

Abstract
Acute respiratory distress syndrome (ARDS) and acute lung injury are among the most frequent reasons for intensive care unit admission, accounting for approximately one-third of admissions. Mortality from ARDS has been estimated as high as 70% in some studies. Until recently, however, no targeted therapy had been found to improve patient outcome, including mortality. With the completion of the National Institutes of Health-sponsored Acute Respiratory Distress Syndrome Network low tidal volume study, clinicians now have convincing evidence that ventilation with tidal volumes lower than those conventionally used in this patient population reduces the relative risk of mortality by 21%. These data confirm the long-held suspicion that the role of mechanical ventilation for acute hypoxemic respiratory failure is more than supportive, in that mechanical ventilation can also actively contribute to lung injury. The mechanisms of the protective effects of low tidal volume ventilation in conjunction with positive end expiratory pressure are incompletely understood and are the focus of ongoing studies. The objective of the present article is to review the potential cellular mechanisms of lung injury attributable to mechanical ventilation in patients with ARDS and acute lung injury.
Keywords: acute lung injury, acute respiratory distress syndrome, alveolar epithelium, mechanical ventilation, ventilator-induced lung injury
Introduction
Since the first description of acute respiratory distress syndrome (ARDS) in 1967 [1] and the first description of the treatment of ARDS with mechanical ventilation in 1971 [2], the only therapeutic invention to convincingly demonstrate a significant reduction in mortality in patients with ARDS and acute lung injury is a lung-protective strategy of mechanical ventilation. No pharmacologic intervention has significantly reduced mortality in a large-scale trial [3]. In the recent National Institutes of Health-sponsored Acute Respiratory Distress Syndrome Network study of 861 patients [4], ventilation with 6 ml/kg (predicted body weight) and a plateau airway pressure limit of 30 cmH2O reduced mortality from 40 to 31% compared with a conventional tidal volume of 12 ml/kg and similar levels of positive end expiratory pressure (PEEP). These data confirm a long-held suspicion of many clinicians that mechanical ventilation has a double role in ARDS: life saving, but also potentially magnifying the severity of lung injury.
Despite the demonstrated benefits of tidal volume reduction, the mechanisms of the protective effect are incompletely understood. Lung injury related to mechanical ventilation ranges from macroscopic air leaks to intracellular changes in protein phosphorylation signaling cascades and gene expression [5]. The focus of the present article is to review these more subtle changes and their roles in the release of proinflammatory mediators, in changes in permeability, and in changes in ion and solute transport in ventilator-induced lung injury (VILI). Because the precise contribution of mechanical ventilation to lung injury can be difficult to discern in patients with pre-existing acute lung injury, the term ventilator-associated lung injury (VALI) is often used in place of VILI, especially in clinical studies [5].
Why are patients with ARDS at risk for VALI?
The incidence of ARDS has been estimated at 5–15/100,000 per year [6,7,8,9], but recent data suggest the incidence may be higher [10]. ARDS is a syndrome characterized by the formation of protein-rich pulmonary edema, hyaline membranes, and the influx of neutrophils into the airspace [3]. Nearly all patients with ARDS require mechanical ventilation and are therefore at risk for VALI. This appears to be due in part to the uneven distribution of lung injury and edema in ARDS.
Studies using computerized tomography scanning have demonstrated that the distribution of air and fluid in the lungs of ARDS patients is not uniform [11]. Heterogeneity in the lung results in the functional reduction of the lung volume and predisposes the lung to mechanical forces not encountered in normal physiology. These potentially pathogenetic forces include excessive tensile strain (stretch) from overdistention and interdependence, and shear stress to the epithelial cells of the airspaces due to the movement of air and fluid during tidal ventilation. The latter might be especially important when collapsed lung units are re-expanded.
Under this paradigm, regions of the injured lung exist in one of three conditions: fluid-filled or collapsed and never inflated; collapsed or fluid containing at end exhalation, but re-expanded with air on end inhalation; or aerated throughout the respiratory cycle, but prone to overdistention due to the uneven distribution of an inflated breath and interdependence. Interdependence refers to the forces exerted on an alveolus by the surrounding alveoli. In a normal lung, the alveolar distending force is equal to the transpulmonary pressure. In the injured lung, local distending forces will differ to oppose heterogeneity and to restore lung expansion [5]. For example, many years ago Mead and colleagues [12] proposed that, at a transpulmonary pressure of 30 cmH2O, the pressure across an atelectatic region surrounded by a fully expanded lung would be approximately 140 cmH2O. In the heterogeneously injured lung, strain may therefore be greater in areas where the inflated lung is adjacent to the atelectatic or fluid-filled lung due to interdependence. The potentially injurious effects of strain and shear force on lung epithelial and endothelial cells are summarized in Figure 1.
Figure 1.
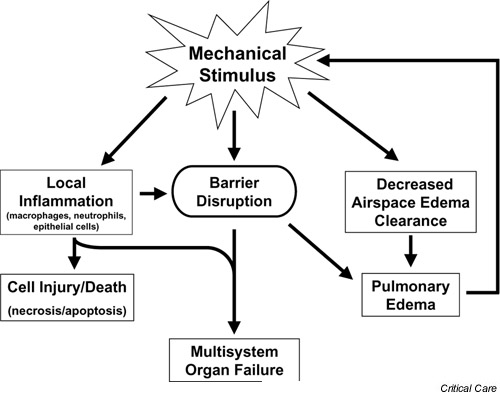
Potential mechanisms of ventilator-induced lung injury. Mechanical ventilation induces tensile strain and shear forces in the lung. These forces result in increased permeability and disruption of the alveolar–capillary barrier. Mechanical forces also induce an increase in the concentrations of proinflammatory mediators (including IL-1β, tumor necrosis factor alpha, IL-8 and IL-6) in the distal airspaces of the lung. The loss of compartmentalization in the lung results in the release of these mediators into the systemic circulation where they may play a role in end organ dysfunction. Mechanical strain also reduces the active sodium transport-dependent clearance of edema fluid from the airspaces. This potentially contributes to increased edema formation, ongoing lung volume loss, and greater ventilator-associated lung injury.
Effects of mechanical forces on lung injury
Inflammation
One potential mechanism of lung injury propagation in VALI is increased local inflammation in response to mechanical stimuli. Ranieri and colleagues [13] measured bronchoalveolar lavage (BAL) and plasma levels of several proinflammatory cytokines in 44 patients with ARDS. At the time of diagnosis, patients with ARDS were randomized to receive mechanical ventilation with a conventional strategy (mean tidal volume, 11.1 ml/kg; mean plateau airway pressure, 31 cmH2O; mean PEEP, 6.5 cmH2O) or to receive a low tidal volume, higher PEEP strategy of ventilation (mean tidal volume, 7.6 ml/kg; mean plateau airway pressure, 24.6 cmH2O; mean PEEP, 14.8 cmH2O). In the lower tidal volume group, the PEEP was set above the lower inflection point of the respiratory system pressure–volume curve (Fig. 2). Plasma and BAL cytokines were then measured serially for 36 hours. BAL fluid from patients in the lower tidal volume, higher PEEP group had significantly fewer neutrophils and lower concentrations of tumor necrosis factor alpha (TNF-α), IL-1β, IL-6, and IL-8. Plasma levels of IL-6 were also significantly lower in the patients that received protective ventilation [13]. Plasma IL-6 levels also declined in patients ventilated with low tidal volume compared with conventional tidal volume in the National Institutes of Health Acute Respiratory Distress Syndrome Network study [4]. In other clinical studies, elevations in proinflamma-tory cytokines correlate with increased patient mortality in ARDS [14,15].
Figure 2.
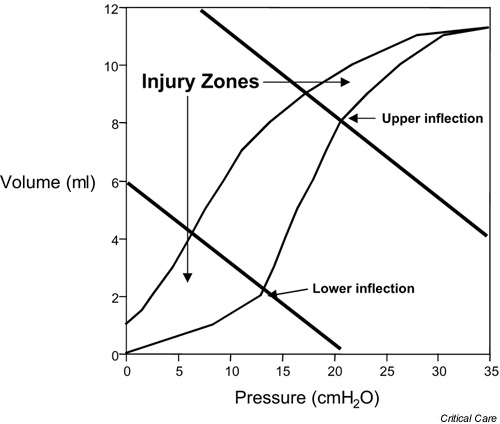
Static pressure–volume curve of a rat with aspiration-induced acute lung injury. The shaded areas indicate lung volumes where ventilator-associated lung injury is likely to be most severe based on data from experimental studies. Some clinical studies of protective ventilation in acute respiratory distress syndrome patients have used the lower inflection point of the inspiratory limb as a guide to set the positive end expiratory pressure (PEEP). The events associated with this inflection at the alveolar level are uncertain, however, and a clear inflection is not always apparent. The recent National Insitutes of Health-sponsored Acute Respiratory Distress Syndrome Network study that demonstrated a reduction in mortality did not use a pressure–volume curve to set the PEEP [4].
In experimental studies, high tidal volume, low PEEP ventilation induces the release of proinflammatory cytokines into the airspaces and bloodstream, neutrophil infiltration into the lung, and the activation of lung macrophages [16]. Tremblay and colleagues [17] found that isolated, nonperfused rat lungs ventilated with a tidal volume of 40 ml/kg without PEEP for 2 hours had large increases in lavage concentrations of TNF-α, IL-1β, IL-6, and macrophage inflammatory peptide 2. Reduction of the tidal volume to 15 ml/kg or lower reduced the lavage concentrations of these mediators, even if the end inspiratory lung volume was similar. The increase in these cytokines was greater if rats were pretreated with endotoxin, but the differences among the groups persisted. High tidal volume ventilation also increased the expression of c-fos mRNA, a transcription factor important in the early stress response [17].
Although others using a similar model have disagreed with these findings [18], the results are consistent with clinical and experimental studies of VALI. Chiumello and colleagues [19] found that injurious ventilatory strategies increased levels of TNF-α and macrophage inflammatory peptide 2 in the lung as well as the systemic circulation in a rat model of acid aspiration-induced lung injury. Ventilation of lungs isolated from rats exposed to cecal ligation-induced sepsis with a tidal volume of 20 ml/kg, without PEEP, resulted in higher BAL levels of TNF-α, IL-1β, and IL-6 compared with unventilated controls and lungs ventilated with a tidal volume of 10 ml/kg and a PEEP level of 3 cmH2O [20]. In a rat model of acid-induced acute lung injury, ventilation with a higher tidal volume (12 ml/kg) resulted in higher plasma concentrations of IL-1β compared with those with lower tidal volume ventilation (Fig. 3).
Figure 3.
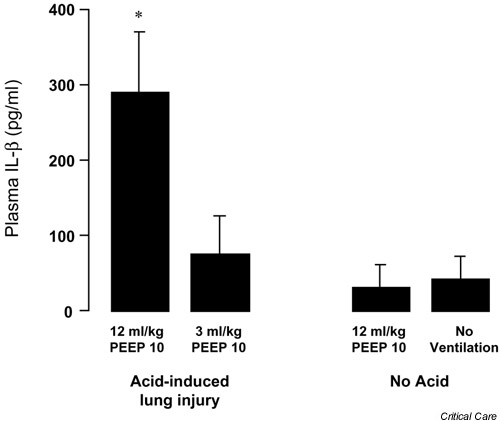
Plasma IL-1β following 4 hours of mechanical ventilation in a rat model of acid aspiration. Ventilation with a tidal volume of 12 ml/kg significantly increased plasma levels of IL-1β compared with rats ventilated with 3 ml/kg and a similar level of positive end expiratory pressure (PEEP; cmH2O) (*P < 0.05 by paired t test, mean ± standard deviation). IL-1β levels in the 3 ml/kg acid-injured group were not different from those in uninjured rats ventilated with 12 ml/kg or from uninjured, never ventilated rats (n = 5 in each acid injured group and n = 3 in the uninjured groups).
The potential importance of proinflammatory mediators in the development of VALI is also supported by data from experimental studies of the effects of anti-TNF-α antibody and IL-1 receptor antagonist on lung injury following surfactant depletion. Imai and colleagues [21] reported that the pretreatment of surfactant-depleted rabbits with anti-TNF-α antibody prior to the initiation of mechanical ventilation resulted in less severe histologic lung injury and preserved oxygenation. In a similar model, IL-1 receptor antagonist pretreatment reduced endothelial albumin permeability and neutrophil infiltration [22].
To identify the cellular source of inflammatory cytokines in VILI, Pugin and colleagues [23] cultured human alveolar macrophages on flexible silastic membranes and exposed the cells to cyclic stretch for up to 32 hours. Cyclic strain induced an increase in the secretion of IL-8. When the macrophages were pretreated with lipopolysaccharide, TNF-α and IL-6 secretion also increased to a greater extent in strained cells compared with static cultures. The authors also noted that there was an increase in nuclear NFκB in macrophages after 30 min of cyclic strain [23].
In another study by the same group, a variety of cell types, including macrophages, A549 cells, two endothelial cell lines, a bronchial epithelial cell line, and primary lung fibroblasts, were exposed to the same cyclic strain. Of these cell types, only macrophages and A549 cells secreted IL-8 in response to mechanical distention. The relative quantity of IL-8 secreted from macrophages was much greater than the amount secreted from A549 cells. It should be noted that A549 cells are a transformed cell line from a patient with bronchioloavleolar cell carcinoma, and may not respond to cyclic stretch in the same way as primary bronchial or alveolar epithelial cells. In the absence of endotoxin stimulation, cytokines were not secreted in significant amounts from any of the other cell types [24]. The importance of this finding is highlighted by clinical data that demonstrate high levels of IL-8 in pulmonary edema fluid from ventilated patients with ARDS [25,26]. The alveolar macrophage may therefore be an important stretch-responsive cell in the initiation of the inflammatory response observed in VILI. This does not, however, rule out a possible role for other cell types in the propagation of early proinflammatory signaling in VILI.
Held and colleagues [27] recently reported that mechanical stimuli mediate the release of inflammatory cytokines by increasing phosphorylation of IκB and translocation of NFκB to the nucleus. Interestingly, initiation of NFκB activation in response to mechanical stimuli may be independent of the TRL-4/lipopolysaccharide receptor and can be inhibited by corticosteroids. This finding raises the possibility that pharmacologic therapies could be targeted at ventilator-induced NFκB activation without completely inhibiting the innate immune response [5].
Barrier disruption
Integral to the current hypothesis of the pathogenesis of VILI is disruption of the alveolar–capillary barrier. Most clinical strategies of protective ventilation have focused on minimizing tensile strain and shear stress by minimizing the end inspiratory lung volume and maintaining a relatively high end expiratory lung volume (Table 1). Data from experimental studies have served as the basis for these clinical studies. In a sentinel study, Webb and Tierney [28] reported that high tidal volume ventilation induced pulmonary edema and diffuse alveolar damage histologically indistinguishable from ARDS in a rat model. They also found that high volume (high inspiratory pressure) ventilation was much less injurious when PEEP was used. Rats ventilated with a peak airway pressure of 45 cmH2O and no PEEP developed significantly more edema than rats ventilated with the same peak inspiratory pressure and a PEEP of 10 cmH2O. Of course, the tidal volume used to achieve a comparable peak pressure was considerably lower when PEEP was added (43 ml/kg compared with 15 ml/kg with PEEP).
Table 1.
Clinical studies of protective ventilation
| Study | Intervention group | Control group | Outcome |
| Acute Respiratory Distress Syndrome Network low tidal volume (861 patients) [4] | VT, 6.2 ± 0.8 ml/kg (PBW); PEEP, 9.4 ± 3.6 cmH2O | VT, 11.8 ± 0.8 ml/kg (PBW); PEEP, 8.6 ± 3.6 cmH2O | Mortality reduced from 40% to 31% with low tidal volume, more ventilator-free days, more organ failure-free days |
| Amato et al. (53 patients) [62] | VT, ~6 ml/kg; PEEP, 14.7 ± 3.9 cmH2O (PEEP set by PVC) | VT, ~12 ml/kg; PEEP, 8.7 ± 0.4 cmH2O | Mortality reduced from 71% to 38% with intervention |
| Stewart et al. (120 patients) [66] | VT, 7.0 ± 0.7 ml/kg; PEEP, 8.6 ± 3.0 cmH2O | VT, 10.7 ± 1.4 ml/kg; PEEP, 7.2 ± 3.3 cmH2O | No difference |
| Brochard et al. (116 patients) [65] | VT, 7.1 ± 1.3 ml/kg; PEEP, 10.7 ± 2.9 cmH2O | VT, 10.3 ± 1.7 ml/kg; PEEP, 10.7 ± 2.3 cmH2O | No difference |
| Brower et al. (52 patients) [64] | VT, 7.3 ± 0.7 ml/kg (PBW); PEEP, 8.3 ± 0.5 cmH2O | VT, 10.2 ± 0.7 ml/kg (PBW); PEEP, 9.5 ± 0.5 cmH2O | No difference |
VT, tidal volume; PEEP, positive end expiratory pressure; PBW, predicted body weight (0.91 [height (cm) – 152.4] + 50 for males or 0.91[height (cm) – 152.4] + 45.5 for females – note that PBW is generally up to 20% lower than dry body weight [used in other studies]); PVC, pressure–volume curve of the respiratory system.
Dreyfuss and colleagues [29] subsequently found that high tidal volume ventilation induced increased permeability edema and that transpulmonary pressure rather than peak airway pressure was the most important determinant of edema formation. Transpulmonary pressure, or the alveolar distending pressure, is analogous to lung volume. These investigators ventilated rats with a peak airway pressure of 45 cmH2O using either positive or negative pressure ventilation, and found similar increases in lung edema and protein permeability. Dreyfuss and colleagues also ventilated rats that had rubber bands applied to the chest and abdomen such that the peak airway pressure was the same but the tidal volume was reduced by roughly one-half, and they found that no edema developed. These findings correlated with scanning electron micrograph studies of lungs exposed to high distending pressures, which reported endothelial and epithelial plasma membrane breaks [29,30].
Parker and Ivey [31] expanded these findings, showing that changes in intracellular signaling also contributed to the increased permeability edema associated with high tidal volume ventilation. In isolated, perfused lungs, the administration of a β-adrenergic agonist or a phosphodiesterase inhibitor to increase intracellular cAMP resulted in significantly less lung edema and lower protein permeability during high tidal volume ventilation. Furthermore, blocking strain-activated calcium channels with gadolinium also reduced the severity of ventilator-induced pulmonary edema and protein permeability [32]. The same group also reported that inhibition of tyrosine kinase, calcium/calmodulin, or inhibition of phosphorylation of myosin light chain kinase also reduces edema and protein permeability in rats ventilated with large tidal volumes [33,34]. Phosphorylation and activation of myosin light chain kinase results in the formation of cytoskeletal stress fibers and in the formation of intercellular gaps. In vitro studies of endothelial cells have demonstrated that shear stress induces a signaling cascade culminating in myosin light chain kinase activation and the formation of stress fibers [35].
As with endothelial permeability, alveolar epithelial permeability increases with increasing lung volume. For example, increasing lung volume by the application of PEEP during mechanical ventilation results in increased clearance of inhaled 99mTc-DPTA (molecular weight, 393Da), in excess of what would be predicted from a change in surface area alone [36,37]. Alveolar epithelial permeability to albumin also increases with increasing lung volume [38,39]. In one study, the epithelium of isolated lung lobes distended with fluid to a pressure of 40 cmH2O became more permeable to albumin [39]. This correlated with an increase in the equivalent pore radius from approximately 1 to 5 nm. When entire lungs rather than isolated lobes were tested, the effect was less pronounced because regional differences in transpulmonary pressure were prevented [38]. Which experimental condition most closely approximates clinical VALI is uncertain; however, lung distention near to or exceeding the limits of normal physiology results in increased epithelial permeability even in uninjured lungs.
Ventilation of injured lungs with tidal volumes within a physiologic range can also exacerbate epithelial permeability changes. In a rat model of acid-induced acute lung injury, Frank and colleagues [40] found that ventilation with 6 ml/kg resulted in less alveolar flooding and less alveolar epithelial injury as measured by plasma levels of a type I cell-specific marker of injury (RTI40) compared with 12 ml/kg and a similar level of PEEP. This finding correlated with histologic and ultrastructural differences in airspace edema and epithelial cell injury. When the tidal volume was further reduced to 3 ml/kg, epithelial injury and airspace edema improved even more. Reducing PEEP during ventilation with a tidal volume of 12 ml/kg, such that the end inspiratory lung volume and mean airway pressures were similar to the 6 ml/kg group, did not prevent epithelial injury or edema [40]. Similar findings have also been reported following surfactant depletion. In this model, tidal volume reduction prevented airspace edema formation and preserved oxygenation, suggesting preserved epithelial barrier function. Interestingly, when surfactant-depleted animals were ventilated with high-frequency oscillatory ventilation (HFOV), edema and histologic injury were further reduced [41,42].
Studies of alveolar epithelial type II cells grown on silastic membranes have helped to characterize the mechanical properties of these cells and have provided insight into the mechanisms of cell injury in VILI. In one study, increasing the duration, amplitude, or frequency of the cyclic strain increased the plasma membrane injury and cell death [43]. Most cell injury occurred within 5 min. If small amplitude deformation was superimposed on basal tonic strain, there was less membrane disruption and cell death compared with a large amplitude stain to same peak level. In this study, the rate of cellular deformation during a single strain did not affect the plasma membrane injury [43]. In another study, plasma membrane disruption induced by cyclic mechanical strain in vitro was dependent on the rate of plasma membrane trafficking to the cell surface. Inhibition of cytoskeletal remodeling had little impact on the cell injury, indicating that mechanical disruption of the cytoskeleton is less important than plasma membrane disruption [44,45]. Although these data do not exclude strain-induced signaling through the cytoskeleton as an important mechanism of VILI, they support the hypothesis that membrane disruption and impaired lipid trafficking may be a major mechanism.
Disruption of the alveolar–capillary barrier is an important mechanism responsible for the formation of alveolar edema, which is characteristic of VILI. This loss of compartmentalization combined with the ventilator-induced amplification of inflammation in acute lung injury may also be an important mechanism of multisystem organ failure, one of the most common causes of death in ARDS (Fig. 1). Several investigators have shown that increased permeability of the alveolar–capillary barrier correlated with the increased levels of proinflammatory mediators in the systemic circulation. von Bethmann and colleagues [46] reported that, in an isolated perfused murine lung model, ventilation with a transpulmonary pressure of 25 cmH2O compared with 10 cmH2O lead to a significant increase in the concentrations of both TNF-α and IL-6 in the perfusate. In patients with ARDS, concentrations of TNF-α, IL-1β and IL-6 were higher in the arterial blood (obtained via a wedged pulmonary artery catheter) compared with mixed venous blood, suggesting that the lungs were a major source of systemic proinflammatory cytokines in these patients [47]. Several recent studies evaluated the influence of mechanical ventilation strategy on the translocation of bacteria from the lung into the bloodstream [48,49,50]. After intra-tracheal instillation of bacteria, animals ventilated with a higher tidal volume and minimal PEEP (0–3 cmH2O) develop more bacteremia more frequently and more rapidly than animals ventilated with protective strategies.
The release of proinflammatory cytokines into the systemic circulation may have important consequences. In the National Institutes of Health Acute Respiratory Distress Syndrome Network low tidal volume study, as already discussed, plasma levels of IL-6 in the 6 ml/kg tidal volume group were significantly lower than in the conventional tidal volume group. This result was associated with a greater number of organ failure-free days, although this outcome variable may not be independent of mortality. In an experimental study of acid aspiration, Imai and colleagues [51] reported that 8 hours of mechanical ventilation with an injurious strategy led to epithelial cell apoptosis in the kidney and small intestine, and to increased plasma creatinine levels. An increase in distal ileal permeability has also been reported in rats ventilated with a tidal volume of 20 ml/kg compared with 10 ml/kg [52]. Taken together, these data suggest a role for VALI in the pathogenesis of multisystem organ failure.
Reduced airspace edema clearance
The presence of edema fluid in the airspaces is both an effect of lung injury and a potential mechanism by which VILI is amplified. Edema fluid fills alveoli and promotes airspace collapse by inactivating surfactant and filling airways [53,54,55]. This loss of lung volume leads to heterogeneity of the lung, resulting in even greater overdistention of the remaining lung units [56]. Therefore, if the clearance of edema fluid from the distal airspaces is reduced, a vicious cycle of airspace edema leading to greater lung overdistention and shear stress will ensue (Fig. 1). For example, flooding distal lung units of rats with saline was found to act synergistically with high tidal volume ventilation to increase endothelial permeability to albumin [57]. In this study, the authors also found that permeability to albumin increased as the respiratory system compliance decreased, suggesting that a smaller lung volume was ventilated. As ventilated lung volume decreased, more injury resulted [57].
The clearance of edema from the airspaces requires the active transport of sodium across the epithelium. Lecuona and colleagues [58] reported that high tidal volume ventilation induced a reduction in energy-dependent sodium transport. Using alveolar type II cells isolated from rats ventilated with a tidal volume of either 30 or 40 ml/kg, these authors found that sodium-potassium ATPase activity was reduced compared with rats ventilated with a lower tidal volume (10 ml/kg). In another study, airspace edema clearance in lungs isolated from rats ventilated for 40 min with a tidal volume of 40 ml/kg was reduced by approximately 50%. Instilling the airspaces of the isolated lungs with a β-adrenergic agonist restored the rate of airspace edema clearance by increasing the activity and quantity of sodium-potassium ATPase in the basolateral membrane. This effect was blocked by disrupting the microtuble assembly with colchicine, suggesting that it is the translocation of sodium-potassium ATPase from intracellular pools to the plasma membrane that accounts for much of the effect [59]. In an in vivo rat model of acute lung injury, tidal volume reduction from 12 to 3 ml/kg resulted in greater preservation of airspace fluid transport (Fig. 3) [40]. In clinical studies of ARDS patients, preserved airspace fluid clearance correlates with improved survival [60,61]. Taken together, these data suggest that pharmacologic therapy targeted at upregulating airspace fluid clearance may have a role in the prevention of VALI, although further study is necessary.
Prevention of VALI
Prospective clinical studies of patients with ARDS and acute lung injury have demonstrated that protective ventilation strategies incorporating relatively high levels of PEEP and low tidal volumes reduce mortality [4,62,63]. It is clear from the most convincing of these studies [4] that excessive end inspiratory lung volume is a critical mediator of VALI (Table 1). In this multicenter study, ventilation with similar levels of PEEP but with a tidal volume of 6 ml/kg (predicted body weight) was associated with 31% patient mortality, while ventilation with the conventional 12 ml/kg was associated with 40% mortality. The plateau airway pressure in the low tidal volume group was required to be less than 30 cmH2O (mean, 25 ± 6 cmH2O), compared with a mean plateau airway pressure of 33 ± 8 cmH2O in the conventional tidal volume group. This highlights the fact that the primary difference between the groups was in end inspiratory lung volume.
Furthermore, the mortality benefit persisted regardless of the initial respiratory system compliance. In a smaller study of 53 patients by Amato and colleagues [62], limiting the tidal volume to less than 6 ml/kg with the PEEP set above the lower inflection point of the static pressure–volume curve (Fig. 2) also reduced mortality, although mortality in the conventional ventilation group in this study was high (71% compared with 38% in the protective ventilation group). Other small studies testing intermediate tidal volumes have not demonstrated a mortality benefit (Table 1) [64,65,66]. These data indicate that limiting the tidal volume to 6 ml/kg in ARDS and acute lung injury patients reduces mortality, but smaller incremental reductions in tidal volume may not. Furthermore, in the Acute Respiratory Distress Syndrome Network study, the mortality benefit appears to be primarily attributable to tidal volume reduction as the PEEP levels were comparable (Table 1).
The other common feature to strategies of protective ventilation is a relatively high level of PEEP. Based on experimental data, the PEEP may minimize VILI by preserving the lung volume, by preserving surfactant function, and by reducing shear forces created by the opening and collapse of airways and alveoli. The best method to select a PEEP level for a given patient with ARDS is not yet known. Although some studies have used the pressure–volume curve of the respiratory system to set the PEEP above the lower inflection point (Fig. 2), others have used arbitrary scales of PEEP. Both strategies, when combined with low tidal volume ventilation, reduce mortality from ARDS [4,62].
In the study of Amato and colleagues [62], a PEEP level greater than the lower inflection point and a recruitment maneuver at the start of the study were used. In the Acute Respiratory Distress Syndrome Network study, the PEEP was set according to a predetermined scale and not according to the pressure–volume curve. A predetermined scale was used because the relationship between the shape of the pressure–volume curve and events at the alveolar level is affected by numerous factors and is not obvious in every patient [67,68,69,70]. In a subsequent study by the Acute Respiratory Distress Syndrome Network that combined the low tidal volume protocol with a scale incorporating higher PEEP levels compared with the previously tested scale [4], no additional mortality benefit was observed [71]. Other methods of setting the PEEP, including adjusting the PEEP based on the shape of a constant flow compliance curve, are the subject of ongoing studies [72].
Based on the recent findings that tidal volume reduction is protective in ARDS, there is renewed interest in HFOV. Combined with a strategy of lung volume maintenance, HFOV would potentially prevent excessive end inspiratory lung volume and would maintain sufficient end expiratory lung volume to a greater degree than conventional ventilation. Preliminary data suggest that this method of ventilation is safe in adults [73,74]. Ongoing studies are comparing HFOV combined with lung volume maintenance to low tidal volume ventilation in children and adults. Previous negative studies of HFOV in children have not always included a protocol for the maintenance of lung volume [75].
Recognition of patients at risk for VALI
Although some workers have criticized the current definition of acute lung injury and ARDS (Table 2) for not including a measure of compliance, or for other reasons, it is of the utmost importance to realize that the current definition was used to select patients for the recent randomized, controlled Acute Respiratory Distress Syndrome Network study. The patients who meet the clinical criteria of the current definition therefore benefited from the Acute Respiratory Distress Syndrome Network protocol of low tidal volume and relatively high PEEP, regardless of lung compliance or the risk factor for ARDS [4,76]. Whether one agrees with the current definition of acute lung injury and ARDS should not affect the decision to initiate this ventilation protocol in a patient meeting the criteria presented in Table 2.
Table 2.
Clinical definition of acute respiratory distress syndrome and acute lung injury [76]
| Clinical feature | Acute respiratory distress syndrome | Acute lung injury |
| Timing | Acute onset | Same |
| Chest radiograph | Bilateral infiltrates | Same |
| Exclusion of alternative diagnosis | Pulmonary artery wedge pressure ≤ 18 mmHg OR absence of clinical evidence for left atrial hypertension | Same |
| Oxygenation | PaO2 : FiO2 ratio ≤ 200 | PaO2 : FiO2 ratio ≤ 300 |
PaO2 : FiO2, ratio of arterial partial pressure for oxygen to the fraction of oxygen in the inspired air.
Summary
For the first time, clinicians have a well-defined therapeutic intervention that reduces patient mortality from acute lung injury and ARDS. Although the precise mechanisms of the protective effect of low tidal volume ventilation are not fully understood, clinical and experimental data suggest that excessive strain and airspace epithelial shear stress amplify lung inflammation, exacerbate barrier disruption, and promote ongoing pulmonary edema formation. Early recognition of patients with acute lung injury and ARDS (Table 2), and the implementation of protective ventilation is critical if the mortality benefits observed in the recent Acute Respiratory Distress Syndrome Network study are to be realized in clinical practice.
Competing interests
None declared.
Abbreviations
ARDS = acute respiratory distress syndrome; BAL = bronchoalveolar lavage; HFOV = high-frequency oscillatory ventilation; IL = interleukin; NF = nuclear factor; PEEP = positive end expiratory pressure; TNF-α = tumor necrosis factor alpha; VALI = ventilator-associated lung injury; VILI = ventilator-induced lung injury.
See related Commentary http://ccforum.com/content/7/3/209
References
- Ashbaugh D, Bigelow D, Petty T, Levine B. Acute respiratory distress syndrome in adults. Lancet. 1967;2:319–323. doi: 10.1016/s0140-6736(67)90168-7. [DOI] [PubMed] [Google Scholar]
- Petty T, Ashbaugh D. The adult respiratory distress syndrome: clinical features, factors influencing prognosis, and principles of management. Chest. 1971;60:233–239. doi: 10.1378/chest.60.3.233. [DOI] [PubMed] [Google Scholar]
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- ARDS Network Investigators Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- Frank J, Imai Y, Slutsky A. Pathogenesis of ventilator-induced lung injury. In: Matthay M, editor. In Acute Respiratory Distress Syndrome. Marcel Dekker, New York; [Google Scholar]
- Thomsen GE, Morris AH. Incidence of the adult respiratory distress syndrome in the state of Utah. Am J Respir Crit Care Med. 1995;152:965–971. doi: 10.1164/ajrccm.152.3.7663811. [DOI] [PubMed] [Google Scholar]
- Luhr OR, Antonsen K, Karlsson M, Aardal S, Thorsteinsson A, Frostell CG, Bonde J. Incidence and mortality after acute respiratory failure and acute respiratory distress syndrome in Sweden, Denmark, and Iceland. The ARF Study Group. Am J Respir Crit Care Med. 1999;159:1849–1861. doi: 10.1164/ajrccm.159.6.9808136. [DOI] [PubMed] [Google Scholar]
- Hudson LD, Steinberg KP. Epidemiology of acute lung injury and ARDS. Chest. 1999;116:74S–82S. doi: 10.1378/chest.116.suppl_1.74s-a. [DOI] [PubMed] [Google Scholar]
- Arroliga AC, Ghamra ZW, Trepichio AP, Trepichio PP, Komara JJ, Jr, Smith A, Wiedemann HP. Incidence of ARDS in an Adult Population of Northeast Ohio. Chest. 2002;121:1972–1976. doi: 10.1378/chest.121.6.1972. [DOI] [PubMed] [Google Scholar]
- Rubenfeld G, Caldwell E, Martin D, Steinberg K, Hudson L. The incidence of acute lung injury in adults in the U.S.: Results of the King County lung injury project [abstract]. Am J Respir Crit Care Med. 2002;165:A219. [Google Scholar]
- Gattinoni L, Caironi P, Pelosi P, Goodman LR. What has computed tomography taught us about the acute respiratory distress syndrome? Am J Respir Crit Care Med. 2001;164:1701–1711. doi: 10.1164/ajrccm.164.9.2103121. [DOI] [PubMed] [Google Scholar]
- Mead J, Takishima T, Leith D. Stress distribution in lungs: a model of pulmonary elasticity. J Appl Physiol. 1970;28:596–608. doi: 10.1152/jappl.1970.28.5.596. [DOI] [PubMed] [Google Scholar]
- Ranieri VM, Suter PM, Tortorella C, De Tullio R, Dayer JM, Brienza A, Bruno F, Slutsky AS. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA. 1999;282:54–61. doi: 10.1001/jama.282.1.54. [DOI] [PubMed] [Google Scholar]
- Meduri GU, Headley S, Kohler G, Stentz F, Tolley E, Umberger R, Leeper K. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest. 1995;107:1062–1073. doi: 10.1378/chest.107.4.1062. [DOI] [PubMed] [Google Scholar]
- Goodman RB, Strieter RM, Martin DP, Steinberg KP, Milberg JA, Maunder RJ, Kunkel SL, Walz A, Hudson LD, Martin TR. Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1996;154:602–611. doi: 10.1164/ajrccm.154.3.8810593. [DOI] [PubMed] [Google Scholar]
- Imanaka H, Shimaoka M, Matsuura N, Nishimura M, Ohta N, Kiyono H. Ventilator-induced lung injury is associated with neutrophil infiltration, macrophage activation, and TGF-beta 1 mRNA upregulation in rat lungs. Anesthesia Analgesia. 2001;92:428–436. doi: 10.1097/00000539-200102000-00029. [DOI] [PubMed] [Google Scholar]
- Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest. 1997;99:944–952. doi: 10.1172/JCI119259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss D, Saumon G. Production of inflammatory cytokines in ventilator-induced lung injury: a reappraisal. Am J Respir Crit Care Med. 2001;163:1176–1180. doi: 10.1164/ajrccm.163.5.2006053. [DOI] [PubMed] [Google Scholar]
- Chiumello D, Pristine G, Slutsky AS. Mechanical ventilation affects local and systemic cytokines in an animal model of acute respiratory distress syndrome. Am J Respir Crit Care Med. 1999;160:109–116. doi: 10.1164/ajrccm.160.1.9803046. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Malloy J, McCaig L, Yao LJ, Joseph M, Lewis J, Veldhuizen R. Mechanical ventilation of isolated septic rat lungs: effects on surfactant and inflammatory cytokines. J Appl Physiol. 2001;91:811–820. doi: 10.1152/jappl.2001.91.2.811. [DOI] [PubMed] [Google Scholar]
- Imai Y, Kawano T, Iwamoto S, Nakagawa S, Takata M, Miyasaka K. Intratracheal anti-tumor necrosis factor-alpha antibody attenuates ventilator-induced lung injury in rabbits. J Appl Physiol. 1999;87:510–515. doi: 10.1152/jappl.1999.87.2.510. [DOI] [PubMed] [Google Scholar]
- Narimanbekov IO, Rozycki HJ. Effect of IL-1 blockade on inflammatory manifestations of acute ventilator-induced lung injury in a rabbit model. Exp Lung Res. 1995;21:239–254. doi: 10.3109/01902149509068830. [DOI] [PubMed] [Google Scholar]
- Pugin J, Dunn I, Jolliet P, Tassaux D, Magnenat JL, Nicod LP, Chevrolet JC. Activation of human macrophages by mechanical ventilation in vitro. Am J Physiol. 1998;275:L1040–L1050. doi: 10.1152/ajplung.1998.275.6.L1040. [DOI] [PubMed] [Google Scholar]
- Dunn I, Pugin J. Mechanical ventilation of various human lung cells in vitro: identification of the macrophage as the main producer of inflammatory mediators. Chest. 1999;116(1 suppl):95S–97S. doi: 10.1378/chest.116.suppl_1.95s. [DOI] [PubMed] [Google Scholar]
- Kurdowska A, Miller EJ, Noble JM, Baughman RP, Matthay MA, Brelsford WG, Cohen AB. Anti-IL-8 autoantibodies in alveolar fluid from patients with the adult respiratory distress syndrome. J Immunol. 1996;157:2699–2706. [PubMed] [Google Scholar]
- Miller EJ, Cohen AB, Matthay MA. Increased interleukin-8 concentrations in the pulmonary edema fluid of patients with acute respiratory distress syndrome from sepsis. Crit Care Med. 1996;24:1448–1454. doi: 10.1097/00003246-199609000-00004. [DOI] [PubMed] [Google Scholar]
- Held HD, Boettcher S, Hamann L, Uhlig S. Ventilation-induced chemokine and cytokine release is associated with activation of nuclear factor-KB and is blocked by steroids. Am J Respir Crit Care Med. 2001;163:711–716. doi: 10.1164/ajrccm.163.3.2003001. [DOI] [PubMed] [Google Scholar]
- Webb H, Tierney D. Experimental pulmonary edema due to intermittent positive pressure ventilation. Protection by positive end-expiratory pressure. Am Rev Respir Dis. 1974;110:556–565. doi: 10.1164/arrd.1974.110.5.556. [DOI] [PubMed] [Google Scholar]
- Dreyfuss D, Soler P, Basset G, Saumon G. High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis. 1988;137:1159–1164. doi: 10.1164/ajrccm/137.5.1159. [DOI] [PubMed] [Google Scholar]
- West JB, Tsukimoto K, Mathieu-Costello O, Prediletto R. Stress failure in pulmonary capillaries. J Appl Physiol. 1991;70:1731–1742. doi: 10.1152/jappl.1991.70.4.1731. [DOI] [PubMed] [Google Scholar]
- Parker JC, Ivey CL. Isoproterenol attenuates high vascular pressure-induced permeability increases in isolated rat lungs. J Appl Physiol. 1997;83:1962–1967. doi: 10.1152/jappl.1997.83.6.1962. [DOI] [PubMed] [Google Scholar]
- Parker JC, Ivey CL, Tucker JA. Gadolinium prevents high airway pressure-induced permeability increases in isolated rat lungs. J Appl Physiol. 1998;84:1113–1118. doi: 10.1063/1.368111. [DOI] [PubMed] [Google Scholar]
- Parker JC, Ivey CL, Tucker A. Phosphotyrosine phosphatase and tyrosine kinase inhibition modulate airway pressure-induced lung injury. J Appl Physiol. 1998;85:1753–1761. doi: 10.1152/jappl.1998.85.5.1753. [DOI] [PubMed] [Google Scholar]
- Parker JC. Inhibitors of myosin light chain kinase and phosphodiesterase reduce ventilator-induced lung injury. J Appl Physiol. 2000;89:2241–2248. doi: 10.1152/jappl.2000.89.6.2241. [DOI] [PubMed] [Google Scholar]
- Wysolmerski RB, Lagunoff D. Involvement of myosin light-chain kinase in endothelial cell retraction. Proc Natl Acad Sci USA. 1990;87:16–20. doi: 10.1073/pnas.87.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA, van der Zee H, Line BR, Malik AB. Relationship of end-expiratory pressure, lung volume, and 99mTc-DTPA clearance. J Appl Physiol. 1987;63:1586–1590. doi: 10.1152/jappl.1987.63.4.1586. [DOI] [PubMed] [Google Scholar]
- Marks JD, Luce JM, Lazar NM, Wu JN, Lipavsky A, Murray JF. Effect of increases in lung volume on clearance of aerosolized solute from human lungs. J Appl Physiol. 1985;59:1242–1248. doi: 10.1152/jappl.1985.59.4.1242. [DOI] [PubMed] [Google Scholar]
- Egan EA. Lung inflation, lung solute permeability, and alveolar edema. J Appl Physiol. 1982;53:121–125. doi: 10.1152/jappl.1982.53.1.121. [DOI] [PubMed] [Google Scholar]
- Egan EA. Responseof alveolar epithelial solute permeability to changes in lung inflation. J Appl Physiol. 1980;49:1032–1036. doi: 10.1152/jappl.1980.49.6.1032. [DOI] [PubMed] [Google Scholar]
- Frank JA, Gutierrez JA, Jones KD, Allen L, Dobbs L, Matthay MA. Low tidal volume reduces epithelial and endothelial injury in acid-injured rat lungs. Am J Respir Crit Care Med. 2002;165:242–249. doi: 10.1164/ajrccm.165.2.2108087. [DOI] [PubMed] [Google Scholar]
- Imai Y, Nakagawa S, Ito Y, Kawano T, Slutsky AS, Miyasaka K. Comparison of lung protection strategies using conventional and high-frequency oscillatory ventilation. J Appl Physiol. 2001;91:1836–1844. doi: 10.1152/jappl.2001.91.4.1836. [DOI] [PubMed] [Google Scholar]
- Simma B, Luz G, Trawoger R, Hormann C, Klima G, Kreczy A, Baum M. Comparison of different modes of high-frequency ventilation in surfactant-deficient rabbits. Pediatr Pulmonol. 1996;22:263–270. doi: 10.1002/(SICI)1099-0496(199610)22:4<263::AID-PPUL6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Tschumperlin DJ, Oswari J, Margulies AS. Deformation-induced injury of alveolar epithelial cells. Effect of frequency, duration, and amplitude. Am J Respir Crit Care Med. 2000;162:357–362. doi: 10.1164/ajrccm.162.2.9807003. [DOI] [PubMed] [Google Scholar]
- Vlahakis NE, Schroeder MA, Pagano RE, Hubmayr RD. Deformation-induced lipid trafficking in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;280:L938–L946. doi: 10.1152/ajplung.2001.280.5.L938. [DOI] [PubMed] [Google Scholar]
- Vlahakis N, Schroeder MA, Pagano RE, Hubmayr RD. The role of the cytoskeleton in deformation induced alveolar epithelial wounding and repair [abstract]. Am J Respir Crit Care Med. 2002;165:A379. [Google Scholar]
- von Bethmann AN, Brasch F, Nusing R, Vogt K, Volk HD, Muller KM, Wendel A, Uhlig S. Hyperventilation induces release of cytokines from perfused mouse lung. Am J Respir Crit Care Med. 1998;157:263–272. doi: 10.1164/ajrccm.157.1.9608052. [DOI] [PubMed] [Google Scholar]
- Douzinas E, Tsidemiadou P, Pitaridis M, Andrianakis I, Bobota-Chloraki A, Katsouyanni K, Sfyras D, Malagari K, Roussos C. The regional production of cytokines and lactate in sepsis-related multiple organ failure. Am J Respir Crit Care Med. 1997;155:53–59. doi: 10.1164/ajrccm.155.1.9001289. [DOI] [PubMed] [Google Scholar]
- Nahum A, Hoyt J, Schmitz L, Moody J, Shapiro R, Marini JJ. Effect of mechanical ventilation strategy on dissemination of intra-tracheally instilled Escherichia coli in dogs. Crit Care Med. 1997;25:1733–1743. doi: 10.1097/00003246-199710000-00026. [DOI] [PubMed] [Google Scholar]
- Verbrugge SJ, Sorm V, van't Veen A, Mouton JW, Gommers D, Lachmann B. Lung overinflation without positive end-expiratory pressure promotes bacteremia after experimental Kleb-siella pneumoniae inoculation. Intensive Care Med. 1998;24:172–177. doi: 10.1007/s001340050541. [DOI] [PubMed] [Google Scholar]
- Savel RH, Yao EC, Gropper MA. Protective effects of low tidal volume ventilation in a rabbit model of Pseudomonas aeruginosa-induced acute lung injury. Crit Care Med. 2001;29:392–398. doi: 10.1097/00003246-200102000-00032. [DOI] [PubMed] [Google Scholar]
- Imai Y, Kajikawa O, Frevert C, de Perrot M, Fischer S, Parodo J, Edwards V, Cutz E, Zhang H, Ranieri M, Liu M, Keshavjee S, Marshall J, Martin T, Slutsky A. Injurious ventilatory strategies enhance organ apoptosis in rabbits [abstract]. Am J Respir Crit Care Med. 2001;163:667A. [Google Scholar]
- Guery B, Neviere R, Fialdes P, Chidiac C, Rousel-Delvallez M, Marchandise X, Beaucaire G. Mechanical ventilation regimen induces intestinal permeability changes in a rat model [abstract]. Am J Respir Crit Care Med. 2002;165:A505. [Google Scholar]
- Yukitake K, Brown CL, Schlueter MA, Clements JA, Hawgood S. Surfactant apoprotein A modifies the inhibitory effect of plasma proteins on surfactant activity in vivo. Pediatr Res. 1995;37:21–25. [PubMed] [Google Scholar]
- Wyszogrodski I, Kyei-Aboagye K, Taeusch HW, Jr, Avery ME. Surfactant inactivation by hyperventilation: conservation by end-expiratory pressure. J Appl Physiol. 1975;38:461–466. doi: 10.1152/jappl.1975.38.3.461. [DOI] [PubMed] [Google Scholar]
- Veldhuizen RA, Welk B, Harbottle R, Hearn S, Nag K, Petersen N, Possmayer F. Mechanical ventilation of isolated rat lungs changes the structure and biophysical properties of surfactant. J Appl Physiol. 2002;92:1169–1175. doi: 10.1152/japplphysiol.00697.2001. [DOI] [PubMed] [Google Scholar]
- Martynowicz MA, Minor TA, Walters BJ, Hubmayr RD. Regional expansion of oleic acid-injured lungs. Am J Respir Crit Care Med. 1999;160:250–258. doi: 10.1164/ajrccm.160.1.9808101. [DOI] [PubMed] [Google Scholar]
- Dreyfuss D, Martin-Lefevre L, Saumon G. Hyperinflation-induced lung injury during alveolar flooding in rats: effect of perfluoro-carbon instillation. Am J Respir Crit Care Med. 1999;159:1752–1757. doi: 10.1164/ajrccm.159.6.9805018. [DOI] [PubMed] [Google Scholar]
- Lecuona E, Saldias F, Comellas A, Ridge K, Guerrero C, Sznajder JI. Ventilator-associated lung injury decreases lung ability to clear edema in rats. Am J Respir Crit Care Med. 1999;159:603–609. doi: 10.1164/ajrccm.159.2.9805050. [DOI] [PubMed] [Google Scholar]
- Saldias FJ, Lecuona E, Comellas AP, Ridge KM, Rutschman DH, Sznajder JI. Beta-adrenergic stimulation restores rat lung ability to clear edema in ventilator-associated lung injury. Am J Respir Crit Care Med. 2000;162:282–287. doi: 10.1164/ajrccm.162.1.9809058. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Wiener-Kronish JP. Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. Am Rev Respir Dis. 1990;142:1250–1257. doi: 10.1164/ajrccm/142.6_Pt_1.1250. [DOI] [PubMed] [Google Scholar]
- Ware L, Matthay M. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- Amato MB, Barbas CS, Medeiros DM, Magaldi RB, Schettino GP, Lorenzi-Filho G, Kairalla RA, Deheinzelin D, Munoz C, Oliveira R, Takagaki TY, Carvalho CR. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med. 1998;338:347–354. doi: 10.1056/NEJM199802053380602. [DOI] [PubMed] [Google Scholar]
- Hickling KG, Walsh J, Henderson S, Jackson R. Low mortality rate in adult respiratory distress syndrome using low-volume, pressure-limited ventilation with permissive hypercapnia: a prospective study. Crit Care Med. 1994;22:1568–1578. doi: 10.1097/00003246-199422100-00011. [DOI] [PubMed] [Google Scholar]
- Brower RG, Shanholtz CB, Fessler HE, Shade DM, White P, Jr, Wiener CM, Teeter JG, Dodd-o JM, Almog Y, Piantadosi S. Prospective, randomized, controlled clinical trial comparing traditional versus reduced tidal volume ventilation in acute respiratory distress syndrome patients. Crit Care Med. 1999;27:1492–1498. doi: 10.1097/00003246-199908000-00015. [DOI] [PubMed] [Google Scholar]
- Brochard L, Roudot-Thoraval F, Roupie E, Delclaux C, Chastre J, Fernandez-Mondejar E, Clementi E, Mancebo J, Factor P, Matamis D, Ranieri M, Blanch L, Rodi G, Mentec H, Dreyfuss D, Ferrer M, Brun-Buisson C, Tobin M, Lemaire F. Tidal volume reduction for prevention of ventilator-induced lung injury in acute respiratory distress syndrome. The Multicenter Trail Group on Tidal Volume reduction in ARDS. Am J Respir Crit Care Med. 1998;158:1831–1838. doi: 10.1164/ajrccm.158.6.9801044. [DOI] [PubMed] [Google Scholar]
- Stewart TE, Meade MO, Cook DJ, Granton JT, Hodder RV, Lapinsky SE, Mazer CD, McLean RF, Rogovein TS, Schouten BD, Todd TR, Slutsky AS. Evaluation of a ventilation strategy to prevent barotrauma in patients at high risk for acute respiratory distress syndrome. Pressure- and Volume-Limited Ventilation Strategy Group. N Engl J Med. 1998;338:355–361. doi: 10.1056/NEJM199802053380603. [DOI] [PubMed] [Google Scholar]
- Adams AB, Cakar N, Marini JJ. Static and dynamic pressure-volume curves reflect different aspects of respiratory system mechanics in experimental acute respiratory distress syndrome. Respir Care. 2001;46:686–693. [PubMed] [Google Scholar]
- Hickling KG. The pressure–volume curve is greatly modified by recruitment. A mathematical model of ARDS lungs. Am J Respir Crit Care Med. 1998;158:194–202. doi: 10.1164/ajrccm.158.1.9708049. [DOI] [PubMed] [Google Scholar]
- Jonson B, Richard JC, Straus C, Mancebo J, Lemaire F, Brochard L. Pressure–volume curves and compliance in acute lung injury: evidence of recruitment above the lower inflection point. Am J Respir Crit Care Med. 1999;159:1172–1178. doi: 10.1164/ajrccm.159.4.9801088. [DOI] [PubMed] [Google Scholar]
- Karason S, Sondergaard S, Lundin S, Wiklund J, Stenqvist O. Evaluation of pressure/volume loops based on intratracheal pressure measurements during dynamic conditions. Acta Anaesthesiol Scand. 2000;44:571–577. doi: 10.1034/j.1399-6576.2000.00515.x. [DOI] [PubMed] [Google Scholar]
- Brower R, for the ARDS Network investigators Update on the ALVEOLI Study. American Thoracic Society International Conference, Atlanta, GA. Monday, May 20, 2002.
- Ranieri VM, Zhang H, Mascia L, Aubin M, Lin CY, Mullen JB, Grasso S, Binnie M, Volgyesi GA, Eng P, Slutsky AS. Pressure-time curve predicts minimally injurious ventilatory strategy in an isolated rat lung model. Anesthesiology. 2000;93:1320–1328. doi: 10.1097/00000542-200011000-00027. [DOI] [PubMed] [Google Scholar]
- Mehta S, Lapinsky SE, Hallett DC, Merker D, Groll RJ, Cooper AB, MacDonald RJ, Stewart TE. Prospective trial of high-frequency oscillation in adults with acute respiratory distress syndrome. Crit Care Med. 2001;29:1360–1369. doi: 10.1097/00003246-200107000-00011. [DOI] [PubMed] [Google Scholar]
- Fort P, Farmer C, Westerman J, Johannigman J, Beninati W, Dolan S, Derdak S. High-frequency oscillatory ventilation for adult respiratory distress syndrome – a pilot study. Crit Care Med. 1997;25:937–947. doi: 10.1097/00003246-199706000-00008. [DOI] [PubMed] [Google Scholar]
- The HIFI Study Group High-frequency oscillatory ventilation compared with conventional intermittent mechanical ventilation in the treatment of respiratory failure in preterm infants: neurodevelopmental status at 16 to 24 months of postterm age. J Pediatr. 1990;117:939–946. doi: 10.1016/s0022-3476(05)80142-8. [DOI] [PubMed] [Google Scholar]
- Eisner MD, Thompson T, Hudson LD, Luce JM, Hayden D, Schoenfeld D, Matthay MA. Efficacy of low tidal volume ventilation in patients with different clinical risk factors for acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;164:231–236. doi: 10.1164/ajrccm.164.2.2011093. [DOI] [PubMed] [Google Scholar]