mock infected or infected with ICP0:lacZ at an MOI of 2 and processed 24 h later for flow cytometry.
To determine whether activation of Chk2 correlated with virus growth, two experiments were performed. In the first, we asked whether virus production was affected by specific inhibition of ATM. AD-293 cells, an adherence-improved cell line derived from HEK293, were infected with HSV-1 KOS at an MOI of 0.2 in the presence of increasing concentrations of KU-55933, and virus yields were determined by plaque assay on Vero cells at 18 hpi. As shown in Fig. 5C, yields decreased with increasing drug concentrations. By cell viability assay on uninfected cultures treated in the same manner, we found that these decreases could not be explained by cytotoxicity of the drug (Fig. 5D). Thus these results were consistent with the suggestion that the growth-promoting effect of Chk2 on wild-type virus is dependent on ATM activity and thus likely involves the activated form of Chk2. In the second experiment, Chk2 phosphorylation was examined in 293T cells infected with ICP0:lacZ or HSV-1 KOS. Although we used a 25-fold higher MOI for ICP0:lacZ here (MOI=5) than in the growth experiment with this virus (Fig. 5B), Chk2 T68 phosphorylation was nearly undetectable (Fig. 5E). In contrast, phospho-T68 Chk2 was readily observed following wild-type virus infection performed at a 10-fold lower MOI (MOI=0.5). Likewise, phosphorylation of the ATM substrate Brca1 on residue S1423, which is required for IR-induced G2/M arrest (Xu et al., 2001), was substantially less pronounced in ICP0:lacZ-infected cells than in wild-type virus-infected cells. Viral ICP0 deficiency had a smaller effect on γ-H2AX induction, which may be attributable to compensatory activation of DNA-PK (Falck et al., 2005; Stiff et al., 2004). Infection and replication were confirmed by examination of viral VP16 levels. VP16 is carried into the cells with the infecting virus particles while de novo VP16 synthesis requires viral DNA replication. Thus the early (4 hpi) detection of VP16 in ICP0:lacZ-infected cells, but not KOS-infected cells, is consistent with the MOI difference between the two viruses, while the higher levels of VP16 at 8 and 16 hpi in KOS-infected cells are indicative of more vigorous replication by the wild-type virus. Cell-cycle analysis of ICP0:lacZ-infected cells showed no evidence of G2/M arrest at 24 hpi (mock, 23% G2/M; infected, 25% G2/M; Fig. 5F), in agreement with the lack of significant Chk2 and Brca1 phosphorylation detected under similar conditions. Together, these results supported the conclusion that ICP0 induces G2/M arrest through activation of Chk2 and that these events are linked to the role of ICP0 in promoting virus growth.
Discussion
Cell cycle arrest in response to DNA damage is one aspect of the cell’s mechanism to preserve the integrity of its genome. Temporary stalling of the cell cycle is thought to facilitate the repair of lesions prior to cell division, thereby preventing the propagation of mutations into daughter cells. Among the growing number of proteins recognized for their involvement in the response to DNA damage, ATM plays a central role in the induction of G2/M arrest following exposure of cells to ionizing radiation. IR-induced ATM activation typically leads to phosphorylation and activation of the kinase function of Chk2, among other targets, which in turn results in phosphorylation of various substrates, including Cdc25C at residue 216 [for recent reviews, see (Lavin and Kozlov, 2007; Shiloh, 2006)]. Unlike other phosphorylated forms of Cdc25C, the S216-phosphorylated version is excluded from the nucleus where the protein is required for G2/M progression. Consequently, cell cycle arrest takes effect.
HSV-1 infection, like infection by certain other viruses, activates a cellular mechanism similar to the IR-induced DNA damage response (Lilley et al., 2005; Shirata et al., 2005). It is well established that damage to the host cell DNA promotes virus reactivation from latency in HSV-1-infected sensory neurons (Fogel et al., 1979). More recently, it has been demonstrated that virus growth is impaired in mutant cells that lack key components of the DNA damage response machinery (Lilley et al., 2005). Such observations support the suggestion that HSV-1 exploits cellular response mechanisms to DNA damage for its own optimal growth (Lilley et al., 2007). Recent studies have mapped significant portions of the HSV-1-induced damage response pathway, showing early activation of ATM followed by modification of downstream targets such as Chk2 and Nbs1 (Lilley et al., 2005; Shirata et al., 2005), the latter a component of the MRN (Mre11) complex which accumulates at sites of viral DNA replication and normally functions in cellular DNA repair (Assenmacher and Hopfner, 2004; Lee and Paull, 2005; Lilley et al., 2005; Taylor and Knipe, 2004; Wilkinson and Weller, 2004). While the precise trigger of this HSV-1-responsive pathway remains unknown, candidates include cellular recognition of stalled viral replication forks or pre-replicative viral genome structures as damaged DNA, or recombination during virus replication (Lilley et al., 2005; Taylor and Knipe, 2004; Wilkinson and Weller, 2004; Wilkinson and Weller, 2005).
Much like cellular DNA damage, HSV-1 infection results in interruption of cell-cycle progression (Ehmann et al., 2000; Song et al., 2001). Hobbs and DeLuca previously reported that expression of ICP0 from a viral genome that is defective for expression of all other viral IE proteins results in p53-independent cell cycle arrest in G1/S and G2/M whereas an isogenic variant that expressed none of the IE genes showed no effect on the cell cycle (Hobbs and DeLuca, 1999). Likewise, Lomonte and Everett have shown that ICP0 plays a key role in HSV-1-mediated inhibition of cell-cycle progression through the G1/S boundary as well as mitosis (Lomonte and Everett, 1999).
Lilley and co-workers have shown that HSV-1 growth is impaired by deficiencies in the virus-induced DNA damage response, such as ATM deficiency or the absence of functional Mre11 protein (Lilley et al., 2005). While these results are at odds with the reported observation that shRNA-mediated ATM knock-down in 293T cells does not affect HSV-2 growth (Shirata et al., 2005), they are consistent with our findings that wild-type HSV-1 yields are reduced on Chk2-deficient cells and in the presence of the ATM-specific inhibitor KU-55933; we have not explored the apparent discrepancy with the HSV-2 results. It is also well established that ICP0-deficient HSV-1 growth is impaired at low multiplicities of infection (Cai and Schaffer, 1992; Hobbs and DeLuca, 1999; Lomonte and Everett, 1999). These observations suggest that HSV-1 at low MOI relies on ICP0 to trigger the damage response cascade to facilitate its growth. Our finding that cellular Chk2 deficiency impairs wild-type virus growth, but does not further impair the growth of an ICP0-deficient mutant virus (ICP0:lacZ; low-MOI infections), is consistent with this proposal. Moreover, we found that ICP0:lacZ, despite its replication competence, failed to induce significant Chk2 phosphorylation or G2/M arrest. These results confirmed that replication competence alone is not sufficient to induce the damage response pathway or pre-mitotic block. In addition, they strengthened the correlation between ICP0-dependent induction of these responses and virus growth at low MOI.
While our results indicate that the role of Chk2 in virus growth is tightly linked to ICP0, other components of the damage response appear to maintain function in the absence of ICP0. For example, it has been shown that cellular Mre11 deficiency, unlike Chk2 deficiency in our study, impairs the growth of ICP0-deficient HSV-1 (Lilley et al., 2005), suggesting that the contribution of Mre11 to virus growth is at least in part ICP0-independent. Likewise, the same study reported ATM activation following infection with replication-competent ICP0-null virus at an MOI of 3 and we observed readily detectable induction of γ-H2AX under similar conditions. Although ATM-independent mechanisms of H2AX phosphorylation have been described (Falck et al., 2005; Stiff et al., 2004), these results suggest that ATM and Chk2 activation are uncoupled in ICP0-null virus infection at elevated MOI; such uncoupling has also been observed in IR-treated cells that were defective in Nbs1-mediated recruitment of ATM to sites of DNA damage (Falck et al., 2005). Together, the evidence suggests that ICP0-null virus infection induces an incomplete damage response whose deficiencies can be overcome by dosing effects.
In sum, we show that ICP0-induced G2/M arrest requires ATM and Chk2 and correlates with activation of the ATM-Chk2-Ccd25C DNA damage response pathway. It can be speculated that cell cycle blockade promotes virus growth by diverting the cellular machinery required for normal cell-cycling processes to virus DNA replication. While it is well known that ICP0 facilitates virus growth, it remains to be shown directly that this is a consequence of cell cycle arrest mediated by ICP0 induction of the ATM-Chk2-Ccd25C pathway.
Materials and methods
Viruses and cells
Mutant viruses were derived from HSV-1 strain KOS. Replication defective recombinant TOZ22R was derived from the previously described vector TOZ.1, which is deleted for the ICP4, ICP27 and ICP22 genes and contains a lacZ expression cassette in UL41 (Krisky et al., 1998), by insertion of a DsRed2 construct at the ICP22 deletion site. QOZ22RHE.1 (referred to in this study as QOZ22R) was derived from TOZ22R by replacement of an ICP0 StuI-HpaI fragment (HSV-1 strain-17 genomic map positions 1554–5901) with a human cytomegalovirus IE promoter (HCMVp)-enhanced green fluorescent protein (eGFP) expression construct [1606-bp AseI-SspI fragment from a BglII-BamHI deleted derivative (unpublished results) of pEGFP-N1 (Clontech, Mountain View, CA)]. TOZ22R was grown on ICP4/ICP27-complementing 7B cells (Krisky et al., 1998). QOZ22R was grown on a new cell line designated 7B0-104 which was derived from 7B cells by introduction of the ICP0 gene under control of the ICP4 promoter (SacII-BamHI fragment, HSV-1 genomic map positions 131590-131399). ICP0:lacZ (kindly provided by S. Silverstein, Columbia University, NY) and HSV-1 KOS were grown and titered on Vero cells.
HCT116 and HCT116 (Chk2−/−) cells were a generous gift from B. Vogelstein (Johns Hopkins University, MD) (Jallepalli et al., 2003), HCT15, HCT15(Chk2+) and HCT15(Chk2-T68A) cells were generously provided by J. Chen (Mayo Clinic, Rochester, NY) (Wu and Chen, 2003), A-T lines GM05849, GM02052 and GM09607 were from Coriell Cell Repositories (Camden, NJ), AD-293 cells were purchased from Stratagene (La Jolla, CA; catalog #240085), and Vero (CCL-81) and 293T cells (CRL-11268) were from ATCC.
Southern blotting
Viral genomic DNA preparations were digested with DraI overnight followed by BamHI digestion for 4 to 24 h. The products were fractionated on a 1% TBE-agarose gel and blotted to a Nytran Supercharge membrane (Schleicher & Schuell BioScience, Keene, NH) by capillary transfer. The membrane was cross-linked in an UV cross-linker (Stratagene) and incubated overnight at 55°C with a probe for the ICP0 locus (Fig. 1B). The probe was prepared using a biotin random-prime labeling kit according to the manufacturer’s instructions (Pierce Chemical Co., Rockford, IL). Following hybridization, the blot was processed using the North2South chemiluminescent hybridization and detection kit (Pierce Chemical Co.).
Virus infections and growth curves
Infections with TOZ22R and QOZ22R were performed in 60-mm dishes at an MOI of 2. The cells were collected at various time points after infection for immunoblotting. Viral growth kinetics were analyzed by infection of HCT116 and HCT116(Chk2−/−) cells with HSV-1 KOS and ICP0:lacZ at MOIs of 0.2. At various times post infection, supernatants and cleared cell lysates were collected, combined, and titered on Vero (HSV-1 KOS) or 7B0-104 cells (ICP0:lacZ).
Transfections
293T cells in 60-mm dishes were transfected with 0.5 μg HCMV promoter-driven ICP0 expression plasmid E110 (kindly provided by Dr. P. Lomonte, Université de Lyon, France) or control plasmid pEGFP-C1 (Clontech) using LipofectAMINE-Plus (Invitrogen, Carlsbad, CA). Cells were collected 24 h later for immunoblotting, as described below.
ATM inhibition experiments
Cells were pretreated for 1 h with 0–30 μM KU-55933 (a kind gift from Dr. G.C.M. Smith, KuDOS Pharmaceuticals, Cambridge, UK) (Hickson et al., 2004) and infected or transfected in the continued presence of drug. Cells were washed and incubated with fresh drug until harvest for plaque assay (18 hpi) or immunoblotting (24 h post transfection). Drug toxicity was assessed by MTS assay using CellTiter96 reagents according to the manufacturer’s instructions (Promega, Madison, WI).
Antibodies
Mouse anti-ICP0 antibody was obtained from Virusys (East Coast Biologics, North Berwick, ME). General and phospho-specific Chk1 (S317) and Chk2 (T68) antibodies were purchased from Cell Signaling (Danvers, MA), ATM antibody from Epitomics (Burlingame, CA), and phospho-S1981-specific ATM antibody from Rockland Immunochemicals (Gilbertsville, PA). VP16, Cdc25A, Cdc25C, and phospho-S216-specific Cdc25C antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies specific for γ-H2AX and phospho-S1423 Brca1 were from Cell Signaling, and β-actin antibodies from Chemicon International (Temecula, CA). ICP0 and β-actin antibodies were used at a dilution of 1:5000. All other primary antibodies were used at a 1:1000 dilution. Peroxidase-conjugated secondary antibodies were obtained from Santa Cruz Biotechnology and used at a dilution of 1:2000.
Immunoblotting
Infected or transfected cells were washed with cold phosphate-buffered saline (PBS; 0.15M NaCl, 0.01M Na2HPO4, 0.01M NaH2PO4, pH 7.4) and lysed by adding 500 μl lysis buffer containing 6M urea, 2% SDS, 10% glycerol, 62.5mM Tris-HCl (pH 6.8), 5% β-mercaptoethanol, and 5 mg/ml bromophenol blue. The lysates were sonicated for 15 seconds using an ultrasonic processor (Heat Systems-Ultrasonics, Farmingdale, NY) and the supernatants were adjusted for equivalent protein concentrations, boiled, and subjected to 10% SDS-PAGE. The proteins were transferred to an Immobilon-P (Millipore Corp, Bedford, MA) membrane using a Bio-Rad Mini Trans-Blot Cell (Bio-Rad Laboratories, Hercules, CA). The membrane was blocked with Tris-buffered saline (TBS-T) containing 5% non-fat dry milk and 0.05% Tween-20 for 1 h and reacted overnight at 4°C with primary antibody. The membrane was then rinsed three times with TBS-T containing 0.5% Tween-20 and reacted with secondary antibody. Immunoblots were developed by enhanced chemi-luminescence detection according to the manufacturer’s instructions (ECL, Amersham Pharmacia Biotech, Piscataway, NJ).
Cell cycle analysis
Cells were seeded in 60-mm dishes and infected at MOIs of 2 (TOZ22R and QOZ22R) or 5 (ICP0:lacZ). Cells were harvested at the indicated time points, washed once with cold PBS, fixed with cold 95% ethanol, and incubated with 50 mg/ml RNase A for 30 minutes. Cells were stained with 50 mg/ml propidium iodide solution for 10 min and cell cycle distribution was determined by flow cytometry (Cell Quest, Becton Dickinson, Mountain View, CA). DNA content and cell cycle stage were established using WinCycle software (Phoenix Flow Systems, San Diego, CA). At least 25,000 cells were counted per experiment. The data are presented as histograms in which cell numbers are plotted against DNA content.
Acknowledgments
We thank Dr. Saul Silverstein (Columbia University) for ICP0:lacZ, Dr. Bert Vogelstein (Johns Hopkins School of Medicine) for HCT116 and HCT116(Chk2−/−) cells, Dr. Junjie Chen (Mayo Clinic, Rochester, MN) for HCT15 cell lines, and Dr. Graeme Smith (KuDOS Pharmaceuticals, Cambridge, UK) for KU-55933. We thank Tom Smithgall and Paul Robbins for critical reading of the manuscript. This work was supported by NIH grants to J.C.G. (CA119298, NS40923, DK044935, AR050733, HL066949) and R.B. (GM60945).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn J, Urist M, Prives C. Questioning the role of checkpoint kinase 2 in the p53 DNA damage response. J Biol Chem. 2003;278:20480–10489. doi: 10.1074/jbc.M213185200. [DOI] [PubMed] [Google Scholar]
- Ahn JY, Schwarz JK, Piwnica-Worms H, Canman CE. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000;60:5934–5936. [PubMed] [Google Scholar]
- Assenmacher N, Hopfner KP. MRE11/RAD50/NBS1: complex activities. Chromosoma. 2004;113:157–166. doi: 10.1007/s00412-004-0306-4. [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Blasina A, de Weyer IV, Laus MC, Luyten WH, Parker AE, McGowan CH. A human homologue of the checkpoint kinase Cds1 directly inhibits Cdc25 phosphatase. Curr Biol. 1999;9:1–10. doi: 10.1016/s0960-9822(99)80041-4. [DOI] [PubMed] [Google Scholar]
- Boutell C, Canning M, Orr A, Everett RD. Reciprocal activities between herpes simplex virus type 1 regulatory protein ICP0, a ubiquitin E3 ligase, and ubiquitin-specific protease USP7. J Virol. 2005;79:12342–12354. doi: 10.1128/JVI.79.19.12342-12354.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutell C, Everett RD. Herpes simplex virus type 1 infection induces the stabilization of p53 in a USP7- and ATM-independent manner. J Virol. 2004;78:8068–8077. doi: 10.1128/JVI.78.15.8068-8077.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Schaffer PA. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J Virol. 1992;66:2904–2915. doi: 10.1128/jvi.66.5.2904-2915.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai WZ, Schaffer PA. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J Virol. 1989;63:4579–4589. doi: 10.1128/jvi.63.11.4579-4589.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Zhang Z, Bower J, Lu Y, Leonard SS, Ding M, Castranova V, Piwnica-Worms H, Shi X. Arsenite-induced Cdc25C degradation is through the KEN-box and ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 2002;99:1990–1995. doi: 10.1073/pnas.032428899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmann GL, McLean TI, Bachenheimer SL. Herpes simplex virus type 1 infection imposes a G(1)/S block in asynchronously growing cells and prevents G(1) entry in quiescent cells. Virology. 2000;267:335–349. doi: 10.1006/viro.1999.0147. [DOI] [PubMed] [Google Scholar]
- Everett RD. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays. 2000;22:761–770. doi: 10.1002/1521-1878(200008)22:8<761::AID-BIES10>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Everett RD, Earnshaw WC, Findlay J, Lomonte P. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J. 1999;18:1526–1538. doi: 10.1093/emboj/18.6.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- Fogel M, Yamanishi K, Rapp F. Enhancement of host cell reactivation of ultraviolet-irradiated Herpes simplex virus by caffeine, hydroxyurea and 5-bromodeoxyuridine. Int J Cancer. 1979;23:657–662. doi: 10.1002/ijc.2910230511. [DOI] [PubMed] [Google Scholar]
- Graves PR, Lovly CM, Uy GL, Piwnica-Worms H. Localization of human Cdc25C is regulated both by nuclear export and 14-3-3 protein binding. Oncogene. 2001;20:1839–1851. doi: 10.1038/sj.onc.1204259. [DOI] [PubMed] [Google Scholar]
- Gu H, Liang Y, Mandel G, Roizman B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci USA. 2005;102:7571–7576. doi: 10.1073/pnas.0502658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Roizman B. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc Natl Acad Sci USA. 2003;100:8963–8968. doi: 10.1073/pnas.1533420100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- Hobbs WE, DeLuca NA. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J Virol. 1999;73:8245–8255. doi: 10.1128/jvi.73.10.8245-8255.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honess RW, Roizman B. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J Virol. 1974;14:8–19. doi: 10.1128/jvi.14.1.8-19.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallepalli PV, Lengauer C, Vogelstein B, Bunz F. The Chk2 tumor suppressor is not required for p53 responses in human cancer cells. J Biol Chem. 2003;278:20475–20479. doi: 10.1074/jbc.M213159200. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–186. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- Krisky DM, Wolfe D, Goins WF, Marconi PC, Ramakrishnan R, Mata M, Rouse RJ, Fink DJ, Glorioso JC. Deletion of multiple immediate-early genes from herpes simplex virus reduces cytotoxicity and permits long-term gene expression in neurons. Gene Ther. 1998;5:1593–1603. doi: 10.1038/sj.gt.3300766. [DOI] [PubMed] [Google Scholar]
- Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6:931–942. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- Lees-Miller SP, Long MC, Kilvert MA, Lam V, Rice SA, Spencer CA. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J Virol. 1996;70:7471–7477. doi: 10.1128/jvi.70.11.7471-7477.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc Natl Acad Sci USA. 2005;102:5844–5849. doi: 10.1073/pnas.0501916102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley CE, Schwartz RA, Weitzman MD. Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol. 2007;15:119–126. doi: 10.1016/j.tim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Lomonte P, Everett RD. Herpes simplex virus type 1 immediate-early protein Vmw110 inhibits progression of cells through mitosis and from G(1) into S phase of the cell cycle. J Virol. 1999;73:9456–9467. doi: 10.1128/jvi.73.11.9456-9467.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomonte P, Sullivan KF, Everett RD. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J Biol Chem. 2001;276:5829–5835. doi: 10.1074/jbc.M008547200. [DOI] [PubMed] [Google Scholar]
- Masrouha N, Yang L, Hijal S, Larochelle S, Suter B. The Drosophila chk2 gene loki is essential for embryonic DNA double-strand-break checkpoints induced in S phase or G2. Genetics. 2003;163:973–982. doi: 10.1093/genetics/163.3.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci USA. 2000;97:10389–10394. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ. ATM and ataxia telangiectasia. EMBO Reports. 2004;5:772–776. doi: 10.1038/sj.embor.7400210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson J, Lees-Miller SP, Everett RD. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol. 1999;73:650–657. doi: 10.1128/jvi.73.1.650-657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- Roizman B, Knipe DM. Herpes simplex viruses and their replication. In: Knipe DM, Howley PM, et al., editors. Fields Virology. 4. Lippincott, Williams & Wilkins; Philadelphia: 2001. pp. 2399–2459. [Google Scholar]
- Sacks WR, Schaffer PA. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J Virol. 1987;61:829–839. doi: 10.1128/jvi.61.3.829-839.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–1501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31:402–410. doi: 10.1016/j.tibs.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Shirata N, Kudoh A, Daikoku T, Tatsumi Y, Fujita M, Kiyono T, Sugaya Y, Isomura H, Ishizaki K, Tsurumi T. Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J Biol Chem. 2005;280:30336–30341. doi: 10.1074/jbc.M500976200. [DOI] [PubMed] [Google Scholar]
- Song B, Liu JJ, Yeh KC, Knipe DM. Herpes simplex virus infection blocks events in the G1 phase of the cell cycle. Virology. 2000;267:326–334. doi: 10.1006/viro.1999.0146. [DOI] [PubMed] [Google Scholar]
- Song B, Yeh KC, Liu J, Knipe DM. Herpes simplex virus gene products required for viral inhibition of expression of G1-phase functions. Virology. 2001;290:320–328. doi: 10.1006/viro.2001.1175. [DOI] [PubMed] [Google Scholar]
- Steiner I, Kennedy PG, Pachner AR. The neurotropic herpes viruses: herpes simplex and varicella-zoster. Lancet Neurol. 2007;6:1015–1028. doi: 10.1016/S1474-4422(07)70267-3. [DOI] [PubMed] [Google Scholar]
- Stevens JG. Human herpesviruses: a consideration of the latent state. Microbiol Rev. 1989;53:318–332. doi: 10.1128/mr.53.3.318-332.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- Strausfeld U, Labbe JC, Fesquet D, Cavadore JC, Picard A, Sadhu K, Russell P, Doree M. Dephosphorylation and activation of a p34cdc2/cyclin B complex in vitro by human CDC25 protein. Nature. 1991;351:242–245. doi: 10.1038/351242a0. [DOI] [PubMed] [Google Scholar]
- Taylor TJ, Knipe DM. Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J Virol. 2004;78:5856–5866. doi: 10.1128/JVI.78.11.5856-5866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XQ, Redpath JL, Fan ST, Stanbridge EJ. ATR dependent activation of Chk2. J Cell Physiol. 2006;208:613–619. doi: 10.1002/jcp.20700. [DOI] [PubMed] [Google Scholar]
- Wilkinson DE, Weller SK. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J Virol. 2004;78:4783–4796. doi: 10.1128/JVI.78.9.4783-4796.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DE, Weller SK. Inhibition of the herpes simplex virus type 1 DNA polymerase induces hyperphosphorylation of replication protein A and its accumulation at S-phase-specific sites of DNA damage during infection. J Virol. 2005;79:7162–7171. doi: 10.1128/JVI.79.11.7162-7171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Chen J. Autophosphorylation of checkpoint kinase 2 at serine 516 is required for radiation-induced apoptosis. J Biol Chem. 2003;278:36163–36168. doi: 10.1074/jbc.M303795200. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Chen Z, Gunasekera AH, Sowin TJ, Rosenberg SH, Fesik S, Zhang H. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem. 2003;278:21767–21773. doi: 10.1074/jbc.M300229200. [DOI] [PubMed] [Google Scholar]
- Xu B, Kim S, Kastan MB. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol Cell Biol. 2001;21:3445–3450. doi: 10.1128/MCB.21.10.3445-3450.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jones C. The bovine herpesvirus 1 immediate-early protein (bICP0) associates with histone deacetylase 1 to activate transcription. J Virol. 2001;75:9571–9578. doi: 10.1128/JVI.75.20.9571-9578.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]